
Abstract
Improved methods employed at Los Alamos National Laboratory for 231Pa/235U radiochronometry are outlined. We present elution curves obtained during 233Pa tracer preparation from 237Np. Additionally, we report model ages for uranium certified reference materials (CRMs) exhibiting a range of 235U enrichments including the first 231Pa/235U model ages for CRMs U200 and U900. Our results enable these CRMs to be used, with increased confidence, as quality control materials during nuclear forensics investigations.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The goal of nuclear forensic investigations is to understand, through physical and chemical analysis, the origin and history of nuclear materials found out of regulatory control [e.g. 1,2,3,4,5,6]. Radiochronometry, or age-dating, is a fundamental research area within nuclear forensics that addresses the question: 'when was a material produced or purified?' [2, 7,8,9,10,11,12,13,14,15,16,17,18,19,20,21]. During production, impurities and progeny isotopes are removed from a nuclear material although the degree of purification can vary depending on the material production methods or the type of impurity or progeny [e.g. 10]. Following production, progeny isotopes are produced at a known rate governed by radioactive decay. The age of a nuclear material can thus be calculated by combining (1) the accurate and precise measurements of the progeny-to-parent atom ratios in the present-day material, (2) the known half-lives of the measured radionuclides, and (3) the model age equation. For the calculated age of a nuclear material to accurately reflect the time since production, the following conditions must be true: (a) progeny isotopes were completely removed during production, and (b) the material has remained a 'closed' system since production with no loss or gain of parent or progeny isotopes. For the purposes of radiochronometry for nuclear forensics, these conditions are assumed to be true, so the calculated age is termed a 'model' age since production.
Ideally, during nuclear forensic investigations, numerous model ages for the same nuclear material will be calculated using different radiochronometers [e.g. 9, 10, 12,13,14, 19, 21]. If the resulting model ages are concordant (agree within an uncertainty coverage factor of two) then there is greater confidence that the model age is an accurate reflection of the time elapsed since purification, and that the model age assumptions are valid. If the model ages are discordant, model age data may not reflect the time since purification but nonetheless may be used to interpret the processing history of the material.
For uranium materials, the 230Th/234U radiochronometer is most widely applied via measurement by isotope dilution mass spectrometry (IDMS) [e.g. 11, 22,23,24,25,26,27,28]. This is due to the relatively high abundance of the 230Th radionuclide, the comparatively easy access to tracers e.g. 229Th and 233U, and the appropriate half-life of 230Th for age-dating nuclear materials.
Recent research has focused on the development, and application to uranium materials, of the 231Pa/235U radiochronometer using IDMS [9, 13,14,15,16, 18,19,20,21]. The measurement of 231Pa is more challenging than 230Th due to (i) the short half-life (~ 27 days) of the 233Pa tracer (requiring regular in-house production and calibration of new tracers), (ii) the lack of reference materials certified for age using the 231Pa/235U chronometer, and (iii) the long half-life of 235U, which leads to a lower concentration of 231Pa. One of the outcomes of previous 231Pa/235U radiochronometry research and development has been the creation of a new 231Pa Reference Material (NFRM Pa-1) [29], which has improved the accuracy and precision of 233Pa tracer calibrations compared to the previous calibration method using rock standards [9, 15]. Additionally, recent research in this area has resulted in publications of 231Pa/235U model ages for the reference materials certified using the 230Th/234U radiochronometer (e.g. New Brunswick Laboratory [NBL] CRM 125-A and NBL CRM U630, as well as Institute for Reference Materials and Measurements [IRMM] IRMM-1000a and IRMM-1000b) as well as other reference materials certified for uranium isotopic composition that have a recorded date of production [9, 10, 12, 15, 16, 18,19,20,21]. These publications have allowed the community to start developing consensus 231Pa/235U model ages for these materials [19, 20].
In this study, we (i) describe improved analytical methods, using only two columns of silica gel for the preparation of a 233Pa spike for 231Pa/235U radiochronometry at Los Alamos National Laboratory (LANL), (ii) present results from a column calibration experiment testing an improved method for the purification of 233Pa from 237Np, and (iii) report the 231Pa/235U model ages and corresponding model purification dates for six certified uranium isotopic reference materials. These results increase the number of published 231Pa/235U model ages for CRMs 125-A, U630, U010, and U100 and provide the first 231Pa/235U model ages for CRMs U200 and U900. The 231Pa/235U model ages presented here are calculated from new analyses of CRMs that span a range of uranium enrichments from 1% 235U to 90% 235U. Until reference materials with certified 231Pa/235U model ages are available, the results reported here will be an invaluable reference that will enable the use of these CRMs as quality control materials during nuclear forensic investigations.
Theory
Model ages for each CRM were calculated by inputting the number of measured atoms of 231Pa and 235U into Eq. 1
where t is the model age, λ is the decay constant (derived from the half life) and N is the number of atoms measured. The decay constants were derived from the following half-lives: 7.04 × 108 ± 0.005 × 108 years for 235U [30], 32,713 ± 110 years for 231Pa [31], and 26.697 ± 0.004 days for 233Pa [32]. By relating the calculated \(t\) to the laboratory analytical reference date the model age can be presented as a model purification date and compared to the known or certified CRM production date.
Experimental
The uranium and protactinium assay measurements were performed by IDMS at LANL in 2021. Sample dissolution, purification, and analysis procedures are described below.
Samples and materials
The certified reference materials analyzed in this study are presented in Table 1. All reagents were prepared with Optima grade acids (Fisher Scientific, Pittsburgh, PA, USA) and Milli-Q water (Millipore Billerica, MA, USA, 18.2 MΩ cm−1). Labware used during radiochemistry was pre-cleaned and acid-leached prior to use.
CRM dissolution
All of the CRMs used for this study were dissolved at Los Alamos National Laboratory in varying quantities. The mass of each CRM dissolved in the 'primary solution' is shown in Table 2. Each CRM was transferred quantitatively to a pre-cleaned quartz crucible, Savillex™ perfluoroalkoxy alkane (PFA), or Nalgene™ fluorinated ethylene propylene (FEP) container and 8 M HNO3 was added for dissolution. The CRMs were heated on a hotplate until dissolution was complete. The solutions were transferred to FEP bottles and the dissolution container rinsed multiple times including at least one wash step with trace HF to achieve complete sample transfer to the primary solution FEP bottle. Each rinse volume was also added to the FEP bottles to ensure complete transfer. Each solution was diluted to approximately 4 M HNO3 and a small volume of HF added. The concentration of uranium in each primary solution is shown in Table 2. Serial dilutions were made gravimetrically from which aliquots were taken for uranium concentration and isotopic measurements. The concentration of uranium in the dilution used for measurement range from 3 to 12 ng U g−1 of dilution.
Uranium assay and isotopic composition measurements
Aliquots containing approximately 10 ng of uranium were taken for 235U assay measurements from the serial dilution of each CRM. Each aliquot was traced with approximately 3 ng of 233U (LANL legacy material, 99.973875 ± 0.000058% 233U). The LANL 233U tracer was calibrated using National Bureau of Standards (NBS) Standard Reference Material (SRM) 960. The sample and tracer were equilibrated by heating, capped, overnight on a hotplate. The uranium was then purified from matrix elements using 1 mL of Eichrom® UTEVA® resin in a Bio-Rad Poly-Prep column. The samples were loaded in 3 M HNO3 and matrix elements were removed using washes of 3 M HNO3, 9 M HCl, and 5 M HCl. The uranium was subsequently eluted in 0.1 M HCl. In order to measure the uranium isotopic composition, aliquots containing approximately 30 ng of uranium were also taken from the same serial dilution. The uranium was purified from matrix elements using the same chemical purification procedure as the aliquots for uranium assay measurements. Process blanks (consisting of 4 M HNO3 + 0.05 M HF solutions) were processed through the same chemistry alongside the samples to quantify background contamination.
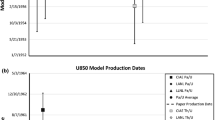
Uranium isotopic composition measurements were performed with a ThermoScientific™ Neptune Plus multi-collector inductively-coupled plasma mass spectrometer (MC-ICP-MS) using static collection routines. Samples were introduced into the mass spectrometer using a CETAC™ Aridus 3 desolvating nebulizer system. For uranium assay measurements, 233U, 235U, and 238U were measured on Faraday detectors with IRMM 074/1 used to determine mass bias corrections and IRMM 074/2 as a quality control standard. For uranium isotopic composition measurements the samples were analyzed in two batches: (1) process blank, CRM U010, CRM U100, CRM U200, and CRM 125A; and (2) CRM U630 and CRM U900. For batch 1, the correction for instrumental mass bias and detector yields for the ion counters on which 234U and 236U were measured was made using the CRM U010 reference material. Instrumental mass bias and detector yields for batch 2 were determined using the CRM U850 reference material. For all CRM samples, 235U and 238U were measured on Faraday detectors and 234U and 236U were measured on ion counters. The same configuration was used with the process blank but with 235U measured on an ion counter. Retarding Potential Quadrupole energy filters (RPQs) were utilized to decrease the tailing on 234U and 236U by 235U and 238U. Tail corrections were calculated by measuring four off-peak masses (at − 0.5, − 0.35, + 0.35, and + 0.5 atomic mass units [amu] away from the peak center), fitting the points to an exponential curve, and subtracting the calculated tail contribution to the measured signal intensity. The reference materials NBS SRM 960, IRMM 185, IRMM 187, NBL CRM U500, and NBL CRM 116-A were measured throughout the analytical sessions as unknown quality control (QC) measurements.
Protactinium tracer production, column calibration, and tracer calibration
In order to measure 231Pa by IDMS a 233Pa tracer is used. Due to the short half life of 233Pa (26.697 ± 0.004 days [32]), a new tracer has to be produced regularly. Previously published methods to produce tracers employ the use of anion resin and quartz wool/silica gel [e.g. 9]. In this study we explored simplifications to this method that only used silica gel and reduced the time for each Pa tracer production by at least one to two days. This shorter method will benefit the radiochronometry and nuclear forensic communities by reducing the time required to produce the necessary tracers for each protactinium measurement campaign.
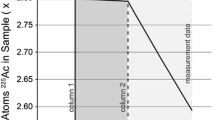
A 233Pa tracer was purified from approximately 7 mg of an in-house 237Np stock solution using silica gel (high purity, 60–200 µm particle size). The 237Np stock solution has been repeatedly purified of 233Pa over approximately five years. The 237Np solution was loaded onto 2 mL of silica gel in 2% HNO3. Protactinium was sorbed to the silica gel and the 237Np was washed from the silica gel using 2% HNO3. The 233Pa was then eluted in 2% HNO3 + 0.1 M HF. This column purification was performed twice (and is summarized in Table 3). In order to further understand the behavior of 233Pa and 237Np, the load, wash and elution fractions from both columns were collected in 2 mL increments into pre-weighed Savillex™ PFA vials. Dilutions of each column fraction were made gravimetrically and the 237Np and 233Pa concentrations were measured using a ThermoScientific™ Element 2 ICP-MS. Four concentrations of the uranium reference material NBS SRM 960 were measured throughout the analytical session to account for potential changes in ion beam intensity via standard sample bracketing throughout the run. These SRM 960 measurements provided an external concentration calibration from which 237Np and 233Pa concentrations were calculated. Given similar atomic masses, it was assumed that 233Pa and 237Np had a similar intensities (counts per second) to 238U at a given concentration. These measurements were also used to confirm adequate Np/Pa separation. For both column fractions and SRM 960 standards, 233Pa, 237Np, 235U, and 238U were measured by peak hopping on a single secondary electron multiplier (SEM) detector. Thorium-232 was also measured in column fractions to monitor hydride interference at mass 233 (232Th + H+). Individual isotopes were measured for 10–30 ms per pass for 30 total passes.
Following the column calibration each Pa elution fraction was combined into a FEP bottle. Each elution fraction vial was rinsed and the rinses were added to the bottle to ensure complete 233Pa transfer. The tracer was subsquently diluted to a concentration of approximately 1 pg 233Pa per g of a 4 M HNO3 + 0.05 M HF solution. The 233Pa tracer was calibrated by reverse IDMS using the NFRM Pa-1 reference material [29]. Aliquots were taken containing ratios of 1:2, 1:1, and 2:1 231Pa:233Pa where 1 represented approximately 1 pg. Prior to the analytical measurement the 233U present from 233Pa decay was removed using a single silica gel column purification described below and the 231Pa to 233Pa ratio was then measured by the MC-ICP-MS method described below. The tracer was calibrated twice during this study, once immediately after preparation and once again approximately three months after preparation. The sample 231Pa assay values were calculated using an average 233Pa concentration from the six individual 233Pa calibration mixes obtained from both tracer calibrations. The uncertainty for the average 233Pa concentration of the tracer was obtained by combining the variance of the six calibration mixes with the relative uncertainty of the NFRM Pa-1 reference material in quadrature.
CRM protactinium assay measurements
The 231Pa concentration in each CRM solution was measured from aliquots containing ~ 5 pg of 231Pa for each CRM except NBL CRM U900. Aliquots of NBL CRM U900 contained ~ 15 pg of 231Pa. Each aliquot was traced with approximately 1 pg of 233Pa. Tracer-sample equilibration was achieved by heating samples overnight in capped Savillex™ PFA vials on a hotplate. Protactinium was purified from matrix elements using a three-column procedure. Firstly, the sample was loaded onto 2 mL of Bio-Rad anion exchange resin (AG1-X8, 100–200 mesh) in 9 M HCl with trace HNO3 and H3BO3. Matrix elements were removed using 9 M HCl column washes and Pa was eluted in 9 M HCl + 0.05 M HF. Next, the sample was loaded onto 2 mL of silica gel in 2% HNO3 with trace H3BO3. Matrix elements were removed using 2% HNO3 column washes and Pa was eluted in 2% HNO3 + 0.05 M HF. The third column was a repeat of the second column designed to remove 233U immediately prior to analysis. Background contamination was quantified by processing a blank (consisting of a 4 M HNO3 + 0.05 M HF solution), alongside the samples, using the same chemistry. Due to the decay of 233Pa (26.697 ± 0.004 days [32]) to 233U, all protactinium mass spectrometry measurements were performed on the same day of the final column purification, to minimize isobaric interference.
Protactinium measurements were performed using a ThermoScientific™ Neptune Plus MC-ICP-MS. Samples were introduced into the mass spectrometer using a CETAC™ Aridus 3 desolvating nebulizer system. A static multi-ion counting routine was employed in which 231Pa and 233Pa were measured on full-size SEMs. Concurrently, 235U (to confirm complete Pa purification) and 232Th (to monitor hydride interference at mass 233) were measured on Faraday detectors. As no certified isotopic Pa reference materials are available, it was assumed that instrumental bias corrections derived from U are appropriate for Pa; NBL CRM U010 was used to determine mass bias and detector gain corrections and NBL CRM U005A was used as a quality control standard. Both CRMs were measured as described above for uranium isotope measurements.
Results and discussion
Protactinium tracer purification column calibration
The mass of 237Np and 233Pa in each fraction of the tracer purification columns is shown in Table 4 and Fig. 1. Approximately 4.7 mg of 237Np—approximately 68.2% of the total 237Np loaded—is collected immediately upon loading as 237Np does not sorb to the resin. All but the remaining 0.01% or 7.3 µg is collected after two rinses of the loading vial. Approximately 117 ng of an isotope with mass 233 is also collected after loading and rinsing of the loading vial and approximately 140 pg of an isotope with mass 233 is measured in the elution. The isotope with mass 233 collected in the load and rinse solution of the first column is hypothesized to be 233U. Uranium does not sorb to silica gel and is produced by the decay of 233Pa. Calculating a minimum solution age of the 237Np solution using this 233U/237Np ratio yields a result that is consistent with the known history of the solution. Accurate age-dating is of course not possible due to recent removal of the intermediary 233Pa isotope during multiple consecutive uses of the sample 237Np stock solution. The isotope in the elution is hypothesized to be 233Pa as protactinium is known to sorb on silica gel in 2% HNO3 and be eluted in 2% HNO3 + 0.1 M HF [e.g. 9]. Approximately 235 pg of 233Pa is expected to be present in a sample containing 7 mg 237Np once secular equilibrium is achieved. The difference between expected and collected 233Pa gives an indication of the chemical recovery of this method—approximately 60%. A reduced yield could be possible due to residual HF in the load solution. Hydrofluoric acid is added to each Np solution in between tracer productions to remove Si that is washed through the column. The added HF is itself removed by the addition of concentrated H3BO3. The HF removal is required because any residual HF in the load solution will result in 233Pa not sorbing to the silica gel. If HF removal is incomplete a small proportion of the 233Pa will be in the load and rinse fractions and result in a reduced chemical yield.

Graphs showing (a) the mass of 237Np in each column fraction, (b) the mass of 237Np < 0.025 mg in each column fraction, (c) the mass of isotopes at atomic mass unit 233 (Pa or U) in each column fraction, and (d) the mass of isotopes < 0.2 ng at atomic mass unit 233 (Pa or U) in each column fraction. The x-axis refers to sample number. Refer to Table 4 for concentration data used to prepare this figure
231Pa/235U Radiochronometry for CRMs
The measured 231Pa/235U atom ratios, calculated model ages and model purification dates are presented in Table 5. The model purification dates for each CRM are shown in Figs. 2, 3, 4. Process blank concentrations for both uranium and protactinium represent < 0.2% of the aliquot sizes and are therefore insignificant relative to reported measurement uncertainties.

Model 231Pa/235U purification dates from this study of (a) CRM 125-A and (b) CRM U630. Also included are previously published 231Pa/235U model purification dates for each CRM (only the MC-ICP-MS data are shown for Treinen et al., 2019 [18]). Error bars represent expanded uncertainties with a coverage factor of two. The solid black horizontal line shows the certified model date of purification using the 230Th/234U radiochronometer, and the grey horizontal lines illustrate the certified expanded uncertainty. Dates are reported in mm/dd/yyyy format

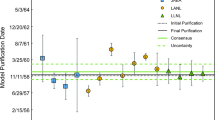
Model 231Pa/235U purification dates from this study of (a) CRM U010 and (b) CRM U100. Also included are previously published 231Pa/235U model purification dates for each CRM. Error bars represent expanded uncertainties with a coverage factor of two. The solid black horizontal line shows the recorded date of final purification [33]. Dates are reported in mm/dd/yyyy format

Model 231Pa/235U purification dates from this study of (a) CRM U200 and (b) CRM U900. Error bars represent expanded uncertainties with a coverage factor of two. The solid black horizontal line shows the recorded date of final purification [33]. Dates are reported in mm/dd/yyyy format. These data are the first published model 231Pa/235U purifcation dates for CRM U200 and CRM U900
The 231Pa/235U model purification dates calculated for CRM 125-A range from May 21, 1994 ± 66 days to June 12, 1994 ± 66 days (Table 5, Fig. 2a). These model purification dates are concordant and agree with the certified model date of purification. Previously reported 231Pa/235U CRM 125-A model purification dates range from April 20, 1994 to January 27, 1995 (Fig. 2a). The results from this study are also concordant with the range of previously published results.
The 231Pa/235U model purification dates calculated for CRM U630 range from December 31, 1988 ± 92 days to January 5, 1989 ± 83 days (Table 5, Fig. 2b). These model purification data are concordant and agree with the certified model date of purification. Previously published 231Pa/235U model purification dates for U630 range from March 13, 1989 to July 14, 1989 (Fig. 2b). Both the model dates reported here are concordant with the range of the previously published results.
The 231Pa/235U model purification dates calculated for CRM U010 range from June 10, 1957 ± 426 days to September 21, 1957 ± 430 days (Table 5, Fig. 3a). Both model dates are concordant with each other and the recorded date of purification [33]. Previously published U010 231Pa/235U model dates range from October 14, 1957 to June 17, 1959 [19]. Both results reported here are concordant with the range of the previously published results.
The 231Pa/235U model purification dates calculated for CRM U100 range from September 4, 1958 ± 188 days to October 6, 1958 ± 195 days (Table 5, Fig. 3b). Both model dates are concordant with each other and with the recorded date of purification [33]. Previously published U100 231Pa/235U model dates range from August 27, 1957 to January 30, 1961 (Fig. 3b). The results from this study are within this range.
The 231Pa/235U model purification dates calculated for CRM U200 range from December 31, 1956 ± 172 days to April 17, 1957 ± 180 days (Table 5, Fig. 4a). These results are concordant with each other but are biased slightly older than the recorded purification date [33]. The 231Pa/235U model purification dates calculated for CRM U900 range from February 3, 1957 ± 162 days to March 13, 1957 ± 165 days (Table 4, Fig. 4b). These results are concordant with each other but are also biased old compared to the recorded date of purification [33]. We are not aware of any previously published model purification dates for U200 or U900 so our results can only be compared with the recorded purification date.
The 231Pa/235U model purification dates presented in this study for CRMs 125-A, U630, U010, and U100 are both concordant with the certified model purification date or recorded purification date and correspond well to the range of previously published results. In addition, these 231Pa/235U model dates of purification are concordant with recently published 230Th/234U model dates of purification [e.g. 9,10,11,12, 18, 21, 26]. These results increase confidence that (a) the LANL 231Pa/235U radiochronometry methods presented in this paper yield accurate results when compared to the international nuclear forensics community and (b) that the radiochronometry model assumptions are valid for these CRMs further validating their use as quality control materials during nuclear forensic investigations. The individual 231Pa/235U model purification dates presented in this study for CRMs U200 and U900 are concordant with each other but are discordant and biased old compared to the recorded purification date. The 231Pa/235U model dates for CRM U900 presented here are, however, concordant with previously published 230Th/234U model dates of purification for CRM U900 [26]. This indicates that the model assumptions of complete Pa purification may not be valid for these CRMs. However, if CRM U200 and U900 meet other requirements such as homogeneity and stability, they may still be useful as CRMs for nuclear forensics investigations.
Conclusions
This study provides an improved, more efficient method of purifying a 233Pa tracer from Np requiring only columns containing silica gel. Elution curves generated during the production of a 233Pa tracer from a 237Np stock solution show that > 99% of 237Np is collected in the load and rinse fractions, along with 233U. Protactinium-233 is collected in the first 4 mL of the elution with 2% HNO3 + 0.01 M HF. Model dates of purification calculated using the 231Pa/235U radiochronometer are reported here for six uranium CRMs. For some CRMs the results of this study add additional data points to a growing multi-laboratory data set, and the results support the validity of the radiochronometry model assumptions for these materials (i.e., complete purification of progeny at the time of material purification and closed-system behavior of the material, with respect to the parent and progeny isotopes, since purification). For two CRMs (U200 and U900) we report the first model dates of purification measured using the 231Pa/235U radiochronometer. For the CRMs with previously published data (CRMs 125-A, U630, U010 and U100) the model dates reported here are concordant with the certified or recorded date of purification and/or previously reported data, which validate LANL’s 231Pa/235U radiochronometry methods. For CRMs U200 and U900 the model dates reported here are biased old compared to the recorded purification date implying the model assumptions may not be valid for these CRMs. These results further demonstrate the utility of the 231Pa/235U radiochronometer to support nuclear forensics measurements. In the absence of reference materials certified for model age using the 231Pa/235U radiochronometer, these data are vital in increasing confidence in the use of these uranium isotopic CRMs as quality control materials for these measurements.
References
Mayer K, Wallenius M, Varga Z (2013) Nuclear forensic science: correlating measurable material parameters to the history of nuclear material. Chem Rev 113:884–900
Sturm M, Richter S, Aregbe Y, Wellum R, Mialle S, Mayer K, Prohaska T (2014) Evaluation of chronometers in plutonium age determination for nuclear forensics: what if the “Pu/U clocks” do not match? J Radioanal Nucl Chem 302:399–411
Mayer K, Wallenius M, Varga Z (2015) Interviewing a silent (radioactive) witness through nuclear forensic analysis. Anal Chem 87:11605–11610
Varga Z, Mayer K, Bonamici CE, Hubert A, Hutcheon I, Kinman W, Kristo M, Pointurier F, Spencer K, Stanley F, Steiner R, Tandon L, William R (2015) Validation of reference materials for uranium radiochronometry in the frame of nuclear forensics investigations. Appl Radiat Isot 102:81–86
Kristo MJ, Gaffney AM, Marks N, Knight K, Cassata WS, Hutcheon ID (2016) Nuclear forensic science: analysis of nuclear material out of regulatory control. Annu Rev Earth Planet Sci 44:555–579
Pastoor KJ, Kemp RS, Jensen MP, Shafer JC (2021) Progress in uranium chemistry: driving advances in front-end nuclear cycle forensics. Inorg Chem 60:8347–8367
Wallenius M, Mayer K (2000) Age determination of plutonium material in nuclear forensics by thermal ionisation mass spectrometry. Fresenius J Anal Chem 366:234–238
Morgenstern A, Apostolidis C, Mayer K (2002) Age determination of highly enriched uranium: separate and analysis of 231Pa. Anal Chem 74:5513–5516
Eppich GR, Williams RW, Gaffney AM, Schorzman KC (2013) 235U–231Pa age dating of uranium materials for nuclear forensic investigations. J Anal At Spectrom 28:666–674
Kayzar TM, Williams RW (2016) Developing 226Ra and 227Ac age-dating techniques for nuclear forensics to gain insight from corcordant and non-concordant radiocrhonometers. J Radioanal Nucl Chem 307:2061–2068
Treinen KC, Kinman WS, Chen Y, Zhu L, Cardon AMR, Steiner RE, Kayzar-Boggs TM, Williams RW, Zhao Y-G (2017) US-DOE and CIAE international cooperation in age-dating uranium standards. J Radioanal Nucl Chem 314:2469–2474
Rolison JM, Treinen KC, McHugh KC, Gaffney AM, Williams RW (2017) Application of the 226Ra-230Th-234U and 227Ac-231Pa-235U radiochronometers to uranium certified reference materials. J Radioanal Nucl Chem 314:2459–2467
Kristo MJ, Williams R, Gaffney AM, Kayzar-Boggs TM, Schorzman KC, Lagerkvist P, Vesterlund A, Ramebäck H, Nelwamondo AN, Kotze D, Song K, Lim SH, Han S-H, Lee C-G, Okubo A, Maloubier D, Cardona D, Samuleev P, Dimayuga I, Varga Z, Wallenius M, Mayer K, Loi E, Keegan E, Harrison J, Thiruvoth S, Stanley FE, Spencer KJ, Tandon L (2018) The application of radiochronometry during the 4th collaborative materials exercise of the nuclear foreniscs international technical working group (ITWG). J Radioanal Nucl Chem 315:425–434
Rolison JM, Williams RW (2018) Application of the 226Ra-230Th-234U and 227Ac-231Pa-235U radiochronometers to UF6 cylinders. J Radioanal Nucl Chem 317:897–905
Treinen KC, Gaffney AM, Rolison JM, Samperton KM, McHugh KC, Miller ML, Williams RW (2018) Improved protactinium spike calibration method applied to 231Pa-235U age-dating of certified reference materials for nuclear forensics. J Radioanal Nucl Chem 318:209–219
Varga Z, Nicholl A, Hrnecek E, Wallenius M, Mayer K (2018) Measurement of the 231Pa/235U ratio for the age determination of uranium materials. J Radioanal Nucl Chem 318:1565–1571
Mathew K, Kayzar-Boggs T, Varga Z, Gaffney A, Denton J, Fulwyler J, Garduno K, Gaunt A, Inglis J, Keller R, Kinman W, Labotka D, Lujan E, Maassen J, Mastren T, May I, Mayer K, Nicholl A, Ottenfeld C, Parsons-Davis T, Porterfield D, Rim J, Rolison J, Stanley F, Steiner R, Tandon L, Thomas M, Torres R, Treinen K, Wallenius M, Wende A, Williams R, Wimpenny J (2019) Intercomparison of the radio-chronometric ages of plutonium-certified reference materials with distinct isotopic compositions. Anal Chem 91:11643–11652
Treinen KC, Samperton KM, Lindvall RE, Wimpenny JB, Gaffney AM, Bavio M, Baransky EJ, Williams RW (2019) Evaluating uranium radiochronometry by single-collector mass spectrometry for nuclear forensics: a multi-instrument investigation. J Radioanal Nucl Chem 322:1627–1640
Denton JS, Treinen KC, Chen Y, Baransky E, Gaffney AM, Huang S-H, Kayzar-Boggs TM, Samperton K, Steiner RE, Wende AM, Williams RW, Zhao Y-G (2020) International cooperation in age-dating uranium standards for nuclear forensics using the 231Pa/235U radiochronometer. J Radioanal Nucl Chem 324:705–714
Kayzar-Boggs TM, Treinen KC, Okubo A, Denton JS, Gaffney AM, Miller M, Steiner RE, Wende AM, Williams RW (2020) An interlaboratory collaboration to determine consensus 231Pa/235U model ages of a uranium certified reference material for nuclear forensics. J Radioanal Nucl Chem 323:1189–1195
Harrison LN, Gaffney AM (2021) 230Th-234U and 231Pa-235U radiochronometry of hydrolyzed uranium hexafluoride gas. J Radioanal Nucl Chem 329:1513–1521
Wallenius M, Morgenstern A, Apostolidis C, Mayer K (2002) Determination of the age of highly enriched uranium. Anal Bioanal Chem 374:379–384
LaMont SP, Hall G (2005) Uranium age determination by measuring the 230Th/234U ratio. J Radioanal Nucl Chem 264:423–427
Varga Z, Surányi G (2007) Production date determination of uranium-oxide materials by inductively coupled plasma mass spectrometry. Anal Chim Acta 599:16–23
Varga Z, Wallenius M, Mayer K (2010) Age determination of uranium samples by inductively coupled plasma mass spectrometry using direct measurement and spectral deconvolution. J Anal At Spectrom 25:1958–1962
Williams RW, Gaffney AM (2011) 230Th-234U model ages of some uranium standard reference materials. Proc Radiochim Acta 1:31–35
Meyers LA, Williams RW, Glover SE, LaMont SP, Stalcup AM, Spitz HB (2013) Radiochronological age of a uranium metal sample from an abandoned facility. J Radioanal Nucl Chem 296:669–674
Gaffney AM, Hubert A, Kinman WS, Magara M, Okubo A, Pointurier F, Schorzman KC, Steiner RE, Williams RW (2016) Round-robin 230Th-234U age dating of bulk uranium for nuclear forensics. J Radioanal Nucl Chem 307:2055–2060
Essex RM, Williams RW, Treinen KC, Collé R, Fitzgerald R, Galea R, Keightley J, LaRosa J, Laureano-Pérez L, Nour S, Pibida L (2019) Preparation and calibration of a 231Pa reference material. J Radioanal Nucl Chem 322:1593–1604
Jaffey AH, Flynn KF, Glendenin LE, Bentley WC, Essling AM (1971) Precision measurement of half-lives and specific activities of 235U and 238U. Phys Rev C 4:1889
Robert J, Miranda CR, Muxart R (1969) Mesure de la periode du protactinium-231 par microcalorimetrie. Radiochim Acta 11:104–108
Jones RT, Merritt JS, Okazaki A (1986) A measurement of the thermal neutron capture cross section of 232Th. Nucl Sci Eng 93:171–180
Petit GS (1960) Preparation of uranium isotopic standards for the National Bureau of Standards. Union Carbide Nuclear Company, Oak Ridge, Tennessee. Oak Ridge National Laboratory, Report#KL-8, Department of Energy/K25 Archives
Acknowledgements
The authors wish to acknowledge the National Nuclear Security Administration's Nuclear Smuggling Detection and Deterrence Office for direct funding. Los Alamos National Laboratory, an affirmative action/equal opportunity employer, is managed by Triad National Security, LLC, for the National Nuclear Security Administration of the US Department of Energy.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
There are no conflicts of interest surrounding this work.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Denton, J.S., Wende, A.M., Kayzar-Boggs, T.M. et al. Improved methods to age-date uranium certified reference materials for nuclear forensics using the 231Pa/235U radiochronometer. J Radioanal Nucl Chem 331, 5753–5762 (2022). https://doi.org/10.1007/s10967-022-08627-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10967-022-08627-0