
Abstract
During 2017–2018 Los Alamos National Laboratory, Lawrence Livermore National Laboratory and the China Institute of Atomic Energy collaborated in an interlaboratory 231Pa/235U radiochronometry exercise. The laboratories used different analytical methods to obtain a consensus model purification date for CRM U010 of December 28, 1958 ± 198 days and for CRM U850 of May 20, 1958 ± 363 days. These results agree with previously reported model dates using the 230Th/234U radiochronometer as well as the production histories of these materials. The concordance of interlaboratory data confirms the ability of laboratories to make reproducible radiochronometry measurements using distinct analytical approaches.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The aim of a nuclear forensics investigation is to establish the origin and history of nuclear materials of unknown origin. This can be achieved through the physical, chemical, and isotopic analysis of material found out of regulatory control or with an unknown history [1,2,3,4,5]. Radiochronometry, or the science of age-dating radioactive materials using parent and progeny isotopes, is a fundamental research area addressing the ‘when was a material produced or purified’ question within the field of nuclear forensics [6,7,8,9,10].
When a nuclear material is purified during production, impurities are removed (including progeny isotopes). Following purification, progeny isotopes will be produced at a rate governed by radioactive decay (the Bateman equations). The time elapsed since purification can be calculated by combining (1) the Bateman equations, with (2) the known half-lives of the radionuclides in question, and (3) accurate and precise measurements of the parent-to-progeny ratios in the present-day material. For the radiochronometric age to be an accurate representation of the production date a number of conditions have to be met: (1) there was complete separation of progeny isotopes from the parent during material production, and (2) the sample has remained a ‘closed’ system since production (i.e. no gain or loss of parent or progeny isotopes except through radioactive decay). In radiochronometry, these conditions are assumed to be true and the resulting age is termed a model age since purification.
There are many potential radiochronometers applicable to nuclear materials and ideally, numerous model purification dates would be obtained from the same sample using multiple radiochronometers. If the model ages determined using different radiochronometers agree (are concordant), then confidence that the model age of the sample represents the purification age is increased. If the model ages disagree (are non-concordant) then this information can be used to interpret a sample’s process history as well as its maximum age [7, 9,10,11].
For uranium materials, 230Th/234U is the most commonly used radiochronometer due to the relatively high abundance of the 230Th radionuclide and the wide availability of tracers e.g. 229Th and 232Th. Recently, many laboratories have focused efforts on developing analytical methods to date nuclear materials using the 231Pa/235U radiochronometer. Protactinium-231 is the daughter of 235U and, depending on the enrichment and age of the uranium material, can be present in measurable quantities (pg level). The use of the 231Pa/235U radiochronometer is, however, complicated by the following:
- (a)
the short half-life (~ 27 days) of the 233Pa tracer rendering each freshly produced tracer obsolete in a few months;
- (b)
until recently there were no protactinium standards against which to calibrate a freshly produced 233Pa tracer;
- (c)
there are no reference materials certified for age using the 231Pa/235U chronometer.
Due to the short half-life of 233Pa, the usefulness of each tracer can be maximized by preparing fresh tracer for each imminent age-dating campaign. The lack of a protactinium standard for 233Pa tracer calibrations (issue (b) above) was remedied recently when a 231Pa Reference Material (NFRM 231Pa) was produced by the National Institute of Standards and Technology (NIST, USA) in collaboration with Lawrence Livermore National Laboratory (LLNL, USA), the National Research Council, (NRC, Canada), and the National Physical Laboratory (NPL, UK) [12]. It is possible to address the lack of reference materials certified for 231Pa/235U radiochronometry [issue (c) above] by the international nuclear forensics community conducting interlaboratory comparison 231Pa/235U measurements. The goal of these exercises is to obtain consensus 231Pa/235U model purification dates of commercially available certified uranium isotopic reference materials as well as those certified for purification date by the 230Th/234U radiochronometer. Interlaboratory studies have been conducted using the 230Th/234U radiochronometer [6, 8] establishing that reproducible ages can be obtained by laboratories around the world.
In this paper we report the results of a 231Pa/235U radiochronometry interlaboratory comparison exercise between Los Alamos National Laboratory (LANL), LLNL and the Chinese Institute of Atomic Energy (CIAE). All three laboratories applied the 231Pa/235U radiochronometer to age date certified reference material (CRM) U010 and CRM U850 produced by New Brunswick National Laboratory (NBL) in the United States. A record of the production of these materials is available providing known purification dates for each material [13]. Data reported here are compared to previously reported results from a 230Th/234U radiochronometry interlaboratory comparison exercise between these laboratories for CRM U010 and CRM U850 [8].
Experimental
LANL, LLNL and CIAE obtained and dissolved CRM U010 and CRM U850 independently. Each laboratory measured 231Pa and 235U assay by isotope dilution mass spectrometry (IDMS) using 233Pa and 233U isotopic tracers. Well-characterized tracers are required for accurate and high-precision measurements and details of the tracers and calibrations can be found in the following sections. Table 1 contains a summary of the analytical methods for each laboratory as well as the half-lives used by each laboratory in the age-dating equation.
LANL analytical methods
CRM dissolution
Primary dissolutions of U010 and U850 were made by dissolving each CRM powder in 8 M HNO3 on a hot plate. Following dissolution each solution was diluted and HF added to generate a final solution for storage of between 4 M HNO3 + 0.01 M HF and 4 M HNO3 + 0.05 M HF. The concentration of uranium in each primary solution was ~ 728 µg U/g for CRM U010 and ~ 285 µg U/g for CRM U850. Two serial dilutions were made in order to measure 235U concentration. These dilutions were made gravimetrically to yield a final secondary dilution uranium concentration of approximately 30 ng U/g for U850 and 3 ng U/g for U010.
Uranium assay and isotope composition measurements
Aliquots containing ~ 2 ng of uranium were taken for 235U assay measurements from the secondary dilution of each CRM and were traced with 233U tracer (LANL legacy material). The LANL 233U tracer was calibrated using National Bureau of Standards (NBS) Standard Reference Material (SRM) 960. Following tracer-sample equilibration the uranium in CRM U850 was purified using 1 mL of UTEVA resin where the samples were loaded in 3 M HNO3 and the uranium eluted in 0.1 M HCl. Aliquots containing ~ 50 ng of uranium were also taken from the secondary dilution to measure the uranium isotope composition. The samples were purified using the same chemistry as the uranium assay samples. To quantify background contamination, process blanks (4 M HNO3 + 0.05 M HF) were processed alongside the samples using the same chemistry. Chemical purification of uranium aliquots was not performed for CRM U010 due to the high-purity of the CRM and tracer materials.
Uranium measurements were performed on a ThermoScientific™ Neptune Plus Multi-collector inductively coupled plasma mass spectrometer (MC-ICP-MS) using static multi-ion counting methods. For uranium assay measurements, 233U, 235U, and 238U were measured on Faraday detectors with IRMM 074/1 used for mass bias corrections and IRMM 074/2 used as a quality control standard. For uranium isotope composition measurements, 238U and 235U were measured on Faraday detectors and 234U and 236U were measured on full-size secondary electron multiplier (SEM) detectors equipped with retarding potential quadrupole (RPQ) lenses. For U010 uranium measurements, CRM U200 was used for mass bias and gain corrections and a separate dissolution of U010 was used as a quality control standard. For U850 uranium measurements, CRM U500 was used for mass bias and gain corrections. The CRMs U930 and IRMM 074/9 were used as quality control standards. Other corrections applied to the final data included tailing, acid blank subtractions, and hydride corrections (235U + 1H on mass 236U).
Protactinium assay measurements
For protactinium isotope dilution measurements LANL purified a 233Pa tracer from ~ 5 mg of 237Np using silica gel (high purity, 63–200 µm particle size). The 237Np solution was loaded onto 2 mL of silica gel in 2% HNO3 and the 237Np was washed from the resin using 2% HNO3. The 233Pa was then eluted in 2% HNO3 + 0.01 M HF. This column purification was performed twice and the resulting elution screened using a ThermoScientific™ Element 2 High Resolution Inductively Coupled Plasma Mass Spectrometer (ICP-MS) to confirm adequate Np/Pa purification. If necessary, the silica gel column was repeated until adequate purification was achieved. The resulting spikes were diluted to a working tracer concentration of ~ 1 pg 233Pa/g solution in 4 M HNO3 + 0.05 M HF. The 233Pa tracers used in this study were calibrated using (a) the NFRM 231Pa reference material and (b) an in-house 231Pa solution, itself characterized by a 233Pa spike calibrated using the NFRM 231Pa reference material. Prior to the calibration analytical session, the 233U present from 233Pa decay was removed using a single silica gel column purification described above. The 231Pa to 233Pa ratio was measured by the MC-ICP-MS method described below. Each tracer was calibrated twice during its lifetime and the average calibration was used for data reduction. For sample 231Pa assay measurements, aliquots containing ~ 5 pg of 231Pa were taken from the primary solution. This represented ~ 100 µg of uranium for U850 and ~ 7300 µg of uranium for U010. These aliquots were traced with ~ 2 pg of 233Pa. Following tracer-sample equilibration the protactinium was purified using a three step process: (1) a column containing 2 mL of BioRad anion exchange resin (AG1-X8, 100–200 mesh) where the sample was loaded in 9 mL HCl with trace HNO3 and H3BO3 and Pa eluted in 9 M HCl + 0.05 M HF, (2) a column containing 2 mL of silica gel where the sample was loaded in 2% HNO3 with trace H3BO3 and Pa eluted in 2% HNO3 + 0.05 M HF, and (3) a repeat of column (2). To quantify background contamination, process blanks (4 M HNO3 + 0.05 M HF) were processed alongside the samples using the same chemistry.
All protactinium measurements were performed the same day of the final column purification to minimize the isobaric interferences from the decay of 233Pa to 233U. Protactinium measurements were performed on a ThermoScientific™ Neptune Plus MC-ICP-MS using static multi-ion counting methods with 231Pa and 233Pa measured on full-size SEM detectors. Faraday detectors are also used to measure 235U (to confirm purification) and 232Th (to monitor hydride interference at mass 233). As no certified isotopic Pa reference materials are available, uranium CRM U010 was used for mass bias and gain corrections and CRM U005A was used as a quality control standard. These CRMs were measured as described above for uranium isotope measurements. It was assumed that the instrumental mass bias correction derived from uranium is appropriate for Pa. Other corrections applied to the final data included tailing and acid blank subtractions as well as a subtraction of 232Th + 1H hydride interference on mass 233.
LLNL analytical methods
CRM dissolution
Primary dissolutions of U010 and U850 were made by dissolving each CRM powder in concentrated HNO3 in pre-cleaned quartz crucibles on a hotplate at 120 °C for at least 4 h. Following dissolution each solution was diluted and HF added to generate a final solution for storage of 2 M HNO3 + 0.05 M HF. The concentration of uranium in each primary solution was ~ 1 mg U/g for U010 and ~ 56 µg U/g for U850. Two serial dilutions were made gravimetrically in order to measure 235U concentration.
Uranium assay and isotope composition measurements
Aliquots containing ~ 40 ng uranium for CRM U010 and ~ 50 ng uranium for CRM U850 were taken for 235U assay measurements from the secondary dilutions of each CRM. Each aliquot was traced with ultra-high purity 233U (LLNL legacy material). The LLNL 233U tracer was calibrated using NBS SRM 960. Following tracer-sample equilibration the uranium was purified using 1 mL of UTEVA resin where the samples were loaded in 4 M HNO3 and the uranium eluted in 0.1 M HCl. Aliquots were taken from the primary solutions and diluted to approximately 10 ng of uranium to measure the uranium isotope composition. The samples were purified using the same chemistry as the uranium assay samples. To quantify background contamination, process blanks were processed alongside the samples through CRM dissolution and chemistry.
Uranium assay measurements for U010 and U850, as well as uranium isotope composition measurements for U850 were performed on a Nu Plasma HR MC-ICP-MS. Uranium isotope composition measurements for U010 were performed on a Nu Plasma 3 MC-ICP-MS. For uranium assay measurements, Faraday detectors were used to measure 238U, 235U, and 233U, 234U was measured on an ion counter. For uranium isotope composition measurements, 238U and 235U were measured on Faraday detectors and 236U, 234U, and 233U were measured on ion counters. In both cases static multi-ion counting methods were employed with a 10 s integration time. CRM U010 was used for mass bias and Faraday-ion counter gain corrections, and the measured isotopic compositions of U010 and U850 were used for IDMS calculations. The CRMs 112A, 129A, and U005-A were used as quality control standards. Other corrections applied to the final data include peak tailing and acid blank subtractions.
Protactinium assay measurements
For protactinium isotope dilution measurements, LLNL purified a 233Pa tracer from a stock solution containing ~ 25 mg 237Np using a three column procedure. The 237Np solution was loaded onto a 2 mL AG MP-1 resin column in 10 M HCl. The 233Pa was eluted in 10 M HCl + 0.05 M HF and the 237Np was recovered using 1 M HCl + 0.5 M HF. This column was repeated using only 1 mL of AG MP-1 resin. Final purification was achieved by loading the 233Pa tracer onto 2 mL of silica gel in 5% HNO3 and eluting the 233Pa in 5% HNO3 + 0.1 M HF. Two separately prepared 233Pa tracers were produced during the course of this study. Final spikes were screened by MC-ICP-MS to verify final Np/Pa ratios of < 1000. The final tracer solutions were diluted to between 16 and 28 pg 233Pa/g solution. The 233Pa tracers used in this study were calibrated using an in-house 231Pa solution. The 231Pa concentration of this in-house solution was previously measured using a 233Pa tracer calibrated using the United States Geological Survey (USGS) BCR-2 rock standard and the Table Mountain Latite (TML) material that has been well-characterized by the geological community [14, 15]. Protactinium-233 tracer calibration methods using geological materials have been previously described [16, 17]. Following tracer-sample equilibration, the 233U present from the decay of 233Pa was removed using a single silica gel column purification described above. The 231Pa to 233Pa ratio was measured by the MC-ICP-MS method described below.
For sample 231Pa assay measurements aliquots containing ~ 6.5–7 pg Pa were taken, which contained ~ 125 µg of uranium for U850 and ~ 12 mg of uranium for U010. These aliquots were traced with ~ 2 pg 233Pa. Protactinium was purified from the bulk uranium matrix using a three column procedure. The first column consisted of a 1 mL BioRad AG1-X8 resin bed. Samples were dried and dissolved in 9 M HCl + trace H3BO3 + trace HNO3 and loaded onto the column. Protactinium was eluted with 9 M HCl + 0.05 M HF. The second column was a smaller resin column volume using the same dissolution, rinsing, and elution solutions. The final column was a 1 mL silica gel (high purity, 75–200 µm particle size) column conditioned with 5% HNO3. The sample was loaded onto the silica gel with 5% HNO3 and protactinium was eluted using 2% HNO3 + 0.05 M HF. To quantify background contamination, process blanks were processed alongside the samples through CRM dissolution and chemistry.
All protactinium measurements were performed the same day of the final column purification to minimize the isobaric interferences from the decay of 233Pa to 233U. Protactinium measurements were performed on a Nu Plasma HR MC-ICP-MS using static multi-ion counting methods with 231Pa and 233Pa collected on ion counters with a 10–15 s integration. CRM U010 was used for mass bias and gain corrections, CRM 005-A was used as a quality control standard. Other corrections applied to the final data include peak tailing and blank subtraction.
CIAE analytical methods
CRM dissolution
Primary dissolutions of U010 and U850 were made by dissolving each CRM powder in 6 M HNO3 on a hotplate at 90 °C for 24 h. Following dissolution each solution was diluted and HF added to generate a final solution for storage of 6 M HNO3 + 0.05 M HF. The concentration of the uranium in each primary solution was 1–4 mg U/g. Two serial dilutions were made in order to measure 235U concentrations.
Uranium assay and isotope composition measurements
Aliquots containing ~ 3 µg (U850) and ~ 27 µg (U010) of uranium were taken for 235U assay measurements from the second dilution. Each aliquot was traced with 233U (IRMM-051). The IRMM-051 233U tracer is certified for 233U concentration by mass, therefore no calibration was necessary. Following tracer-sample equilibration, no chemical purification was performed prior to mass spectrometry due to the high-purity of the CRM and tracer materials. Similar aliquot sizes of uranium were also taken for uranium isotope composition measurement. Again, no chemical purification of the sample aliquots was performed. To quantify background contamination, blank acid samples were processed alongside the samples through CRM dissolution and chemistry.
Uranium measurements were performed on a ThermoScientific™ Element XR. Dynamic ion counting mode was used with an SEM detector peak-jumping between each isotope with a 2 min integration time. The method involved three runs with twenty passes. Twenty lines were used per peak with the central ten lines used for data calculation. No abundance filter tune was used. CRM IRMM-199 was used for mass bias corrections and the Chinese natural uranium standard reference material GBW04205 was used as a quality control standard.
Protactinium assay measurements
For protactinium isotope dilution measurements CIAE purified a 233Pa tracer from tributyl phosphate waste containing 19.7 mg of 237Np using a five column purification with Eichrom TRU resin and silica gel. This more lengthy purification process was due to the more complex matrix of the CIAE 237Np material than that used by LANL and LLNL. The 237Np solution was loaded onto 1.8 mL TRU resin in 1 M HNO3 + 0.1 M NaNO2. The resin was rinsed with 1 M HNO3 + 0.1 M HCl to recover the 237Np and then the 233Pa was eluted in 3 M HCl + 2 M HF. Any residual 237Np was then eluted with high purity water. This column was then repeated. Following elution from the second column, the 233Pa was reconstituted in 3 M HNO3 and loaded onto a column containing 1.8 mL of silica gel. Any remaining 237Np was recovered with washes of 5% HNO3 and high purity water. The 233Pa was eluted in 5% HNO3 + 0.1 M HF. This silica gel column was repeated twice to obtain the 233Pa tracer.
In the absence of a 231Pa standard CIAE calibrated the 233Pa spike using CRM U100. Aliquots of CRM U100 were taken to contain ~ 58 and ~ 53 pg 231Pa and these were traced with ~ 70 pg and ~ 100 pg of 233Pa respectively. Following tracer-sample equilibration for 1 h the samples were reconstituted in 9 M HCl and loaded onto a column containing 1.8 mL BioRad AG1-X8 anion exchange resin. Matrix elements were eluted in 9 M HCl and then Pa was eluted with 9 M HCl + 0.05 M HF. This column was repeated with the subsequent eluted Pa re-constituted in 3 M HNO3 and loaded onto a column containing 1.8 mL silica gel (high-purity, 70–230 µm particle size). Matrix elements were eluted with 5% HNO3 and high-purity water. Finally, protactinium was eluted in 5% HNO3 + 0.05 M HF. The 231Pa to 233Pa ratio was measured by the ICP-MS method described below. For sample 231Pa assay measurements, aliquots containing 2–15 pg of 231Pa were taken which contained ~ 300 µg of uranium for U850 and 1000 µg of uranium for U010. These aliquots were traced with picograms of 233Pa. Following tracer-sample equilibration for an hour the same chemical purification method was performed as described above for the 233Pa tracer calibration. To quantify background contamination, blank acid samples were processed alongside the samples using the same chemistry.
All protactinium measurements were performed within 5 days of the final column purification using a ThermoScientific™ Element XR with the same method and mass bias standards as described above for uranium.
Radiochronometry
The number of atoms of 231Pa and 235U measured by the isotope dilution mass spectrometry methods described above were used in Eq. 1 to calculate the model age of CRMs U010 and U850
where t is the model age, λ is the decay constant (derived from the half life, Table 1) and N is the number of atoms measured. The model age can be presented as a model purification date relative to a laboratory reference date. This model purification date can then be compared to the known production date of the CRM to assess the validity of the original model assumptions detailed in Sect. 1. The uncertainty on these measurements represents a full error propagation including, but not limited to, components such as half-life uncertainties, spike calibration uncertainties, measurement and weighing uncertainties.
Results and discussion
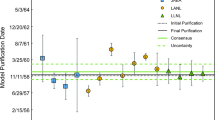
The measured 231Pa/235U atom ratios, resulting model ages, and model purification dates (with associated uncertainties) are presented in Table 2. The model purification dates are shown in Fig. 1. For reference the 230Th/234U model purification dates reported by LANL, LLNL and CIAE are also plotted in Fig. 1 [8]. Process blank concentrations for both uranium and protactinium for all three laboratories represent < 0.2% of the aliquot sizes and are therefore insignificant relative to reported measurement uncertainties.

Model purification dates calculated for a CRM U010, b CRM U850. The black symbols are 231Pa/235U model dates calculated in this study by CIAE, LANL, and LLNL. Gray symbols are 230Th/234U model dates previously reported by [8]. The horizontal dashed lines represent the start and end dates of production: April 16, 1958 and June 5, 1958 for U010 and December 3, 1957 and December 31, 1957 for U850 [13]. Error bars represent expanded uncertainties with a coverage factor of 2. The average model purification dates (circle data points) are calculated mean/consensus values of the model purification dates for each radiochronometer. The expanded uncertainty of this consensus value is calculated using Eq. 2
The 231Pa/235U model purification dates calculated by LANL, LLNL and CIAE for CRM U010 range from October 14, 1957 to June 17, 1959. These model purification dates are concordant (agree) within analytical uncertainty. However, all model purification dates from this study, with the exception of one CIAE data point, are biased younger than the June 5, 1958 date in the historical record marking the end of production [13]. CRM U010 is not certified for radiochronometry but there is a detailed record of production published by the NBS and Union Carbide/Oak Ridge [13]. The recorded production of CRM U010 began on April 16, 1958 and concluded on June 5, 1958 [13]. No details of the chemical purification procedure are available in the CRM production history so it is possible that Th and Pa were purified from the bulk uranium at different times. The younger (relative to historical record) calculated model ages measured in this study could result from loss of 231Pa or gain of 235U over time through CRM handling. Conversely there may also be a systematic bias in either tracer calibrations or corrections during mass spectrometry. If systematic biases were the cause they would need to be consistent across the diverse range of analytical methods used.
Using the independent measurements reported from each laboratory, an average, or mean, model purification date can be calculated. The external uncertainty (k = 2) provided for the mean is calculated as the uncertainty of the mean using Eq. 2 below:
where \(N\) is the number of replicates, \(t_{{\left( {1 - \frac{\alpha }{2}} \right)}}\) is the \(100_{{\left( {1 - \frac{\alpha }{2}} \right)}}\)th percentile of the student’s t-distribution corresponding to a probability \(\alpha = 0.05\), and \(v = N_{r} - 1\) degrees of freedom. This calculation provides the uncertainty of the mean of the replicates at the 95% confidence level. It does not incorporate the uncertainty on each individual measurements and therefore represents the variability within the data set. The calculated average 231Pa/235U model purification date for CRM U010 is December 28, 1958 ± 198 days. This 231Pa/235U interlaboratory consensus value for CRM U010 is almost 7 months younger than, but consistent within analytical uncertainty with, the CRM U010 historical production records [13]. As noted by [8], the model purification dates calculated using the 230Th/234U radiochronometer are generally biased older than June 5, 1958 suggesting that perhaps 230Th was not completely purified during the production of CRM U010. However, many of the 230Th/234U model purification dates reported previously are concordant with the 231Pa/235U model purification dates within analytical uncertainty. Additionally, the calculated mean/consensus 230Th/234U model purification date of November 12, 1957 ± 303 days for U010 is concordant with both the known end of production as well as the consensus 231Pa/235U model purification date. The concordant nature of the 230Th/234U and 231Pa/235U results thus give confidence that the radiochronometry model assumptions and recorded production history of CRM U010 are valid.
The interlaboratory model purification dates calculated for CRM U850 range from April 8, 1957 to December 4, 1961. The calculated model purification dates for CRM U850 show larger variation between laboratories than the CRM U010 231Pa/235U model purification dates, which may result from the calibration methods, materials and instrumentation used. In the case of CRM U850 not all of the model purification dates determined by the laboratories are concordant. There is however, no systematic bias younger or older within these laboratory data than the recorded CRM production date range in the historical record. Similarly to CRM U010, there is a detailed record of production for this CRM published by the NBS and Union Carbide/Oak Ridge [13]. The recorded production began on December 3, 1957 and ended on December 31, 1957 [13] but no details on the chemical purification procedure are available. The calculated mean 231Pa/235U model purification date for CRM U850 is May 20, 1958 ± 363 days. This 231Pa/235U interlaboratory consensus value is approximately 4.5 months younger than but consistent within analytical uncertainty of the recorded end of production. Similarly to CRM U010, the CRM U850 model purification dates calculated using the 230Th/234U radiochronometer are biased older than the known end of production [8]. However, like CRM U010, many of the reported 230Th/234U model purification dates are concordant with the 231Pa/235U model purification dates, within analytical uncertainty. Additionally, the calculated mean/consensus 230Th/234U model purification date for CRM U850 is concordant with the consensus 231Pa/235U model purification date. Therefore these results give confidence that the radiochronometry model assumptions and recorded production history of CRM U850 are also valid. The CIAE U850-2 result is much younger than other model ages. This is due to each U850 model date being derived from different spike calibrations. The lower precision achieved by CIAE is likely due to the use of a single collector mass spectrometer compared to the use of multi-collector instruments at both LANL and LLNL.
Conclusions
The 231Pa/235U model purification dates calculated for CRMs U010 (October 14, 1957 to June 17, 1959) and U850 (April 8, 1957 to December 4, 1961) are generally concordant between laboratories and agree within analytical uncertainty with the production histories of both CRMs. The consistency between the measurements from three different laboratories using different tracers, tracer calibration protocols, purification chemistries and mass spectrometry methods demonstrates the validity of each laboratory’s methods. The concordance observed between the 231Pa/235U model purification dates reported by this study and previous 230Th/234U model purification dates for CRM U010 and CRM U850 increases confidence that both 230Th and 231Pa decay products were well-purified during the production of both CRM U010 and CRM U850. Therefore, both CRMs represent good candidate materials to be used for quality control during radiochronometry measurements of unknown uranium materials for nuclear forensics. Through such international collaborations, we validate methods and establish that reproducible 231Pa/235U ages can be obtained by laboratories around the world. This study therefore validates the use of the 231Pa/235U radiochronometer to support radiochronometry measurements in future nuclear forensics investigations and supports the establishment of a global radiochronometry capability that deters nuclear proliferation.
References
Mayer K, Wallenius M, Varga Z (2013) Nuclear forensic science: correlating measurable material parameters to the history of nuclear material. Chem Rev 113:884–900
Mayer K, Wallenius M, Varga Z (2015) Interviewing a silent (radioactive) witness through nuclear forensic analysis. Anal Chem 87:11605–11610
Sturm M, Richter S, Aregbe Y, Wellum R, Mialle S, Mayer K, Prohaska T (2014) Evaluation of chronometers in plutonium age determination for nuclear forensics: what if the ‘Pu/U clocks’ do not match? J Radioanal Nucl Chem 302:399–411
Varga Z, Mayer K, Bonamici CE, Hubert A, Hutcheon I, Kinman W, Kristo M, Pointurier F, Spencer K, Stanley F, Steiner R, Tandon L, Williams R (2015) Validation of reference materials for uranium radiochronometry in the frame of nuclear forensics investigations. Appl Radiat Isot 102:81–86
Kristo MJ, Gaffney AM, Marks N, Knight K, Cassata WS, Hutcheon ID (2016) Nuclear forensic science: analysis of nuclear material out of regulatory control. Annu Rev Earth Planet Sci 44:555–579
Gaffney AM, Hubert A, Kinman WS, Magara M, Okubo A, Pointurier F, Schorzman KC, Steiner RE, Williams RW (2016) Round-robin 230Th–234U age dating of bulk uranium for nuclear forensics. J Radioanal Nucl Chem 307:2055–2060
Kayzar TM, Williams RW (2016) Developing 226Ra and 227Ac age-dating techniques for nuclear forensics to gain insight from concordant and non-concordant radiochronometers. J Radioanal Nucl Chem 307:2061–2068
Treinen KC, Kinman WS, Chen Y, Zhu L, Cardon AMR, Steiner RE, Kayzar-Boggs TM, Williams RW, Zhao Y-G (2017) J Radioanal Nucl Chem 314:2469–2474
Rolison JM, Williams RW (2018) Application of the 226Ra–230Th–234U and 227Ac–231Pa–235U radiochronometers to UF6 cylinders. J Radioanal Nucl Chem 317:897–905
Kristo MJ, Williams R, Gaffney AM, Kayzar-Boggs TM, Schorzman KC, Lagerkvist P, Vesterlund A, Ramebäck H, Nelwamondo AN, Kotze D, Song K, Lim SH, Lee C-G, Okubo A, Maloubier D, Cardona D, Samuleev P, Dimayuga I, Varga Z, Wallenius M, Mayer K, Loi E, Keegan E, Harrsion J, Thiruvoth S, Stanley FE, Spencer KJ, Tandon L (2018) The application of radiochronometry during the 4th collaborative materials exercise of the nuclear forensics international technical working group (ITWG). J Radioanal Nucl Chem 315:425–434
Rolison JM, Treinen KC, McHugh KC, Gaffney AM, Williams RW (2017) Application of the 226Ra–230Th–234U and 227Ac–231Pa–235U radiochronometers to uranium certified reference materials. J Radioanal Nucl Chem 314:2459–2467
Essex RM, Williams RW, Treinen KC, Collé R, Fitzgerald R, Galea R, Keightley J, LaRosa J, Laureano-Pérez L, Nour S, Pibida L (2019) J Radioanal Nucl Chem. https://doi.org/10.1007/s10967-019-06711-6
Petit GS (1960) Preparation of uranium isotopic standards for the National Bureau of Standards. Union Carbide Nuclear Company, Oak Ridge, Tennessee. Oak Ridge National Laboratory, Report#KL-8, Department of Energy/K25 Archives
Sims KWW, Gill JB, Dosseto A, Hoffmann DL, Lundstrom CC, Williams RW, Ball L, Tollstrup D, Turner S, Prytulak J, Glessner JJG, Standish JJ, Elliott T (2008) An inter-laboratory assessment of the thorium isotopic composition of synthetic and rock reference materials. Geostand Geoanal Res 32(1):65–91
Geological and Environmental Reference Materials (GeoReM). http://georem.mpch-mainz.gwdg.de/. Accessed 1 Sept 2019
Eppich GR, Williams RW, Gaffney AM, Schorzman KC (2013) 235U–231Pa age dating of uranium materials for nuclear forensic investigations. J Anal At Spectrom 28:666–674
Treinen KC, Gaffney AM, Rolison JM, Samperton KM, McHugh KC, Miller ML, Williams RW (2018) Improved protactinium spike calibration method applied to 231Pa–235U age-dating of certified reference materials for nuclear forensics. J Radioanal Nucl Chem 318(1):209–219
Jaffey AH, Flynn KF, Glendenin LE, Bentley WC, Essling AM (1971) Precision measurement of half-lives and specific activities of 235U and 238U. Phys Rev C 4:1889
National Nuclear Data Center, Brookhaven National Laboratory. http://www.nndc.bnl.gov. Accessed 19 Oct 2018
Robert J, Miranda CR, Muxart R (1969) Mesure de la periode du protactinium-231 par microcalorimetrie. Radiochim Acta 11(2):104–108
National Nuclear Data Center, Brookhaven National Laboratory. http://www.nndc.bnl.gov. Accessed May 2018
Jones RT, Merritt JS, Okazaki A (1986) A measurement of the thermal neutron capture cross section of 232Th. Nucl Sci Eng 93:171–180
Acknowledgements
The authors wish to acknowledge the National Nuclear Security Administration’s Nuclear Smuggling Detection and Deterrence office for direct funding. Los Alamos National Laboratory, an affirmative action/equal opportunity employer, is managed by Triad National Security, LLC, for the National Nuclear Security Administration of the US Department of Energy Under Contract 89233218CNA000001. Lawrence Livermore National Laboratory performed this work under the auspices of the US Department of Energy under Contract DE-AC52-07NA27344. The China Institute of Atomic Energy wish to thank colleagues at the State Nuclear Security Technology Center in China for assistance with sample ICP-MS measurements.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Denton, J.S., Treinen, K.C., Chen, Y. et al. International cooperation in age-dating uranium standards for nuclear forensics using the 231Pa/235U radiochronometer. J Radioanal Nucl Chem 324, 705–714 (2020). https://doi.org/10.1007/s10967-020-07084-x
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10967-020-07084-x