
Abstract
Radiochronometric data, a key signature in evaluating the provenance and process history of nuclear material out of regulatory control, are conventionally acquired via multi-collector mass spectrometry. Here we explore the potential of age-dating by single-collector mass spectrometry. To evaluate model age accuracy/precision across different instrument designs, we performed 230Th–234U and 231Pa–235U radiochronometry of CRM 125-A using two single-collector and one multi-collector plasma source mass spectrometers. Single-collector instruments produce accurate model ages for this uranium standard and thus hold promise for nuclear forensics radiochronometry. Increased acquisition of age information via multiple instrument designs will bolster the global response to nuclear interdictions.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Radiochronometry can provide powerful constraints on the history of nuclear material during a nuclear forensic examination. The application of isotope radiochronometers (e.g., 230Th–234U and 231Pa–235U) for characterizing bulk uranium can assist investigators in determining the date of sample production, purification and/or processing (see [1] and references therein). Well-characterized reference materials with known purification ages serve a critical quality control role in validating chemical purification and mass spectrometric methods for radiochronometry [i.e., 2,3,4,5,6,7,8,9,10]. We have previously developed a streamlined method for purification of protactinium (Pa) and thorium (Th) from bulk uranium (U) and have applied this method to analyze U radiochronometry certified reference materials (CRMs), including CRM 125-A, U630 and IRMM-1000 [2, 3, 5].
Nuclear forensic laboratories around the world have a variety of isotope dilution spike materials and mass spectrometers (both single-collector and/or multi-collector instruments) that may in theory be utilized for age-dating. In general, single-collector plasma source mass spectrometers are less expensive, with a wider range of applications, and therefore are more common than multi-collector mass spectrometers. While there are a number of technical considerations that make multi-collector–inductively coupled plasma–mass spectrometers (MC-ICP-MS) ideal for isotope ratio measurements required for radiochronometry, single-collector ICP-MS designs may also be capable of accurate and sufficiently-precise U, Th and Pa isotope ratio characterization. If this can be demonstrated, single collector ICP-MS may represent a robust and widely-available alternative to MC-ICP-MS for radiochronometry, thereby expanding significantly the number of potential radiochronometry practitioners.
To evaluate the feasibility of single-collector ICP-MS radiochronometry analyses, we performed a multi-instrument intercomparison experiment in which single- and multi-collector ICP-MS instruments were used to characterize radiochronometry reference material CRM 125-A. We performed 230Th–234U and 231Pa–235U analyses on CRM 125-A using three different instruments at Lawrence Livermore National Laboratory (LLNL): (1) ThermoScientific iCAP-Q quadrupole single-collector inductively coupled plasma–mass spectrometer (Q-ICP-MS), (2) ThermoScientific Element XR high resolution single-collector ICP-MS (HR-ICP-MS), and (3) Nu Plasma HR multi-collector ICP-MS (MC-ICP-MS). The same sample aliquots, isotopic tracers, QC reference materials, and data reduction algorithms were employed in the generation and processing of data generated by each instrument, allowing for direct comparison of age-dating results amongst the various mass spectrometer designs.
While single-collector instruments have previously been used to measure parent and daughter isotope concentrations in certified radiochronometry standards [10], the current study is the first to directly compare age-dating results across different ICP-MS platforms. The goal of this multi-instrument approach is to evaluate whether the entire radiochronometric analysis process can be performed using a single-collector plasma source instrument, including: (1) isotopic composition determination, (2) spike calibrations for isotope dilution, and (3) parent and daughter isotopic assay quantification, for an independent and stand-alone single-collector instrument technique. Successful demonstration of radiochonometry analyses by single-collector ICP-MS is of benefit to the international nuclear forensic community as it would permit age-dating measurements to be performed by countries that do not have access to MC-ICP-MS for nuclear forensics.
Sample purification
Radiochronometry standard CRM 125-A is an enriched uranium dioxide pellet (UO2; 4.2% enrichment) that is certified for uranium isotopic composition, assay, and model purification date based on 230Th–234U chronometry [11]. In order to evaluate the accuracy and precision of age-dating results from the two single-collector and one multi-collector instruments used in this study, CRM 125-A aliquots were prepared following the sample workflow displayed in Fig. 1. Three aliquots of CRM 125-A were dissolved, chemically separated and purified, and split to provide chemically-identical aliquots for each mass spectrometric technique. Calculation of 230Th–234U and 231Pa–235U model ages requires analyses of the isotopic composition of uranium in the CRM 125-A aliquots, in conjunction with 234U, 235U, 230Th and 231Pa assay (concentration) determination by isotope dilution mass spectrometry (IDMS), referred to hereafter as UID, ThID and PaID, respectively.

CRM 125-A aliquot preparation and splitting workflow for multi-instrument radiochronometric intercomparison study
First, three solid CRM 125-A UO2 pellet aliquots, labeled CRM 125-A-A, -B and -C, were dissolved following LLNL’s standard operating procedure for U, Th, and Pa determination by IDMS [2]. Dissolved CRM 125-A aliquots, or ‘primary solutions’, were prepared in clean, dry 30-mL Savillex® Teflon vials and stored in 4 M HNO3 + 0.05 M HF for further processing.
Following digestion, gravimetric dilution series of the aforementioned primary solutions were prepared for parent isotopic assay determination (i.e., 234U and 235U). Dilution aliquots were spiked with an in-house, ultra-high purity, calibrated 233U isotope dilution spike (see spike calibration section below). These UID dilution series were made to target a final U concentration of approximately 80–100 ppb. Approximately 160–200 ng total U of each solution (CRM 125-A-A, - B, -C) were weighed into clean Savillex® Teflon vials and spiked with approximately 10–15 ng 233U spike. Each UID cross was then split into separate aliquots for analysis using each mass spectrometer.
In this project CRM 125-A was analyzed as if it were an unknown sample; hence, it was necessary to characterize its uranium isotopic composition (UIC). This value and associated uncertainty were included in all age-dating calculations, and thus affect the final results from each instrument. The corresponding 235U/238U ratios influence the final age-dating results through isotope dilution calculations for each instrument. Some isotopic composition results are outside expanded uncertainty envelope of the certified isotopic composition (Table 1). While this may reflect residual instrumental bias, we note that we have previously observed uranium isotopic heterogeneity between individually dissolved CRM 125-A pellets.
Small aliquots (i.e., ~ 50 µg U) of dissolved primary solutions for CRM 125-A-A, -B, and -C were dissolved in 3 mL 2% HNO3. Each mass spectrometric technique required different dilutions of these prepared UIC fractions for isotopic composition determination, contingent upon operating conditions (e.g., sensitivity) of each instrument at the time of analysis.
Three individual (CRM 125-A-A, -B, -C) aliquots were spiked with both 229Th and 233Pa spikes, and then chemically separated and purified for a ‘paired’ daughter (230Th and 231Pa) assay determination by IDMS (ThID and PaID; Fig. 1). All aliquots (30–40 mg bulk U) were spiked with approximately 30 pg of freshly-prepared and calibrated 233Pa spike (see spike calibration section below) and 5–10 ng of NFRM 229Th spike [12]. These ‘paired’ aliquots were separated into ThID and PaID fractions by performing a bulk U separation using Eichrom® AG 1-X8 (100–200 mesh) anion exchange resin. ThID aliquots were further purified with Eichrom® TEVA resin and another Eichrom® AG 1-X8 anion resin column. PaID aliquots were further purified from bulk U and ingrown 233U with a second Eichrom® AG 1-X8 anion exchange resin column, followed by a final purification using Supelco® Silica Gel Resin [2, 5].
Final purified aliquots of Th, Pa, and U were dissolved and split volumetrically for each instrument. All U fractions (both UID and UIC aliquots) were dissolved in 2% HNO3, fluxed on a hoplate to equilibrate, and analyzed soon after preparation. Spiked Pa fractions (PaID) were eluted from the final Si-gel purification column in 2% HNO3 + 0.05 M HF and analyzed the same day as final purification chemistry [2]. Spiked Th fractions (ThID) were dissolved in 2% HNO3 + 0.005 HF, fluxed to equilibrate, and analyzed.
Multi-instrument spike calibrations
Accurate model ages require properly calibrated isotopic tracers, or “spikes” (e.g., 229Th, 233U and 233Pa), for concentration determination by isotope dilution. All spiked aliquots for isotopic assay determination were prepared using a freshly prepared and calibrated 233Pa spike; nuclear forensics reference material (NFRM) 229Th spike [12]; and a very-high purity (99.9877%) 233U spike. Each instrument was used to determine an indepedent 233U and 233Pa spike calibration by IDMS, with the resulting spike concentrations and uncertainties being incorporated into final age calculations. The 229Th spike was not independently calibrated for each instrument, as this solution is certified by mass [12]; as such, the certificate NFRM 229Th calibration was used in all Th IDMS calculations (Fig. 2).

Multi-instrument uranium isotopic composition results (235U/238U) for the three plasma source ICP-MS instruments. A) Nu Plasma HR (MC-ICP-MS) UIC results. B) Element XR (ICP-MS) UIC results. C) iCAP-Q (ICP-MS) UIC results. The horizontal dark grey line and dashed lines are the certified 235U/238U isotopic composition and associated uncertainty [11]. Uncertainties are displayed at the k = 2 level
LLNL’s in-house, ultra-high purity 233U spike was calibrated using two separately prepared gravimetric standard solutions of CRM 112-A (SRM 960), a U isotopic and assay standard, in 4 M HNO3 and 0.05 M HF. Eight spike-sample crosses were prepared, each consisting of approximately 10 ng of 233U spike and 200–800 ng CRM 112-A. Spike calibration crosses 1–4 were prepared with a CRM 112-A solution dissolved in October 2016. Spike calibration crosses 5–8 were prepared with another CRM 112-A solution dissolved in November 2017. The calibration crosses were designed to have a large dynamic range in 238U/233U and 235U/233U, with these ratios being measured subsequently by mass spectrometry. The known concentration of 238U and 235U from CRM 112-A was used to calibrate the unknown 233U concentration in the spike, for a final calculated average value and uncertainty (Table 2). Results for all three instrument approaches are summarized in Fig. 3.

233U spike calibration; Panel A: Nu Plasma HR MC-ICP-MS, Panel B: Element XR ICP-MS, and Panel C: iCAP-Q Q-ICP-MS. Error bars, k = 2, represent the combined uncertainty for each independent cross and analysis. The shaded colored bars in each panel represent the weighted mean and 2RSE for the calculated average
A weighted-mean 233U spike calibration was determined using data produced by each instrument independently and the numerical algorithm of Lyons et al. (1988). As shown in Fig. 3, all three independently-calculated 233U spike calibrations agree within uncertainty (k = 2); the Nu Plasma HR yielded the most precise spike calibration, followed by the Element XR and iCAP-Q.
Calibration analyses of the 233Pa spike were also performed using data from each instrument independently. Analyses were made on the same day with all three instruments, directly following final purification chemistry. In total, five calibration crosses and a chemistry blank were prepared using an in-house 231Pa calibration solution of known concentration, spiking each calibration cross with approximately 10 pg 231Pa. The calibration crosses had a range of concentrations, from 10 to 35 pg 233Pa per sample. Methods used for the 233Pa spike preparation and calibration are described in [2]. The 233Pa spike used for this study was initially calibrated in February 2019. The initial calibration analyses of the 233Pa spike were performed using the Nu Plasma HR MC-ICP-MS (calibration samples 1–3). One week later, these three calibration crosses were re-prepared for analysis by drying the crosses and performing a purification column to remove any ingrown 233U before reanalysis by MC-ICP-MS. These calibration crosses (1–3) were then dried down and stored for future additional calibration measurements by single-collector instruments. The original three calibration crosses were purified a third time for analysis by the Element XR and iCAP-Q ICP-MS in March 2019. Two additional 233Pa spike calibration crosses were prepared in April 2019 and analyzed by all three instruments on the same day (calibration samples 4–5). The results are shown in Fig. 4, with symbols representing individual, gravimetrically prepared calibration crosses. Results for 233U and 233Pa spike calibrations by each instrument and the corresponding relative standard errors are summarized in Table 3. As with the 233U spike calibration, the 233Pa spike calibration was determined for each instrument independently using the numerical algorithm of Lyons et al., 1988 [13].

Multi-instrument 233Pa spike calibration; Panel A: Nu Plasma HR MC-ICP-MS, Panel B: Element XR ICP-MS, and Panel C: iCAP-Q Q-ICP-MS. Numbered data points indicate the same measured spike calibration aliquots. Error bars, k = 2, represent the combined uncertainty for each independent cross and analysis. The shaded colored bars in each panel represent the weighted mean and 2RSE for the calculated average
Results from the Nu Plasma represent two analytical sessions for Pa-233-Cal-1, -2, -3 and one analaytical session for Pa-233-Cal-4, -5. The iCAP-Q and Element XR each analyzed Pa-233 Cal-1-5 once during two analytical sessions each. The average of the results is given with expanded uncertainties, k = 2.
Mass spectrometry methods
Each mass spectrometric technique used in this study is described in detail below: (1) ThermoScientific iCAP-Q quadrupole single-collector ICP-MS, (2) ThermoScientific Element XR single-collector ICP-MS, and (3) Nu Plasma HR multi-collector ICP-MS. For all instruments the reference material CRM U010 was used to monitor instrumental mass-dependent fractionation in each analytical session (see “Mass Spectrometry Data Reduction and Assessment” below). In addition, for all instruments CRM U005A was analyzed over the course of each analytical session as a QC material to ensure the accuracy of isotope ratio measurement and data reduction corrections (e.g., blank, mass bias, etc.). The U isotopic results for CRM U005A for all analytical sessions across all instrument platforms were consistent with certificate values.
The protactinium (PaID) aliquot for both single-collector instruments was first centrifuged at 3000 rpm for 15 min and the supernate was split into two separate fractions (one for each instrument). The decision to centrifuge was based on a previous clogging event of the sample introduction system with SiO2 particles from silica gel resin used to purify the sample immediately prior to analysis. In the March 2019 analytical session, each single-collector instrument had a total of 1 mL purified sample solution with appoximately 3–6 pg 233Pa available for the Pa spike calibrations. The prepared CRM 125-A aliquots, in 2 mL of solution had approximately 25 pg Pa was available for analysis. In the April 2019 analytical session, the Pa spike calibration crosses contained approximately 6 pg 233Pa. The 233Pa spike calibration and samples were analyzed in the eluted 2% HNO3 + 0.05 M HF. The U010 mass bias solution was prepared in 2% HNO3.
ThermoScientific iCAP-Q quadrupole single-collector ICP-MS
The Pa, Th and U analyses were performed using a ThermoScientific iCAP-Q quadrupole ICP-MS with a single secondary electron multiplier (Bremen, Germany). The chosen sample introduction system components varied depending on the required sensitivity for a given analysis.
Protactinium (PaID) and thorium (ThID) analyses were measured using an APEX IR Desolvating Nebulizer (Elemental Scientific) with a self-aspirating APEX PFA ST-1970 variable flow nebulizer (flow rate 0.4 mL/min) and high-sensitivity cones. The combination of the APEX with the high-sensitivty cones provided an approximate 10-fold improvement in sensitivity for a total of ~ 7 × 106 counts per second per ppb (cps/ppb) 238U with an oxide production < 2%. Samples were introduced in the eluted matrix of 2% HNO3 and 0.05 M HF. The mass bias solution, CRM U010, and the QA/QC solution, CRM U005A, were prepared at a concentration of 100–150 pg/g in 2% HNO3 and 0.005 M HF. Data were collected using time resolved analysis and solely within counting mode of the detector. Standard-sample bracketing was utilized in all analysis sequences.
The March 2019 analytical session included 229Th, 230Th, 231Pa, 232Th, 233Pa, 234U, 235U and 238U intensities. Dwell times of 50 ms and 10 ms were assigned to Pa and U masses, respectively. Total analysis time for each pass was 0.16 s; 400 passes were collected for each sample/standard corresponding to a measurement time of approximately 1.5 min. Average total Pa measured for both isotopes was ~ 0.35 pg in the spike calibration solutions and 0.9 pg in the CRM 125-A-A-C sample solutions. The U005 QA/QC solution had a sensitivity of ~ 7.4 × 106 cps/ppb U.
In the April analytical session, 228Th was added to the acquired masses, and a dwell time of 100 ms was assigned to all masses to produce 0.90 s per pass. For the Pa spike calibration analyses, 99 passes were collected for a total measurement time of 1.5 min for each calibration analysis. Average total Pa measured for both isotopes was 2.5 pg. In the Th ID analysis, 198 passes were collected for a total measurement time of 3 min per analysis. Average total Th measured in CRM125-A-C for 229Th and 230Th was 13 pg and 50 pg, respectively.
Sample UID, UIC and 233U spike calibration analyses were performed with a traditional “wet plasma” using a standard quartz introduction system, including a cyclonic spray chamber; a self-aspirating Sea Spray U series nebulizer, 0.5 mm autosampler probe (flow rate ~ 0.36 mL/min); and standard extraction cones. Instrument optimization yielded a sensitivity of 520,000 cps/ppb U and cerium oxide production of < 2%. Data were collected using time resolved analysis and standard-sample bracketing was utilized in all analysis sequences. The mass bias solution (CRM U010) and QC solution (CRM U005A) were prepared in 2% HNO3 at concentrations to match the concentration of U in the samples. UID samples were measured at ~ 2 ng U/g solution to keep both the 233U and 238U signals in the pulse counting mode of the detector to avoid any uncertainty that could result from crossing into analog mode. The 233U spike calibration analyses had different 233:238 ratios and concentrations and were measured at approximately 2 ng U/g solution and 8 ng U/g solution to have sufficient 233U signal for measurement. UIC samples were analysed at ~ 75 ng U/g solution in order to have a 234U signal greater than 10,000 cps (Table 4).
Critical Limit Lc was calculated based on uranium senstivity and using the standard deviation of 3 process blanks and multiplied by the student t factor 9.925 for 2 degrees of freedom at the 99% CI. Only 2 process blanks were analyzed in the 15-April-2019 run and therefore the standard deviation was multipled by 63.657 for 1 degree of freedom at the 99% CI.
ThermoScientific Element XR single-collector ICP-MS
The Element XR is a magnetic sector single-collector ICP-MS instrument with high resolving power designed for trace element concentration measurements. Here, it was utilized to make Pa, Th and U isotope ratio measurements following the procedures outlined below.
Protactinium (PaID) and ThID: Samples were dissolved in 2% HNO3 + trace HF and introduced to the plasma using an ESI ST variable flow Teflon nebulizer connected to an ESI Apex IR introduction system, which partially desolvates the sample and reduces oxide formation. The Element XR was fitted with the standard ‘H’ skimmer cone and high sensitivity ‘Jet’ sample cone, achieving a sensitivity of 1.2 × 107 cps/ppb for 238U and an oxide production rate of < 2%. The analytical method involved collecting data for 230Th, 231Pa, 232Th, 233Pa, 234U, 235U and 238U intensities. During each pass the total sampling time for each isotope was between 5 and 20 ms. Each individual sample or standard measurement consisted of a total of 200 passes, which corresponds to a sampling time of approximately 7 min. Mass bias standard CRM U010 was measured at a concentration of 50 ppt, which produced a 238U signal of ~ 500 k cps. All isotopes in standards and samples were analyzed using counting mode on the secondary electron multipliers, preventing complications associated with detector cross-calibration. To assess the accuracy of these measurements, the standard U005A was analyzed alongside the samples as an unknown.
Uranium (UID) and UIC: Samples and standards were diluted in 2% HNO3 and introduced to the instrument in ‘wet plasma’ mode using a cyclonic spray chamber. Typical sensitivity using this setup was 1.7 × 106 cps/ppb 238U. Both UIC and UID samples were bracketed by a 10 ppb dilution of the U010 standard, with 238U analyzed in analog mode and 233U, 234U, 235U and 236U analyzed in counting mode. Uranium was measured at relatively high level in order to obtain a high 234U signal. Typical 234U intensities for unknowns were ~ 8000 cps. The standard U005A was analyzed alongside the uranium samples for QC purposes and, as mentioned above, results are consistent with certified values [14].
Nu Plasma HR multi-collector ICP-MS
The U, Th and Pa mass spectrometry methods by Nu Plasma multi-collector ICP-MS are described below. UID and spike calibration samples were analyzed by measuring the 233U, 235U, and 238U isotopes simultaneously on Faraday collectors (Fig. 5, routine ‘U-ID-234-IC0’). UIC fractions were measured with the 235U and 238U beams on Faraday collectors, and 234U on an ion counter (Fig. 5, routine “U-Static”). The Nu Plasma HR MC-ICP-MS sample introduction consisted of a CETAC® Aridus II desolvating nebulizer system and autosampler equipped with a Savillex® PFA C-Flow nebulizer (100 µL/min). Average instrument sensitivity over the course of this study was equivalent to ~ 0.5 V/ppb. This setup was used for all U, Th and Pa isotope ratio analyses, as a “dry plasma” optimizes Nu Plamsa HR sensitivity for such actinide measurements. Given this sensitivity and instrument configuration, the 233U spike calibration samples ran betwen 4–6 V 238U and between 30 and 135 mV 233U. CRM 125-A samples were measured with a 238U signal intensity between 300 and 500 mV and 25–35 mV 233U.

Schematic of collector–isotope analysis routines used for Nu Plasma HR MC-ICP-MS U (blue), Th (pink) and Pa (blue) radiochronometric measurements. ``Jump” indicates a two-cycle (C1 and C2) dynamic analysis routine. Italicized isotopic masses indicate Faraday measurements; bold isotopic masses indicate ion counter (IC) measurements
ThID aliquots were analyzed using a two-cycle, dynamic multi-collection routine in which 232Th is measured continously on a Faraday collector and 229Th and 230Th are sequentially analyzed on the same ion counter by peak-jumping (Fig. 5, routine “Th-Jump”). Because 229Th and 230Th analyses are performed contemporaneous with Faraday 232Th measurements, the 229Th/230Th ratio can be calculated by crossing 229Th/232Th (cycle #1) with 230Th/232Th (cycle #2). This approach results in more precise 229Th/230Th data, as the uncertainties associated with Faraday/IC gains are eliminated. ThID samples were measured at a 229Th ion beam intensity between 55,000 and 110,000 cps and a 230Th signal intensity between 125,000 and 450,000 cps. High-purity 232Th spike was added to each sample prior to mass spectrometry to yield an intensity of approximately 30 mV 232Th on a Faraday collector.
Protactinium (PaID) aliquots were analyzed using a one-cycle, static multi-collection routine in which 231Pa and 233Pa are simultaneously measured on separate ion counters (Fig. 5, routine “Pa-Pulse”).). Pa-233 intensities for CRM 125-A-A-C were between 10,500 and 32,000 cps; corresponding 231Pa intensitites ranged 47,000–54,000 cps.
Instrumental mass-dependent fractionation and Faraday/IC gain corrections for U measurements were made using CRM U010 in the same analytical batch run by standard–sample–standard bracketing (see below). For complementary analytical details, see also [2,3,4,5,6,7,8].
Mass spectrometry data reduction and assessment
For all instruments, mass spectrometric data reduction/evaluation; isotopic assay and model age calculations; figure generation; and uncertainty propagation/uncertainty budget construction (see below) were performed using the R statistical programming environment [15]. The application of a common data reduction approach to all mass spectrometric analyses removes potential complications arising from disparate, instrument-specific data reduction algorithms, thereby permitting direct comparison of radiochronometric results from these instruments. As described earlier, instrumental mass-dependent fractionation (mass bias) was quantified on all instruments by repeat monitoring of CRM U010 238U/235U during each analytical session, with applied mass bias factors (β) determined by (1) interpolation of bracketing CRM U010 analyses, and (2) assuming an exponential mass fractionation law [e.g., 16]. A conservative expansion of uncertainties affiliated with interpolated correction factors was applied to yield more robust isotope ratio results. Quantitative blank corrections were made for all instruments; additional quantitative corrections for detector baseline and Faraday/ion counter (IC) gain (as needed) were made for Nu Plasma HR analyses.
Uncertainty propagation, budgets and evaluation
As described above, uncertainty propagation and uncertainty budget construction for all radiochronometric model ages were implemented in the R statistical programming environment (Figs. 6, 7). Uncertainty propagation was performed via the Monte Carlo method [17, 18], a computational alternative to the more common, partial derivative-based linear uncertainty propagation method [19]. One million (106) samplings were performed for each Monte Carlo calculation, which was more than sufficient to obtain convergence of all calculated model ages to the level of two significant figures in the uncertainty associated with each age.

Comprehensive uncertainty budgets for 230Th-234U CRM 125-A model ages for the Nu Plasma HR (a), Element XR (b) and iCAP-Q (c), using results from Aliquot A. Blue and pink uncertainty components correspond to contributions from U and Th analyses, respectively, for all instrument platforms. Yellow uncertainty component for the Nu Plasma HR correspond to Faraday/ion counter (IC) gain factor determination. Model age uncertainties are stated at the k = 2 level
As visualized in Figs. 6 and 7, both similarities and differences are observed amongst the various ICP-MS platforms regarding their major radiochronometric uncertainty contributors. Firstly, uncertainties arising from decay constants, gravimetric masses (i.e., weights of primary solutions and spikes), and blank and baseline corrections are minor contributors for both the multi-collector and single collector instruments. However, for the Nu Plasma HR MC-ICP-MS the most important uncertainty contributors arise from two distinct sources (Figs. 6, 7). The first source is daughter isotope-related measurement uncertainties: for the 230Th–234U chronometer, it is the measured ThID 230Th/229Th (sample/spike isotope) value; for the 231Pa–235U chronometer, it is the 233Pa spike calibration, which is ultimately rooted in the 233Pa/231Pa measurement precision of the protactinium standard-spike aliquot “crosses” and the assay uncertainty for the 231Pa reference material. The second source is the Faraday/IC gain factor uncertainties: these are tied to the certified 236U/235U and 234U/235U values of CRM U010, which are used to empirically determine Faraday/IC0 and Faraday/IC1 gain, respectively, during each analytical session. Notably, these gain intercalibration uncertainties are unique to the Nu Plasma HR in the current study, as the Element XR and iCAP-Q are equipped with only a single detector. Comparatively, the dominant uncertainty for both the Element XR and iCAP-Q arises from the measurement of CRM U010 238U/235U for the mass bias correction (Figs. 6 and 7). This observation is partly explained by true day-to-day variability in the precision at which CRM U010 238U/235U was measured. However, it is also due to the fact that the mass bias determination using CRM U010 238U/235U is an interpolated correction using standard–sample bracketing. Thus, while individual CRM U010 238U/235U measurements on the single-collector instruments have relative uncertainties generally on par with sample/spike isotope ratio measurements (e.g., 230Th/229Th), our data reduction algorithm conservatively expands the uncertainty associated with the mass bias correction applied to samples so as to account for drift in instrumental fractionation over an analytical session. Taken together, the precision of single-collector ICP-MS model ages is expected to improve if subsequent steps can be taken to optimize the measurement of the primary mass bias standard.

Comprehensive uncertainty budgets for 231Pa-235U CRM 125-A model ages for the Nu Plasma HR (a), Element XR (b) and iCAP-Q (c), using results from Aliquot A. Blue and green uncertainty components correspond to contributions from U and Pa analyses, respectively, for all instrument platforms. Yellow uncertainty components for the Nu Plasma HR correspond to Faraday/ion counter (IC) gain factor determinations. Model age uncertainties are stated at the k = 2 level
Results and discussion
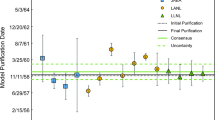
The certified model purification date of CRM 125-A, August 18, 1994 (± 116 days), was determined using the 230Th–234U chronometer [11]. Results of the multi-instrument approach are summarized in Table 5 and Fig. 8. The summary of results to follow are average model dates calculated from the three aliquots CRM 125-A-A, -B, -C for each instrument; the expanded (k = 2) combined uncertainties are given in days. The average 230Th–234U model date from the iCAP-Q is September 5, 1994 (± 202 days); the nominal date is less than 1 month younger than the certified age. The average 231Pa–235U model date from the iCAP-Q analyses is May 9, 1994 (± 356 days), approximately 3 months older than the certified age but in agreement well within the associated uncertainties. The average 230Th–234U model date result from the Element XR is February 12, 1994 (± 221 days), older than the certified model date, but still within uncertainty. The average 231Pa–235U model date result from the Element XR is September 21, 1994 (± 178 days), less than 1 month younger than the certified model date. The average 230Th–234U model date for the Nu Plasma HR is August 18, 1994 (± 69 days). The average 231Pa–235U model date for the Nu Plasma HR is September 27, 1994 (± 76 days), less than 1 month younger than the certified model date. Paired 231Pa–235U and 230Th–234U model ages produced by all instruments are concordant, both internally and with the certified age of CRM 125-A, within stated uncertainties. Certified radiochronometry standard ages and isotopic data from single collector instruments are thus consistent with those from a multi-collector instrument.

Final model age results for multi-instrument approach. Panel A displays results from the Nu Plasma HR; Panel B displays results from the Element XR; Panel C displays results from the iCAP-Q. The average percent relative standard error (%RSE) for 230Th/234U and 231Pa/235U radiochronometers for each instruments results are also displayed. All model age results are calculated with a reference date of 15-April-2019. The certified Th-U model purification date of CRM 125-A is August 18th, 1994 (± 116 days). All expanded uncertainties are k = 2
The reference date for all model age calculations is 15-April-2019. Expanded uncertainties are all 2-sigma. The daughter-parent isotope age-dating (Bateman) equation is used to calculate the age, t, of the material (see Treinen et al., 2018 for equation and discussion). Decay constants (λ) used for calculating model ages are from [20,21,22,23]. The 230Th half-life = 7.569 × 104 ± 115 years, and the 234U half-life = 2.4525 × 105 years. The 231Pa half-life = 32,713 ± 110 years, and the 235U half-life = 7.0381 × 108 ± 4.8 × 105.
Future considerations
While results from the present work are promising insofar as the practical utility of single-collector ICP-MS is concerned, we note that subsequent research and analyses are required to thoroughly evaluate the “real-world” feasibility of this approach to nuclear forensics. Considerations for the application of this approach should focus on the type of sample being examined, i.e., the uranium concentration and isotopic composition, age, and availability of sample under examination. In addition, the methods presented here require complete chemical purification of the trace quantities of Th and Pa from a bulk U sample. Future method development may also explore the level of allowable residual U in the Th or Pa isotope dilution aliquots.
Future method development work for single-collector analysis may also focus on optimizing the analysis sequence. In particular, the measurement method for the mass bias calibration standard may potentially be optimized by modifying dwell times. For UIC analyses, this would also include utilizing the 234U/235U ratio instead of the 234U/238U ratio in order maintain ion beam signal intensites below the threshold of the analog region of the detector, and thereby avoid the additional uncertainty associated with crossing between detector modes on the single-collector ICP-MS platforms. As discussed previously, additional research to stabilize and improve single-collector ICP-MS mass bias measurement would produce more precise radiochronometric results, as mass bias correction uncertainty dominates single-collector model ages (Figs. 6, 7). Future work could also explore the use of a desolvating nebulizer that is designed for the relatively high HF concentration (0.05 M HF) necessitated by the current Pa purification and analysis method.
Additional multi-instrument age-dating comparison work may explore complimentary techniques of isotopic determination for age-dating, including alpha spectrometry, as was explored in [24]. Improvements in alpha spectrometry instrumentation and chemical purfication techniques could allow this method to be a viable choice for laboratories for radiochronometry.
Conclusions
In this study, single collector ICP-mass spectrometry was used to perform calibrations of isotope dilution tracers, as well as to perform isotope dilution analyses of the samples. Additionally, spiked Th and Pa sample aliquots were purified from the bulk U sample matrix prior to analysis, thus minimizing sample matrix effects and isobaric interferences. While multi-collector mass spectrometers ultimately generate the most precise radiochronometric and isotopic data, single collector mass spectrometers can be used to determine the model age of bulk uranium with respectable precision. Because single-collector mass spectrometers are more widely available than multi-collector mass spectrometers, this approach may enable a greater number of nuclear forensic practitioners to perform radiochronometry analyses.
References
Kristo MJ, Gaffney AM, Marks N, Knight K, Cassata WS, Hutcheon ID (2016) Nuclear forensic science: analysis of nuclear material out of regulatory control. Annu Rev Earth Planet Sci 44(1):555–579. https://doi.org/10.1146/annurev-earth-060115-012309
Treinen KC, Gaffney AM, Rolison JM et al (2018) J Radioanal Nucl Chem 318:209. https://doi.org/10.1007/s10967-018-6149-x
Rolison JM, Treinen KC, McHugh KC, Gaffney AM, Williams RW (2017) Application of the 226Ra-230Th-234U and 227Ac-231 Pa-235U radiochronometers to uranium certified reference materials. J Radioanal Nucl Chem 314:2459–2467
Treinen KC, Kinman WS, Yen C, Liuchao Z, Cardon AMR, Steiner RE, Kayzar-Boggs TM, Wililams RW, Yong-Gang Z (2017) US-DOE and CIAE international cooperation in age-dating uranium standards. J Radioanal Nucl Chem 314:2469–2474
Eppich GR, Williams RW, Gaffney AM, Schorzman KC (2013) 235U-231Pa age dating of uranium materials for nuclear forensic investigations. J Anal At Spectrom. https://doi.org/10.1039/c3ja50041a
Gaffney AM, Hubert A, Kinman WS, Magara M, Okubo A, Pointurier F, Schorzman KS, Steiner RE, Williams RW (2016) Round-robin 230Th-234U age dating of bulk uranium for nuclear forensics. J Radioanal Nucl Chem 307:2060–2065
Kayzar TM, Williams RW (2015) Developing 226Ra and 227Ac age-dating techniques for nuclear forensics to gain insight from concordant and non-concordant radiochronometers. J Radioanl Chem 307:2061–2068
Williams RW, Gaffney AM (2011) 230Th–234U model ages of some uranium standard reference materials. Radiochim Acta 1:31–35
Varga Z, Wallenius M, Klaus M (2010) Age determination of uranium samples by inductively coupled plasma mass spectrometry using direct measurement and spectral deconvolution. J Anal Spectrom 25:1958–1962
Varga Z, Nicholl A, Hrnecek E et al (2018) J Radioanal Nucl Chem 318:1565. https://doi.org/10.1007/s10967-018-6247-9
New Brunswick Laboraotory, Department of Energy, Certified Reference Material 125-A. 1997
Essex RM, Mann JL, Williams RW, Kinman WS, Hubert A, Bennet ME, Gourgiotis A (2018) A new thorium-229 reference material. Appl Radiat Isot 134:23–31
Lyons L, Gibaut D, Clifford P (1998) How to combine correlated estimates of a single physical quantity. Nucl Instrum Methods Phys Res A 270(1):110–117. https://doi.org/10.1016/0168-9002(88)90018-6
Richter S, Kühn H, Aregbe Y, Hedberg M, Horta-Domenech J, Mayer K, Poths J (2011) Improvements in routine uranium isotope ratio measurements using the modified total evaporation method for multi-collector thermal ionization mass spectrometry. J Anal At Spectrom 26(3):550–564
R Core Team (2017) R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria. URL https://www.R-project.org/
Russell WA, Papanastassiou DA, Tombrello TA (1978) Ca isotope fractiona- tion on the Earth and other solar system materials. Geochimica et Cosmochimica Acta 42(8):1075–1090. https://doi.org/10.1016/0016-7037(78)90105-9
Metropolis N, Ulam S (1949) The Monte Carlo method. J Am Stat Assoc 44(247):335–341. https://doi.org/10.2307/2280232
JCGM 101:2008. Evaluation of measurement data—Supplement 1 to the “Guide to the expression of uncertainty in measurement”—Propagation of distributions using a Monte Carlo method
JCGM 100:2008. GUM 1995 with minor corrections. Evaluation of measurement data—“Guide to the expression of uncertainty in measurement”
Cheng H, Edwards RL, Hoff J, Gallup CD, Richards DA, Asmerom Y (2000) The half-lives of uranium-234 and thorium-230. Chem Geol 169:17
Robert PJ, Miranda CF, Muxart R (1968) Mesure de la periode du protactinium 231 par microcalorimetrie. Radiochim Acta 11(2):104–108
Jaffey AH, Flynn KF, Glendenin LE, Bentley WC, Essling AM (1971) Precision measurement of half-lives and specific activities of 235U and 238U. Phys Rev C 4:1889–1906
Jones RT, Merritt JS, Okazaki A (1986) A measurement of the thermal neutron capture cross-section of 232Th. Nucl Sci Eng 93:171–180
Morgenstern A, Apostolidis C, Mayer K (2002) Age determination of highly enriched uranium: separation and analysis of 231Pa. Anal Chem 74:5513–5516
Acknowledgements
This work was performed under the auspices of the U.S. Department of Energy by Lawrence Livermore National Laboratory under Contract DE-AC52-07NA27344. Releasable to external audiences, LLNL-CONF-784319. The authors would like to acknowledge the contributions made by LLNL team members Victoria D. Genetti, Dana L. Drew, and Sam R. Shipman.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Treinen, K.C., Samperton, K.M., Lindvall, R.E. et al. Evaluating uranium radiochronometry by single-collector mass spectrometry for nuclear forensics: a multi-instrument investigation. J Radioanal Nucl Chem 322, 1627–1640 (2019). https://doi.org/10.1007/s10967-019-06832-y
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10967-019-06832-y