
Abstract
Application of the 231Pa/235U radiochronometer for nuclear forensic investigations is challenged by a lack of certified reference materials with 231Pa/235U model purification dates. The Japan Atomic Energy Agency, Los Alamos National Laboratory, and Lawrence Livermore National Laboratory completed an interlaboratory study measuring 231Pa/235U model ages of New Brunswick Laboratory CRM U100. Results from independent laboratories were combined to calculate a consensus 231Pa/235U model purification date for CRM U100 of March 26, 1959 ± 237 days. This 231Pa/235U consensus date for CRM U100 may be used by the nuclear forensic community for quality control of 231Pa/235U radiochronometry measurements of unknown materials.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Radiochronometry, or the science of age dating a nuclear material using the radioactive decay of parent isotopes to daughter isotopes in a closed system, can provide predictive signatures that may be used during a law enforcement investigation of nuclear or other radioactive material found out of regulatory control [1]. During the application of radiochronometry, a model age for a radioactive material is calculated which represents the time that has passed since the material was last purified of any decay products. This age may also be used to calculate a “model purification date” or “model production date” by assuming that the timing of purification corresponds to the time of production of the material. In the case of uranium (U) materials, the most commonly used radiochronometer for age dating is the 230Th/234U chronometer where the parent isotope, 234U, decays to produce 230Th over time [2,3,4,5,6,7,8,9]. However, in recent years, the nuclear forensics community has demonstrated interest in using more than one chronometer during the characterization of the age or the time of production of uranium materials [10,11,12,13,14]. The use of multiple chronometers may provide more confidence in measured model ages of nuclear material and/or may also provide more information about the production history of an unknown material.
A second chronometer that has been used for uranium radiochronometry is the 231Pa/235U (daughter/parent) chronometer [10,11,12,13,14,15]. Several primary challenges associated with 231Pa/235U radiochronometry arise from a lack of certified reference materials available for commercial purchase to use for method validation and quality control as well as to support isotope dilution mass spectrometry measurements of 231Pa. For example, there is no commercially available 233Pa single isotope spike for isotope dilution measurements of 231Pa due to the short half-life of 233Pa (~ 26.97 days [16, 17]). There are also no protactinium (Pa) reference materials certified for isotope composition that can be used during mass spectrometry analysis to correct for analytical artefacts such as instrumental mass bias. Finally, there are no certified reference materials that are certified for 231Pa/235U model ages or model purification dates that can be used as quality control standards during 231Pa/235U radiochronometry measurements of unknown materials. In the absence of certified Pa standards, U certified reference materials have been used to ensure quality control and correct for instrumental bias. Until metrology laboratories can produce and certify reference materials for 231Pa/235U radiochronometry, one approach that may be used to address the gap in certified reference materials is for laboratories with 231Pa/235U chronometry capabilities to produce consensus ages of commercially available U certified reference materials [11,12,13, 15, 18].
To date, most studies that have measured 231Pa/235U model ages for commercially available U certified reference materials have presented data that were generated from single laboratories [11,12,13, 15, 18]. When single laboratories measure different certified reference materials, it is impossible to assess if laboratory separation methods, spike calibration methods, and analytical methods result in 231Pa/235U model age biases. However, if single laboratories are independently measuring the same certified reference material with a known production history, measured model ages can be compiled to calculate consensus ages for the forensic community. In this study, we present results from a unique interlaboratory study in which the Japan Atomic Energy Agency (JAEA) partnered with the United States Department of Energy (US-DOE) laboratories, Los Alamos National Laboratory (LANL) and Lawrence Livermore National Laboratory (LLNL), to independently measure model ages of a low-enriched uranium certified reference material—New Brunswick Laboratory CRM U100. This interlaboratory study compares data from laboratories using different radiochemistry and analytical methods to examine the magnitude of model age reproducibility between laboratories with 231Pa/235U age dating capabilities. Results from independent measurements made by each laboratory are combined to provide the community with an interlaboratory 231Pa/235U consensus model purification date for CRM U100 that may be used by the radiochronometry community for quality control of future 231Pa/235U measurements.
Theory
Model ages reported in this study are calculated using a standard age dating equation provided as Eq. (1) below,
where t = calculated model age, λ235U and λ231Pa are the decay constants for the parent isotope 235U and the daughter decay product 231Pa respectively, and N231Pa/N235U is the measured 231Pa/235U atom ratio. The half-lives used for calculations were the following: 235U t1/2 = 7.0381 × 108 ± 4.8 × 105 years [19]; 231Pa t1/2 = 32,713 ± 110 years [20]; 233Pa t1/2 = 26.967 ± 0.002 days ([16], used by JAEA and LANL) and 26.98 ± 0.02 days (Bureau International des Poids et Mesures [21], used by LLNL).
Experimental
Sample description
New Brunswick Laboratory (NBL) certified reference material CRM U100 was chosen for this study for interlaboratory comparative age dating. The certificate for CRM U100 was originally issued by the United States National Bureau of Standards (NBS) in 1970 as standard reference material (SRM) U-100. Descriptions of the production of CRM U100 are available in NBS Special Publication 260-27 [22] and Petit [23]. Based on production documents, CRM U100 was purified between December 3, 1958 and January 8, 1959. The well-defined production history of this material provides an opportunity for testing if CRM U100 was effectively purified of 231Pa at the time of production and for testing the accuracy of the 231Pa/235U chronometer. Units of CRM U100 distributed by NBL consist of 10 mg of triuranium octoxide (U3O8) powder. The isotope abundance of CRM U100 is 10.190 ± 0.010 atom percent 235U; therefore, the material is a low-enriched uranium oxide powder. This material was chosen for this study not only for its production history, but also because it is representative of material that many countries have access to for nuclear power purposes and typifies material with the potential to be discovered out of regulatory control.
Methods
The methods used for spike production, spike calibration, sample digestion, sample purification, and analyses differed between participating laboratories. Individual laboratory methods are summarized in Table 1 and are described briefly here.
Sample digestion
All participating laboratories used a CRM U100 U3O8 powder as their starting sample material and digested the powder using hotplate digestions with HNO3 acid. At JAEA, 13 mg of CRM U100 powder was digested with 1 mL of 8 M HNO3 in a Teflon vial on a hotplate at 90°C. Once dissolved, the sample solution was diluted to produce a 4 mL approximately 3250 ppm U primary solution in 4 M HNO3 + 0.05 M HF in a Teflon vial. At LANL, 100 mg of CRM U100 powder (from SRM U-100 unit) was digested with 20 mL of 8 M HNO3 in a pre-cleaned and weighed quartz crucible on a hotplate at 80 °C with a heat lamp. The dissolved sample was transferred to a PTFE bottle and was diluted to produce a 200 mL approximately 380 ppm U primary solution in 3 M HNO3 + 0.05 M HF. Sample preparation methods at LLNL involved the digestion of CRM U100 powder in a pre-cleaned and weighed quartz crucible on a hotplate at 120 °C. The dissolved sample was transferred to a clean FEP bottle and was diluted to produce a 130 ppm U primary solution in 2 M HNO3 + 0.01 M HF.
Protactinium isotope dilution methods
For this study, participating laboratories determined the concentration of 231Pa in CRM U100 via isotope dilution with a 233Pa spike. There are no commercially available 233Pa spikes due to the short half-life of 233Pa (~ 27 days). All laboratories separated their 233Pa spike from a 237Np source wherein 237Np decays by alpha-decay to produce 233Pa. Neptunium-237 materials that have not been purified within the timeframe of a year contain 233Pa in secular equilibrium with the 237Np.
The spike produced by JAEA was purified from 0.71 mg of an Eckert and Ziegler 237Np source with greater than 99% purity (source number 1649-19). The 233Pa was purified using four ion-exchange columns. The first column consisted of a 1 mL anion exchange resin bed conditioned with 9.46 M HCl. Protactinium and U adsorb to the resin providing efficient separation from neptunium (Np). The Pa fraction was then eluted from the column using 9.46 M HCl + 0.05 M HF. The second column used the same resin and acids but consisted of a smaller 0.3 mL resin volume. The third purification was completed using silica gel conditioned in 3% HNO3. Silica gel allows for the purification of 233U (decay product of 233Pa) from Pa [10]. Protactinium was eluted from the silica gel using 3% HNO3 + 0.05 M HF. The final purification by JAEA was the same as the second anion column. During production of the 233Pa spike at JAEA, it was noted that the Eckert and Ziegler 237Np source contained 231Pa which is a complication for 231Pa assay measurements. The original 237Np was recovered during the separation of 233Pa and was allowed to decay again to ingrow new 233Pa, which resulted in a higher purity 233Pa without 231Pa contamination that was used for this work.
The spike produced at LANL was purified from 5 mg of legacy 237Np material available at LANL. Protactinium-233 was purified from 237Np using two 2 mL silica gel columns. The silica gel was pre-cleaned with 6 M HCl + 0.05 M HF, Milli-Q H2O, and 6 M HCl batch rinses to remove 232Th which forms a hydride and isobaric interference during mass spectrometry. The first 2 mL column was conditioned with 2% HNO3 and the 237Np was loaded in 2% HNO3 during which Pa sorbed to the column and an efficient purification from Np and U was possible. The Pa was eluted using 2% HNO3 + 0.1 M HF, dried, redissolved in 2% HNO3, and the column was repeated a second time. The purity of the 233Pa was evaluated using a ThermoScientific™ Element 2 ICP-MS instrument prior to use.
The spike produced at LLNL was purified from 25 mg of legacy 237Np material available at LLNL. Purification of the 233Pa was achieved using a combination of BioRad AG1-X8 anion resin and silica gel exchange columns. The first column used was a 2 mL resin volume of AG1-X8 conditioned with 10 M HCl where 233Pa was eluted using 10 M HCl + 0.05 M HF. The second column was the same as the first but used a smaller 1 mL resin volume. The 237Np material was recovered from these two initial columns for future use. The final purification was done using a 1.8 mL silica gel column conditioned with 5% HNO3. Protactinium was eluted using 5% HNO3 + 0.1 M HF. Once purified, the 233Pa spike was diluted and screened using a Nu Instruments Nu Plasma HR MC-ICP-MS to evaluate the Np:Pa separation factor and to ensure that the 233Pa spike was pure enough for use.
Calibrations of the 233Pa spikes produced at JAEA, LANL, and LLNL were done independently by all laboratories using a United States-produced 231Pa nuclear forensics reference material (231Pa NFRM [24]). The 231Pa NFRM is certified by mass and allows for accurate and precise determinations of 233Pa concentration by reverse isotope dilution [25]. Because all laboratories used the 231Pa NFRM, the results of this study will be dependent on the certification values of this reference material. Mixtures containing pg-levels of 233Pa and the 231Pa NFRM were produced by each laboratory for calibration. At JAEA, the mixtures were equilibrated and purified using anion resin prior to analysis. At LANL and LLNL, the mixtures were equilibrated and purified using silica gel prior to analysis.
After 233Pa production and spike calibration, each laboratory spiked aliquots of CRM U100 for 231Pa concentration determination. At JAEA, three separate aliquots of CRM U100 providing approximately 7.6 pg of Pa were taken and spiked with 0.3 pg of 233Pa. The spiked CRM U100 solutions were purified twice using 0.3 mL anion exchange columns (MCl GEL, CA08P, Mitsubishi Chemical Corporation). The sample solutions were dried, dissolved in 10 μL of concentrated HNO3, and prepared in 0.5 mL 9.46 M HCl + 25 μL H3BO3. The anion column was conditioned with 9.46 M HCl, the sample was loaded and washed, and Pa was eluted with 9.46 M HCl + 0.05 M HF. At LANL, six separate aliquots of CRM U100 providing 2–5 pg of Pa were taken and spiked with 2 pg of 233Pa. The spiked CRM U100 solutions were purified using a three column procedure. The first column was a 2 mL BioRad AG1-X8 column conditioned with 9 M HCl. Samples were loaded in 9 M HCl + trace H3BO3 + trace HNO3. The resin was washed with 9 M HCl and Pa was eluted with 9 M HCl + 0.05 M HF. The samples were dried and reconstituted in 2% HNO3 + trace H3BO3 and were loaded onto a 2 mL silica gel column conditioned with 2% HNO3. The resin was washed with 2% HNO3 and Pa was eluted with 2% HNO3 + 0.05 M HF. The samples were dried and reconstituted again in 2% HNO3 + trace H3BO3 for the final third column. The final column was the same as the second column; however, this column purification was conducted immediately prior to analysis to remove ingrown 233U isobaric interferences. Following purification, the eluted Pa in 2% HNO3 + 0.05 M HF was analyzed immediately by MC-ICP-MS. At LLNL, three separate aliquots of CRM U100 providing approximately 4 pg of Pa were taken and spiked with 2 pg of 233Pa. Protactinium was purified from the bulk U matrix using a three column procedure. The first column consisted of a 1 mL BioRad AG1-X8 resin bed. Samples were dried and dissolved in 9 M HCl + trace H3BO3 + trace HNO3 and loaded onto the column. Protactinium was eluted with 9 M HCl + 0.05 M HF. Samples were dried and prepared for the second column which was a repeat of the first column. The final column used for purification was a 1 mL silica gel column conditioned with 5% HNO3. The sample was loaded onto the silica gel with 5% HNO3 and Pa was eluted using 2% HNO3 + 0.05 M HF. Similar to procedures used by LANL, the Pa fractions were immediately analyzed by MC-ICP-MS prior to ingrowth of 233U from 233Pa decay.
Uranium isotope dilution and isotope composition methods
All participating laboratories determined 235U concentrations in CRM U100 through isotope dilution mass spectrometry (IDMS) with a 233U spike. Each laboratory used a commercially available certified reference material to calibrate the concentration of their individual 233U spike. At JAEA, an in-house 233U sspike was calibrated with a high-purity uranium metal standard certified by the Japan Atomic Energy Research Institute, JAERI-U4. At both US-DOE laboratories (LANL and LLNL), in-house 233U spikes were also calibrated using a high-purity uranium metal—National Bureau of Standards Standard Reference Material 960 (SRM 960). In order to take sample aliquots for U assay determination, all laboratories made gravimetrically prepared serial dilutions of their primary CRM U100 solutions. At JAEA, two serial dilutions of the primary solution were made and three aliquots containing 200 ng of total U were removed for assay measurements. At LANL, two serial dilutions of the primary solution were also made and three aliquots containing 2 ng of total U were removed for assay measurements. At LLNL, one dilution of the primary solution was made and three aliquots containing 75 ng of total U were removed for assay measurements. All laboratories also took aliquots of CRM U100 for U isotope composition determination. Aliquot sizes for U isotope composition were approximately 200 ng, 50 ng, and 50 ng of total U at JAEA, LANL, and LLNL respectively.
The uranium fractions taken by each laboratory were purified prior to analysis by mass spectrometry using Eichrom UTEVA resin. At JAEA, a 0.3 mL UTEVA resin bed was used, samples were loaded in 3 M HNO3, and U was eluted with 0.5 M HCl. At LANL, a 1 mL UTEVA resin bed was used, samples were loaded in 3 M HNO3, and U was eluted with 0.1 M HCl. Finally, LLNL utilized a 1 mL UTEVA column, samples were loaded in 4 M HNO3, and U was eluted with 0.1 M HCl. At LLNL, only traced IDMS U aliquots were purified prior to analysis, and U isotope concentration aliquots were analyzed without prior purification due to the high-purity of CRM U100.
Mass spectrometry methods
The mass spectrometry methods used to analyze U and Pa differed between all laboratories. At JAEA, U and Pa measurements were made using a Thermo Scientific™ Triton Plus Multicollector Thermal Ionization Mass Spectrometer (TIMS). Uranium was measured by JAEA using a total evaporation method with each isotope measured on Faraday collectors. Protactinium was measured in a peak-jumping mode on the secondary electron multiplier (SEM) equipped with a retarding potential quadrupole lens (RPQ) using four second integrations. Mass bias corrections for JAEA measurements were made using NBL CRM U050. Gain calibrations were performed prior to analysis and blank subtractions were made to Pa measurements using the process blank generated from chemical separation of Pa. The process blank represented 0.03% of the CRM U100 sample.
At LANL, U and Pa measurements were made using a Thermo Scientific™ Neptune Plus Multicollector Inductively-Coupled Plasma Mass Spectrometer (MC-ICP-MS). Uranium IDMS measurements were made using a static routine with 233U, 235U, and 238U measured on Faraday collectors using eight second integrations. Uranium isotope composition measurements were made using a static routine with 235U and 238U on Faraday detectors and 234U and 236U on SEMs with RPQs using four second integrations. Certified reference materials IRMM 074/1 and NBL CRM U200 were used as mass bias correction standards for assay and isotope composition measurements respectively, and IRMM 074/2 and NBL CRM U050 were used for quality control. Protactinium measurements at LANL were made using static multicollection with 231Pa and 233Pa measured on SEMs. A U standard, NBL CRM U010 was used for mass bias corrections and NBL CRM U005-A was used for quality control. All data were corrected for mass bias, peak tailing, acid blank contributions, instrument background, Faraday-ion counting gain corrections, and hydride interferences (235U + 1H on 236U and 232Th + 1H on 233Pa).
At LLNL, U and Pa measurements were made using a Nu Plasma HR MC-ICP-MS. Uranium IDMS measurements were made using a static routine with 233U, 235U, and 238U measured on Faraday collectors. Uranium isotope composition measurements were made using a static routine with 235U and 238U on Faraday detectors and 233U, 234U, and 236U on ion counters. Mass bias corrections for all measurements were made with NBL CRM U010, and NBL CRMs U005-A, 129-A, and 112-A were used for quality control. Protactinium measurements were made using static multi-collection with 231Pa and 233Pa on ion counters. Mass bias corrections for Pa measurements were made using U standard CRM U010 and quality control was done using CRM U005-A. All measurements were corrected for mass bias, peak tailing, Faraday-ion counting gain corrections, and acid blank contributions.
Results and discussion
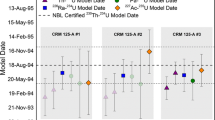
Individual CRM U100 231Pa/235U ratios measured by each laboratory and associated model purification dates are reported in Table 2 and are shown graphically in Fig. 1. The average 231Pa/235U model purification dates measured by JAEA, LANL, and LLNL for CRM U100 were December 15, 1958 ± 1106 days, June 12, 1959 ± 487 days, and January 28, 1959 ± 228 days respectively (Fig. 1). Given the small number of replicate measurements made by each laboratory (n = 3 to 6), the 95% (k = 2) external uncertainties provided for the average model purification date for each laboratory were calculated using the following:
where Nr is the number of replicates, t(1−α/2) is the 100(1 − α/2)th percentile of the t-distribution corresponding to a probability α = 0.05 and v = Nr − 1 degrees of freedom. This calculation provides the uncertainty of the mean of the replicates at the 95% confidence level. These average model purification dates are consistent within analytical uncertainty between laboratories. The model 231Pa/235U purification dates reported by participating laboratories are also consistent with the known production history of CRM U100, which according to production documents, was purified between December 3, 1958 and January 8, 1959 [23] (solid and dashed lines in Fig. 1). The model purification dates measured in this study also agree with prior published measurements of 230Th/234U and 231Pa/235U model ages for CRM U100 [11, 12, 25]. Model ages measured by JAEA and LANL display the largest internal laboratory variation with model purification dates ranging between 1958 and 1960. Measurements made by LLNL were more internally consistent with model purification dates ranging between October 1958 and April 18, 1959. The observed agreement between 231Pa/235U model ages reported from three laboratories using different chemical purification and mass spectrometry methods demonstrates that the methods used by participating laboratories are valid for Pa-U age dating of bulk U materials. These results suggest that laboratories interested in Pa-U age dating of bulk U materials can make 231Pa/235U measurements using a variety of different spikes, resins, certified reference materials and instrumentation.

Interlaboratory model purification date results for CRM U100. Individual measurements from each laboratory are shown as blue squares (JAEA), yellow circles (LANL), and green triangles). The calculated average model purification date from each laboratory is shown using the same symbol with a bold outline and is denoted with the letter ‘A’. The consensus model purification date of March 26, 1959 ± 237 days calculated from the average of all individual measurements (n = 12) is represented with a bold green line. (Color figure online)
Assuming that the interlaboratory variation in measured 231Pa/235U atom ratios for CRM U100 is representative of variation that might occur between forensic laboratories, one can calculate an average consensus 231Pa/235U model purification date of CRM U100. The consensus model purification date was calculated by taking the average of all twelve independent measurements reported by JAEA, LANL, and LLNL (Table 2 model purification dates). The expanded uncertainty on the consensus value was calculated using Eq. (2). The calculated consensus model purification date of CRM U100 based on data from this study is March 26, 1959 ± 237 days. The calculated 237 day expanded uncertainty on the consensus date is assumed to be representative of expected interlaboratory variation during Pa-U age dating by experienced radiochronometry laboratories. This consensus purification date agrees within analytical uncertainty with the full production history of CRM U100 [23], but is approximately 2 months younger than the last date of purification from production records. Given the lack of certified reference materials that are certified for 231Pa/235U radiochronometry, the consensus 231Pa/235U model purification date of CRM U100 from this study may be used for quality control of future 231Pa/235U measurements of bulk low-enriched U materials.
References
Kristo MJ, Gaffney AM, Marks N, Knight K, Cassata WS, Hutcheon ID (2016) Nuclear forensic science: analysis of nuclear material out of regulatory control. Ann Rev Earth Planet Sci 44:555–579
Wallenius M, Morgenstern A, Apostolidis C, Mayer K (2002) Determination of the age of highly enriched uranium. Anal Bioanal Chem 374(3):379–384
LaMont SP, Hall G (2005) Uranium age determination by measuring the 230Th/234U ratio. J Radioanal Nucl Chem 264(2):423–427
Varga Z, Suranyi G (2007) Production date determination of uranium-oxide materials by inductively coupled plasma mass spectrometry. Anal Chim Acta 599:16–23
Varga Z, Wallenius M, Mayer K (2010) Age determination of uranium samples by inductively coupled plasma mass spectrometry using direct measurement and spectral deconvolution. J Anal At Spectrom 25:1958–1962
Williams RW, Gaffney AM (2011) 230Th–234U model ages of some uranium standard reference materials. Proc Radiochim Acta 1:31–35
Pointurier F, Hubert A, Roger G (2013) A method for dating small amounts of uranium. J Radioanal Nucl Chem 296:593–598
Gaffney AM, Hubert A, Kinman WS, Magara M, Okubo A (2015) Round-robin 230Th–234U age dating of bulk uranium for nuclear forensics. J Radioanal Nucl Chem 235:129–132
Treinen KC, Kinman WS, Chen Y, Zhu L, Cardon AMR, Steiner RE, Kayzar-Boggs TM, Williams RW, Zhao YG (2017) US-DOE and CIAE international cooperation in age-dating uranium standards. J Radioanal Nucl Chem 314(3):2469–2474
Morgenstern A, Apostolidis C, Mayer K (2002) Age determination of highly enriched uranium: separation and analysis of 231Pa. Anal Chem 74:5513–5516
Eppich GR, Williams RW, Gaffney AM, Schorzman KC (2013) 235U–231Pa age dating of uranium materials for nuclear forensic investigations. J Anal At Spectrom 28:666–674
Kayzar TM, Williams RW (2016) Developing Ra-226 and Ac-227 age-dating techniques for nuclear forensics to gain insight from concordant and non-concordant radiochronometers. J Radioanal Nucl Chem 307(3):2061–2068
Rolison JM, Treinen KC, McHugh KC, Gaffney AM, Williams RW (2017) Application of the 226Ra–230Th–234U and 227Ac–231Pa–235U radiochronometers to uranium certified reference materials. J Radioanal Nucl Chem 314:2459–2467
Higginson M, Gilligan C, Taylor F, Knight D, Kaye P, Shaw T, Thompson P (2018) Development of rapid methodologies for uranium age dating. J Radioanal Nucl Chem 318:157–164
Varga Z, Nicholl A, Hrnecek E, Wallenius M, Mayer K (2018) Measurement of the 231Pa/235U ratio for the age determination of uranium materials. J Radioanal Nucl Chem 318(3):1565–1571
Jones RT, Merritt JS, Okazaki A (1986) A measurement of the thermal neutron capture cross section of 232Th. Nucl Sci Eng 93:171–180
Usman K, MacMahon TD (2000) Determination of the half-life of 233Pa. App Radiat Isotopes 52:585–589
Treinen KC, Samperton KM, Lindvall RE, Wimpenny JB, Gaffney AM, Bavio M, Baransky EJ, Williams RW (2019) Evaluating uranium radiochronometry by single-collector mass spectrometry for nuclear forensics: a multi-instrument investigation. J Radioanal Nucl Chem. https://doi.org/10.1007/s10967-019-06832-y
Jaffey AH, Flynn KF, Glendenin LE, Bentley WC, Essling AM (1971) Precision measurement of half-lives and specific activities of 235U and 238U. Phys Rev C 4:1889
Robert J, Miranda CF, Muxart R (1969) Mesure de la periode du protactinium-231 par microcalorimetrie. Radiochim Acta 11(2):104–108
Bureau International des Poids et Mesures (BIPM) (2010) Monograph 5: table of radionuclides, 5-A: 22:244. 465. http://www.nucleide.org/DDEP_WG/DDEPdata.htm. Accessed 11 March 2019
National Bureau of Standards Special Publication 260-27 (1971) Standard reference materials: Uranium isotopic standard reference materials. US Department of Commerce, US Government Printing Office, Washington DC
Petit GS (1960) Preparation of uranium isotopic standards for the National Bureau of Standards. Report Number KL-8 Addendum-18. Union Carbide Nuclear Company, Oak Ridge Tennessee
Essex RM, Williams RW, Treinen KC, Colle R, Fitzgerald R, Galea R, Keightley J, LaRosa J, Laureano-Perez L, Nour S, Pibida L (2019) Preparation and calibration of a 231Pa reference material. J Radioanal Nucl Chem. https://doi.org/10.1007/s10967-019-06711-6
Treinen KC, Gaffney AM, Rolison JM, Samperton KM, McHugh KC, Miller ML, Williams RW (2018) Improved protactinium spike calibration method applied to 231Pa–235U age-dating of certified reference materials for nuclear forensics. J Radioanal Nucl Chem 318(1):209–219
Funding
Funding was provided by U.S. Department of Energy.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Kayzar-Boggs, T.M., Treinen, K.C., Okubo, A. et al. An interlaboratory collaboration to determine consensus 231Pa/235U model ages of a uranium certified reference material for nuclear forensics. J Radioanal Nucl Chem 323, 1189–1195 (2020). https://doi.org/10.1007/s10967-020-07030-x
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10967-020-07030-x