
Abstract
Surface carbohydrate-containing molecules, such as glycoproteins and glycolipids, have been shown to play crucial regulatory roles in the normal physiological process as well as in pathological conditions including tumor progression. Those glycans which are overexpressed on the surface of tumor cells, but not detected or only weakly expressed in some limited normal tissues, are designated as tumor-associated carbohydrate antigens (TACAs). These TACAs may serve as potential targets for immunotherapy. The biological functions of TACAs and therapeutic strategies against TACAs will be addressed in this review.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
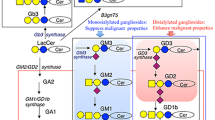
Glycosylation is an important posttranslational modification process to produce diverse glycans that are frequently attached to proteins and lipids. These glycoconjugates play a key role in cells, including receptor activation, cell adhesion, signal transduction, endocytosis, molecular trafficking, and clearance (Ohtsubo and Marth 2006). Altered glycosylation on glycoproteins and glycolipids is a prominent feature of cancer cells (Reis et al. 2010). These abnormal glycoconjugates are involved in tumor proliferation, invasion, angiogenesis, and metastasis. Patient with altered glycoconjugates in tumor tissue usually has poor prognosis (Miyake et al. 1992). Changes in glycosylation, including over-, under-, and neo-expression of sugar moieties, might result from the upregulation/downregulation of some glycosyltransferases and glycosidases. Increased N-glycosylation, such as 1,6-branched N-glycans, was observed in breast cancer and colon cancer (Dennis et al. 1987; Fernandes et al. 1991; Seelentag et al. 1998; Murata et al. 2004), which was mediated by GnT-V. On the other hand, O-glycosylation is often reduced resulting in the accumulation of core 1-based O-glycan during the tumorigenesis. The most common O-glycan epitopes were TF (Galβ1, 3GalNAc), Tn (GalNAc), Lewisx, Lewisa, and their sialylated counterparts (Springer 1984; Yuan et al. 1986; Itzkowitz et al. 1989; Itzkowitz et al. 1986; Tozawa et al. 2005). They were reported to enhance the intravasation of cancer cells, binding of circulating cancer cells to endothelium, and extravasation and colonization at the distant sites (Rosen and Bertozzi 1994; Borsig et al. 2001, 2002). For instance, increased expression of sialyl-Lex (sLex) and sialyl-Lea (sLea) was shown to assist the invasion and metastasis of tumor (Hoff et al. 1989; Kannagi 1997) and was associated with poor survival of patients (Makino et al. 2001). So far, a long list of TACAs has been identified, such as Tn, sialyl-Tn, TF, Lewisy, sialyl Lewisx, sialyl Lewisa, Lewisx, Globo H, stage-specific embryonic antigen-3 (SSEA-3), GD2, GD3, GM2, fucosyl GM1, Neu5Gc GM3, and polysialic acid. Some of these have been exploited as targets for immunotherapy of cancers.
Conventional treatments of cancer, including radiation, surgery, and chemotherapy, are not cancer-specific. Immunotherapy, on the other hand, provides a strategy to target specific cancer cells. Cancer immunotherapy can be categorized into active and passive immunotherapy. An active cancer immunotherapy is to activate the immune system of patients to attack cancer cells, which can trigger immunological memory. On the other hand, passive immunotherapy is to deliver tumor antigen-specific monoclonal antibodies to kill cancer cells through complement-dependent cytotoxicity (CDC) or antibody-dependent cell-mediated cytotoxicity (ADCC). Since the initial approval of anti-CD20 (rituximab) for the treatment of lymphoma in 1994, more than 10 monoclonal antibodies have been approved for passive immunotherapy of cancer and all of them target protein antigens. On the other hand, the first and only approved active immunotherapy is sipuleucel-T (Provenge, Dendreon), for the treatment of metastatic prostate cancer (Kantoff et al. 2010). Sipuleucel-T is an autologous cellular vaccine activated ex vivo by recombinant prostate acid phosphatase (PAP) fused to GM-CSF. Although a number of clinical trials of cancer immunotherapy targeting TACAs have been conducted over the past two decades, majority of the trials did not proceed beyond early phase I/II studies. Only three TACAs have reached clinical phase III development: sialyl-Tn, GM2, and GD2. Unfortunately, randomized phase III clinical trials of sialyl-Tn-KLH vaccine (Theratope) in metastatic breast cancer (Miles et al. 2011a) and GM2-KLH vaccine in melanoma (Eggermont et al. 2013) failed to demonstrate any benefit of the vaccine, although subsequent subgroup analysis did demonstrate survival benefit of Theratope in metastatic breast cancer patients on endocrine therapy (Ibrahim et al. 2013). On the other hand, passive immunotherapy with dinutuximab, a chimeric anti-GD2, has demonstrated a significant improvement in event-free survival and overall survival in patients with high-risk neuroblastoma (Yu et al. 2010) which led to regulatory approval in the USA and Europe in 2015. Thus, GD2 is the first TACA proven to be an effective target antigen for cancer immunotherapy.
The approval of ipilimumab (anti-CTLA-4) for the treatment of melanoma in 2011 as the first monoclonal antibody targeting an immune checkpoint molecule (Hodi et al. 2010) heralded a new era of cancer immunotherapy. The success of ipilimumab was closely followed by the development of additional immune checkpoint inhibitors, including nivolumab (Robert et al. 2015) and pembrolizumab (Robert et al. 2014), which target PD-1 and ensure the emergence of many more immune checkpoint blockers on the horizon. Such breakthroughs beg the question whether TACAs may act as immune checkpoint molecules. Indeed, several cancer-associated gangliosides were shown to inhibit immune cell responses, including antigen processing and presentation (Peguet-Navarro et al. 2003), T-cell proliferation (Biswas et al. 2009; Chu and Sharom 1993; Morioka et al. 1991), and cytokine production, such as IFN-γ and IL-4 (Biswas et al. 2006; Irani et al. 1996). Purified gangliosides from cancer cells displayed immunosuppressive activities which aided cancers to escape from host immune surveillance (Ladisch et al. 1992; Wolfl et al. 2002), which were mediated by hampering the interaction of IL-2 with its receptor (Lu and Sharom 1996), inducing apoptotic cell death (Das et al. 2008) and deviation toward Th2 response (Crespo et al. 2006). Such ganglioside-induced T-cell dysfunction involved NF-kappa B inhibition (Uzzo et al. 1999) through degradation of RelA and p50 proteins (Thornton et al. 2004). In contrast, there are relatively few studies on the functions of Globo-series TACAs. The core structure of Globo-series glycosphingolipids is Galα1,4-Galβ1,4Glc-ceramide (Gb3), which is catalyzed by α1,4-galactosyltransferase (A4galt) through the transfer of a galactose to lactosylceramide (Kojima et al. 2000). Gb3 has been found in Burkitt lymphoma (Wiels et al. 1981) and germ cell-derived tumors (Murray et al. 1985) and also in a subpopulation of B cells in germinal centers (Klein et al. 1983) and on kidney proximal tubules and intestinal epithelial cells (Fujii et al. 2005). In addition, Gb3 could be induced on the surface of human monocytes by LPS (van Setten et al. 1996) or on endothelial cells by interleukin-1 and tumor necrosis factor-alpha (van de Kar et al. 1992). Besides, Gb3 could serve as a receptor on endothelial cells for verotoxins produced by Escherichia coli O157 (Jacewicz et al. 1986), which was further confirmed by increased sensitivity to LPS-induced lethal shock in A4galt knockout mice (Okuda et al. 2006) (Kondo et al. 2013). Intriguingly, in wild-type mice, injection of LPS did not increase the expression of Gb3 on the surface of endothelial cells although it induced the expression of A4galt RNA and globotetraosylceramide (Gb4), one of the Globo-series glycolipids generated by b1,3-N-acetylgalactosaminyltransferase (B3galnt1) through the transfer of galactosamine to Gb3, suggesting that Gb4, but not Gb3, might play a role in LPS-induced lethal shock. Indeed, administration of Gb4 increased the survival rate of mice injected with LPS. The protective effect of Gb4 on LPS-challenged mice was mediated by the binding of Gb4 to the complex of toll-like receptor-4 and myeloid differentiation factor 2 on the endothelia, thereby interfering with the binding of LPS to this complex (Kondo et al. 2013). However, the effects of Globo-series glycolipids on immune cells have remained unclear until Tsai et al. reported the immunosuppressive activity of Globo H ceramide (Tsai et al. 2013). Globo H ceramides released from the surface of tumor cells were taken up by T and B lymphocytes, with ensuing inhibition of activation of lymphocytes. Tumor-infiltrating lymphocytes in close proximity to the Globo H-expressing tumor cells showed positive staining of anti-Globo H antibody by IHC, consistent with the in vitro observation of uptake of Globo H ceramide released from tumor cells by lymphocytes. Treatment of lymphoid cells with Globo H ceramide did not induce apoptosis nor expand regulatory T cells. The molecular mechanisms of Globo H ceramide induced immunosuppression involved upregulation of id3 and itch via upregulation of egr2/3, leading to diminished expression of Notch, which is crucial for T-cell activation (Palaga et al. 2003). These results provide the first evidence that Globo H ceramide acts as an immune checkpoint molecule to facilitate the escape of cancer cells from immune surveillance.
In addition to the function of TACAs as immune checkpoints, several gangliosides have been reported to exhibit angiogenic activities. Tumor cells with GM2 synthase/GM3 synthase deficiency formed avascular tumor on mice (Liu et al. 2014a), whereas upregulation of GM1, GM2, and GD1a enhanced blood vessel density in tumors (Manfredi et al. 1999). On the other hand, GM3 blocked the dimerization of vascular endothelial growth factor receptor 2 (VEGFR2) to inhibit the signaling transduced by VEGF (Chung et al. 2009). The effects of Globo-series TACA on angiogenesis were first addressed in a report showing angiogenic activity of Globo H ceramide (Cheng et al. 2014). Globo H ceramide induced tube formation of endothelial cell in vitro and angiogenesis in vivo. When Globo H-positive tumor cells were sorted into two subpopulations based on Globo H expression, the Globo Hhi tumor cells grew faster with greater vessel density than Globo Hlow tumor cells in vivo. Consistent with this was the observation of higher vessel density in Globo H+ than Globo H− breast cancer specimens. Mechanistic investigations linked the angiogenic effects of Globo H ceramide to its endocytosis and binding to TRAX, with consequent release of PLCβ1 from TRAX to trigger Ca2+ mobilization. This is the first globoside shown to display angiogenic activity, along with elucidation of its mechanisms. On the other hand, Globo H has been identified as one of the glycans that bind human RNase 1, facilitating the internalization of the RNase 1 which induced cell death. Blocking the interaction of Globo H and RNase 1 with anti-Globo H antibody partially rescued the cells from RNase-induced cell lysis (Eller et al. 2015). These findings suggested multifaceted roles of Globo H in tumor biology.
The findings of certain TACAs acting as immune checkpoint molecules and angiogenic factors further strengthen the scientific rationales for immunotherapy targeting TACAs. The following sections will address various strategies for developing TACA-targeted cancer immunotherapies.
2 Disialoganglioside (GD2)-Targeted Cancer Immunotherapies
Disialoganglioside (GD2), a b-series ganglioside, is a sialic acid-containing surface glycolipid that generated from precursor GM2 by GD3 synthase and GD2 synthase. It is expressed by neuroblastoma (>98 %), melanoma, glioma, small-cell lung cancer, sarcomas (Schulz et al. 1984; Cheung et al. 1987), breast cancer stem cell (Liang et al. 2013; Battula et al. 2012), as well as some normal neuroectodermal (Yanagisawa et al. 2011), and mesenchymal stem cells (MSCs) (Martinez et al. 2007; Jin et al. 2010). GD2 plays an important role in the proliferation and invasiveness of tumor cells (Yoshida et al. 2001; Shibuya et al. 2012). It could directly induce activation of the proto-oncogene c-Met to enhance proliferation of triple-negative breast cancer cells (Cazet et al. 2012). It upregulates integrin α2β1-mediated tyrosine phosphorylation of p125FAK, which enhances platelet adhesion to extracellular matrix collagen, thereby promoting metastasis of neuroblastoma cells (Chen et al. 2013). Furthermore, GD2+ murine bone marrow MSCs (mBM-MSC) possessed not only much greater clonogenic and proliferative capabilities but also stronger differentiation potential to adipocytes and osteoblasts, as compared to unsorted mBM-MSCs (Xu et al. 2013). Moreover, in human osteosarcoma cell lines, a murine anti-GD2 antibody, mAb 14G2a, effectively inhibits cell invasiveness, MMP-2 activity, and cell viability (Liu et al. 2014b). On the other hand, in human tissues, only weak expression of GD2 is observed in neurons, skin melanocytes, and peripheral pain fibers (Svennerholm et al. 1994). Therefore, GD2 is an ideal glycan antigen target for immunotherapy. Three immunotherapeutic strategies have been developed so far, including GD2-specific monoclonal antibodies, GD2-specific chimeric antigen receptor T cells, and GD2 vaccines.
2.1 GD2-Specific Monoclonal Antibodies
2.1.1 3F8
3F8 is a murine IgG3 monoclonal antibody which binds to GD2-expressing tumor cells and mediates cytotoxicity by activating human complement system (Cheung et al. 1985). 131I-labeled 3F8 has been used for neuroblastoma imaging (Miraldi et al. 1986) and shown to eradicate human NB xenografts (Cheung et al. 1986). A phase I clinical trial of 3F8 was conducted in 17 patients with relapsed or refractory neuroblastoma. Significant toxicities including neuropathic pain, tachycardia, hypotension, hypertension, fever, and urticaria were observed. Antitumor activities were noted in some patients, ranging from complete clinical remissions to mixed responses. All patients developed human anti-mouse antibodies (HAMA) to 3F8. A phase II study in 16 patients with stage 4 neuroblastoma showed clinical responses in bony lesions and marrow diseases (Cheung et al. 1998a). Subsequently, the effect of 3F8 on minimal residual disease of stage 4 neuroblastoma was evaluated in 34 patients in first or subsequent response, and 13/34 patients remained progression-free for 53–143 months (Cheung et al. 2000). A series of sequential phase II studies in 139 patients showed an overall 5-year EFS of 62 % for stage 4 patients in first remission who received 3F8 + GM-CSF + cis-retinoic acid (Cheung et al. 2012) and a correlation of a better outcome for patients with the FCGR2A (R/R) genotype which favored the binding of the IgG3 antibody (Cheung et al. 2006). Moreover, a better survival correlated with a transient anti-mouse response or completion of 4 cycles of 3F8 treatment (Cheung et al. 1998b). Humanized 3F8 has been generated along with the generation of hu3F8/IL-2 and hu3F8/GM-CSF, which are undergoing phase I clinical trials in patients with high-risk neuroblastoma: hu3F8 (NCT01419834) and hu3F8/IL-2 (NCT01662804 and NCT01757626).
2.1.2 14G2a
MAb 14G2a was generated from murine 14.18 IgG3 anti-GD2 by class switch to IgG2a antibody (Mujoo et al. 1987, 1989). Three phase I trials of mAb 14G2a were conducted in patients with melanoma, neuroblastoma, and osteosarcoma. The dose of mAb 14G2a was escalated up to 500 mg/m2/course, with significant dose and infusion rate-dependent toxicities, including pain, tachycardia, hypotension, hypertension, fever, hyponatremia, and urticaria (Uttenreuther-Fischer et al. 1995a; Murray et al. 1994; Saleh et al. 1992a). Pain was thought to be due to binding of antibody to peripheral nerve fibers expressing GD2 (Svennerholm et al. 1994). Clinical benefits were observed in some patients even in these early phase trials. To enhance the ADCC effect, 14G2a was combined with IL-2 and a maximum tolerated dose (MTD) of 14G2a plus IL-2 was 15 mg/m2/day. Similar side effects were observed although IL-2 might have contributed to some of the toxicities, such as fever. One patient with neuroblastoma had a partial response (PR), and one patient with osteosarcoma had a complete response (CR) (Frost et al. 1997).
2.1.3 Ch14.18
A human-mouse chimeric anti-GD2 monoclonal antibody, ch14.18, was constructed by combining the variable regions of 14G2a and the constant regions of human IgG1-k (Gillies et al. 1989). MAb ch14.18 could activate complement system (Zeng et al. 2005) and mediate ADCC through neutrophils, natural killer (NK) cells, and lymphokine-activated killer (LAK) cells (Barker et al. 1991) with an efficiency 50–100 times greater than the murine mAb 14G2a (Mueller et al. 1990). Investigational New Drug (IND) application for ch14.18 was filed in 1989, marking the first IND application for mAb generated by recombinant DNA technology. Two phase I clinical trials of ch14.18 in relapsed/refractory neuroblastoma revealed similar toxicity profile as 14G2a (Yu et al. 1998; Handgretinger et al. 1995). As expected, the half-life of ch14.18 was longer than 14G2a, with a beta t 1/2 of 66.6 ± 27.4 h for ch14.18 and 18.3 ± 11.8 h for 14G2a (Handgretinger et al. 1995; Uttenreuther-Fischer et al. 1995b). Among a total of 19 neuroblastoma patients, 2 CR and 3 PR were observed, although another phase I trial in 13 adult patients with metastatic melanoma showed no clinical responses (Saleh et al. 1992b). Based on the in vitro findings that GM-CSF not only raised the number of leukocytes but also enhanced their anti-GD2-mediated ADCC (Barker et al. 1991), a pilot study of ch14.18 + GM-CSF was conducted, which showed 5 CRs and 3 stable diseases (SDs) in 17 refractory/recurrent neuroblastoma (Yu et al. 1995). This was subsequently confirmed by a phase II Pediatric Oncology Group study showing 2 CR, 2 PR, and 1 mixed response in 32 neuroblastoma patients (Yu et al. 1997). In these early phase trials, most clinical responses occurred in patients with small disease burden, esp. bone marrow metastasis. Thus, anti-GD2 immunotherapy was subsequently developed to target neuroblastoma in the setting of minimal residual disease (MRD). The feasibility of administering ch14.18 in combination with GM-CSF, IL-2, and isotretinoin after high-dose chemotherapy and stem cell transplant period was demonstrated in 2 pilot phase I studies, and the maximum tolerated dose (MTD) of ch14.18 in combination with cytokines was 25 mg/m2/d for 4 days (Gilman et al. 2009; Ozkaynak et al. 2000). These studies paved the way for the pivotal phase III randomized clinical trial of ch14.18 + IL-2/GM-CSF. Patients with high-risk neuroblastoma who achieved at least PR to induction therapy and received stem cell transplantation and posttransplant radiotherapy were randomly assigned, in a 1:1 ratio, to receive standard therapy with six cycles of isotretinoin or immunotherapy with six cycles of isotretinoin and five concomitant cycles of ch14.18 in combination with alternating GM-CSF and IL-2. Randomization was stopped early because interim analysis of 226 eligible patients revealed a significant 2-year overall survival (86 ± 4 % versus 75 ± 5 %, p = 0.02 without adjustment for interim analyses) and event-free survival (66 ± 5 % versus 46 ± 5 % at 2 years, p = 0.01) advantage for 113 patients receiving immunotherapy versus those 113 receiving standard therapy (ClinicalTrials.gov number NCT00026312) (Yu et al. 2010). This major breakthrough has now been considered as a standard treatment for high-risk neuroblastoma. It also marks the first successful immunotherapy to target a nonprotein antigen.
2.1.4 Hu14.18K322A
Ch14.18 was further humanized by CDR grafting of 14.18 V regions to generate humanized 14.18 (hu14.18) antibody (Metelitsa et al. 2002). Since anti-GD2-induced neuropathic pain is complement-dependent, a K322A mutation of the C region of IgG1 in hu14.18 was made to limit the ability of complement fixation of hu14.18. Preclinical studies in rats confirmed that hu14.18K322A elicited significantly less allodynia than ch14.18 while maintaining its ADCC activity (Sorkin et al. 2010). A phase I clinical trial of hu14.18K322A in 38 neuroblastoma showed the MTD, and recommended phase II dose, of hu14.18K322A to be 60 mg/m2 per day for 4 days (Navid et al. 2014). Adverse effects, predominately pain, were manageable and improved with subsequent courses. Median hu14.18K322A α (initial phase) and β (terminal phase) half-lives were 1.74 and 21.1 days, respectively. Objective responses (four complete responses; two partial responses) were noted in 6 of 31 patients evaluable for response by iodine-123 metaiodobenzylguanidine score. Several early phase trials in patients with GD2+ tumors are in progress (ClinicalTrials.gov numbers NCT01576692 and NCT00743496).
2.1.5 Hu14.18-IL-2
Another strategy to enhance the antitumor efficacy of an antibody is to link the antibody with cytokine to generate immunocytokine fusion proteins that accumulate high cytokine concentrations in the tumor microenvironment and thereby stimulate cellular immune responses against cancer cell.
Hu14.18-IL-2 is a fusion protein of hu14.18 and IL-2 (Neal et al. 2004a, b). A phase I trial of hu14.18-IL-2 in recurrent/refractory neuroblastoma (n = 27) and melanoma (n = 1) patients showed the MTD to be 12 mg/m2/day, with similar toxicities as anti-GD2 combined with IL-2. No measurable CRs or PRs to hu14.18-IL-2 were observed; however, evidence of antitumor activity was noted in three neuroblastoma patients (Osenga et al. 2006). A phase II study showed 5 CR in 23 neuroblastoma patients with evaluable disease only by MIBG and/or bone marrow histology, but no responses for patients with measurable disease (Shusterman et al. 2010). In this study, patients with KIR-ligand mismatch seemed to be associated with better clinical response (Delgado et al. 2010). Another phase I trial of hu14.18-IL-2 in adults with melanoma (n = 33) showed MTD to be 7.5 mg/m2/day, with the dose-limiting toxicities of hypoxia, hypotension, and elevations of AST and ALT, which were reversible (King et al. 2004). Subsequently, a phase II study was conducted in metastatic melanoma patients (n = 14) who received hu14.18-IL-2 at 6 mg/m2/day as 4-h intravenous infusions on days 1, 2, and 3 of each 28-day cycle. All patients received 2 cycles of treatment, and one patient had a PR (7.1 %, 1/14) and 4 patients had SD (28.5 %, 4/14). The toxicities were reversible, including grade 3 hypotension (n = 2) and grade 2 renal insufficiency with oliguria (n = 1). The accrual was held due to limited availability of hu14.18-IL-2 (Albertini et al. 2012).
2.1.6 Anti-O-Acetyl GD2 Monoclonal Antibody 8B6
Although the therapeutic efficacy of anti-GD2 has been well-documented, neuropathic pain can limit its application. O-acetyl GD2 is an analog of GD2 with an acetyl group linked to oxygen at the 9 position of NeuAc. An O-acetyl GD2-specific antibody 8B6 was shown to bind to neuroblastoma and other neuroectodermal tumors, but not peripheral pain fibers (Alvarez-Rueda et al. 2011). Thus, antibodies against O-acetyl GD2 may have the advantage over anti-GD2 which is dose-limited by neuropathic pain. Indeed, a mouse-human chimeric antibody c.8B6 was reported to display potent antitumor activity without inducing allodynia in preclinical studies (Terme et al. 2014). Clinical trials of mAbc.8B6 are eagerly awaited.
2.1.7 Bispecific Antibody
Bispecific antibody which binds to two different types of antigen by combining fragments of two different monoclonal antibodies is an attractive alternative to immunocytokine. A GD2-targeting bispecific antibody 3F8 × CD3 (3F8BiAb) has been developed. It could redirect activated T cells to GD2-expressing murine neuroblastoma (Yankelevich et al. 2012). A phase I/II clinical trial of 3F8BiAb in children and young adults with neuroblastoma and osteosarcoma is under development (NCT02173093).
2.2 GD2 Chimeric Antigen Receptor
T lymphocytes can be engineered to express chimeric antigen receptors (CARs), which can bind to tumor antigens, leading to antitumor activity in an MHC-independent manner. CARs are generated by joining a single-chain variable fragment (scFv) of monoclonal antibody with the transmembrane and cytoplasmic portions of T-cell receptor (TCR) ζ-chain, via a flexible hinge region, to form a functional CAR (Savoldo and Dotti 2013). Louis et al. generated GD2-CAR-expressing T lymphocytes for the treatment of 19 patients with neuroblastoma. Persistence of GD2-CAR T lymphocytes beyond 6 weeks was associated with better clinical outcome, and three patients with active disease achieved complete remission. Thus, the GD2-CAR T lymphocytes might provide an alternative strategy for immunotherapy of neuroblastoma (Louis et al. 2011).
2.3 GD2-Specific Vaccines
2.3.1 GD2-KLH
The main challenge for developing carbohydrate vaccines is their poor immunogenicity. Chemical conjugation of glycans to a highly immunogenic protein scaffold, such as keyhole limpet hemocyanin (KLH), may enhance the immune responses to glycans. GD2-KLH is a synthetic GD2 conjugated to KLH. A phase I clinical trial of GD2-KLH using monophosphoryl lipid A (MPL-A) as an adjuvant in seven patients with recurrent or progressive gliomas showed no adverse effects. However, neither anti-GD2 antibody nor clinical response was observed (Becker et al. 2002). Another phase I clinical trial of GD2-KLH using OPT-821 combined with oral beta-glucan as adjuvants was conducted in neuroblastoma. Anti-GD2 antibody was induced in 12 of 15 patients. Importantly, disappearance of MRD was observed in 6 of 10 patients (Kushner et al. 2014). A subsequent phase I study of combined GM2-KLH and GD2-KLH mixed with QS-21 adjuvant in 31 patients with melanoma or sarcoma showed successful induction of IgM/IgG anti-GM2 and anti-GD2 in 97 % and 73 % of patients, respectively (Chapman et al. 2000). These encouraging findings suggest that adjuvants may play an important role in glycan-based vaccine.
2.3.2 Anti-GD2 Idiotype Monoclonal Antibody 1A7
mAb1A7 is an anti-idiotype antibody mimicking GD2 antigen which was generated by immunizing mice with anti-GD2, mAb 14G2a (Saleh et al. 1993). Active immunotherapy with anti-idiotype antibody is anticipated to induce a gradual release of anti-GD2 via humoral antibody response, which may be beneath the threshold of anti-GD2-induced toxicities. In preclinical study, immunization of C57BL/6 mice and rabbits with mAb1A7 induced anti-GD2 antibodies of IgG isotype that recognized GD2 by ELISA and flow cytometry. These antisera specifically lysed GD2-positive target cells in an ADCC assay (Sen et al. 1998). Foon et al. initiated a clinical trial for anti-GD2 idiotype antibody (1A7) in patients with advanced melanoma. Patients (n = 47) received 1A7 (TriGem) at dose of 1, 2, 4, or 8 mg mixed with QS-21 (100 μg) weekly for 4 weeks and then monthly until disease progression. A majority of patients (40/47, 85.1 %) generated an anti-1A7 response. The isotypic specificity of the anti-1A7 antibody was predominantly IgG, with minimal IgM, and these antibodies reacted specifically with tumor cells expressing GD2 by flow cytometry. Immune sera from five patients tested displayed ADCC activity. Complete response lasting for 24 months was noted in one patient and stable disease (14+ to 37+ months) in 12 patients. Disease progression occurred in 32 patients (1–17 months) and 21 had died (1–16 months). The Kaplan-Meier-derived overall median survival was not reached. Toxicities were mild, including local reaction at the site of the injection, with mild fever and chills (Foon et al. 1998; Foon et al. 2000). In addition, a clinical trial of mAb1A7 as a GD2 vaccine was conducted in high-risk neuroblastoma patients (n = 31, 26 stage IV, 5 stage III) who achieved first or subsequent complete remission or very good partial remission (Yu et al. 2001). Patients received subcutaneous injection of 1A7 mixed with QS-21 as adjuvant every 2 weeks for 4 weeks and then monthly for 11 months thereafter and switched to 1A7 in aluminum hydroxide gel during the second year. After treatment, all patients had local reactions, four developed transient fever and chills, and one patient had serum sickness. All patients generated anti-1A7 antiserum, and immune sera from some patients displayed CDC and ADCC activities against neuroblastoma. At a median of 6.8 years from study entry, 76.1 % (16/21) patients who enrolled during first remission have no evidence of disease progression, whereas only one of ten patients who enrolled during second or subsequent remission remains progression-free. Thus, active immunotherapy with anti-idiotypic antibody-based GD2 vaccine may offer therapeutic advantage over passive immunotherapy with reduced infusion-related toxicities.
3 Sialyl-Tn-Targeted Cancer Vaccine
Sialyl-Tn, Neu5Acα2,6-N-acetylgalactosamine (STn), is a carcinoma-associated carbohydrate determinant expressed on cancer-associated mucins, while it is weakly expressed in fetal and restricted normal adult tissues (Kjeldsen et al. 1988). Circulating STn has been detected in patients with gastrointestinal (Motoo et al. 1991) and ovarian (Kobayashi et al. 1992) malignancies. Expression of STn in colorectal carcinoma (Itzkowitz et al. 1990), gastric carcinoma (Ma et al. 1993), and breast cancer (Leivonen et al. 2001) correlates with poor prognosis and predicts a poor response to chemotherapy (Miles et al. 1994). In endometrial cancer, overexpression of STn correlated with overexpression of cyclooxygenase 2 (Ohno et al. 2006), which is linked to angiogenesis, tumor growth (Ohno et al. 2005a), and inhibition of the infiltration of CD8 T cell (Ohno et al. 2005b). STn has been reported to be involved in cell-cell aggregation, ECM adhesion, and migration and invasion of tumor cells, as shown in STn-overexpressing gastric cancer cells transfected with ST6Gal I transferase (Pinho et al. 2007). Moreover, STn on the tumor cells could interact with Siglec-15 expressed on tumor-associated macrophages to enhance the production of transforming growth factor-β through spleen tyrosine kinase (Syk) pathway (Takamiya et al. 2013). These findings suggest that STn may be a good candidate target for cancer immunotherapy.
A synthetic STn-keyhole limpet hemocyanin (KLH) vaccine (Theratope) was evaluated in clinical trials as an active specific immunotherapy in the treatment of advanced cancer. One of the first studies of Theratope was conducted by MacLean and colleagues in patients with metastatic breast cancer, ovarian cancer, and colon cancer (MacLean et al. 1996). They reported that 51 patients who produced anti-STn + mucin IgG titers higher than the median value survived longer than 46 patients who generated lower titers. Based on promising results of STn-KLH vaccine in early clinical trials, a phase III randomized trial was conducted in patients with metastatic breast cancer who had nonprogressive disease after first-line chemotherapy. A total of 1028 patients were randomly assigned to either STn-KLH plus Detox as adjuvant or KLH plus Detox (control group). The vaccine was well tolerated, with mild to moderate injection-site reactions and reversible flu-like symptoms. Specific IgG and IgM antibodies were detected at week 12. Unfortunately, there were no significant differences in the time to progression (TTP) and overall survival (OS) between STn-KLH vaccine group (3.4 and 23.1 months, respectively) and control group (3 and 22.3 months, respectively) (Miles et al. 2011b), although a post hoc analysis suggested benefit of concurrent endocrine therapy and STn-KLH vaccine for women with metastatic breast cancer (Ibrahim et al. 2013). Several factors may have contributed to the lack of overall clinical efficacy of this vaccine. First, STn is not expressed uniformly in all breast cancer specimens. It ranges from low 20 % to high 80 % in various reports (Julien et al. 2012). In this phase III study, STn expression was not determined nor used as enrollment criteria, which might mask any benefit from the vaccine due to heterogeneity in STn expression among patients. Second, significant titers of anti-KLH IgM and IgG antibodies were observed in control group, which may have conferred some anticancer benefits. Nonetheless, lessons learned from this failed large randomized clinical trial may serve as stepping stones to the ultimate success by modifying the clinical design and patient selection.
4 GM2-Targeted Cancer Vaccines
While GM3 is the predominant ganglioside in normal melanocytes (Carubia et al. 1984), in malignant melanoma, activation of glycosylating enzymes leads to increased expression of GD3, GD2, GM2, and 9-O-acetyl GD3 (Tsuchida et al. 1987). GM2 is also expressed on metastatic prostate cancer specimens (Zhang et al. 1998) and adult T-cell leukemia (Suzuki et al. 1987). Antibodies against GM2 were able to induce apoptosis (Retter et al. 2005; Nakamura et al. 1999) or necrosis (Bjerkvig et al. 1991) of GM2-expressing cancer cell lines. Furthermore, GM2 was found to inhibit immunoglobulin production of human B cell lines through impeding the production of IL-10 and TNF-α (Kimata and Yoshida 1996). In addition, complex of GM2 and GM3 was shown to associate with cMet-CD82 to regulate hepatocyte growth factor-induced motility of HCV29 cells (Todeschini et al. 2008). These findings suggest that GM2 is an attractive target for immunotherapy.
In 1994, 122 patients with stage III melanoma (N = 122) were treated with unconjugated GM2 and bacillus Calmette-Guerin (BCG) or BGC alone. The OS and DFS were not statistically significant between patients treated with GM2/BCG and BCG, although DFS was greater in patients producing anti-GM2 antibody (Livingston et al. 1994a). Most anti-GM2 antibodies induced by GM2/BCG vaccine in patients were IgM, suggesting that BCG adjuvant in glycan vaccine could not efficiently trigger antibody isotype switch to GM2-specific IgG antibody, which is an important mediator of ADCC. Subsequently, potent carrier protein, KLH, and adjuvant, QS-21, were used to generate GM2-KLH/QS-21 vaccine which induced higher titers of IgM anti-GM2 antibody and more IgG anti-GM2 antibody responses than GM2/BCG vaccine (Helling et al. 1995). A phase I trial of GM2-KLH vaccine plus QS-21 as an adjuvant in 22 patients with AJCC stage III/IV melanoma showed the induction of IgM and IgG antibodies against GM2 in patients treated with 100 or 200 μg of QS-21 (Livingston et al. 1994b). This led to two randomized phase III trials. One was conducted in 880 patients with resected high-risk melanoma (AJCC stages IIB and III) comparing the therapeutic efficacy of GM2-KLH/QS-21 (GMK) vaccine with standard therapy, high-dose interferon alfa-2b (HDI) (Kirkwood et al. 2001). The trial was closed after interim analysis showing inferiority of GMK compared with HDI, although patients with higher antibody responses to GM2 had a trend toward improved RFS and OS (p = 0.068 at day 29). Another phase III trial was conducted in 1314 patients with stage II melanoma to evaluate the efficacy and toxicity of GMK vaccine as compared to observation. Unfortunately, GM2-KLH/QS-21 failed to improve RFS, distant metastasis-free survival, and overall survival (Eggermont et al. 2013). In view of the impressive response of melanoma to immune checkpoint blockade therapy (Hodi et al. 2010), it is possible that clinical benefit of GM2 vaccine may become evident when combined with inhibitors of immune checkpoint.
5 Globo H-Targeted Cancer Vaccines
Globo H, a hexasaccharide (Fucα1 → 2Galβ1 → 3GalNAcβ1 → 3Galα1 → 4Galβ1 → 4Glcβ1), was initially identified as a ceramide-linked glycolipid in human breast cancer cell line MCF-7 (Kannagi et al. 1983) and subsequently found to be expressed on a variety of epithelial cancers including breast, colon, ovarian, gastric, pancreatic, lung, and prostate cancers (Zhang et al. 1997, 1998).
Examination of Globo H expression in breast cancer stem cells (BCSCs) by flow cytometry revealed Globo H expression in 61 % (25/41) of breast cancer specimens and in 20 % (8/40) of BCSC-enriched subpopulation (CD44+/CD24−). The expression of Globo H precursor, stage-specific embryonic antigen 3 (SSEA3), was 77.5 % (31/40) in breast cancer tissues and 62.5 % (25/40) in BCSCs. Like Globo H, SSEA3 expression in normal tissues was predominately at the secretory borders of epithelium, where access to the immune system is restricted. Immunization of mice with Globo H-KLH and alpha-GalCer induced antibodies reactive with Globo H and SSEA3, suggesting that a Globo H-based vaccine will target tumor cells expressing Globo H or SSEA3, including BCSCs (Chang et al. 2008).
The overexpression of Globo H in cancer with limited expression in normal tissues makes Globo H a potential target for cancer immunotherapy. The findings of Globo H ceramide as stem cell markers (Chang et al. 2008), immune checkpoint molecules (Tsai et al. 2013), and angiogenic factors (Cheng et al. 2014) provide further impetus for Globo H-targeted immunotherapy (Sabbatini et al. 2007). Two phase I clinical trials of Globo H-KLH/QS-21 vaccine were conducted in patients with relapsed prostate cancer (n = 18) (Slovin et al. 1999) and metastatic breast cancer (n = 27), respectively (Gilewski et al. 2001). The treatment schedules consisted of injection of Globo H-KLH at weeks 1, 2, 3, 7, and 19. In general, the vaccine was well tolerated, with only local reactions and occasional fever and chills. Humoral responses to Globo H-KLH vaccine were observed. In the trial of relapsed prostate cancer, the highest median IgM antibody titer was around 300 at the dose level of 10, 30, and 100 μg of Globo H-KLH, and peak response was observed at weeks 34, 3, and 9, respectively. For the dose level of 3 μg of Globo H-KLH, the peak titer was 150 at week 7. Interestingly, the production of Globo H-specific IgG antibody showed a different pattern from IgM responses. There were two obvious peak titers at the dose level of 10 μg and 30 μg of Globo H-KLH. At 30 μg Globo H-KLH, the IgG antibody titer reached to the maximal titer of 160 at weeks 9 and 34. At 10 μg Globo H-KLH, the peak IgG antibody titer was 80 at week 3 and week 35. In patients treated with 100 μg or 3 μg of Globo H-KLH, the titers of Globo H-specific IgG was only 100 (around week 31) or less than 20 (week 26), respectively. In the trial of metastatic breast cancer, Globo H-specific IgM peaked around weeks 5–7. Antisera in several patients of both trials displayed CDC activity. Both trials demonstrated that the Globo H-KLH vaccine was safe and effective in inducing humoral antibody response with moderate Globo H-specific IgM antibody titers in most patients, but only minimal IgG antibody. Recently, a multinational randomized phase II/III clinical trial of Globo H-KLH vaccine vs. placebo in patients with metastatic breast cancer has completed accrual of 349 patients and is awaiting further follow-up for outcome analysis (NCT01516307). Another phase II clinical trial of this vaccine in ovarian cancer is ongoing. In addition, a new generation of Globo H vaccine consisting of Globo H conjugated to diphtheria toxin as a carrier protein was shown to elicit more desirable IgG anti-Globo H, when combined with a novel analog of NKT-stimulatory alpha-galactosylceramide (α-GalCer) as an adjuvant. The efficacy of this promising Globo H vaccine awaits clinical trial in the near future (Huang et al. 2013).
6 Lewisy-Targeted Cancer Vaccine
In ovarian cancer, Lewisy is overexpressed which promotes metastasis through epididymis protein 4 (Zhuang et al. 2013, 2014). The expression of Lewisy antigen was considered as an independent, drug resistance-related risk factors (Gao et al. 2014). A clinical trial of Lewisy pentasaccharide conjugated with KLH together with immunological adjuvant QS-21 in ovarian cancer patients (n = 25) showed that the majority of the patients (16/24) produced anti-Lewisy antibodies with significant antitumor cell reactivity as assessed by CDC in some patients. The vaccine was well tolerated without any gastrointestinal, hematologic, renal, or hepatic toxicity (Sabbatini et al. 2000). Another phase II trial was conducted with a doxorubicin-conjugated chimeric variant of anti-Lewisy monoclonal antibody, BMS-182248-01, in patients (n = 15) with advanced gastric carcinoma (Ajani et al. 2000). However, BMS-182248-01 vaccine appeared to be ineffective in patients with gastric carcinoma with 10 patients progressed on study.
7 Polysialic Acid-Targeted Cancer Vaccine
Polysialic acid (polySA), a carbohydrate polymer of negatively charged sialic acid attached to the neural cell adhesion molecule (NCAM), is overexpressed on the surface of various cancers including small-cell lung cancer (SCLC) (Tanaka et al. 2001), Wilms’ tumor (Roth et al. 1988a, b), neuroblastoma (Gluer et al. 1998), and neuroectodermal tumors (Figarella-Branger et al. 1990). A clinical trial of polySA-KLH (30 μg) vaccine in small-cell lung cancer (n = 13) did not induce immune response, but N-propionylated (NP)-polySA (30 μg) developed high-titer anti-SA antibody along with peripheral neuropathy and ataxia in several patients (Krug et al. 2004). Another trial of lower dose of NP-polySA vaccine (10 μg) resulted in the induction of IgM antibodies against polySA antigen in all 18 patients, with self-limited grade 3 ataxia of unclear etiology in 1 of 18 patients (Krug et al. 2012).
8 Polyvalent Glycan Vaccine
A hexavalent vaccine, including GM2, Globo H, Lewisy, glycosylated MUC-1-32mer, Tn, and TF in a clustered formation conjugated to KLH, mixed with QS-21 was administered in a phase II setting to 30 patients with relapsed prostate cancer. All 30 patients showed increased antibody titers to at least two of the six antigens, but these serologic responses were lower than those seen previously with the respective monovalent vaccines (Slovin et al. 2007). In another study, GPI-0100, a semisynthetic low toxicity saponin, was used as adjuvant at doses ranging between 100 and 5000 μg for a bivalent vaccine containing the Globo H and the mucin MUC2 conjugated to KLH with in groups of five prostate cancer patients who had no evidence of disease except for rising PSA levels. All doses of GPI-0100 were well tolerated with dose-dependent increases in antibody titers against Globo H and MUC2. At the 5000 μg dose level, toxicity remained minimal with only occasional grade II local toxicity at vaccination sites and occasional sporadic grade I elevations in ALT. Compared with a subsequent trial with the same bivalent vaccine plus QS-21 at the maximal tolerated dose of 100 μg, the 5000 μg dose of GPI-0100 induced comparable antibody titers (Slovin et al. 2005).
9 Conclusion
Tumor-associated carbohydrate antigens are attractive targets for cancer therapy. Glycan-targeted immunotherapy holds the promise to have less side effects and greater specificity compared to conventional cancer therapy. To date, passive immunotherapy with anti-GD2 antibody in patients with neuroblastoma is the first successful glycan-targeted immunotherapy, which has documented that targeting TACA is a feasible strategy for cancer immunotherapy. On the other hand, carbohydrate-based vaccines for active immunotherapy have yet to be proven effective in phase III randomized trials, although encouraging results were noted in early clinical trials. New strategies are needed for enhancing the potency of carbohydrate-based cancer vaccine by improving the design of vaccine. Designs with better adjuvants that effectively boost IgG humoral and/or cellular immune response against TACAs are also critically needed.
References
Ajani JA et al (2000) A multi-institutional phase II study of BMS-182248-01 (BR96-doxorubicin conjugate) administered every 21 days in patients with advanced gastric adenocarcinoma. Cancer J 6(2):78–81
Albertini MR et al (2012) Phase II trial of hu14.18-IL2 for patients with metastatic melanoma. Cancer Immunol Immunother 61(12):2261–2271
Alvarez-Rueda N et al (2011) A monoclonal antibody to O-acetyl-GD2 ganglioside and not to GD2 shows potent anti-tumor activity without peripheral nervous system cross-reactivity. PLoS ONE 6(9), e25220
Barker E et al (1991) Effect of a chimeric anti-ganglioside GD2 antibody on cell-mediated lysis of human neuroblastoma cells. Cancer Res 51(1):144–149
Battula VL et al (2012) Ganglioside GD2 identifies breast cancer stem cells and promotes tumorigenesis. J Clin Invest 122(6):2066–2078
Becker R et al (2002) Phase I clinical trial on adjuvant active immunotherapy of human gliomas with GD2-conjugate. Br J Neurosurg 16(3):269–275
Biswas K et al (2006) GM2 expression in renal cell carcinoma: potential role in tumor-induced T-cell dysfunction. Cancer Res 66(13):6816–6825
Biswas S et al (2009) Elevated levels of select gangliosides in T cells from renal cell carcinoma patients is associated with T cell dysfunction. J Immunol 183(8):5050–5058
Bjerkvig R et al (1991) Anti-GM2 monoclonal antibodies induce necrosis in GM2-rich cultures of a human glioma cell line. Cancer Res 51(17):4643–4648
Borsig L et al (2001) Heparin and cancer revisited: mechanistic connections involving platelets, P-selectin, carcinoma mucins, and tumor metastasis. Proc Natl Acad Sci USA 98(6):3352–3357
Borsig L et al (2002) Synergistic effects of L- and P-selectin in facilitating tumor metastasis can involve non-mucin ligands and implicate leukocytes as enhancers of metastasis. Proc Natl Acad Sci USA 99(4):2193–2198
Carubia JM et al (1984) Gangliosides of normal and neoplastic human melanocytes. Biochem Biophys Res Commun 120(2):500–504
Cazet A et al (2012) The ganglioside G(D2) induces the constitutive activation of c-Met in MDA-MB-231 breast cancer cells expressing the G(D3) synthase. Glycobiology 22(6):806–816
Chang WW et al (2008) Expression of Globo H and SSEA3 in breast cancer stem cells and the involvement of fucosyltransferases 1 and 2 in Globo H synthesis. Proc Natl Acad Sci USA 105(33):11667–11672
Chapman PB et al (2000) Vaccination with a bivalent G(M2) and G(D2) ganglioside conjugate vaccine: a trial comparing doses of G(D2)-keyhole limpet hemocyanin. Clin Cancer Res 6(12):4658–4662
Chen YX et al (2013) Effect of tumor gangliosides on tyrosine phosphorylation of p125FAK in platelet adhesion to collagen. Oncol Rep 29(1):343–348
Cheng JY et al (2014) Globo-H ceramide shed from cancer cells triggers translin-associated factor X-dependent angiogenesis. Cancer Res 74:6856–6866
Cheung NK et al (1985) Monoclonal antibodies to a glycolipid antigen on human neuroblastoma cells. Cancer Res 45(6):2642–2649
Cheung NK et al (1986) Complete tumor ablation with iodine 131-radiolabeled disialoganglioside GD2-specific monoclonal antibody against human neuroblastoma xenografted in nude mice. J Natl Cancer Inst 77(3):739–745
Cheung NK et al (1987) Ganglioside GD2 specific monoclonal antibody 3F8: a phase I study in patients with neuroblastoma and malignant melanoma. J Clin Oncol 5(9):1430–1440
Cheung NK et al (1998a) 3F8 monoclonal antibody treatment of patients with stage 4 neuroblastoma: a phase II study. Int J Oncol 12(6):1299–1306
Cheung NK et al (1998b) Anti-G(D2) antibody treatment of minimal residual stage 4 neuroblastoma diagnosed at more than 1 year of age. J Clin Oncol 16(9):3053–3060
Cheung NK et al (2000) Induction of Ab3 and Ab3’ antibody was associated with long-term survival after anti-G(D2) antibody therapy of stage 4 neuroblastoma. Clin Cancer Res 6(7):2653–2660
Cheung NK et al (2006) FCGR2A polymorphism is correlated with clinical outcome after immunotherapy of neuroblastoma with anti-GD2 antibody and granulocyte macrophage colony-stimulating factor. J Clin Oncol 24(18):2885–2890
Cheung NK et al (2012) Murine anti-GD2 monoclonal antibody 3F8 combined with granulocyte-macrophage colony-stimulating factor and 13-cis-retinoic acid in high-risk patients with stage 4 neuroblastoma in first remission. J Clin Oncol 30(26):3264–3270
Chu JW, Sharom FJ (1993) Gangliosides inhibit T-lymphocyte proliferation by preventing the interaction of interleukin-2 with its cell surface receptors. Immunology 79(1):10–17
Chung TW et al (2009) Ganglioside GM3 inhibits VEGF/VEGFR-2-mediated angiogenesis: direct interaction of GM3 with VEGFR-2. Glycobiology 19(3):229–239
Crespo FA et al (2006) The immunoregulatory effects of gangliosides involve immune deviation favoring type-2 T cell responses. J Leukoc Biol 79(3):586–595
Das T et al (2008) GM1 and tumor necrosis factor-alpha, overexpressed in renal cell carcinoma, synergize to induce T-cell apoptosis. Cancer Res 68(6):2014–2023
Delgado DC et al (2010) Genotypes of NK cell KIR receptors, their ligands, and Fcgamma receptors in the response of neuroblastoma patients to Hu14.18-IL2 immunotherapy. Cancer Res 70(23):9554–9561
Dennis JW et al (1987) Beta 1–6 branching of Asn-linked oligosaccharides is directly associated with metastasis. Science 236(4801):582–585
Eggermont AM et al (2013) Adjuvant ganglioside GM2-KLH/QS-21 vaccination versus observation after resection of primary tumor > 1.5 mm in patients with stage II melanoma: results of the EORTC 18961 randomized phase III trial. J Clin Oncol 31(30):3831–3837
Eller CH et al (2015) Human cancer antigen globo H is a cell-surface ligand for human ribonuclease 1. ACS Cen Sci 1(4):181–190
Fernandes B et al (1991) Beta 1–6 branched oligosaccharides as a marker of tumor progression in human breast and colon neoplasia. Cancer Res 51(2):718–723
Figarella-Branger DF, Durbec PL, Rougon GN (1990) Differential spectrum of expression of neural cell adhesion molecule isoforms and L1 adhesion molecules on human neuroectodermal tumors. Cancer Res 50(19):6364–6370
Foon KA et al (1998) Antibody responses in melanoma patients immunized with an anti-idiotype antibody mimicking disialoganglioside GD2. Clin Cancer Res 4(5):1117–1124
Foon KA et al (2000) Clinical and immune responses in advanced melanoma patients immunized with an anti-idiotype antibody mimicking disialoganglioside GD2. J Clin Oncol 18(2):376–384
Frost JD et al (1997) A phase I/IB trial of murine monoclonal anti-GD2 antibody 14.G2a plus interleukin-2 in children with refractory neuroblastoma: a report of the Children’s Cancer Group. Cancer 80(2):317–333
Fujii Y et al (2005) Murine glycosyltransferases responsible for the expression of globo-series glycolipids: cDNA structures, mRNA expression, and distribution of their products. Glycobiology 15(12):1257–1267
Gao J et al (2014) Expression of CD147 and Lewis y antigen in ovarian cancer and their relationship to drug resistance. Med Oncol 31(5):920
Gilewski T et al (2001) Immunization of metastatic breast cancer patients with a fully synthetic globo H conjugate: a phase I trial. Proc Natl Acad Sci USA 98(6):3270–3275
Gillies SD, Lo KM, Wesolowski J (1989) High-level expression of chimeric antibodies using adapted cDNA variable region cassettes. J Immunol Methods 125(1–2):191–202
Gilman AL et al (2009) Phase I study of ch14.18 with granulocyte-macrophage colony-stimulating factor and interleukin-2 in children with neuroblastoma after autologous bone marrow transplantation or stem-cell rescue: a report from the Children’s Oncology Group. J Clin Oncol 27(1):85–91
Gluer S et al (1998) Polysialylated neural cell adhesion molecule in childhood ganglioneuroma and neuroblastoma of different histological grade and clinical stage. Langenbecks Arch Surg 383(5):340–344
Handgretinger R et al (1995) A phase I study of human/mouse chimeric antiganglioside GD2 antibody ch14.18 in patients with neuroblastoma. Eur J Cancer 31A(2):261–267
Helling F et al (1995) GM2-KLH conjugate vaccine: increased immunogenicity in melanoma patients after administration with immunological adjuvant QS-21. Cancer Res 55(13):2783–2788
Hodi FS et al (2010) Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 363(8):711–723
Hoff SD et al (1989) Increased expression of sialyl-dimeric LeX antigen in liver metastases of human colorectal carcinoma. Cancer Res 49(24 Pt 1):6883–6888
Huang YL et al (2013) Carbohydrate-based vaccines with a glycolipid adjuvant for breast cancer. Proc Natl Acad Sci USA 110(7):2517–2522
Ibrahim NK et al (2013) Survival advantage in patients with metastatic breast cancer receiving endocrine therapy plus sialyl Tn-KLH vaccine: post Hoc analysis of a large randomized trial. J Cancer 4(7):577–584
Irani DN, Lin KI, Griffin DE (1996) Brain-derived gangliosides regulate the cytokine production and proliferation of activated T cells. J Immunol 157(10):4333–4340
Itzkowitz SH et al (1986) Lewisx- and sialylated Lewisx-related antigen expression in human malignant and nonmalignant colonic tissues. Cancer Res 46(5):2627–2632
Itzkowitz SH et al (1989) Expression of Tn, sialosyl-Tn, and T antigens in human colon cancer. Cancer Res 49(1):197–204
Itzkowitz SH et al (1990) Sialosyl-Tn. A novel mucin antigen associated with prognosis in colorectal cancer patients. Cancer 66(9):1960–1966
Jacewicz M et al (1986) Pathogenesis of shigella diarrhea. XI. Isolation of a shigella toxin-binding glycolipid from rabbit jejunum and HeLa cells and its identification as globotriaosylceramide. J Exp Med 163(6):1391–1404
Jin HJ et al (2010) GD2 expression is closely associated with neuronal differentiation of human umbilical cord blood-derived mesenchymal stem cells. Cell Mol Life Sci 67(11):1845–1858
Julien S, Videira PA, Delannoy P (2012) Sialyl-tn in cancer: (how) did we miss the target? Biomolecules 2(4):435–466
Kannagi R (1997) Carbohydrate-mediated cell adhesion involved in hematogenous metastasis of cancer. Glycoconj J 14(5):577–584
Kannagi R et al (1983) New globoseries glycosphingolipids in human teratocarcinoma reactive with the monoclonal antibody directed to a developmentally regulated antigen, stage-specific embryonic antigen 3. J Biol Chem 258(14):8934–8942
Kantoff PW et al (2010) Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med 363(5):411–422
Kimata H, Yoshida A (1996) Inhibition of spontaneous immunoglobulin production by ganglioside GM2 in human B cells. Clin Immunol Immunopathol 79(2):197–202
King DM et al (2004) Phase I clinical trial of the immunocytokine EMD 273063 in melanoma patients. J Clin Oncol 22(22):4463–4473
Kirkwood JM et al (2001) High-dose interferon alfa-2b significantly prolongs relapse-free and overall survival compared with the GM2-KLH/QS-21 vaccine in patients with resected stage IIB-III melanoma: results of intergroup trial E1694/S9512/C509801. J Clin Oncol 19(9):2370–2380
Kjeldsen T et al (1988) Preparation and characterization of monoclonal antibodies directed to the tumor-associated O-linked sialosyl-2----6 alpha-N-acetylgalactosaminyl (sialosyl-Tn) epitope. Cancer Res 48(8):2214–2220
Klein G et al (1983) Expression of the BLA antigen, defined by the monoclonal 38.13 antibody, on Burkitt lymphoma lines, lymphoblastoid cell lines, their hybrids and other B-cell lymphomas and leukemias. Int J Cancer 31(5):535–542
Kobayashi H, Terao T, Kawashima Y (1992) Serum sialyl Tn as an independent predictor of poor prognosis in patients with epithelial ovarian cancer. J Clin Oncol 10(1):95–101
Kojima Y et al (2000) Molecular cloning of globotriaosylceramide/CD77 synthase, a glycosyltransferase that initiates the synthesis of globo series glycosphingolipids. J Biol Chem 275(20):15152–15156
Kondo Y et al (2013) TLR4-MD-2 complex is negatively regulated by an endogenous ligand, globotetraosylceramide. Proc Natl Acad Sci USA 110(12):4714–4719
Krug LM et al (2004) Vaccination of small cell lung cancer patients with polysialic acid or N-propionylated polysialic acid conjugated to keyhole limpet hemocyanin. Clin Cancer Res 10(3):916–923
Krug LM et al (2012) Immunization with N-propionyl polysialic acid-KLH conjugate in patients with small cell lung cancer is safe and induces IgM antibodies reactive with SCLC cells and bactericidal against group B meningococci. Cancer Immunol Immunother 61(1):9–18
Kushner BH et al (2014) Phase I trial of a bivalent gangliosides vaccine in combination with beta-glucan for high-risk neuroblastoma in second or later remission. Clin Cancer Res 20(5):1375–1382
Ladisch S, Becker H, Ulsh L (1992) Immunosuppression by human gangliosides: I. Relationship of carbohydrate structure to the inhibition of T cell responses. Biochim Biophys Acta 1125(2):180–188
Leivonen M et al (2001) STn and prognosis in breast cancer. Oncology 61(4):299–305
Liang YJ et al (2013) Differential expression profiles of glycosphingolipids in human breast cancer stem cells vs. cancer non-stem cells. Proc Natl Acad Sci USA 110(13):4968–4973
Liu Y et al (2014a) Tumor gangliosides accelerate murine tumor angiogenesis. Angiogenesis 17(3):563–571
Liu B et al (2014b) Endothelin A receptor antagonism enhances inhibitory effects of anti-ganglioside GD2 monoclonal antibody on invasiveness and viability of human osteosarcoma cells. PLoS ONE 9(4), e93576
Livingston PO et al (1994a) Improved survival in stage III melanoma patients with GM2 antibodies: a randomized trial of adjuvant vaccination with GM2 ganglioside. J Clin Oncol 12(5):1036–1044
Livingston PO et al (1994b) Phase 1 trial of immunological adjuvant QS-21 with a GM2 ganglioside-keyhole limpet haemocyanin conjugate vaccine in patients with malignant melanoma. Vaccine 12(14):1275–1280
Louis CU et al (2011) Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood 118(23):6050–6056
Lu P, Sharom FJ (1996) Immunosuppression by YAC-1 lymphoma: role of shed gangliosides. Cell Immunol 173(1):22–32
Ma XC et al (1993) Expression of sialyl-Tn antigen is correlated with survival time of patients with gastric carcinomas. Eur J Cancer 29A(13):1820–1823
MacLean GD et al (1996) Antibodies against mucin-associated sialyl-Tn epitopes correlate with survival of metastatic adenocarcinoma patients undergoing active specific immunotherapy with synthetic STn vaccine. J Immunother Emphasis Tumor Immunol 19(1):59–68
Makino T et al (2001) Carbohydrate antigens as a risk factor for hematogenous recurrence of esophageal squamous cell carcinoma patients. Oncol Rep 8(5):981–985
Manfredi MG et al (1999) Gangliosides influence angiogenesis in an experimental mouse brain tumor. Cancer Res 59(20):5392–5397
Martinez C et al (2007) Human bone marrow mesenchymal stromal cells express the neural ganglioside GD2: a novel surface marker for the identification of MSCs. Blood 109(10):4245–4248
Metelitsa LS et al (2002) Antidisialoganglioside/granulocyte macrophage-colony-stimulating factor fusion protein facilitates neutrophil antibody-dependent cellular cytotoxicity and depends on FcgammaRII (CD32) and Mac-1 (CD11b/CD18) for enhanced effector cell adhesion and azurophil granule exocytosis. Blood 99(11):4166–4173
Miles DW et al (1994) Expression of sialyl-Tn predicts the effect of adjuvant chemotherapy in node-positive breast cancer. Br J Cancer 70(6):1272–1275
Miles D et al (2011) Phase III multicenter clinical trial of the sialyl-TN (STn)-keyhole limpet hemocyanin (KLH) vaccine for metastatic breast cancer. Oncologist 16(8):1092–1100
Miraldi FD et al (1986) Diagnostic imaging of human neuroblastoma with radiolabeled antibody. Radiology 161(2):413–418
Miyake M et al (1992) Correlation of expression of H/Le(y)/Le(b) antigens with survival in patients with carcinoma of the lung. N Engl J Med 327(1):14–18
Morioka N et al (1991) Gangliosides inhibit the proliferation of human T cells stimulated with interleukin-4 or interleukin-2. J Dermatol 18(8):447–453
Motoo Y et al (1991) Serum sialyl-Tn antigen levels in patients with digestive cancers. Oncology 48(4):321–326
Mueller BM et al (1990) Enhancement of antibody-dependent cytotoxicity with a chimeric anti-GD2 antibody. J Immunol 144(4):1382–1386
Mujoo K et al (1987) Disialoganglioside GD2 on human neuroblastoma cells: target antigen for monoclonal antibody-mediated cytolysis and suppression of tumor growth. Cancer Res 47(4):1098–1104
Mujoo K et al (1989) Functional properties and effect on growth suppression of human neuroblastoma tumors by isotype switch variants of monoclonal antiganglioside GD2 antibody 14.18. Cancer Res 49(11):2857–2861
Murata K et al (2004) Attachment of human colon cancer cells to vascular endothelium is enhanced by N-acetylglucosaminyltransferase V. Oncology 66(6):492–501
Murray LJ et al (1985) Expression of Burkitt lymphoma-associated antigen (defined by the monoclonal antibody 38.13) on both normal and malignant germinal-centre B cells. Int J Cancer 36(5):561–565
Murray JL et al (1994) Phase I trial of murine monoclonal antibody 14G2a administered by prolonged intravenous infusion in patients with neuroectodermal tumors. J Clin Oncol 12(1):184–193
Nakamura K et al (1999) Apoptosis induction of human lung cancer cell line in multicellular heterospheroids with humanized antiganglioside GM2 monoclonal antibody. Cancer Res 59(20):5323–5330
Navid F et al (2014) Phase I trial of a novel anti-GD2 monoclonal antibody, Hu14.18K322A, designed to decrease toxicity in children with refractory or recurrent neuroblastoma. J Clin Oncol 32(14):1445–1452
Neal ZC et al (2004a) Enhanced activity of hu14.18-IL2 immunocytokine against murine NXS2 neuroblastoma when combined with interleukin 2 therapy. Clin Cancer Res 10(14):4839–4847
Neal ZC et al (2004b) NXS2 murine neuroblastomas express increased levels of MHC class I antigens upon recurrence following NK-dependent immunotherapy. Cancer Immunol Immunother 53(1):41–52
Ohno S et al (2005a) Multiple roles of cyclooxygenase-2 in endometrial cancer. Anticancer Res 25(6A):3679–3687
Ohno Y et al (2005b) Role of cyclooxygenase-2 in immunomodulation and prognosis of endometrial carcinoma. Int J Cancer 114(5):696–701
Ohno S et al (2006) Expression of Tn and sialyl-Tn antigens in endometrial cancer: its relationship with tumor-produced cyclooxygenase-2, tumor-infiltrated lymphocytes and patient prognosis. Anticancer Res 26(6A):4047–4053
Ohtsubo K, Marth JD (2006) Glycosylation in cellular mechanisms of health and disease. Cell 126(5):855–867
Okuda T et al (2006) Targeted disruption of Gb3/CD77 synthase gene resulted in the complete deletion of globo-series glycosphingolipids and loss of sensitivity to verotoxins. J Biol Chem 281(15):10230–10235
Osenga KL et al (2006) A phase I clinical trial of the hu14.18-IL2 (EMD 273063) as a treatment for children with refractory or recurrent neuroblastoma and melanoma: a study of the Children’s Oncology Group. Clin Cancer Res 12(6):1750–1759
Ozkaynak MF et al (2000) Phase I study of chimeric human/murine anti-ganglioside G(D2) monoclonal antibody (ch14.18) with granulocyte-macrophage colony-stimulating factor in children with neuroblastoma immediately after hematopoietic stem-cell transplantation: a children’s cancer group study. J Clin Oncol 18(24):4077–4085
Palaga T et al (2003) TCR-mediated Notch signaling regulates proliferation and IFN-gamma production in peripheral T cells. J Immunol 171(6):3019–3024
Peguet-Navarro J et al (2003) Gangliosides from human melanoma tumors impair dendritic cell differentiation from monocytes and induce their apoptosis. J Immunol 170(7):3488–3494
Pinho S et al (2007) Biological significance of cancer-associated sialyl-Tn antigen: modulation of malignant phenotype in gastric carcinoma cells. Cancer Lett 249(2):157–170
Reis CA et al (2010) Alterations in glycosylation as biomarkers for cancer detection. J Clin Pathol 63(4):322–329
Retter MW et al (2005) Characterization of a proapoptotic antiganglioside GM2 monoclonal antibody and evaluation of its therapeutic effect on melanoma and small cell lung carcinoma xenografts. Cancer Res 65(14):6425–6434
Robert C et al (2015) Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med 372(4):320–330
Robert C et al (2014) Anti-programmed-death-receptor-1 treatment with pembrolizumab in ipilimumab-refractory advanced melanoma: a randomised dose-comparison cohort of a phase 1 trial. Lancet 384(9948):1109–1117
Rosen SD, Bertozzi CR (1994) The selectins and their ligands. Curr Opin Cell Biol 6(5):663–673
Roth J et al (1988a) Presence of the long chain form of polysialic acid of the neural cell adhesion molecule in Wilms’ tumor. Identification of a cell adhesion molecule as an oncodevelopmental antigen and implications for tumor histogenesis. Am J Pathol 133(2):227–240
Roth J et al (1988b) Reexpression of poly(sialic acid) units of the neural cell adhesion molecule in Wilms tumor. Proc Natl Acad Sci USA 85(9):2999–3003
Sabbatini PJ et al (2000) Immunization of ovarian cancer patients with a synthetic Lewis(y)-protein conjugate vaccine: a phase 1 trial. Int J Cancer 87(1):79–85
Sabbatini PJ et al (2007) Pilot study of a heptavalent vaccine-keyhole limpet hemocyanin conjugate plus QS21 in patients with epithelial ovarian, fallopian tube, or peritoneal cancer. Clin Cancer Res 13(14):4170–4177
Saleh MN et al (1992a) Phase I trial of the murine monoclonal anti-GD2 antibody 14G2a in metastatic melanoma. Cancer Res 52(16):4342–4347
Saleh MN et al (1992b) Phase I trial of the chimeric anti-GD2 monoclonal antibody ch14.18 in patients with malignant melanoma. Hum Antibodies Hybridomas 3(1):19–24
Saleh MN et al (1993) Generation of a human anti-idiotypic antibody that mimics the GD2 antigen. J Immunol 151(6):3390–3398
Savoldo B, Dotti G (2013) Chimeric antigen receptors (CARs) from bench-to-bedside. Immunol Lett 155(1–2):40–42
Schulz G et al (1984) Detection of ganglioside GD2 in tumor tissues and sera of neuroblastoma patients. Cancer Res 44(12 Pt 1):5914–5920
Seelentag WK et al (1998) Prognostic value of beta1,6-branched oligosaccharides in human colorectal carcinoma. Cancer Res 58(23):5559–5564
Sen G et al (1998) Induction of IgG antibodies by an anti-idiotype antibody mimicking disialoganglioside GD2. J Immunother 21(1):75–83
Shibuya H et al (2012) Enhancement of malignant properties of human osteosarcoma cells with disialyl gangliosides GD2/GD3. Cancer Sci 103(9):1656–1664
Shusterman S et al (2010) Antitumor activity of hu14.18-IL2 in patients with relapsed/refractory neuroblastoma: a Children’s Oncology Group (COG) phase II study. J Clin Oncol 28(33):4969–4975
Slovin SF et al (1999) Carbohydrate vaccines in cancer: immunogenicity of a fully synthetic globo H hexasaccharide conjugate in man. Proc Natl Acad Sci USA 96(10):5710–5715
Slovin SF et al (2005) A bivalent conjugate vaccine in the treatment of biochemically relapsed prostate cancer: a study of glycosylated MUC-2-KLH and Globo H-KLH conjugate vaccines given with the new semi-synthetic saponin immunological adjuvant GPI-0100 OR QS-21. Vaccine 23(24):3114–3122
Slovin SF et al (2007) A polyvalent vaccine for high-risk prostate patients: “are more antigens better?”. Cancer Immunol Immunother 56(12):1921–1930
Sorkin LS et al (2010) Anti-GD(2) with an FC point mutation reduces complement fixation and decreases antibody-induced allodynia. Pain 149(1):135–142
Springer GF (1984) T and Tn, general carcinoma autoantigens. Science 224(4654):1198–1206
Suzuki Y et al (1987) Aberrant expression of ganglioside and asialoglycosphingolipid antigens in adult T-cell leukemia cells. Jpn J Cancer Res 78(10):1112–1120
Svennerholm L et al (1994) Gangliosides and allied glycosphingolipids in human peripheral nerve and spinal cord. Biochim Biophys Acta 1214(2):115–123
Takamiya R et al (2013) The interaction between Siglec-15 and tumor-associated sialyl-Tn antigen enhances TGF-beta secretion from monocytes/macrophages through the DAP12-Syk pathway. Glycobiology 23(2):178–187
Tanaka F et al (2001) Prognostic significance of polysialic acid expression in resected non-small cell lung cancer. Cancer Res 61(4):1666–1670
Terme M et al (2014) Chimeric antibody c.8B6 to O-acetyl-GD2 mediates the same efficient anti-neuroblastoma effects as therapeutic ch14.18 antibody to GD2 without antibody induced allodynia. PLoS ONE 9(2):e87210
Thornton MV et al (2004) Degradation of NF-kappa B in T cells by gangliosides expressed on renal cell carcinomas. J Immunol 172(6):3480–3490
Todeschini AR et al (2008) Ganglioside GM2/GM3 complex affixed on silica nanospheres strongly inhibits cell motility through CD82/cMet-mediated pathway. Proc Natl Acad Sci USA 105(6):1925–1930
Tozawa K et al (2005) Positive correlation between sialyl Lewis X expression and pathologic findings in renal cell carcinoma. Kidney Int 67(4):1391–1396
Tsai YC et al (2013) A prevalent cancer associated glycan, globo H ceramide, induces immunosuppression by reducing Notch1 signaling. J Cancer Sci Ther 5(7):264–270
Tsuchida T et al (1987) Gangliosides of human melanoma. J Natl Cancer Inst 78(1):45–54
Uttenreuther-Fischer MM et al (1995a) Pharmacokinetics of anti-ganglioside GD2 mAb 14G2a in a phase I trial in pediatric cancer patients. Cancer Immunol Immunother 41(1):29–36
Uttenreuther-Fischer MM, Huang CS, Yu AL (1995b) Pharmacokinetics of human-mouse chimeric anti-GD2 mAb ch14.18 in a phase I trial in neuroblastoma patients. Cancer Immunol Immunother 41(6):331–338
Uzzo RG et al (1999) Renal cell carcinoma-derived gangliosides suppress nuclear factor-kappaB activation in T cells. J Clin Invest 104(6):769–776
van de Kar NC et al (1992) Tumor necrosis factor and interleukin-1 induce expression of the verocytotoxin receptor globotriaosylceramide on human endothelial cells: implications for the pathogenesis of the hemolytic uremic syndrome. Blood 80(11):2755–2764
van Setten PA et al (1996) Effects of verocytotoxin-1 on nonadherent human monocytes: binding characteristics, protein synthesis, and induction of cytokine release. Blood 88(1):174–183
Wiels J, Fellous M, Tursz T (1981) Monoclonal antibody against a lymphoma-associated antigen. Proc Natl Acad Sci USA 78(10):6485–6488
Wolfl M et al (2002) Gangliosides inhibit the development from monocytes to dendritic cells. Clin Exp Immunol 130(3):441–448
Xu J et al (2013) Neural ganglioside GD2(+) cells define a subpopulation of mesenchymal stem cells in adult murine bone marrow. Cell Physiol Biochem 32(4):889–898
Yanagisawa M, Yoshimura S, Yu RK (2011) Expression of GD2 and GD3 gangliosides in human embryonic neural stem cells. ASN Neuro 3(2):e00054
Yankelevich M et al (2012) Anti-CD3 x anti-GD2 bispecific antibody redirects T-cell cytolytic activity to neuroblastoma targets. Pediatr Blood Cancer 59(7):1198–1205
Yoshida S et al (2001) Ganglioside G(D2) in small cell lung cancer cell lines: enhancement of cell proliferation and mediation of apoptosis. Cancer Res 61(10):4244–4252
Yu AL, Uttenreuther-Fischer MM, Kamps A (1995) Combined use of a human mouse chimeric anti-GD2 (ch14.18) and GM-CSF in the treatment of refractory neuroblastoma. Antibody Immunocon Radiopharm 8:12
Yu AL et al (1997) Usefulness of a chimeric anti-GD2 (ch14.18) and GM-CSF for refractory neuroblastoma: A POG phase II study. Proc ASCO 16:1846
Yu AL et al (1998) Phase I trial of a human-mouse chimeric anti-disialoganglioside monoclonal antibody ch14.18 in patients with refractory neuroblastoma and osteosarcoma. J Clin Oncol 16(6):2169–2180
Yu AL, Eskenazi A, Strother D (2001) A pilot study of anti-idiotype monoclonal antibody as tumor vaccine in patients with high risk neuroblastoma. Proc Am Soc Clin Oncol 20:1470
Yu AL et al (2010) Anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. N Engl J Med 363(14):1324–1334
Yuan M et al (1986) Comparison of T-antigen expression in normal, premalignant, and malignant human colonic tissue using lectin and antibody immunohistochemistry. Cancer Res 46(9):4841–4847
Zeng Y et al (2005) Anti-neuroblastoma effect of ch14.18 antibody produced in CHO cells is mediated by NK-cells in mice. Mol Immunol 42(11):1311–1319
Zhang S et al (1997) Selection of tumor antigens as targets for immune attack using immunohistochemistry: II. Blood group-related antigens. Int J Cancer 73(1):50–56
Zhang S et al (1998) Expression of potential target antigens for immunotherapy on primary and metastatic prostate cancers. Clin Cancer Res 4(2):295–302
Zhuang H et al (2013) Co-expression of Lewis y antigen with human epididymis protein 4 in ovarian epithelial carcinoma. PLoS ONE 8(7), e68994
Zhuang H et al (2014) Overexpression of Lewis y antigen promotes human epididymis protein 4-mediated invasion and metastasis of ovarian cancer cells. Biochimie 105:91–98
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Japan
About this chapter
Cite this chapter
Hung, JT., Yu, A.L. (2016). Targeting Glycans for Immunotherapy of Human Cancers. In: Furukawa, K., Fukuda, M. (eds) Glycosignals in Cancer: Mechanisms of Malignant Phenotypes . Springer, Tokyo. https://doi.org/10.1007/978-4-431-55939-9_11
Download citation
DOI: https://doi.org/10.1007/978-4-431-55939-9_11
Published:
Publisher Name: Springer, Tokyo
Print ISBN: 978-4-431-55937-5
Online ISBN: 978-4-431-55939-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)