
Abstract
GD2 ganglioside has been identified as a key determinant of bone marrow-derived mesenchymal stem cells (BM-MSCs). Here, we characterized GD2 ganglioside expression and its implications in umbilical cord blood-derived MSCs (UCB-MSCs). Using immune-selection analysis, we showed that both GD2-positive and GD2-negative UCB-MSCs expressed general stem cell markers and possessed mesodermal lineage differentiation potential. Although the fraction of GD2-expressing cells was lower in UCB-MSC than in BM-MSC populations, inhibition of GD2 synthesis in UCB-MSCs suppressed neuronal differentiation and down-regulated basic helix-loop-helix (bHLH) transcription factors, which are involved in early stage neuronal differentiation. In addition, the levels of bHLH factors in neuronally induced UCB-MSCs were significantly higher in GD2-positive than GD2-negative cells. Our data demonstrate that GD2 ganglioside expression is associated with regulation of bHLH factors and identify neurogenic-capable UCB-MSCs, providing new insights into the potential clinical applications of MSC-based therapy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Mesenchymal stem cells (MSCs) undergo self-renewing divisions that enable cells to differentiate along various lineages [1, 2]. Importantly, it has been proposed that MSCs act as “trophic mediators,” which act through secretion of bioactive factors to serve either as immunosuppressors or regeneration promoters [3–5]. MSCs can be easily obtained, typically from bone marrow, but also from other sources such as umbilical cord blood, adipose tissue, and placenta [6–8]. These attributes make MSCs an attractive cell source for use in tissue engineering and cell-based therapies.
Umbilical cord blood-derived MSCs (UCB-MSCs) are less mature than adult MSCs and they expand readily in vitro [6]. Recently, several groups reported that UCB-MSCs have a number of advantages over bone marrow-derived MSCs (BM-MSCs) in terms of availability, accessibility, multi-potency, and ability to migrate toward cancer [9, 10]. These characteristics make UCB-MSCs more suitable for clinical application than BM-MSCs [10, 11].
The process for isolating MSCs is quite simple, and depends on the adherence of these cells to plastic ware used to culture cells, and the exclusion of non-adherent cells that express hematopoietic lineage markers. The simplicity of MSC isolation procedures, however, comes with concerns about the heterogeneity of the resulting cell population. That MSC populations are clonally heterogeneous is evidenced by the study of CFU-Fs (colony forming units-fibroblasts), of which differentiation potential remains restricted to the mesenchymal lineages [12, 13]. This clonal heterogeneity remains a major obstacle, not only for gaining a general understanding of the mechanism by which MSCs maintain their proliferation potential and undergo differentiation toward specific lineage characteristics but also with respect to achieving better outcomes in cell-based therapy. In this context, researchers have attempted to establish a standard set of criteria for attaining more uniform populations of MSCs. As a part of these efforts, recent studies have exploited the pattern of membrane protein antigen expression to identify and define stem cells, although the expression of these antigens likely varies with MSC origin and culture conditions. These efforts notwithstanding, no specific marker capable of distinguishing a homogeneous MSC population has thus far been identified.
A number of cell markers have been suggested to be selective for cells isolated from mononuclear cells (MNCs) or BM-MSCs, including SSEA-4, CD271, fibroblast activation protein α (FAP), CD146, and neural ganglioside (GD2) [14–18]. Gangliosides are a family of glycosphingolipids (GSLs) that are ubiquitous components of eukaryotic cell membranes [19]. Among these, ganglioside GD2 is abundant in vertebrate brain tissue, where it appears to play a role in coordinating the expression of simple and complex gangliosides on the cell surface [20]. Recent studies have confirmed that ganglioside type and expression levels are developmentally controlled in a cell type-specific manner [21, 22].
Here, we focused on identifying markers of stem/progenitor cells that can be effectively used to assess the purity of MSCs. We first tested several markers to confirm their expression in UCB-MSCs and then analyzed these markers for their association with the stem cell characteristics of UCB-MSCs. Among the markers tested, GD2 was shown to be expressed in both UCB-MSCs and BM-MSCs, although these two populations differed with respect to the percentage of GD2-positive cells. More intriguingly, inhibition of GD2 synthesis caused suppression of neuronal differentiation in both BM-MSCs and UCB-MSCs in association with down-regulation of several basic helix loop helix (bHLH) transcription factors known to be involved in neuronal differentiation. To the best of our knowledge, this is the first report to verify the expression of GD2 in UCB-MSCs and suggest a possible role for GD2 in determining the neuronal fate of MSCs. Defining the mechanistic relationship between GD2 expression and the process of neuronal fate determination may pave the way to the development of further cell-based therapy applications of MSCs.
Materials and methods
Cell culture
This study was approved by the Institutional Review Board of Medipost Inc., Seoul, Korea. UCB-MSCs were separated and maintained as described previously [23]. Umbilical cord blood was collected from umbilical veins after neonatal delivery with informed consent of the pregnant mothers. MSCs were isolated by separating MNCs using a Ficoll-Hypaque solution (d = 1.077 g/cm3; Sigma, St. Louis, MO, USA). After transferring into α-minimum essential medium (α-MEM; Gibco, Carlsbad, CA, USA) supplemented with 10% (v/v) fetal bovine serum (FBS; Gibco). MNCs were seeded at 5 × 105 cells/cm2 into culture flasks. Cells maintained in a humidified 5% CO2 at 37°C formed colonies of spindle-shaped cells. At 50% confluence, cells were harvested after treatment with 0.25% (w/v) trypsin–EDTA (Gibco) and were reseeded for expansion. BM-MSCs (Cambrex, Walkersville, MD, USA) and HS68 human fibroblast cells (ATCC, Rockville, MD, USA) were cultured under the same conditions.
Immunotyping and sorting of MSCs
For cytometric analysis of cultured cell phenotypes, cells were stained with antibodies against human CD14-FITC, CD34-FITC, CD45-FITC, HLA DR-FITC (Becton–Dickinson, San Jose, CA, USA), CD29-PE, CD44-PE, CD73-PE, CD90-PE, CD146-PE, CD271-PE (Pharmingen, Los Angeles, CA, USA), and CD105-PE (Serotec, Kidlington, UK) for 15 min at room temperature. The respective mouse isotype antibodies were used as controls. For indirect assays, cells were stained with anti-human GD2 (BD Biosciences), SSEA-4 (BD Bioscience), and FAP (Santa Cruz Biotechnology, Santa Cruz, CA, USA) primary antibodies and then incubated with the corresponding FITC-conjugated anti-mouse IgG secondary antibody (Jackson ImmunoResearch, West Grove, PA, USA). The cells were washed with phosphate buffered saline (PBS) and fixed with 1% (v/v) paraformaldehyde. MSC immunotype was determined by flow cytometry using FACSCaliber (BD Biosciences) and the percentage of expressed cell surface antigen was calculated for 10,000 gated-cell events.
For sorting analyses, MSCs were incubated with a murine monoclonal primary antibody against GD2 (BD Biosciences) at concentration of 1 μg antibody/106 cells for 40 min at 4°C, followed by incubation with goat anti-mouse IgG secondary antibody (Jackson ImmunoResearch) for 30 min at 4°C. Both GD2-positive and GD2-negative cells were sorted to 95% purity using a FACSVantage cell-sorting system. The sorted populations were used in the following experiments.
CFU-F (colony forming unit-fibroblast) and cell proliferation assays
CFU-F assays were performed by seeding cells at 3 cells/cm2 into 60 × 15 mm-dishes (BD Biosciences) and incubating in a humidified 5% CO2 at 37°C; culture medium was exchanged every 3 days. After 14 days in culture, the dishes were washed twice with PBS, fixed with 100% methanol, and stained with 3% Crystal violet. The number of colonies was counted. To investigate cell proliferation, cells (2.5 × 104) were plated in T-25 flasks (Nunc, Roskilde, Denmark) and incubated for 5 days. Cells were harvested daily and total cell counts were obtained using a hemocytometer.
In vitro multi-lineage differentiation
UCB-MSCs and BM-MSCs were examined for their multi-lineage potential by incubating cells under specialized conditions to induce differentiation into osteoblasts, chondrocytes, and adipocytes [23]. UCB-MSCs were also committed into respiratory epithelial cells or neurogenic cells [23, 24]. Prior to neurogenic induction, cells were pretreated for 3 days with 20 ng/ml basic fibroblast growth factor (bFGF; R&D Systems, Minneapolis, MN, USA) in DMEM containing 10% FBS. Neuronal differentiation was induced by changing cultured medium to neurogenic medium composed of DMEM/F12 supplemented with 200 μM butylated hydroxyanisole (BHA; Sigma), 25 μg/ml insulin (Sigma), 25 μM KCl, 2 μM valproic acid (Sigma), 10 μM forskolin, and 1 μM hydrocortisone (Sigma). After the differentiation under specific induction conditions, the multi-lineage potentials of UCB-MSCs and BM-MSCs were evaluated as previously reported [6, 24]. Briefly, osteoblast formation was assessed by measuring the level of von Kossa (Sigma) and ALP (Sigma) staining and the activity of ALP; chondrocyte formation was determined by safranin O (Sigma) staining and the positive area to safranin O and oil red O staining were measured using i-solution software (IMTech-nology, Doosan, Daejeon, Korea); adipocytes formation was assessed based on staining of accumulated lipid vacuoles oil Red O; and formation of respiratory epithelium was analyzed by measuring the level of surfactant protein C (SP-C; Chemicon, Temecula, CA, USA) using fluorescence detection, flow cytometry, and real-time PCR. On day 6 of induction, cells grown under neurogenesis-inducing conditions were assessed by staining for microtubule-associated protein 2 (MAP-2; Chemicon) and Western blotting against MAP-2 and Neu-N.
Reverse transcription-polymerase chain reaction (RT-PCR) and real-time PCR
Total RNA was isolated using an RNeasy Mini Kit (Qiagen, Crawley, UK) according to the manufacturer’s instructions. One microgram of total RNA was reversed transcribed to cDNA using oligo dT primers (Invitrogen, La Jolla, CA, USA) and superscript II polymerase (Invitrogen) in a 40-μl reaction. The primers used to amplify target mRNAs were as follows: GD2 synthase, (forward) 5′-GAC AAG CCA GAG CGC GTT A-3′ and (reverse) 5′-TAC TTG AGA CAC GGC CAG GTT-3′ (99-bp product); nestin, (forward) 5′-ATG CTC CTC TCT CTC TGC TCC A-3′ and (reverse) 5′-GCT TCA ACG GCA AAG TTC TC-3′ (480-bp product); vimentin, (forward) 5′-GGC TCA GAT TCA GGA ACA GC-3′ and (reverse) 5′ GCT ACG GCA AAC TTC TC-3′ (408-bp product); and β-actin, (forward) 5′-TCC CTC CCT GGA GAA GCT A-3′ and (reverse) 5′-AGG AGG AGC AAT GAT CTT GAT C-3′ (300-bp product). PCR cycling conditions were 95°C for 12 min (initial denaturation), then 35 cycles of 95°C for 30 s (denaturation), 58°C for 45 s (annealing), and 72°C for 30 s (extension), followed by a final extension at 72°C for 5 min.
Real-time PCR was performed using a LightCycler TM 480 (Roche, Mannheim, Germany) and TaqMan to quantitatively detect mRNA for the following genes: MASH1 (NM_004316.3), MATH-1 (NM_005172.1), neurogenin-1 (NM_006161.2), MAP-2 (NM_001039538.1), SP-C (NM_003018.3), and β-actin (NM_001101.3). The primers and probes were designed using the Universal Probe Library Assay design Center (Roche). Relative quantities of the mRNA of interest were calculated by the comparative threshold cycle (2−ΔΔCt) method and normalized to β-actin [25]. Values are expressed as fold-increases relative to the levels in uninduced cells, defined as 1.
Transfection of small interfering RNAs (siRNAs)
siRNAs for GD2 synthase and GD3 synthase, and a non-specific control siRNA were designed by Dharmacon (Chicago, IL, USA). An siRNA pool targeting GD2 synthase consisted of the following four duplexes: (1) 5′-GGA AGU GAC UGG AGU UAC U-3′; (2) 5′-CAA CAU CAG CGC UCU AGU UC-3′; (3) 5′-UCA CCG ACG GCG UGG UUAA-3′; and (4) 5′-GGA CUG ACA AGG UGC GCG A-3′. An siRNA pool targeting GD3 consisted of the following four duplexes: (1) 5′-CUA CUA UGA CAA CGU CUU A-3′; (2) 5′-CCG GCU GCC CAA CGA GAA A-3′; (3) 5′-GCA UAA UUC GGC AAA GGU U-3′; and (4) 5′-GAG CCA UCU UUG AGG GUU U-3. Control siRNAs consisted of the following four duplexes: (1) 5′-UGG UUU ACA UGU CGA CUA A-3′: (2) 5′-UGG UUU ACA UGU UGU GUG A-3′; (3) 5′-UGG UUU UUC ACA UGU UUU CUG A-3′; and (4) 5′-UGG UUU ACA UGU UUU CCU A-3′. For siRNA experiments, MSCs were seeded on 60-mm dishes at 70% confluence and, after 24 h, were transfected with GD2 synthase siRNA and GD3 synthase siRNA, or control siRNA using the Dharmafect reagent (Dharmacon) according to the manufacturer’s protocol. Transfection efficiency was assessed by labeling siRNA with SiGLO red fluorescent dye (Dharmacon). Because de novo synthesis of ganglioside GD2 occurs as a set of stepwise glycosylations in which the glycosyltransferases, GD3 synthase (α-2,8-sialytransferase) and GD2/GM2 synthase (β1,4-N-acetyl-galactosaminlytransferase), participate in series; equal amounts of siRNA mixtures targeting GD2 synthase and GD3 synthase were added in these experiments to ensure optimal conditions for GD2 ganglioside suppression The cells in each plate were transfected with a mixture of siRNAs targeting GD2 synthase (50 μΜ) and GD3 synthase (50 μΜ), or control siRNA (100 μΜ). After 24 h, cells were trypsinized, and the expression levels of GD2 synthase were analysis by fluorescence microscopy and FACs. To verify the intra-cellular uptake of the treated siRNAs, the cells transfected with siRNA-fluorescent oligo were suspended in Cytofix/Cytoperm fixation/permeabilization solution (BD Biosciences) and underwent flow cytometric analyses.
Alkaline phosphatase (ALP) activity assay
ALP activity was measured using an ALP kit from Takara Inc. (Shiga, Japan). Whole-cell lysates were prepared by extracting proteins with lysis buffer. Protein concentrations were quantified using the bicinchoninic acid (BCA) protein assay, and then 10 μg protein was incubated with ALP substrate solution for 30 min at 37°C. The reactions were terminated by the addition of a stop solution (0.9 N NaOH), and the absorbance was measured at 405 nm.
Glycosaminoglycan (GAG) analysis
A Blyscan GAG Assay Kit (Biocolor, Belfast, UK), based on the quantitative, specific binding of the cationic dye, 1,9-dim-ethylmethylene blue, was used to analyze the glycosaminoglycan (GAG) components of sulfated proteoglycans, as described by the manufacturer. Briefly, Blyscan dye reagent was added to test samples and GAG standard solutions prepared at different concentrations (0, 1, 2, 3, 4, and 5 μg/ml), and the mixtures were shaken on a mechanical shaker for 30 min. After removing the solution containing unbound dye, the mixture was resuspended in 1 ml of dissociation buffer. Sample absorbance was measured at 656 nm.
Statistical analysis
Statistical significance was determined using paired Student’s t-tests. The data are reported as the mean ± standard error (SE) of at least three independent experiments. A p-value < 0.05, determined using the SIGMAPLOT program and SASS (ver. 9.1) software based on a linear-mixed model, was considered statistically significant.
Results
UCB-MSCs with multi-lineage differentiation potential express GD2 ganglioside on the cell surface
UCB-MSCs adhered to plastic culture dishes and exhibited a spindle-shaped fibroblast-like morphology. UCB-MSCs were positive for the expression of CD29, CD44, CD73, CD90, CD105, and HLA-ABC, but did not express CD14, CD34, CD45, or HLA-DR (data not shown), consistent with previous reports. There were no obvious differences in the expression of these surface antigens between UCB-MSCs and fibroblasts.
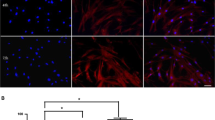
The multi-lineage differentiation capacity of UCB-MSCs was demonstrated by incubating cells in medium suitable for mesodermal (osteogenic, chondrogenic, and adipogenic), endodermal (respiratory epithelial cells) or ectodermal (neurogenic) induction. Osteogenesis was confirmed by the presence of von Kossa-staining calcium deposits; chondrogenesis, characterized by an increase in proteoglycans, was demonstrated by safranin O staining; adipogenesis was evidenced by staining of cytoplasmic lipid vacuoles by oil Red O; and to quantitate and compare the magnitude of multi-lineage differentiations of UCB-MSCs and BM-MSCs, we adopted alkaline phosphatase activity (UCB: 0.68 ± 0.01 O.D., BM: 0.61 ± 0.01 O.D.), safranin O positivity (UCB: 83.4 ± 24.8%, BM: 68.8 ± 31.2%) and oil Red O positivity (UCB: 9.0 ± 6.6%, BM: 41.2 ± 23.9%) as positive markers for osteogenic, chondrogenic, and adipogenic differentiations, respectively. Direct comparison of UCB-MSCs to BM-MSCs demonstrated the same pattern of extent and level of multi-lineage differentiation except for the adipogenic potential (Fig. 1a). Moreover, formation of respiratory epithelium in UCB-MSCs cultured in small airway growth medium (SAGM, Lonza) for 5 days was confirmed by expression of the type II pneumocyte-specific marker, surfactant C (70.7% of the cells; 14.2-fold increase in SP-C mRNA) (Fig. 1b). Cells that differentiated along the neuronal lineage after culture in induction medium for 6 days were positive for MAP-2, a mature neuron marker. Similarly, expressions of MAP-2 and Neu-N proteins were up-regulated in the cells under neurogenic induction (Fig. 1c). These results confirmed that UCB-MSCs are capable of giving rise to multiple cell types of the three germ layers.

Differentiation characteristics of human UCB-MSCs and BM-MSCs. a Left after incubation in specialized induction media, UCB-MSCs or BM-MSCs showed multi-lineage differentiation based on staining for typical stem cell markers. Osteogenic cells formed a calcium matrix stained with von Kossa after 3 weeks. Chondrogenic cells accumulated sulfated proteoglycan that was stained with safranin O after 3 weeks. Adipogenic cells accumulated lipid vacuoles within the cytoplasm that was stained with oil Red O after 4 weeks. Right level of differentiation to each lineage was quantified by measuring the activity of ALP and analyzing the percentage of the cells positive to each staining in both MSCs derived from UCB and BM. No differences in differentiation potential of UCBs and BMs (to osteo, chondro) were detected, while oil Red O staining positive to adipogenic cells were less obvious in UCB-MSCs (*p < 0.05). b Respiratory epithelial cells were observed by immunostaining, FACS, and real-time PCR for surfactant C after 5 days of respiratory induction. c Neuronal cells were observed by staining for MAP-2 and Western blotting for MAP-2 and Neu-N after 6 days of the neurogenic induction. d Flow cytometry showed GD2 expression in HS68 fibroblasts, BM-MSCs, and UCB-MSCs. Plots were analyzed by GD2 staining profile (filled graph) versus isotype-control staining profile (empty graph). The percentages indicate the mean values of %-positive cells ± SE. (scale bars in a, b, and c 50 μm)
A number of well-studied cell surface markers, including GD2 (nervous system), CD146 (Me1-CAM), SSEA-4 (embryonic stem cell marker), FAP (fibroblast protein), and CD271 (nerve growth factor), have been associated with BM-derived MSCs [14–18]. We tested whether expression of these antigens is also characteristic of UCB-MSCs. Of these antigens, only GD2 was significantly expressed in UCB-MSCs, although the percentage of GD2-positive cells in UCB-MSC populations (33.5% ± 7.0%) was less than that in BM-MSC populations (89.5% ± 8.5%; Fig. 1d).
GD2 expression has no effect on the proliferative status of UCB-MSCs
To investigate whether GD2 expression is associated with specific UCB-MSC properties, including proliferation and differentiation potential, we first sorted cells (passage 4) with respect to GD2 expression. An analysis of both sorted populations (GD2+ and GD2−) by flow cytometry showed that each was homogeneous in terms of GD2 expression (n = 3; Fig. 2a).

Immunophenotyping results and features of UCB-MSCs sorted based on GD2 expression. a Unsorted, GD2+, and GD2− cells adhered to culture dishes and showed a fibroblast-like morphology (scale bars 50 μm). The percentage of cells positive for GD2 was analyzed for unsorted, and GD2+ and GD2− sorted populations by flow cytometry. Plots indicate GD2 staining profile (filled graph) versus isotype-control staining profile (empty graph). b Top clonogenic capacity of unsorted, GD2+ and GD2− cells was measured by CFU-F assay and the colony number was counted. GD2+ cells formed fewer colonies than unsorted or GD2− cells. Bottom cell proliferation was examined in cell lines from three different UCB-MSCs donors. GD2− cells showed significantly higher cell growth than GD2+ cells (p < 0.05). c Immunophenotypic characteristics of GD2+ and GD2− cells were examined by flow cytometry. Both GD2+ and GD2− cells were strongly positive for the MSC-specific surface markers, CD73 and CD105, and were negative for CD34 and HLA-DR
A comparison of the proliferative capacity of these populations by CFU-F analysis showed that more colonies formed from GD2− cells (71.5 ± 2.1 colonies/20 cm2) than from GD2+ cells (27.5 ± 3.5 colonies/20 cm2) (Fig. 2b). Consistent with this, the growth rate of GD2− cells was significantly faster than that of GD2+ cells, based on daily cell counts made over the course of 5 days. Collectively, these results indicate that GD2 expression is not correlated with an increase in the proliferative capacity of UCB-MSCs (p < 0.05; Fig. 2b).
To investigate whether surface antigens characteristic of MSCs are associated with GD2 expression, we tested the expression of these antigens (HLD-ABC, HLA-DR, CD14, CD29, CD34, CD44, CD45, CD73, CD90, and CD105) in sorted populations. The antigen expression pattern was similar in both GD2− and GD2+ cells (data not shown): representatively, both populations were positive for CD73 and CD105 and negative for CD34 and HLA-DR (Fig. 2c). There was also no difference between the two populations in the percentage of cells positive for SSEA-4, a pluripotency marker (data not shown).
The functional importance of GD2 in the multi-lineage differentiation of UCB-MSCs was verified by culturing the unsorted GD2+ and GD2− cells under osteogenic-, chondrogenic-, and adipogenic-inducing conditions. Each population differentiated into its respective mesodermal-lineage and displayed the corresponding phenotype, regardless of GD2 expression (Fig. 3). Taken together, these results demonstrate that both GD2+ and GD2− cell populations possess attributes consistent with the characteristics of UCB-MSCs defined by minimal MSC criteria [26].

Mesodermal lineage differentiation of unsorted, GD2+, and GD2− cells. UCB-MSC populations sorted by GD2 expression were cultured under specialized induction conditions. In each population, osteogenic, chondrogenic, and adipogenic differentiation was examined by staining for von Kossa, safranin O, and oil Red O, respectively. No difference in mesodermal lineage differentiation potential was detected between GD2+ and GD2− cells (scale bar 50 μm)
Silencing of GD2 expression attenuates neuronal differentiation of UCB-MSCs
The expression of ganglioside GD2 is closely associated with neurogenesis and the development of the nervous system [27]. GD2 ganglioside is also reported to be expressed in a broad range of vertebrate tissues and organs, and is up-regulated in the majority of lung cancer cells [28]. These and other observations have led to the suggestion that GD2, acting as a recognition molecule or signaling modulator, plays a pivotal role in the regulation of developmental processes [19]. Our preliminary data show that uninduced UCB-MSCs express the neuronal precursor-specific genes, nestin and vimentin; interestingly, we found that the higher the expression of GD2, the easier it was to induce neuronal differentiation in vitro (data not shown).
To extend these observations and confirm that GD2 functionally contributes to determining the neuronal fate of UCB-MSCs, we silenced GD2 and GD3 synthase using a siRNA mixture targeting both enzymes, as described in Materials and methods. At a concentration of 100 μM, fluorescently labeled siRNAs were taken up by more than 90% of cells under the conditions used and caused no morphological changes or obvious toxicity, indicating that these transfection conditions are optimal for effective siRNA delivery (Fig. 4a). Control experiments showed that treatment with the target siRNA mixture also effectively down-regulated GD2 expression at both the mRNA and protein levels, as shown by RT-PCR (Fig. 4c) and flow cytometry analyses (3.0%; Fig. 4b), respectively, and maintained suppression of GD2 expression for up to 7 days (Fig. 4c). As was the case with sorted GD2+ and GD2− subpopulations, silencing of GD2 expression did not disturb development into mesodermal osteoblasts or chondrocytes, or endodermal respiratory epithelium. Thus, despite GD2 suppression, osteogenic, chondrogenic, and respiratory epithelial cell developmental potential was well maintained, as assessed by ALP activity (3 weeks), GAG content (3 weeks), and SP-C expression (5 days), respectively (Fig. 5a). The corresponding phenotypic morphologies were also well preserved in the differentiated cells (data not shown). In contrast, the suppression of GD2 synthase significantly diminished the development of the neuron-shaped phenotype in both neuronally induced UCB-MSCs and BM-MSCs, and attenuated up-regulation of the mature neuronal marker, MAP-2. In BM-MSCs, the increase in MAP-2 levels following neurogenic induction was reduced approximately threefold by GD2 suppression (146.3 ± 116.3-fold increase relative to uninduced control in cells treated with GD2-targeting siRNAs versus 450.7 ± 201.6-fold increase in cells treated with control siRNA); in UCB-MSCs, GD2 suppression reduced MAP-2 up-regulation by more than fourfold (35.5 ± 7.7-fold increase in cells treated with GD2-targeting siRNAs versus 158.2 ± 34.3-fold increase in cells treated with control siRNA) (Fig. 5b, c). Restoration of GD2 synthase expression was not detected in target siRNAs-treated cells incubated in neurogenic induction media for up to 6 days (12.9 ± 2.53% of uninduced no siRNA-treated cells) (Fig. 5d). Taken together, these results suggest that down-regulation of GD2 synthase disturbs the processes involved in neuronal fate determination in both BM-MSCs and UCB-MSCs.

Silencing of GD2 expression in UCB-MSCs. a Cells transfected with control siRNA and GD2-targeting siRNAs were homogeneous and spindled-shaped. Fluorescence image shows effective siRNA delivery without cytotoxicity (scale bar = 50 μm). b Down-regulation of GD2 synthase was confirmed by flow cytometry in cells treated with GD2-targeting siRNAs. c siRNA-mediated suppression of GD2 synthase expression was maintained up to 7 days after the transfection. β-actin was used as a control

Quantitative analysis of multi-lineage specific markers in GD2-silenced cells. a Cells transfected with control siRNA and GD2-targeting siRNAs were similar in their capacity to differentiate into osteoblasts (ALP activity), chondrocytes (GAG content), and respiratory epithelial cells (SP-C expression) under each induction condition. b After 6 days of neurogenic induction, the cells treated with GD2-targeting siRNAs showed a decrease in the neuron-shaped phenotype (scale bar 50 μm). c Quantitative real-time PCR analysis revealed that the expression of the mature neuronal marker, MAP-2, was significantly down-regulated in cells from BM-MSC and UCB-MSC cultures transfected with GD2-targeting siRNAs (*p < 0.05). d Silencing of GD2 synthase expression was maintained up to 6 days after neurogenic induction. Each band was analyzed by densitometry. Graphs indicate the level of gene expression normalized to β-actin values (*p < 0.05)
To further examine the mechanistic relationship between GD2 expression and early neuronal development, we tested whether expression of the bHLH transcription factors, MASH1, MATH-1, and neurogenin-1, known to be involved in the early stage of neuronal development [29], were altered by GD2 silencing. Strikingly, we found that treatment of UCB-MSCs with target siRNAs totally abrogated the neurogenic-induced up-regulation of these transcriptional factors, reducing their levels to below those in uninduced control cells (Fig. 6a). In target siRNAs-treated UCB-MSCs, MASH1, which has been shown to appear during the initial development of GABAergic cells [30], was decreased to a level 0.2 ± 0.1 that of uninduced cells compared with a 6.2 ± 2.4-fold increase in induced cells not treated with siRNA and a 6.1 ± 1.7-fold increase in control siRNA-treated cells. In UCB-MSCs, suppression of GD2 decreased the level of neurogenin-1, which is known to be stably expressed in BM-MSCs upon neurogenic induction, to 0.4 ± 0.2 that of control values; in contrast, neurogenin-1 was highly up-regulated in induced cells not treated with siRNA (27.3 ± 4.1-fold) and in those treated with control siRNAs (27.3 ± 3.9-fold) (Fig. 6a). Similar results were obtained in BM-MSCs, except with respect to MATH-1 expression. MATH-1, which is important in regulating the early phase of neural plate formation, was decreased only in target siRNAs-treated UCB-MSCs, and not in BM-MSCs (Fig. 6a). In addition, the expressions of RUNX2, a well-known osteogenic transcription factor [31], and Sox-9, a chondrogenic transcription factor [23, 32], were not altered through any challenge of siRNA treatment upon osteogenic and chondrogenic inductions, respectively, which strengthen our speculation about close relationship between GD2 expression and up-regulation of the neurogenic factors (data not shown).

Quantitative real-time PCR analysis of bHLH transcription factors associated with neurogenesis in human UCB-MSCs. a UCB-MSCs and BM-MSCs were transfected with control siRNA or GD2-targeting siRNAs and exposed to neurogenic-inducing conditions. After 6 days, the levels of MASH1 and neurogenin-1 were dramatically decreased in GD2-targeting siRNAs-treated cells from both UCB-MSC and BM-MSC cultures compared to siRNA-untreated cells and control siRNA-treated cells; MATH-1 was also decreased in UCB-MSCs but not in BM-MSCs. b The GD2+ and GD2− cells sorted from UCB-MSCs were examined for MASH1, MATH-1, and neurogenin-1 expression by real-time PCR following neurogenic inductions. Three bHLH transcription factors were expressed at high levels in GD2+ cells compared to unsorted and GD2− cells (*p < 0.05, **p < 0.01). The levels of all gene transcripts were normalized to those of β-actin, and expressed relative to the levels in unsorted, induced cells, defined as 1
To extend these observations, we also examined the expression of bHLH factors in both GD2+ and GD2− sorted populations of UCB-MSCs following neuronal fate induction (Fig. 6b). As expected, the fold-increase in MASH1 mRNA levels following induction was significantly higher in GD2+ cells (15.4 ± 2.9-fold) than in unsorted (5.3 ± 1.6-fold) or GD2− (1.7 ± 0.1-fold) cells (p < 0.05 for both). Interestingly, MASH1 expression was up-regulated to an even greater extent by induction in GD2+ cells than in unsorted cells. MATH-1 expression was also significantly higher in GD2+ (2.5 ± 0.3-fold increase) and unsorted cells (2.3 ± 0.1-fold increase) than in GD2− cells (0.9 ± 0.1 that of unsorted, uninduced cells; p < 0.05). A similar pattern of up-regulation after neurogenic induction was observed for neurogenin-1, which was increased 3.7 ± 0.2-fold in unsorted cells and 3.1 ± 0.3-fold in the GD2+ population, and was reduced to a level 0.9 ± 0.1 that of unsorted, uninduced cells in GD2− cells (p < 0.01). Collectively, these data imply that GD2+ cells have a higher potential for neuronal differentiation than do GD2− cells, and suggest that GD2 expression might be involved in the up-regulation of neurogenic bHLH transcription factors.
Discussion
Multiple efforts by different laboratories aimed at identifying authentic stem/progenitor cell markers from BM-MNCs or MSCs [14–18] have led to the identification of GD2 ganglioside as a key cellular determinant in adipose tissue- and BM-derived MSCs. Recently, the GD2-positive population of BM-MNCs was shown to possess the capacity for mesoderm-lineage differentiation [15]. However, no reports have established if UCB-MSCs express GD2 on their membrane surfaces and, if so, whether such expression might be functionally associated with the stem cell characteristics of UCB-MSCs.
In the present report, we provide the first demonstration that GD2 ganglioside is present on the cell surface of UCB-MSCs. Moreover, we found that UCB-MSCs in which GD2 expression was reduced, either through siRNA-mediated knockdown or by sorting for a GD2 down-regulated cell population (GD2−), retained the same morphological and immunological phenotypes and mesodermal lineage-differentiation capacity as GD2-expressing cells, indicating that both GD2+ and GD2− UCB-MSC populations meet the per se criteria of MSC defined by the International Society of Cell Therapy (ISCT) [26]. Intriguingly, however, the expression of GD2 was found to be closely associated with up-regulation of neurogenic bHLH transcription factors, which ultimately affect the outcome of neurogenic induction of UCB-MSCs. These data imply that GD2 may be functionally relevant in the early phase of neuronal fate determination of UCB-MSCs. Collectively, our results suggest that GD2 may represent an authentic marker capable of predicting the neurogenic capacity rather than the general stem cell characteristics of UCB-MSCs. Furthermore, GD2− cells showed a higher proliferation rate than GD2+ cells, which might indicate that the GD2− population contains more immature and self-renewing cells than the GD2+ population. These intriguing features could heighten the potential value of UCB-MSCs as candidates for cell-based therapy.
Compared with previous reports to characterize GD2 in MSCs, the UCB-MSC population had the lower fraction of GD2-expressing cells than the BM-MSC population [15]. Also, the GD2+ cells derived from umbilical cord (UC-MSCs) possessed higher proliferation capacity and stronger multi-differentiation potentials compared with the unsorted or GD2− cells [33], which is inconsistent with our results. UCB-MSCs have conceivable features that are distinct from UC-MSCs and BM-MSCs, such as a somewhat restricted or at least delayed adipocyte differentiation capacity [34]. Furthermore, the MSCs from UCB and UC exhibited dissimilar protein-coding gene expression profiles, which might be due to the distinct local environment where they reside [35]. In addition, UC-MSCs have been reported to be comparable to BM-MSCs with respect to their proliferation and differentiation capacities and the two MSCs were proven to share a very similar gene expression profile [36, 37]. Together, despite previously reported biological similarities, UCB-MSCs have features distinguishable to MSCs of different origins, which might support our distinct findings of GD2 as a marker of neurogenic capacity in UC-MSCs.
Gangliosides are ubiquitously expressed in many tissues, including the central nervous system [20], where GD2 plays a modulatory role in balancing the expression both of simple and complex ganglioside on the cell surface [20]. GD2 may also be involved in cell context-specific cellular functions [21, 22]. For example, GD2 ganglioside was reported to be associated with cell proliferation and regulation of apoptosis in small-cell lung cancer cells [28]. With regard to neuronal system development, there are several reports that expression of gangliosides is related to neuronal differentiation and that a ganglioside deficiency leads to down-regulation of genes involved in brain development [38]. Consistent with these observations, recent studies have demonstrated that MSCs spontaneously express neural proteins in culture and are neurogenic after transplantation [39, 40]; it has also been suggested that the expression of gangliosides is closely related to neuronal differentiation of embryonic stem cells in vitro [41]. However, whether GD2 plays a functional role in neuronal developmental processes is not yet clear. Adding to the complexity of establishing the functional relevance of gangliosides in mammalian systems is the existence of hundreds of different ganglioside structures, reflecting the diversity of glycan moieties and variations in acyl lipids linked to the amide group of ceramide. De novo synthesis of ganglioside GD2 occurs as a set of stepwise glycosylations in which the glycosyltransferases, GD3 synthase (α-2,8-sialytransferase) and GD2/GM2 synthase (β1,4-N-acetyl-galactosaminlytransferase), participate in series; the latter enzyme catalyzes the transfer of 1,4-N-acetyl-galactosamine to the reaction moiety of the preceding ganglioside GD3/GM3 [38, 42]. Because of this tandem synthesis mechanism, we achieved more effective suppression of GD2 expression using a mixture of siRNAs consisting of siRNAs targeting GD2 synthase (B4GALNT1) and siRNAs against GD3 synthase (ST9SLA1) than we did using either siRNA alone (data not shown).
Neurogenic bHLH transcription factors are expressed in both central and peripheral nervous systems during development and govern the early phase of neuronal differentiation [29, 43]. Here, we report that GD2 expression was closely associated with up-regulation of several neurogenic bHLH transcriptional factors, including MASH1, MATH-1, and neurogenin-1 upon neurogenic induction. To our knowledge, this is the first report to describe a relationship between GD2 expression and neurogenic transcription factors in UCB-MSCs. Previous investigations have shown that MASH1 acts via sonic hedgehog signaling to enhance the survival of neural precursor cells after transplantation, and neurogenin-1, a proneural gene, is sufficient to convert the mesodermal fate of MSCs to a neuronal fate [44, 45]. Both MASH1 and neurogenin-1 are up-regulated in GABAergic cells derived from primary fetal hippocampal neurons (unpublished observations), and MASH1 has been reported to selectively induce GABAergic cells via the Akt pathway [30]. GABAergic cells emerge at the very early stage of neuronal development. Here, all neurogenic transcriptional factors tested were up-regulated following neurogenic stimulation in both UCB-MSCs and BM-MSCs. With the exception of MATH-1 in BM-MSCs, the expression of these factors was highly responsive to GD2 suppression, which reduced their levels remarkably, even to levels below those in uninduced control cells. Notably, MASH1 was more up-regulated upon neurogenic induction in the GD2+ enriched population of UCB-MSCs than in the unsorted population (Fig. 6b). Taken together, these results allow us to speculate that, given the proper conditions, UCB-MSCs are more predisposed to differentiate toward certain neuronal fates, such as GABAergic cells, than are other MSCs, and GD2 is necessarily involved in the early processes of GABAergic neuronal fate determination.
A number of fundamental issues should command attention in formulating future research directions. First is the likelihood that GD2− cells exert regulatory actions that counter those of GD2+ cells, a factor that may account for the high susceptibility of the GD2+-enriched population to neurogenic induction. Second, why the expression of MASH1 is even higher in UCB-MSCs than in BM-MSCs upon neurogenic stimulation despite the fact that the percentage of GD2+ cells in UCB-MSCs is less than that in BM-MSCs is not clear and should be addressed. Third, it is important to determine the specific fate of cells committed to the neuronal lineage, for example, whether they are GABAergic or glutaminergic neurons. Additionally, it will be interesting to determine which types of neurons the GD2+ enriched population is capable of differentiating into in vivo, and whether GD2 governs the expression of neurogenic transcription factors in neural stem cells at an early developmental stage. Finally, studies designed to elucidate the mechanism by which GD2 ganglioside leads MSCs to neuronal predisposition are warranted.
In conclusion, the expression of GD2 ganglioside is closely associated with up-regulation of early neurogenic transcriptional factors and may serve as an authentic marker for the neurogenic capacity, but not the general stem cell characteristics, of UCB-MSCs. These findings may expand the range of potential clinical applications of MSC-based therapy in neurodegenerative diseases and suggest that enrichment for GD2+ cells may be a prerequisite for successful clinical outcome.
References
Jiang Y, Jahagirdar BN, Reinhardt RL, Schwartz RE, Keene CD, Ortiz-Gonzalez XR, Reyes M, Lenvik T, Lund T, Blackstad M, Du J, Aldrich S, Lisberg A, Low WC, Largaespada DA, Verfaillie CM (2002) Pluripotency of mesenchymal stem cells derived from adult marrow. Nature 418:41–49
Lee OK, Kuo TK, Chen WM, Lee KD, Hsieh SL, Chen TH (2004) Isolation of multipotent mesenchymal stem cells from umbilical cord blood. Blood 103:1669–1675
Caplan AI, Dennis JE (2006) Mesenchymal stem cells as trophic mediators. J Cell Biochem 98:1076–1084
Gnecchi M, He H, Noiseux N, Liang OD, Zhang L, Morello F, Mu H, Melo LG, Pratt RE, Ingwall JS, Dzau VJ (2006) Evidence supporting paracrine hypothesis for Akt-modified mesenchymal stem cell-mediated cardiac protection and functional improvement. Faseb J 20:661–669
Parekkadan B, van Poll D, Megeed Z, Kobayashi N, Tilles AW, Berthiaume F, Yarmush ML (2007) Immunomodulation of activated hepatic stellate cells by mesenchymal stem cells. Biochem Biophys Res Commun 363:247–252
Kern S, Eichler H, Stoeve J, Kluter H, Bieback K (2006) Comparative analysis of mesenchymal stem cells from bone marrow, umbilical cord blood, or adipose tissue. Stem Cells 24:1294–1301
Fukuchi Y, Nakajima H, Sugiyama D, Hirose I, Kitamura T, Tsuji K (2004) Human placenta-derived cells have mesenchymal stem/progenitor cell potential. Stem Cells 22:649–658
Gronthos S, Mankani M, Brahim J, Robey PG, Shi S (2000) Postnatal human dental pulp stem cells (DPSCs) in vitro and in vivo. Proc Natl Acad Sci USA 97:13625–13630
Flynn A, Barry F, O’Brien T (2007) UC blood-derived mesenchymal stromal cells: an overview. Cytotherapy 9:717–726
Kim DS, Kim JH, Lee JK, Choi SJ, Kim JS, Jeun SS, Oh W, Yang YS, Chang JW (2009) Overexpression of CXC chemokine receptors is required for the superior glioma-tracking property of umbilical cord blood-derived mesenchymal stem cells. Stem Cells Dev 18:511–519
Kim SW, Han H, Chae GT, Lee SH, Bo S, Yoon JH, Lee YS, Lee KS, Park HK, Kang KS (2006) Successful stem cell therapy using umbilical cord blood-derived multipotent stem cells for Buerger’s disease and ischemic limb disease animal model. Stem Cells 24:1620–1626
Bruder SP, Jaiswal N, Haynesworth SE (1997) Growth kinetics, self-renewal, and the osteogenic potential of purified human mesenchymal stem cells during extensive subcultivation and following cryopreservation. J Cell Biochem 64:278–294
Owen ME, Cave J, Joyner CJ (1987) Clonal analysis in vitro of osteogenic differentiation of marrow CFU-F. J Cell Sci 87(Pt 5):731–738
Gang EJ, Bosnakovski D, Figueiredo CA, Visser JW, Perlingeiro RC (2007) SSEA-4 identifies mesenchymal stem cells from bone marrow. Blood 109:1743–1751
Martinez C, Hofmann TJ, Marino R, Dominici M, Horwitz EM (2007) Human bone marrow mesenchymal stromal cells express the neural ganglioside GD2: a novel surface marker for the identification of MSCs. Blood 109:4245–4248
Baksh D, Yao R, Tuan RS (2007) Comparison of proliferative and multilineage differentiation potential of human mesenchymal stem cells derived from umbilical cord and bone marrow. Stem Cells 25:1384–1392
Buhring HJ, Battula VL, Treml S, Schewe B, Kanz L, Vogel W (2007) Novel markers for the prospective isolation of human MSC. Ann N Y Acad Sci 1106:262–271
Bae S, Park CW, Son HK, Ju HK, Paik D, Jeon CJ, Koh GY, Kim J, Kim H (2008) Fibroblast activation protein alpha identifies mesenchymal stromal cells from human bone marrow. Br J Haematol 142:827–830
Hakomori S (1990) Bifunctional role of glycosphingolipids. Modulators for transmembrane signaling and mediators for cellular interactions. J Biol Chem 265:18713–18716
Giraudo CG, Rosales Fritz VM, Maccioni HJ (1999) GA2/GM2/GD2 synthase localizes to the trans-golgi network of CHO-K1 cells. Biochem J 342(Pt 3):633–640
Yu RK, Macala LJ, Taki T, Weinfield HM, Yu FS (1988) Developmental changes in ganglioside composition and synthesis in embryonic rat brain. J Neurochem 50:1825–1829
Yu RK (1994) Development regulation of ganglioside metabolism. Prog Brain Res 101:31–44
Lee JK, Lee MK, Jin HJ, Kim DS, Yang YS, Oh W, Yang SE, Park TS, Lee SY, Kim BS, Jeun SS (2007) Efficient intracytoplasmic labeling of human umbilical cord blood mesenchymal stromal cells with ferumoxides. Cell Transplant 16:849–857
Jin HJ, Park SK, Oh W, Yang YS, Kim SW, Choi SJ (2009) Down-regulation of CD105 is associated with multi-lineage differentiation in human umbilical cord blood-derived mesenchymal stem cells. Biochem Biophys Res Commun 381:676–681
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25:402–408
Dominici M, Le Blanc K, Mueller I, Slaper-Cortenbach I, Marini F, Krause D, Deans R, Keating A, Prockop D, Horwitz E (2006) Minimal criteria for defining multipotent mesenchymal stromal cells, The International Society for Cellular Therapy position statement. Cytotherapy 8:315–317
Lopez PH, Schnaar RL (2009) Gangliosides in cell recognition and membrane protein regulation. Curr Opin Struct Biol 19:549–557
Yoshida S, Fukumoto S, Kawaguchi H, Sato S, Ueda R, Furukawa K (2001) Ganglioside G(D2) in small cell lung cancer cell lines: enhancement of cell proliferation and mediation of apoptosis. Cancer Res 61:4244–4252
Bertrand N, Castro DS, Guillemot F (2002) Proneural genes and the specification of neural cell types. Nat Rev Neurosci 3:517–530
Oishi K, Watatani K, Itoh Y, Okano H, Guillemot F, Nakajima K, Gotoh Y (2009) Selective induction of neocortical GABAergic neurons by the PDK1-Akt pathway through activation of Mash1. Proc Natl Acad Sci USA 106:13064–13069
Zhao Z, Zhao M, Xiao G, Franceschi RT (2005) Gene transfer of the Runx2 transcription factor enhances osteogenic activity of bone marrow stromal cells in vitro and in vivo. Mol Ther 12:247–253
Heng BC, Cao T, Lee EH (2004) Directing stem cell differentiation into the chondrogenic lineage in vitro. Stem Cells 22:1152–1167
Xu J, Liao W, Gu D, Liang L, Liu M, Du W, Liu P, Zhang L, Lu S, Dong C, Zhou B, Han Z (2009) Neural ganglioside GD2 identifies a subpopulation of mesenchymal stem cells in umbilical cord. Cell Physiol Biochem 23:415–424
Rebelatto CK, Aguiar AM, Moretao MP, Senegaglia AC, Hansen P, Barchiki F, Oliveira J, Martins J, Kuligovski C, Mansur F, Christofis A, Amaral VF, Brofman PS, Goldenberg S, Nakao LS, Correa A (2008) Dissimilar differentiation of mesenchymal stem cells from bone marrow, umbilical cord blood, and adipose tissue. Exp Biol Med (Maywood) 233:901–913
Secco M, Moreira YB, Zucconi E, Vieira NM, Jazedje T, Muotri AR, Okamoto OK, Verjovski-Almeida S, Zatz M (2009) Gene expression profile of mesenchymal stem cells from paired umbilical cord units: cord is different from blood. Stem Cell Rev Rep 5:387–401
Panepucci RA, Siufi JL, Silva WA Jr, Proto-Siquiera R, Neder L, Orellana M, Rocha V, Covas DT, Zago MA (2004) Comparison of gene expression of umbilical cord vein and bone marrow-derived mesenchymal stem cells. Stem Cells 22:1263–1278
Secco M, Zucconi E, Vieira NM, Fogaca LL, Cerqueira A, Carvalho MD, Jazedje T, Okamoto OK, Muotri AR, Zatz M (2008) Multipotent stem cells from umbilical cord: cord is richer than blood!. Stem Cells 26:146–150
Takamiya K, Yamamoto A, Furukawa K, Yamashiro S, Shin M, Okada M, Fukumoto S, Haraguchi M, Takeda N, Fujimura K, Sakae M, Kishikawa M, Shiku H, Furukawa K, Aizawa S (1996) Mice with disrupted GM2/GD2 synthase gene lack complex gangliosides but exhibit only subtle defects in their nervous system. Proc Natl Acad Sci USA 93:10662–10667
Blondheim NR, Levy YS, Ben-Zur T, Burshtein A, Cherlow T, Kan I, Barzilai R, Bahat-Stromza M, Barhum Y, Bulvik S, Melamed E, Offen D (2006) Human mesenchymal stem cells express neural genes, suggesting a neural predisposition. Stem Cells Dev 15:141–164
Deng J, Petersen BE, Steindler DA, Jorgensen ML, Laywell ED (2006) Mesenchymal stem cells spontaneously express neural proteins in culture and are neurogenic after transplantation. Stem Cells 24:1054–1064
Lee DH, Koo DB, Ko K, Ko K, Kim SM, Jung JU, Ryu JS, Jin JW, Yang HJ, Do SI, Jung KY, Choo YK (2007) Effects of daunorubicin on ganglioside expression and neuronal differentiation of mouse embryonic stem cells. Biochem Biophys Res Commun 362:313–318
Furukawa K, Takamiya K, Furukawa K (2002) Beta1, 4-N-acetylgalactosaminyltransferase–GM2/GD2 synthase: a key enzyme to control the synthesis of brain-enriched complex gangliosides. Biochim Biophys Acta 1573:356–362
Ross SE, Greenberg ME, Stiles CD (2003) Basic helix-loop-helix factors in cortical development. Neuron 39:13–25
Yi SH, Jo AY, Park CH, Koh HC, Park RH, Suh-Kim H, Shin I, Lee YS, Kim J, Lee SH (2008) Mash1 and neurogenin 2 enhance survival and differentiation of neural precursor cells after transplantation to rat brains via distinct modes of action. Mol Ther 16:1873–1882
Kim SS, Yoo SW, Park TS, Ahn SC, Jeong HS, Kim JW, Chang DY, Cho KG, Kim SU, Huh Y, Lee JE, Lee SY, Lee YD, Suh-Kim H (2008) Neural induction with neurogenin1 increases the therapeutic effects of mesenchymal stem cells in the ischemic brain. Stem Cells 26:2217–2228
Acknowledgments
This work was supported in part by a grant from the Korea Health 21 R&D Project, Ministry of Health and Welfare, Republic of Korea (A084036), and by Basic Science Research Program through the National Research Foundation of Korea funded by the Ministry of Education, Science and Technology (2009-0089589 to SW Kim).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Jin, H.J., Nam, H.Y., Bae, Y.K. et al. GD2 expression is closely associated with neuronal differentiation of human umbilical cord blood-derived mesenchymal stem cells. Cell. Mol. Life Sci. 67, 1845–1858 (2010). https://doi.org/10.1007/s00018-010-0292-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-010-0292-z