
Abstract
Alzheimer’s disease (AD) should be regarded as a degenerative metabolic disease caused by brain insulin resistance and deficiency, and overlapping with the molecular, biochemical, pathophysiological, and metabolic dysfunctions in diabetes mellitus, non-alcoholic fatty liver disease, and metabolic syndrome. Although most of the diagnostic and therapeutic approaches over the past several decades have focused on amyloid-beta (Aβ42) and aberrantly phosphorylated tau, which could be caused by consequences of brain insulin resistance, the broader array of pathologies including white matter atrophy with loss of myelinated fibrils and leukoaraiosis, non-Aβ42 microvascular disease, dysregulated lipid metabolism, mitochondrial dysfunction, astrocytic gliosis, neuro-inflammation, and loss of synapses vis-à-vis growth of dystrophic neurites, is not readily accounted for by Aβ42 accumulations, but could be explained by dysregulated insulin/IGF-1 signaling with attendant impairments in signal transduction and gene expression. This review covers the diverse range of brain abnormalities in AD and discusses how insulins, incretins, and insulin sensitizers could be utilized to treat at different stages of neurodegeneration.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
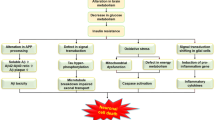
Alzheimer’s disease should be regarded as an insulin-resistance-mediated neurodegenerative disorder that has the same fundamental abnormalities that occur in diabetes mellitus, metabolic syndrome, and non-alcoholic fatty liver disease. |
Contrary to popular perception, Alzheimer’s disease is associated with a number of major abnormalities in the brain which are not attended to by diagnostic and therapeutic approaches that specifically target amyloid-beta and phospho-tau accumulation. |
Disease remediation for Alzheimer’s and probably many other neurodegenerative diseases should be approached by attacking underlying impairments in the actions of insulin and insulin-like growth factors. Orchestrating the repertoire of drugs that support their multifaceted functions in the brain by efficiently and safely delivering insulins (short-, long- and ultralong-acting forms), incretins, and insulin sensitizers for disease-stage intervention could slow or halt progression of neurodegeneration. |
1 Introduction
Alzheimer’s disease (AD) is clinically manifested by progressive behavioral changes, loss of recent, i.e. short-term memory, declines in executive functions, and deficits in cognition [1]. Through structured longitudinal neuropsychological assessments of memory, intellectual function, and language skills, a diagnosis of possible or probable AD can be rendered. However, higher levels of diagnostic accuracy can be achieved using laboratory tests such as paired cerebrospinal fluid (CSF) and serum assays of amyloid precursor protein-amyloid beta 1-42 peptide (Aβ42) and phospho-tau (pTau231) [2, 3], and neuroimaging [1], particularly magnetic resonance imaging (MRI) of the brain [4, 5], functional MRI (fMRI), diffusion tensor imaging (DTI) [6], single-photon emission computed tomography (SPECT), positron emission tomography (PET), and magnetic resonance spectroscopy (MRS) [7, 8].
1.1 Characteristic Neuropathology
Typically, neurodegeneration begins before it becomes clinically manifested as the typical neuropathological changes are detectable by postmortem examination of asymptomatic individuals. The brain structures hit earliest by AD include medial temporal and orbitofrontal regions, which are linked to neuronal plasticity needed for learning and memory. Over time, neurodegeneration grows in severity and distribution, with initial involvement of corticolimbic structures, followed by progressive destruction of other regions within the cerebral hemispheres.
Neurodegeneration is manifested by atrophy of cortical, white matter, and medial temporal structures with loss of neurons and synaptic terminals, neuro-inflammation, reactive astrocytosis, micro-vascular disease, accumulations of hyper-phosphorylated tau (pTau)-containing cytoskeletal lesion, increased amyloid-beta (Aβ42) deposits in plaques, vessels, and neurons, and increased ubiquitin immunoreactivity in degenerating neurons [9–11]. Aberrant phosphorylation of tau via inappropriate activation of kinases causes Tau fibrillization, aggregation, and ubiquitination, followed by the stress-activated unfolded protein response (UPR), and ultimately cell death. Insoluble, fibrillar aggregates of hyper-phosphorylated and ubiquitinated Tau produce characteristic paired-helical filaments (PHFs) that are detectable by transmission electron microscopy and immunohistochemical staining. PHFs are major components of neurofibrillary tangles, dystrophic neurites, and neuropil threads which are signature AD lesions [12].
Aβ42 is a ~4 kD peptide generated by secretase cleavage of amyloid beta precursor protein (AβPP). Under normal circumstances, Aβ42 is continuously cleared from the brain by transport into the general circulation [13]. In aging and AD, Aβ42 accumulates in cortical and leptomeningeal vessel walls, cortical and sub-cortical perivascular spaces, and plaques, and as neurotoxic, oligomeric soluble diffusible ligands (ADDLs) [14, 15].
1.2 AD Pathologies Unrelated to Aβ42 and pTau
Although a neuropathologic diagnosis of AD is rendered by assessing the distribution and abundance of neurofibrillary tangles and senile plaques [10], the features of neurodegeneration are far broader and include: neuronal loss; neuro-inflammation; gliosis; white matter degeneration; and vascular degeneration, particularly in white matter [16, 17]. In addition, impairments in brain metabolism (glucose and oxygen utilization), although recognized for decades and frequently assessed, has not been incorporated into the cluster AD biomarkers. Failure to consider these additional aspects of AD limits opportunity to fully understand the nature of disease and therapeutically target its underlying basis. For example, significant degenerative changes emerge in cerebral white matter and micro-vessels early in AD; these abnormalities can be more conspicuous than neurofibrillary tangles and plaques. Restricting therapeutic interventions to Aβ42 and pTau accumulations would not attend to other major pathologies that impair function and worsen with AD severity.
2 Neuro-Inflammation
Inflammatory responses in brains with neurodegeneration have been recognized for years but only recently have gained renewed attention as potential mediators of AD. Neuro-inflammation refers to the presence of activated microglia and astrocytes which cause injury through expression and release of pro-inflammatory cytokines, chemokines, and complement, increased generation of membrane fatty acids, eicosanoids, lipid peroxidation products, and reactive oxygen and reactive nitrogen species [18–20].
2.1 Neuro-Inflammation as a Mediator of Neurodegeneration
Neuro-inflammation is an early and consistent feature of many neurodegenerative diseases, including AD. Increased expression of pro-inflammatory cytokines such as interleukin-1β (IL-1β), IL-6, interferon-gamma, and macrophage migration inhibitory factor in the vicinities of Aβ42 plaques supports the concept that neuro-inflammation is an important mediator or propagator of AD neurodegeneration [21, 22]. In addition, neuro-inflammation promotes neuronal injury and cholinergic dysfunction [23]. Downstream effects include oxidative stress with increased production of reactive oxygen and reactive nitrogen species, which can damage nerve terminals, causing synaptic dysfunction and attendant cognitive impairment [19]. Furthermore, since chronic inflammation is known to exacerbate insulin resistance associated with systemic disease-states [24–28], neuro-inflammation could also have an important etiopathic role in the brain insulin and insulin-like growth factor-1 (IGF-1) resistances that occur in AD [29–34] and Parkinson’s disease [35]. Furthermore, inflammation has inhibitory effects on incretin (orexin) expression and function, while reduced insulin resistance afforded by activation of incretin signaling inhibits pro-inflammatory cytokines such as tumor necrosis factor-alpha (TNF-α) [36].
2.2 Role of Insulin Resistance in Neuro-Inflammation
Despite compelling evidence for a possible causal role of neuro-inflammation in neurodegeneration, its common presence in many other central nervous system (CNS) diseases including multiple sclerosis, malignant brain tumors, infections, traumatic injury, ischemic injury and stroke, perinatal leukoencephalopathy, and toxic-metabolic disorders suggests that such responses may be reactive rather than causal. In that regard, neuro-inflammation may produce secondary bystander injury rather than operate as the primary driver of neurodegeneration. In humans with peripheral insulin resistance diseases, including diabetes mellitus and metabolic syndrome, cognitive impairment and AD are partly driven by brain insulin resistance, together with neuro-inflammation and vasculopathy [37–39].
One of the brain abnormalities in AD that can be linked to both insulin resistance and neuroinflammation is down-regulation of peroxisome proliferator activator receptor (PPAR) delta [40]. In brain, PPAR-delta is the most abundantly expressed of the three isoforms of PPAR nuclear hormone receptors (alpha, beta/delta, and gamma) [41]. PPARs play critical roles in modulating insulin-stimulated gene expression in response to signals transmitted from surface membranes [42]. However, in addition to their insulin sensitizing actions, ligand activation of PPARs has potent anti-inflammatory effects [42–44]. The findings that, (1) PPAR-delta expression is reduced in AD brains [40]; (2) experimental depletion of PPAR-delta increases neuro-inflammation, astrogliosis, oxidative stress, Aβ42 deposition, and PHF tau [45]; (3) PPAR-delta agonists are neuroprotective [46]; and (4) PPAR-delta agonists reduce neuro-inflammation and Aβ42 deposition [44, 47, 48], convincingly support the notion that impairments in insulin signaling could account for many major abnormalities in AD, including neuro-inflammation. However, the fact that neuro-inflammation exacerbates insulin resistance, neurotoxic and oxidation-mediated cell death, gliosis, Aβ42 toxicity, and PHF pathology means that the association between insulin resistance and neuro-inflammation is tight. Another possible interpretation of this scenario is that insulin resistance and neuro-inflammatory pathologies co-conspire in a positive feedback loop to mediate neurodegeneration.
2.3 Oxidative Stress Contributes to Neurodegeneration
Neurodegeneration is consistently associated with oxidative stress resulting from increased generation of reactive oxygen and reactive nitrogen species [49]. These products exert their neurotoxic effects by reacting with macromolecules including lipids, nucleic acids (RNA and DNA), and proteins, causing their dysfunction [49, 50]. Oxidative damage occurs at very early stages of neurodegeneration [51] and has been linked to mitochondrial dysfunction in AD and other neurodegenerative diseases [52, 53]. Like neuro-inflammation, oxidative stress and free radical injury are not specific to neurodegeneration. Although oxidative stress can be caused by a broad range of exposures including hypoxia, ischemia, and insulin resistance, the resulting molecular, biochemical and cellular abnormalities overlap extensively with related pathologies in AD [54].
A long-standing hypothesis is that free radical stress and injury in AD are mediated by Fenton-type reactions [55] linked to excess iron accumulation in the brain [56]. This concept is supported by experimental evidence that impairments in iron metabolism increase levels of neuronal iron and lipid-peroxidation and protein carbonyl adducts in the brain [57]. In AD and many other neurodegenerative diseases, heme accumulation is regarded as a common and important mediator of oxidative stress [58]. In the brain, the deleterious effects of heme-induced free radical damage are broad and include inhibition of the muscarinic acetylcholine receptor [59], which likely contributes to cognitive decline in AD. Correspondingly, heme-associated impairments in muscarinic acetylcholine receptor function can be prevented by anti-oxidant treatment [59]. Heme oxygenases function by either dynamically or constitutively degrading heme to ferrous iron, carbon monoxide and biliverdin-IX-alpha. Heme oxygenases’ anti-oxidant responses protect cells from injury [49] and activate the alpha7 nicotinic receptor [60]. Correspondingly, they exert their neuroprotective actions in part by inducing of heme-oxygenase 1 (HO-1) and reducing the levels of redox active iron in the brain.
Further evidence that iron accumulation and oxidative stress have etiopathic roles in AD stems from studies showing that deferoxamine treatment of P301L transgenic mice normalized performance in the radial arm water maze [61]. The P301L AD model is associated with phospho-Tau accumulations in the brain. Mechanistically, deferoxamine treatments reduced neuroinflammation, protein oxidation, and GSk-3β activation without altering brain levels of phospho-Tau [61]. Similarly, in the APP/PS1 mouse model, intranasal deferoxamine significantly improved performance in spatial learning and memory [62] while reducing GSK-3β activation, oxidative stress, and levels of soluble Aβ40 and Aβ42 in the brain [63]. Despite these compelling insights, the ability to extend this logic to human diseases is limited by the uncommon and uncharacteristic nature of the experimental mouse genetic profiles relative to most human cases, and the fact that the causes of brain iron accumulation in these models are not obvious.
2.4 The Insulin Resistance, Oxidative Stress, Iron Overload, Neurodegeneration Network
Insulin resistance promotes to oxidative stress by dysregulating carbohydrate and lipid metabolism, increasing GSK-3β activation, and impairing cell survival/anti-apoptotic signaling, energy balance, mitochondrial function [54], and choline acetyltransferase and neurotrophin gene expression [64]. Brain insulin resistance is also associated with increased levels of phospho-Tau and Aβ42 [54]. Iron accumulation occurs in both AD and type 2 diabetes mellitus [65]. Reducing iron load in the body enhances glycemic control in type 2 diabetes [65], just as deferoxamine treatment reverses or prevents AD-type abnormalities in experimental models [62, 63]. Paradoxically, HO-1 expression is substantially elevated in AD brains [66] and peripheral blood [67, 68]. Since HO-1 degrades heme, the up-regulated expression in AD suggests that endogenous neuroprotective mechanisms may become activated as a compensatory response to neurodegeneration. However, this explanation has been called into question by studies showing that in postmortem brains of patients with AD or mild cognitive impairment (MCI), HO-1 protein is post-translationally modified by serine phosphorylation, which increases its activity, and lipid peroxidation adducts, which would impair its function [69]. Thus, it is uncertain whether the aberrantly elevated levels of HO-1 in AD brains represent responses to neurodegeneration or oxidative damage and attendant inhibition of enzyme activity.
The findings in several studies that various aspects of neurodegeneration were abrogated by reducing heme make it difficult to refute the argument that neurodegeneration in insulin resistance diseases is mediated by dysregulation of iron metabolism, oxidative stress, and free radical damage [65, 68]. However, further exploration of this concept was enabled by review of neuro-cognitive abnormalities associated with genetic disorders of iron metabolism that lead to iron overload. Of particular interest is that hemochromatosis has been linked to glucose intolerance, insulin resistance, cognitive-motor impairments, and neurodegeneration [70–73], and studies have shown that the glucose intolerance and insulin resistance improve with iron chelation therapy [74]. Unfortunately, these observations are also not entirely conclusive since people with hemochromatosis also have liver disease, and hepatic dysfunction can also cause cognitive-motor deficits and neurodegeneration.
Further studies addressed the same question using transgenic mouse models of hemochromatosis (HFE). One study found no evidence to support a direct and causal role for dysregulated iron metabolism in the pathogenesis of AD since the associated alterations in brain mRNA levels did not confer increased risk for AD-type neurodegeneration [75]. Additionally relevant findings in HFE models were that, (1) brain iron accumulations were most abundant in regions that regulate motor rather than cognitive functions; and (2) HFE hemochromatosis gene expression was mainly localized in choroid plexus epithelial cells, vascular endothelial cells, and ependymal lining cells rather than neurons and glia [70]. Despite early studies demonstrating probable links between the mutant hemochromatosis gene (HFE H63D) and increased risk for AD [76], other groups observed equivocal associations [77], and a later meta-analysis revealed that mutant HFE (H63D) most likely plays a protective role in reducing AD risk [78]. Additional research is needed to better understand how dysregulated iron metabolism contributes to neurodegeneration and cognitive impairment. One consideration is that iron accumulation may impair mitochondrial function and thereby cause oxidative stress and mitochondrial DNA damage, and metabolic dysfunction [79–82]. Mitochondrial DNA damage and dysfunction are well-recognized features of AD [53, 83] and other neurodegenerative diseases [84].
3 Brain Metabolic Dysfunction in AD
3.1 Deficits in Brain Glucose Utilization
The constellation of progressive neuropathological abnormalities in AD illustrates the need to conceptualize pathogenic mechanisms in ways that accommodate all aspects of disease rather than focus on just neurofibrillary tangles and senile plaques. The fact that most aspects of AD are fairly consistent from case to case suggests that the underlying basis of the seemingly unrelated pathologies may be shared. Deficits in brain energy metabolism, particularly with respect to glucose utilization in AD have been recognized for years. PET imaging with 18F-fluorodeoxyglucose (18F-FDG) is the standard approach for detecting early impairments in brain glucose metabolism [85, 86]. The most significant finding across multiple studies is that AD is associated with global reductions in brain glucose metabolism relative to normal healthy control brains [87–90].
3.2 Insulin Functions in the Brain
Insulin and insulin-like growth factor, type 1 (IGF-1) polypeptides (growth factors) and receptors are expressed in the brain, most abundantly in regions that are most vulnerable to AD neurodegeneration [91]. Insulin regulates neuronal and oligodendroglial cell survival and neuronal plasticity [91, 92]. Experimental models have shown that brain insulin resistance or deficiency impairs learning and memory [64]. In early or intermediate stages of AD, brain and CSF levels of insulin are decreased [34], while Aβ42 and advanced glycation end-products are increased [34, 54, 93]. High levels of Aβ42 in brain and CSF are associated with low levels in serum due to decreased clearance [94]. Insulin administration improves working memory and cognition [95–98] and enhances Aβ42 clearance from the brain [98].
3.3 Primary Brain Insulin and IGF Deficiency and Resistance in AD—Type 3 Diabetes
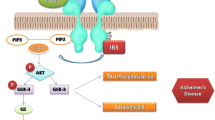
To validate the roles of insulin and IGF deficiencies and resistances in AD, human postmortem brains with different Braak stage severities of AD were used to measure expression levels of the corresponding trophic factors and receptors, ligand-receptor binding, and constitutive activation of downstream signaling [31, 99, 100]. Those investigations revealed significant AD stage-dependent declines in the expression of molecules needed to relay insulin and IGF-1 signaling including ligands, receptors and insulin-receptor substrate, type 1 (IRS-1), reduced insulin and IGF-1 binding to their cognate receptors, impairments in signaling through PI3K-Akt pathways needed for neuronal survival, plasticity, and metabolism, together with increased GSK-3β activation [31, 99, 100]. Of further significance was the finding that impairments in brain insulin, IGF-1, and IGF-2 signaling mechanisms were correlated with reduced expression of choline acetyltransferase, which is needed to generate acetylcholine [31, 99, 100]. Subsequent studies confirmed that insulin and IGF-1 resistance [101] and impaired signaling through IRS proteins with attendant increased activation of GSK-3β and suppression of PI3K-Akt [32] were fundamental features of AD in human brains.
The concept that brain insulin/IGF resistance and deficiency have significant roles in the pathogenesis of AD is supported by data showing that in the early stages of disease, CSF levels of insulin, IGF-1, nerve growth factor, and glial-derived neurotrophic factor levels were significantly decreased while neuroinflammatory indices were increased relative to aged controls [34]. That study was highly significant because a postmortem diagnosis of AD was confirmed in all patients [34]. Together, these observations suggest that besides pTau and Aβ42, biomarkers of brain metabolic and neurotrophin dysfunction should be included in CSF panels designed to aid in early detection of AD.
Because the human AD-associated abnormalities in insulin and IGF-1 signaling are highly reminiscent of what occurs in both type 1 (T1DM) and type 2 diabetes mellitus (T2DM), yet they selectively involve the brain, we coined the term ‘type 3 diabetes’ [99]. The objective was to convey the concept that AD is a brain form of diabetes in which both ligand (insulin and IGF-1) deficiencies and receptor resistances account for functional impairments in activating downstream signaling pathways. In addition to the deficits in insulin and IGF-1 signaling, cognitive impairment and AD are associated with reduced sensitivity (resistance) or ligand deficiencies pertaining to leptin and neurotrophins [34, 102, 103], and therapeutic responsiveness to incretins [104–106]; these abnormalities also occur in obesity, T2DM and other insulin-resistance diseases [107–113]. Therefore, in both type 3 diabetes and peripheral insulin resistance diseases, the mediators and correlates of metabolic dysfunction extend beyond impairments in insulin and IGF-1 signaling and more broadly include deficits in neurotrophin functions. For ‘type 3 diabetes’, although we hypothesized that the brain is the main target of metabolic dysfunction, since none of the subjects in our initial studies had clinical evidence of peripheral insulin resistance, emerging evidence suggests that the CNS regulates glucose metabolism in the body [114, 115] and that CNS impairments in metabolic signaling may drive obesity and peripheral insulin resistance [116–119]. Altogether, these concepts point toward the use of insulin and/or insulin sensitizer drugs for remediating brain diabetes early in the course of disease to prevent its progression and potential precipitation or exacerbation of systemic insulin resistance diseases.
3.4 Secondary Systemic Insulin Resistance Diseases and Cognitive Impairment
Analysis of epidemiological trends over several decades shows that the rates of AD increased sharply within all age groups, 50 years and above [120]. Parallel trends occurred for diabetes mellitus [120], suggesting that the underlying factors might be related. Equally important, the rapid shifts in disease prevalence correspond with effects of exposures rather than genetic factors. Correspondingly, data generated from multiple laboratories and institutions have shown that overweight or obese people and diabetics are at increased risk for developing cognitive impairment and dementia [121–125]. Furthermore, individuals with inadequately controlled T1DM and T2DM also have higher rates of cognitive impairment [126–131] and a two-fold or greater risk of developing AD relative to people without diabetes [132]. Other studies have linked various forms of peripheral insulin resistance [133, 134] including pre-diabetes [127], metabolic syndrome [135], high fat diet-induced obesity [136, 137], and non-alcoholic fatty liver disease [138–140] to AD-type pathology and cognitive decline. Finally, multivariate analysis of a late onset AD international, multicenter cohort identified gene clusters associated with inflammation, diabetes, and obesity as pathologic processes linked to neurodegeneration [141].
In light of the evidence that various peripheral insulin resistance diseases can negatively impact brain structure and function and cause brain insulin resistance, it must be acknowledged that cognitive impairment and AD-type neurodegeneration can emerge either as primary or secondary CNS disease processes. In other words, ‘type 3 diabetes’ can arise in isolation (primary) with selective involvement of the brain, or as a consequence of systemic insulin resistance (secondary) due to obesity, diabetes mellitus, non-alcoholic fatty liver disease, or metabolic syndrome. Yet a third way to conceptualize the problem is to regard insulin resistance diseases as one process that can afflict one or multiple organs and tissues in the same way that atherosclerosis can target one or more vessels and produce distinct manifestations of disease. Importantly, the explanation for the rapid increase in rates of cognitive impairment and AD, even adjusting for age, probably is rooted in the parallel epidemics of obesity and other insulin resistance diseases rather than genetics per se. The good news is that the exposure-induced excesses in rates of neurodegeneration are probably preventable and treatable. The strategies are likely to be similar to those used and continuously under development to manage diabetes mellitus.
4 Therapeutic Strategies for Abrogating Brain Insulin Deficiency and Insulin Resistance in AD
4.1 Rational Therapeutic Options
AD neurodegeneration is associated with energy imbalance, dysregulated lipid and carbohydrate metabolism, cytokine-mediated inflammation, increased oxidative and other types of cellular stresses, on-going cell death, and vascular degeneration. The finding that these abnormalities are also present in T2DM, metabolic syndrome, and non-alcoholic fatty liver disease supports the concept that insulin-resistance diseases are all inter-related, could have the same root causes, and may be managed by similar if not identical therapeutic strategies. Therefore, in designing treatment approaches, it would be prudent to remain mindful of the full range of molecular and cellular pathologies that must be addressed, and determine if the treatment responses are short-term or long-term, cell- or tissue-type specific, and broadly evident across different aspects of disease. Furthermore, non-invasive approaches are needed to assess and monitor the impact of various treatments on each component of neurodegeneration.
Regarding AD, any drug that can enhance brain glucose utilization and insulin signaling through the PI3K-Akt pathway, and thereby support neuronal survival, plasticity, and metabolism, should be regarded as a first-line therapeutic agent. Insulins should be given high-level consideration because besides stimulating insulin pathways, inflammation, cellular stress and neurotransmitter deficits are responsive to insulin. In contrast, anti-inflammatory agents, anti-oxidants and cholinesterase inhibitor drugs have narrower targeted effects, although they should be included with multi-pronged therapeutic regimens. Most of the chronic and progressive abnormalities, including white matter and vascular degeneration, can be attributed at least in part to brain insulin resistance; therefore, those aspects of AD should also respond to insulin and/or insulin sensitizers. Therapeutic options for treating AD as a metabolic disorder can be grouped under three headings: lifestyle changes, anti-inflammatory/anti-oxidant measures, and insulin signaling support.
4.2 Lifestyle Interventions
Lifestyle measures, including aerobic physical exercise, weight training and adopting a healthful diet are established strategies for lowering disease risk and severity in states of insulin resistance, including cognitive impairment and AD, and particularly in their preclinical and early stages [142, 143]. A meta-analysis study showed that reduced rates of physical exercise increase risk for developing AD [144]. This suggests that, in addition to protecting against cardiovascular disease, diabetes, obesity, and hypertension, healthful diets and regular physical exercise are neuroprotective and guard against mild cognitive impairment and AD [97, 145], and also positively impact neuronal plasticity [146]. Epidemiological data further suggest that the “Mediterranean diet” which is rich in fruits, vegetables and extra virgin olive oil, provides neuroprotection with aging [147], and that tight regulation of glycemia in diabetics helps preserve cognitive function [148]. In contrast, diets rich in simple sugars, particularly fructose, increase rates of T2DM, which in turn, enhances risk for AD [149]. Smoking is yet another avoidable risk factor for AD neurodegeneration as it increases neuro-inflammation, tau phosphorylation and Aβ42 deposition [150]. Other potential exposure-mediated causes of neurodegeneration linked to insulin resistance include neurotoxins [151], particulate matter present in polluted air [152, 153], and nitrosamines incorporated into processed and preserved foods and tobacco [154–158].
Nearly all protective lifestyle measures drive the body toward reducing insulin resistance, whereas accelerators of aging and cognitive impairment worsen insulin responsiveness. Aging is the dominant risk factor for cognitive impairment and neurodegeneration, including AD. Insulin resistance increases with aging. Lifestyle measures can reduce aging-associated insulin resistance. The protective effects of caloric restriction on aging demonstrated in earlier studies were likely due to concomitant reductions in insulin resistance. Therefore, lifestyle measures that preserve insulin responsiveness would likely protect against cognitive impairment and neurodegeneration [159]. From a public health perspective, although adopting healthy active lifestyles is the most logical and economically feasible approach to resolving the insulin resistance diseases epidemic, compliance has proven stubbornly difficult to sustain over prolonged periods. An additional consideration is whether lifestyle measures would even have significant neuroprotective effects in the intermediate or late stages of AD. If not, more definitive therapeutic strategies must be considered.
4.3 Anti-Inflammatory/Anti-Oxidant Agents
Epidemiological studies suggested that individuals who were chronically maintained on anti-inflammatory or anti-oxidant drugs for unrelated conditions had lower risks for developing cognitive impairment and AD [160]. Moreover, pre-clinical studies provided encouraging results showing that anti-inflammatory agents decreased neurofibrillary tangle burden [161, 162], tau phosphorylation [163], neurobehavioral (memory) deficits [164], Aβ42 burden [165, 166], synaptic degeneration [48, 167, 168], and mitochondrial dysfunction [169]. Therefore, anti-oxidants with neuroprotective actions may be beneficial in the treatment of neurodegeneration, including AD [170, 171].
Curcumin has received considerable attention as a natural anti-oxidant and anti-inflammatory agent for treating AD due to its inhibitory effects on Aβ42 deposition, Aβ42 aggregation, and Tau phosphorylation, and positive effects on neurobehavioral function in preclinical AD models [166]. Other anti-oxidants and anti-inflammatory agents that may provide neuroprotection for AD include alpha-lipoic acid [172–174], Vitamin E [175, 176], resveratrol [170], phytonutrients [177], cyclooxygenase 2 inhibitors [178], Ginkgo [179], and melatonin [180]. However, thus far, formal clinical trials designed to treat or prevent AD by targeting neuro-inflammation and oxidative stress [181, 182] have yielded disappointing results with respect to neuroprotection and preservation of cognitive function [166, 168, 183–185]. On the other hand, since neuro-inflammation emerges early and in pre-symptomatic stages of AD [186], relatively delayed treatment may not be effective because the peak period of related injury would have passed. The same phenomenon may hold with respect to Aβ42 accumulations. Although clinical trials designed to remove plaques and increase Aβ42 clearance failed to restore cognitive function, prevent cognitive decline, and reduce critical AD dementia-associated neuropathological abnormalities such as neuropil threads, dystrophic neurites, and neurofibrillary tangles [187–190], in order for this mode of therapy to be effective, it may have to be administered early, i.e. within the pre-symptomatic stages of disease. The same concept may hold with respect to therapeutic targeting of neuro-inflammation and oxidative stress [186] to prevent, reduce severity, or slow the rate of AD neurodegeneration. In support of this concept, in a recent publication of a randomized clinical pilot study, combined early intervention with omega-3 fatty acids, aerobic exercise, and cognitive stimulation significantly reduced progression of frontal, parietal and cingulate cortex atrophy in subjects with mild cognitive impairment [191].
4.4 Insulins
Insulin polypeptide has 51 amino acids and a molecular weight of 5808 Daltons. Insulin is first synthesized as a pre-prohormone in which its signal peptide directs the protein to the ER for cleavage and subsequent formation of proinsulin. Folding, with the addition of three disulfide bonds, and subsequent enzymatic processing by prohormone convertases and carboxypeptidase E in the trans-Golgi network releases disulfide bond-linked A- and B- chains of insulin. Mature insulin is stored in mature granules as an inactive hexamer. Active monomeric insulin is released on demand in response to elevated blood glucose and food intake. In T1DM, insulin production is reduced or absent due to auto-immune destruction of the pancreatic islet beta cells. In T2DM, insulin production is elevated due to insulin receptor resistance in target organs, including skeletal muscle and adipose tissue. Higher levels of insulin are needed to achieve cellular responses to dietary glucose and energy metabolism. In addition, as T2DM progresses, states of insulin deficiency can emerge, further increasing the need for insulin therapy.
4.4.1 Physiological and Therapeutic Effects of Brain Insulin and IGF-1: Rationale for Insulin/IGF-1 Therapy in Neurodegenerative Diseases
In addition to stimulating glucose metabolism, insulin and IGF-1 support neuronal and oligodendroglial cell survival [91, 92, 192], abundance and integrity of synapses [193], synaptic plasticity [91, 92, 193], and working memory and cognition [64, 95–98]. Other mechanistic actions of insulin that pertain to its potential use in targeting neurodegeneration include its anti-inflammatory effects [194], enhancement of Aβ42 clearance from the brain [98, 195], and stimulation of brain energy metabolism [196, 197], cerebral blood flow, including to regions damaged in AD [198], and acetylcholine production [196]. It is noteworthy that insulin enhancement of Aβ42 clearance was found not to be associated with altered expression of insulin degrading enzyme [195], which degrades both Aβ42 and insulin [199], and therefore other mechanisms must be involved.
4.4.2 Pharmacokinetics of Insulins used for Therapy
Insulins vary with respect to their onsets of action, which correlate with the time interval required for their detection in peripheral blood after injection, peak periods of therapeutic response, i.e. maximum effectiveness in lowering blood glucose, and duration of action (time period of detection in peripheral blood). In addition, insulin’s strength, conveyed in international units (U), ranges from 20 to 500, although the most common human dosage is 100 U. The fastest acting insulins are termed ‘rapid-acting’. They are detectable in peripheral blood within 15 min of injection; their levels peak within 1 h; and blood levels are sustained for 2–4 h. Regular insulins are similar to natural pancreatic islet-derived insulin as they are short acting, detectable in peripheral blood 30 min after injection, with peak blood levels achieved within 2 or 3 h, and a therapeutic window that lasts 3–6 h.
Newer synthetic or recombinant insulins tend to have longer half-lives, later peak periods of maximum strength for lowering blood glucose, and longer durations of action relative to rapid- and short-acting insulins, and thus their therapeutic windows are considerably broader than with rapid- and short-acting insulins. Intermediate-acting insulins are detectable in peripheral blood 2–4 h after injection, produce peak levels 4 and 12 h later, and their therapeutic effects are sustained for 12–18 h. Long-acting insulins produce relatively stable peripheral blood insulin levels over a 24-h period, enabling once-per-day treatments. Finally, inhaled insulins were developed to administer rapid-acting insulin after meals and avoid additional injections in T1DM or T2DM patients who were already being managed with long-acting insulin. Inhaled insulin’s effects are evident within 12–15 min and peak by 30 min. An advantage of this approach is that the “piggy backed” inhaled insulin has very short-term effects as it is not detectable 3 h after administration.
4.5 Insulin Therapy, Including Intranasal Delivery for Cognitive Impairment and AD
The brain needs insulin to support metabolism, neuronal survival, synaptic plasticity, myelin maintenance, growth and repair, and neuroprotection. Insulin deficiency and resistance emerge early in AD, and the resulting molecular, pathological, and biochemical abnormalities are essentially indistinguishable from the effects of other insulin resistance diseases such as diabetes mellitus and metabolic syndrome. This concept led to the term, ‘type 3 diabetes’ which conveys that the underlying problems in AD can be explained as a brain form of diabetes, and therefore, the therapeutic interventions could be the same or closely related. Insulin can be administered subcutaneous, transdermal, oral, sublingual, buccal, rectal, vaginal, intramuscular, intraperitoneal, or intranasal. Since insulin normally crosses the blood–brain barrier, systemic administration by various routes can modulate brain metabolism. Furthermore, in AD, compromise of the blood-barrier renders the brain even more accessible to insulin delivery from the periphery circulation.
Although clinical and experimental evidence suggests that insulin could be used to remediate brain metabolic dysfunction in AD, concerns about using peripherally injected insulin to treat the brain include off-target systemic effects such as hypoglycemia, and uncertainty about the efficiency of drug delivery across the blood–brain barrier. Intranasal delivery provides a realistic solution to these problems because with this non-invasive approach, insulin and other neurotrophins can enter the CNS and bypass the blood–brain barrier [96, 200, 201] while avoiding systemic effects. In contrast to inhalation, intranasal administration efficiently delivers drugs to ventromedial corticolimbic structures in the brain via olfactory and trigeminal nerves innervating the nasal cavity (olfactory) mucosa [202]. Due to connections with corticolimbic structures, intranasal insulin can to be used to treat the most vulnerable targets of AD. Intranasal delivery approaches provide virtually unrestricted transfer of therapeutic compounds across the blood–brain barrier to the brain or CSF for disease-specific targeting at high levels of safety [200, 201].
4.6 Clinical Trials of Insulin Therapy for Cognitive Impairment and Early AD
A number of studies have demonstrated that insulin administration improves cognition and brain energy metabolism in people with mild cognitive impairment or early AD. In addition, insulin enhances clearance of Aβ42, decreases activity of kinases that promote tau hyper-phosphorylation, and enhances signaling through pathways needed for synaptic plasticity [92, 203–205]. Furthermore, results from several clinical trials demonstrated that intranasal insulin improves working memory and cognition in individuals with mild cognitive impairment or early AD [98]. However, such responses are not fully generalizable as they appear to have been modulated by genetic background, particularly APOE-ε4 genotype. APOE-ε4 is the most prevalent genetic risk factor for AD and is present in 18–20% of the population. APOE-ε4-negative subjects were reported to exhibit greater insulin-mediated improvements in cognition [203, 206, 207] while APOE-ε4+ individuals either had no significant responses or their manifestations of AD worsened [207].
After a series of small trials, the multicenter 12-month SNIFF trial was conducted to examine responses to intranasal long-acting insulin (detemir) in subjects with amnestic mild cognitive impairment or probable mild AD [208]. Intranasal detemir improved cognition, verbal and visuospatial memory, and activities of daily living. Responses were most robust in subjects with the worst baseline performances, and they were modulated with respect to APOE-ε4 carrier status [208]. However, in contrast to the earlier study from the same group [207], the SNIFF trial results revealed that significant improvements in verbal memory occurred in APOE-ε4 positive carriers whereas significant declines were associated with APOE-ε4 negative status [208]. Conceivably, the discrepant findings were related to insufficient statistical power of the earlier clinical trials. Nonetheless, the findings in multiple studies provide ample evidence that insulin therapy, particularly via intranasal delivery, slows progression of cognitive impairment and AD, although the role of genetic factor regulation of treatment responsiveness requires further investigation. An additional caveat is that the data could reflect the need to go beyond monotherapy and extend the therapeutic targets to mediators of insulin resistance, as well as other abnormalities including neuroinflammation and deficits in neurotrophin and incretin signaling in AD.
4.6.1 Challenges Facing Intranasal Insulin Therapy for Cognitive Impairment and AD
Although cognitive impairment and AD can be treated with either systemic or intranasal insulin, proper administration of the latter has the potential to selectively target the brain with generally high levels of safety due to the low rates of off-target side effects such as hypoglycemia [96]. Furthermore, intranasal therapy avoids the need for multiple daily injections. Finally, as the priority dates of patents for intranasal agent delivery, including insulin to treat AD have expired (US5624898 A and US6313093 B1), opportunities exist for expanding this mode of therapy, including to the use of long-acting insulins to address a pressing need due to the large number of patients with AD throughout the world.
Despite its compelling strengths, intranasal therapy faces several challenges. First, delivery protocols must be standardized and simplified to enable routine management by elderly individuals; otherwise dose variability will lead to suboptimum and inconsistent responses. A second concern is the need to avoid off-target effects due to inadvertent systemic delivery and attendant hypoglycemia. This concern is serious and potentially devastating, particularly in elderly patients because the brain regions damaged by hypoglycemia are the very ones that require treatment for AD. The third point is that therapeutic agents, including insulin, delivered via the intranasal route should be structurally stable, relatively resistant to degradation, and able to penetrate the nasal mucosa. Finally, the potential for developing nasal transport resistance and reduced absorbance could limit the effectiveness of intranasal therapy [96]. Therefore, to further the use of intranasal insulin and other neurotrophins to treat CNS diseases, improvements in delivery systems, means of monitoring CNS delivery and brain responses, and assessments of nasal transport resistance are needed. On the other hand, the emergence of newer, ultra-long-acting insulins with predictably sustained release and bioactivity profiles, together with substantial margins of safety and efficacy [209–211], could represent the next generation of insulin compounds for treating cognitive impairment and AD.
4.7 Incretins
Incretins, including glucose-dependent insulinotropic polypeptide (GIP), glucagon-like peptide-1 (GLP-1), are metabolic hormones that signal reductions in blood glucose levels by stimulating insulin secretion, insulin gene expression, and pancreatic beta cell proliferation [212]. In addition, long-acting liraglutide was demonstrated to ameliorate impairments in insulin signaling in an AD mouse model (APPSWE/PS1dE9) by normalizing cell membrane distributions and localizations of the insulin receptor, reducing expression of pS(616) IRS-1, which inhibits downstream insulin signaling, neuroinflammation, and astrogliosis [213]. Incretin enhancement of nutrient-induced insulin release has potentially broad therapeutic applications, including for the treatment of cognitive impairment, osteoporosis, T2DM, and obesity [214, 215]. However, since GLP-1 and GIP are rapidly degraded by dipeptidyl peptidase-4 (DPP-4) following secretion by K and L cells of the gastrointestinal track [119, 212], they have minimal exogenous therapeutic utility. Instead, synthetic long-acting analogs with insulinotropic activity, including liraglutide and Exendin-4, are more suitable for in vivo experimentation and human clinical trials. Alternatively, inhibitors of DPP-4 could potentially be used to prolong the half-life of GLP-1 and GIP and regulate blood glucose in T2DM [107].
Although the effects of liraglutide could have been mediated by systemically increased insulin secretion, GLP-1 is expressed in brainstem neurons [216] that project widely throughout the CNS [217], and its receptors are expressed in ventral tegmental area, paraventricular nucleus and nucleus accumbens neurons [218] which receive projections from GLP-1 expressing brainstem neurons [218, 219]. The peripherally administered GLP-1 analog effects on food intake and body weight are likely mediated through CNS actions [220, 221], as these compounds can cross the blood–brain barrier [222, 223]. However, there is also evidence that CNS-mediated GLP-1 receptor activation has peripheral effects in regulating blood glucose [224], lipid metabolism [225], cardiovascular function [226].
In the CNS, incretins and incretin analogues are neuroprotective [227] because they enhance synaptic plasticity, cell proliferation, and memory, and they reduce Aβ42 plaques, oxidative stress, neuro-inflammation, and gliosis without causing adverse cardiovascular effects [228–232]. In light of the significant and progressive diabetes-like impairments in insulin signaling that occur with increasing AD stage [31, 32, 54], translational and clinical research to determine if incretin therapy can be extended to AD and other neurodegenerative diseases has been conducted.
The long-lasting GIP incretin hormone analogue, D-Ala(2)GIP, was found to protect memory formation and synaptic plasticity, normalize proliferation of stem cells, and reduce Aβ42 plaques and activation of microglia and astrocytes (neuroinflammation and oxidative stress) in the APPswe/PS1ΔE9 mouse model of AD [228]. In addition, Aβ42-inhibition of insulin signaling in neuronal cells was reversed by treatment with a DPP-4 inhibitor [233]. Exendin-4 (Ex-4), an incretin mimetic long-acting GLP-1 receptor agonist approved for T2DM, has neurotrophic and neuroprotective activity in cellular and animal models of stroke, Alzheimer’s and Parkinson’s diseases [229–231]. Correspondingly, encouraging results were obtained from a small pilot study in which liraglutide was found to have modest neuroprotective effects manifested by improvements in brain glucose metabolism in subjects with AD [234].
One potential disadvantage of incretins is that they must be administered by injection and they must be long-acting to be effective for human therapy. Moreover, since brain insulin resistance worsens as AD progresses to intermediate and late stages, the therapeutic window for targeting AD with insulins may narrow and possibly vanish over time. Additional research is needed to determine if incretin receptor resistance also develops with advancement of AD stage of neurodegeneration.
4.8 Metformin
The potential use of anti-diabetes drugs apart from insulins to manage cognitive impairment and AD is very exciting due to the large body of data from clinical trials and clinical experience. Evidence is emerging that cognitive function can be preserved by managing diabetes, including with metformin [132, 204, 235–237]. Thus far, just two major metformin clinical trial results have been published, one from Australia and the other from Singapore [132, 204]. However, the findings were somewhat contradictory. The Australian study reported an increased risk for cognitive impairment among patients with T2DM following long-term treatment with metformin [236], whereas the Singapore study showed that metformin provided neuroprotection in older adults with T2DM [238]. The confounding factor in the Australian study was that the adverse effects of metformin were largely due to concomitant Vitamin B12 deficiencies, which when corrected, abrogated the apparently detrimental effects of metformin. In a later independent study, metformin treatment of overweight or obese, previously untreated diabetics with amnestic mild cognitive impairment had a statistical trend for positive performance effects on the selective reminding test [239]. Given the widespread use of metformin and its potential effects on cognition, additional research including execution of a large multinational longitudinal study and meta-analysis of smaller studies are warranted.
4.9 Thiazolidinediones: Nuclear Hormone Receptor (Peroxisome-Proliferator Activated Receptor) Agonists
Peroxisome-proliferator activated receptors (PPARs) are nuclear hormone receptors of which there are three main types: alpha, beta/delta, and gamma. In addition, subtypes of PPAR-gamma have been identified [240]. Differential expression of one or more PPAR type and subtype enables relatively selective modulation of gene expression in tissues. Once activated, PPAR protein heterodimerizes with retinoid X receptor (RXR) and the complex then binds to target sequences that contain peroxisome proliferator hormone response elements within DNA promoters and function as transcription factors to regulate target gene expression, including in the brain [240]. Since PPARs have pivotal roles in regulating carbohydrate, protein, and lipid metabolism, as well as inflammatory responses [241–245], their agonists offer excellent opportunities for treating various aspects of brain diabetes while circumventing problems stemming from insulin resistance [46, 47]. In addition, recent evidence suggests that PPAR agonists can also activate IGF-1-regulated pathways in the brain [47].
In one early but small clinical trial, PPAR-gamma agonist treatment of subjects with mild cognitive impairment or early AD was demonstrated to enhance cognitive function [246]. In a later study, responses to metformin, rosiglitazone and dual treatments were examined. The results indicated that rosiglitazone treatment of diabetics was more effective than metformin in stabilizing long-term cognitive function [247].
Pioglitazone is the second PPAR-gamma agonist to gain attention for potential treatment of AD. In preclinical models, pioglitazone was shown to restore deficits in synaptic transmission and enhance long-term potentiation [248], reduce cerebellar dysfunction [249], and restore dendritic spine densities and adaptive plasticity responses damaged by Aβ42 [250]. Currently, in an on-going Phase III clinical trial, low-dose pioglitazone is being tested to determine if it can delay the onset of MCI and its progression to AD in normal subjects [251]. Importantly, the experimental treatment dose is considerably lower and therefore less toxic than the one used to treat T2DM because dose optimization was achieved by determining the pioglitazone dose that was needed to increase oxygen consumption in the brain based on blood-oxygen-level-dependent contrast imaging with fMRI [251]. Correspondingly, in preclinical experiments, low doses of PPAR agonists proved to be more effective and less toxic that high doses for reversing or preventing AD pathologies in experimental models of sporadic AD [46, 47, 252].
Effective brain targeting to enhance brain insulin/IGF signaling and thereby slow or prevent neurodegeneration and cognitive impairment with PPAR agonists will most likely require the use of mainly PPAR-delta, followed by PPAR-gamma agonists since most of the PPARs expressed in brain are delta [41]. Furthermore, PPAR-delta agonists are neuroprotective [253, 254], they inhibit neuro-inflammation [44, 255], reduce Aβ42 accumulation [44], support function in vascular endothelial [256] and smooth muscle [257] cells, promote oligodendrocyte maturation [258], which is needed for myelin maintenance, may negatively regulate CNS neuronal circuitry that drives obesity [259, 260], support cognitive function [261], and prevent neurodegeneration, including AD-type [262] in experimental models [46, 48]. However, since PPAR-gamma agonists also have neuroprotective effects, in may be beneficial to therapeutically target both PPAR-delta and PPAR-gamma for disease remediation in AD. In this regard, preclinical studies have shown that T3D-959, a novel hybrid PPAR-delta/PPAR-gamma agonist [263–265] can effectively restore cognitive function, integrity of temporal lobe/hippocampal structure, and insulin/IGF-1 sensitivity and inhibit neuro-inflammation in a sporadic model of AD [47, 48, 266]. PPAR delta/gamma agonists could be most effective in early to intermediate stages of AD, coinciding with emergence and progression of brain insulin/IGF-1 resistance, and either in concert or tandem with early insulin and/or incretin therapy for mild cognitive impairment and AD neurodegeneration.
5 Conclusions
AD should be regarded as a brain form of diabetes in which insulin resistance and deficiency develop either primarily in the brain, or due to systemic insulin resistance disease with secondary involvement of the brain. Nearly all pathologies in AD, including the typical Aβ42 and phospho-tau containing, PHF-associated structural lesions, metabolic dysfunction, neuro-inflammation, cellular stress, synaptic disconnection with proliferation of dystrophic neurites (reflecting loss of neuronal plasticity), cell death, white matter atrophy and degeneration, and microvascular disease, could be attributed to impairments in insulin and IGF signaling. Therefore, apart from reducing burdens of Aβ42 and pTau, therapeutic interventions must be designed to ameliorate the broader and multifaceted components of AD neurodegeneration. To that end, therapeutic interventions should include measures that increase insulin supply, enhance insulin/IGF-receptor responsiveness, and modulate downstream signaling through IRS and insulin/IGF target gene expression.
References
McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR Jr, Kawas CH, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7(3):263–9.
Blennow K, Dubois B, Fagan AM, Lewczuk P, de Leon MJ, Hampel H. Clinical utility of cerebrospinal fluid biomarkers in the diagnosis of early Alzheimer’s disease. Alzheimers Dement. 2015;11(1):58–69.
Olsson B, Lautner R, Andreasson U, Ohrfelt A, Portelius E, Bjerke M, et al. CSF and blood biomarkers for the diagnosis of Alzheimer’s disease: a systematic review and meta-analysis. Lancet Neurol. 2016;15(7):673–84.
Duncan GW, Firbank MJ, O’Brien JT, Burn DJ. Magnetic resonance imaging: a biomarker for cognitive impairment in Parkinson’s disease? Mov Disord. 2013;28(4):425–38.
Pantano P, Caramia F, Pierallini A. The role of MRI in dementia. Ital J Neurol Sci. 1999;20(5 Suppl):S250–3.
Amlien IK, Fjell AM. Diffusion tensor imaging of white matter degeneration in Alzheimer’s disease and mild cognitive impairment. Neuroscience. 2014;12(276):206–15.
Jones RS, Waldman AD. 1H-MRS evaluation of metabolism in Alzheimer’s disease and vascular dementia. Neurol Res. 2004;26(5):488–95.
Ewers M, Cheng X, Zhong Z, Nural HF, Walsh C, Meindl T, et al. Increased CSF-BACE1 activity associated with decreased hippocampus volume in Alzheimer’s disease. J Alzheimers Dis. 2011;25(2):373–81.
Nelson PT, Alafuzoff I, Bigio EH, Bouras C, Braak H, Cairns NJ, et al. Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J Neuropathol Exp Neurol. 2012;71(5):362–81.
Hyman BT, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Carrillo MC, et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement. 2012;8(1):1–13.
Montine TJ, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Dickson DW, et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta Neuropathol. 2012;123(1):1–11.
Serrano-Pozo A, Frosch MP, Masliah E, Hyman BT. Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med. 2011;1(1):a006189.
Ueno M, Chiba Y, Matsumoto K, Nakagawa T, Miyanaka H. Clearance of beta-amyloid in the brain. Curr Med Chem. 2014;21(35):4085–90.
Kalaria RN, Ballard C. Overlap between pathology of Alzheimer disease and vascular dementia. Alzheimer Dis Assoc Disord. 1999;13(Suppl 3):S115–23.
Viola KL, Klein WL. Amyloid beta oligomers in Alzheimer’s disease pathogenesis, treatment, and diagnosis. Acta Neuropathol. 2015;129(2):183–206.
Brun A, Liu X, Erikson C. Synapse loss and gliosis in the molecular layer of the cerebral cortex in Alzheimer’s disease and in frontal lobe degeneration. Neurodegeneration. 1995;4(2):171–7.
Vinters HV. Emerging concepts in Alzheimer’s disease. Annu Rev Pathol. 2015;10:291–319.
Piro JR, Benjamin DI, Duerr JM, Pi Y, Gonzales C, Wood KM, et al. A dysregulated endocannabinoid-eicosanoid network supports pathogenesis in a mouse model of Alzheimer’s disease. Cell Rep. 2012;1(6):617–23.
Agostinho P, Cunha RA, Oliveira C. Neuroinflammation, oxidative stress and the pathogenesis of Alzheimer’s disease. Curr Pharm Des. 2010;16(25):2766–78.
Singhal G, Jaehne EJ, Corrigan F, Toben C, Baune BT. Inflammasomes in neuroinflammation and changes in brain function: a focused review. Front Neurosci. 2014;8:315.
Mehlhorn G, Hollborn M, Schliebs R. Induction of cytokines in glial cells surrounding cortical beta-amyloid plaques in transgenic Tg2576 mice with Alzheimer pathology. Int J Dev Neurosci. 2000;18(4–5):423–31.
Dandrea MR, Reiser PA, Gumula NA, Hertzog BM, Andrade-Gordon P. Application of triple immunohistochemistry to characterize amyloid plaque-associated inflammation in brains with Alzheimer’s disease. Biotech Histochem. 2001;76(2):97–106.
Giovannini MG, Scali C, Prosperi C, Bellucci A, Vannucchi MG, Rosi S, et al. Beta-amyloid-induced inflammation and cholinergic hypofunction in the rat brain in vivo: involvement of the p38MAPK pathway. Neurobiol Dis. 2002;11(2):257–74.
Rose DP, Gracheck PJ, Vona-Davis L. The interactions of obesity, inflammation and insulin resistance in breast cancer. Cancers (Basel). 2015;7(4):2147–68.
Juhan-Vague I, Morange PE, Alessi MC. The insulin resistance syndrome: implications for thrombosis and cardiovascular disease. Pathophysiol Haemost Thromb. 2002;32(5–6):269–73.
You T, Nicklas BJ. Chronic inflammation: role of adipose tissue and modulation by weight loss. Curr Diabetes Rev. 2006;2(1):29–37.
Holvoet P. Relations between metabolic syndrome, oxidative stress and inflammation and cardiovascular disease. Verh K Acad Geneeskd Belg. 2008;70(3):193–219.
Vykoukal D, Davies MG. Vascular biology of metabolic syndrome. J Vasc Surg. 2011;54(3):819–31.
de la Monte SM. Insulin resistance and Alzheimer’s disease. BMB Rep. 2009;42(8):475–81.
Messier C, Teutenberg K. The role of insulin, insulin growth factor, and insulin-degrading enzyme in brain aging and Alzheimer’s disease. Neural Plast. 2005;12(4):311–28.
Rivera EJ, Goldin A, Fulmer N, Tavares R, Wands JR, de la Monte SM. Insulin and insulin-like growth factor expression and function deteriorate with progression of Alzheimer’s disease: link to brain reductions in acetylcholine. J Alzheimers Dis. 2005;8(3):247–68.
Talbot K, Wang HY, Kazi H, Han LY, Bakshi KP, Stucky A, et al. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J Clin Invest. 2012;122(4):1316–38.
Talbot K. Brain insulin resistance in Alzheimer’s disease and its potential treatment with GLP-1 analogs. Neurodegener Dis Manag. 2014;4(1):31–40.
Lee S, Tong M, Hang S, Deochand C, de la Monte S. CSF and Brain Indices of Insulin Resistance, Oxidative Stress and Neuro-Inflammation in Early versus Late Alzheimer’s Disease. J Alzheimers Dis Parkinsonism. 2013;31(3):128.
Tong M, Dong M, de la Monte SM. Brain insulin-like growth factor and neurotrophin resistance in Parkinson’s disease and dementia with Lewy bodies: potential role of manganese neurotoxicity. J Alzheimers Dis. 2009;16(3):585–99.
Clark IA, Vissel B. Inflammation-sleep interface in brain disease: TNF, insulin, orexin. J Neuroinflammation. 2014;21(11):51.
Misiak B, Leszek J, Kiejna A. Metabolic syndrome, mild cognitive impairment and Alzheimer’s disease–the emerging role of systemic low-grade inflammation and adiposity. Brain Res Bull. 2012;89(3–4):144–9.
Samaras K, Sachdev PS. Diabetes and the elderly brain: sweet memories? Ther Adv Endocrinol Metab. 2012;3(6):189–96.
Gaspar JM, Baptista FI, Macedo MP, Ambrosio AF. Inside the diabetic brain: role of different players involved in cognitive decline. ACS Chem Neurosci. 2016;7(2):131–42.
de la Monte SM, Wands JR. Molecular indices of oxidative stress and mitochondrial dysfunction occur early and often progress with severity of Alzheimer’s disease. J Alzheimers Dis. 2006;9(2):167–81.
Cimini A, Benedetti E, Cristiano L, Sebastiani P, D’Amico MA, D’Angelo B, et al. Expression of peroxisome proliferator-activated receptors (PPARs) and retinoic acid receptors (RXRs) in rat cortical neurons. Neuroscience. 2005;130(2):325–37.
Collino M, Patel NS, Thiemermann C. PPARs as new therapeutic targets for the treatment of cerebral ischemia/reperfusion injury. Ther Adv Cardiovasc Dis. 2008;2(3):179–97.
Dunn SE, Bhat R, Straus DS, Sobel RA, Axtell R, Johnson A, et al. Peroxisome proliferator-activated receptor delta limits the expansion of pathogenic Th cells during central nervous system autoimmunity. J Exp Med. 2010;207(8):1599–608.
Kalinin S, Richardson JC, Feinstein DL. A PPARdelta agonist reduces amyloid burden and brain inflammation in a transgenic mouse model of Alzheimer’s disease. Curr Alzheimer Res. 2009;6(5):431–7.
Barroso E, del Valle J, Porquet D, Vieira Santos AM, Salvado L, Rodriguez-Rodriguez R, et al. Tau hyperphosphorylation and increased BACE1 and RAGE levels in the cortex of PPARbeta/delta-null mice. Biochim Biophys Acta. 2013;1832(8):1241–8.
de la Monte SM, Tong M, Lester-Coll N, Plater M Jr, Wands JR. Therapeutic rescue of neurodegeneration in experimental type 3 diabetes: relevance to Alzheimer’s disease. J Alzheimers Dis. 2006;10(1):89–109.
de la Monte SM, Tong M, Schiano I, Didsbury J. Improved brain insulin/IGF signaling and reduced neuro-inflammation with T3D–959 in an experimental model of sporadic Alzheimer’s disease. J Alzheimers Dis. 2017;55(2):849–64.
Malm T, Mariani M, Donovan LJ, Neilson L, Landreth GE. Activation of the nuclear receptor PPARdelta is neuroprotective in a transgenic mouse model of Alzheimer’s disease through inhibition of inflammation. J Neuroinflamm. 2015;12:7.
Barone E, Di Domenico F, Mancuso C, Butterfield DA. The Janus face of the heme oxygenase/biliverdin reductase system in Alzheimer disease: it’s time for reconciliation. Neurobiol Dis. 2014;62:144–59.
Nunomura A, Moreira PI, Castellani RJ, Lee HG, Zhu X, Smith MA, et al. Oxidative damage to RNA in aging and neurodegenerative disorders. Neurotox Res. 2012;22(3):231–48.
Nunomura A, Perry G, Aliev G, Hirai K, Takeda A, Balraj EK, et al. Oxidative damage is the earliest event in Alzheimer disease. J Neuropathol Exp Neurol. 2001;60(8):759–67.
Bonda DJ, Wang X, Lee HG, Smith MA, Perry G, Zhu X. Neuronal failure in Alzheimer’s disease: a view through the oxidative stress looking-glass. Neurosci Bull. 2014;30(2):243–52.
de la Monte SM, Luong T, Neely TR, Robinson D, Wands JR. Mitochondrial DNA damage as a mechanism of cell loss in Alzheimer’s disease. Lab Invest. 2000;80(8):1323–35.
de la Monte SM, Tong M. Brain metabolic dysfunction at the core of Alzheimer’s disease. Biochem Pharmacol. 2014;88(4):548–59.
Smith MA, Perry G. Free radical damage, iron, and Alzheimer’s disease. J Neurol Sci. 1995;134(Suppl):92–4.
Bishop GM, Robinson SR, Liu Q, Perry G, Atwood CS, Smith MA. Iron: a pathological mediator of Alzheimer disease? Dev Neurosci. 2002;24(2–3):184–7.
Barbeito AG, Garringer HJ, Baraibar MA, Gao X, Arredondo M, Nunez MT, et al. Abnormal iron metabolism and oxidative stress in mice expressing a mutant form of the ferritin light polypeptide gene. J Neurochem. 2009;109(4):1067–78.
Castellani R, Smith MA, Richey PL, Kalaria R, Gambetti P, Perry G. Evidence for oxidative stress in Pick disease and corticobasal degeneration. Brain Res. 1995;696(1–2):268–71.
Fawcett JR, Bordayo EZ, Jackson K, Liu H, Peterson J, Svitak A, et al. Inactivation of the human brain muscarinic acetylcholine receptor by oxidative damage catalyzed by a low molecular weight endogenous inhibitor from Alzheimer’s brain is prevented by pyrophosphate analogs, bioflavonoids and other antioxidants. Brain Res. 2002;950(1–2):10–20.
Navarro E, Buendia I, Parada E, Leon R, Jansen-Duerr P, Pircher H, et al. Alpha7 nicotinic receptor activation protects against oxidative stress via heme-oxygenase I induction. Biochem Pharmacol. 2015;97(4):473–81.
Fine JM, Baillargeon AM, Renner DB, Hoerster NS, Tokarev J, Colton S, et al. Intranasal deferoxamine improves performance in radial arm water maze, stabilizes HIF-1alpha, and phosphorylates GSK3beta in P301L tau transgenic mice. Exp Brain Res. 2012;219(3):381–90.
Hanson LR, Fine JM, Renner DB, Svitak AL, Burns RB, Nguyen TM, et al. Intranasal delivery of deferoxamine reduces spatial memory loss in APP/PS1 mice. Drug Deliv Transl Res. 2012;2(3):160–8.
Fine JM, Renner DB, Forsberg AC, Cameron RA, Galick BT, Le C, et al. Intranasal deferoxamine engages multiple pathways to decrease memory loss in the APP/PS1 model of amyloid accumulation. Neurosci Lett. 2015;1(584):362–7.
de la Monte SM, Tong M, Bowling N, Moskal P. si-RNA inhibition of brain insulin or insulin-like growth factor receptors causes developmental cerebellar abnormalities: relevance to fetal alcohol spectrum disorder. Mol Brain. 2011;4:13.
Grunblatt E, Bartl J, Riederer P. The link between iron, metabolic syndrome, and Alzheimer’s disease. J Neural Transm (Vienna). 2011;118(3):371–9.
Schipper HM, Cisse S, Stopa EG. Expression of heme oxygenase-1 in the senescent and Alzheimer-diseased brain. Ann Neurol. 1995;37(6):758–68.
Di Domenico F, Barone E, Mancuso C, Perluigi M, Cocciolo A, Mecocci P, et al. HO-1/BVR-a system analysis in plasma from probable Alzheimer’s disease and mild cognitive impairment subjects: a potential biochemical marker for the prediction of the disease. J Alzheimers Dis. 2012;32(2):277–89.
Barone E, Butterfield DA. Insulin resistance in Alzheimer disease: is heme oxygenase-1 an Achille’s heel? Neurobiol Dis. 2015;84:69–77.
Barone E, Di Domenico F, Sultana R, Coccia R, Mancuso C, Perluigi M, et al. Heme oxygenase-1 posttranslational modifications in the brain of subjects with Alzheimer disease and mild cognitive impairment. Free Radic Biol Med. 2012;52(11–12):2292–301.
Nandar W, Connor JR. HFE gene variants affect iron in the brain. J Nutr. 2011;141(4):729S–39S.
Nielsen JE, Jensen LN, Krabbe K. Hereditary haemochromatosis: a case of iron accumulation in the basal ganglia associated with a parkinsonian syndrome. J Neurol Neurosurg Psychiatry. 1995;59(3):318–21.
Giambattistelli F, Bucossi S, Salustri C, Panetta V, Mariani S, Siotto M, et al. Effects of hemochromatosis and transferrin gene mutations on iron dyshomeostasis, liver dysfunction and on the risk of Alzheimer’s disease. Neurobiol Aging. 2012;33(8):1633–41.
Percy M, Somerville MJ, Hicks M, Garcia A, Colelli T, Wright E, et al. Risk factors for development of dementia in a unique six-year cohort study. I. An exploratory, pilot study of involvement of the E4 allele of apolipoprotein E, mutations of the hemochromatosis-HFE gene, type 2 diabetes, and stroke. J Alzheimers Dis. 2014;38(4):907–22.
Stremmel W, Niederau C, Berger M, Kley HK, Kruskemper HL, Strohmeyer G. Abnormalities in estrogen, androgen, and insulin metabolism in idiopathic hemochromatosis. Ann N Y Acad Sci. 1988;526:209–23.
Johnstone DM, Graham RM, Trinder D, Riveros C, Olynyk JK, Scott RJ, et al. Changes in brain transcripts related to Alzheimer’s disease in a model of HFE hemochromatosis are not consistent with increased Alzheimer’s disease risk. J Alzheimers Dis. 2012;30(4):791–803.
Connor JR, Lee SY. HFE mutations and Alzheimer’s disease. J Alzheimers Dis. 2006;10(2–3):267–76.
Lehmann DJ, Worwood M, Ellis R, Wimhurst VL, Merryweather-Clarke AT, Warden DR, et al. Iron genes, iron load and risk of Alzheimer’s disease. J Med Genet. 2006;43(10):e52.
Lin M, Zhao L, Fan J, Lian XG, Ye JX, Wu L, et al. Association between HFE polymorphisms and susceptibility to Alzheimer’s disease: a meta-analysis of 22 studies including 4,365 cases and 8,652 controls. Mol Biol Rep. 2012;39(3):3089–95.
Horowitz MP, Greenamyre JT. Mitochondrial iron metabolism and its role in neurodegeneration. J Alzheimers Dis. 2010;20(Suppl 2):S551–68.
Santambrogio P, Dusi S, Guaraldo M, Rotundo LI, Broccoli V, Garavaglia B, et al. Mitochondrial iron and energetic dysfunction distinguish fibroblasts and induced neurons from pantothenate kinase-associated neurodegeneration patients. Neurobiol Dis. 2015;81:144–53.
Dusi S, Valletta L, Haack TB, Tsuchiya Y, Venco P, Pasqualato S, et al. Exome sequence reveals mutations in CoA synthase as a cause of neurodegeneration with brain iron accumulation. Am J Hum Genet. 2014;94(1):11–22.
Matthes T, Rustin P, Trachsel H, Darbellay R, Costaridou S, Xaidara A, et al. Different pathophysiological mechanisms of intramitochondrial iron accumulation in acquired and congenital sideroblastic anemia caused by mitochondrial DNA deletion. Eur J Haematol. 2006;77(2):169–74.
Bonda DJ, Wang X, Perry G, Smith MA, Zhu X. Mitochondrial dynamics in Alzheimer’s disease: opportunities for future treatment strategies. Drugs Aging. 2010;27(3):181–92.
Sangchot P, Sharma S, Chetsawang B, Porter J, Govitrapong P, Ebadi M. Deferoxamine attenuates iron-induced oxidative stress and prevents mitochondrial aggregation and alpha-synuclein translocation in SK-N-SH cells in culture. Dev Neurosci. 2002;24(2–3):143–53.
Daulatzai MA. Quintessential risk factors: their role in promoting cognitive dysfunction and Alzheimer’s disease. Neurochem Res. 2012;37(12):2627–58.
Schaffer C, Sarad N, DeCrumpe A, Goswami D, Herrmann S, Morales J, et al. Biomarkers in the diagnosis and prognosis of Alzheimer’s disease. J Lab Autom. 2015;20(5):589–600.
de Leon MJ, George AE, Ferris SH, Rosenbloom S, Christman DR, Gentes CI, et al. Regional correlation of PET and CT in senile dementia of the Alzheimer type. AJNR Am J Neuroradiol. 1983;4(3):553–6.
Faulstich ME. Brain imaging in dementia of the Alzheimer type. Int J Neurosci. 1991;57(1–2):39–49.
Waldron AM, Wintmolders C, Bottelbergs A, Kelley JB, Schmidt ME, Stroobants S, et al. In vivo molecular neuroimaging of glucose utilization and its association with fibrillar amyloid-beta load in aged APPPS1-21 mice. Alzheimers Res Ther. 2015;7(1):76.
Wurtman R. Biomarkers in the diagnosis and management of Alzheimer’s disease. Metabolism. 2015;64(3 Suppl 1):S47–50.
de la Monte SM, Wands JR. Review of insulin and insulin-like growth factor expression, signaling, and malfunction in the central nervous system: relevance to Alzheimer’s disease. J Alzheimers Dis. 2005;7(1):45–61.
de la Monte SM. Contributions of brain insulin resistance and deficiency in amyloid-related neurodegeneration in Alzheimer’s disease. Drugs. 2012;72(1):49–66.
Shuvaev VV, Laffont I, Serot JM, Fujii J, Taniguchi N, Siest G. Increased protein glycation in cerebrospinal fluid of Alzheimer’s disease. Neurobiol Aging. 2001;22(3):397–402.
Zafari S, Backes C, Meese E, Keller A. Circulating biomarker panels in Alzheimer’s disease. Gerontology. 2015;61(6):497–503.
Benedict C, Frey WH 2nd, Schioth HB, Schultes B, Born J, Hallschmid M. Intranasal insulin as a therapeutic option in the treatment of cognitive impairments. Exp Gerontol. 2011;46(2–3):112–5.
de la Monte SM. Intranasal insulin therapy for cognitive impairment and neurodegeneration: current state of the art. Expert Opin Drug Deliv. 2013;10(12):1699–709.
Kidd PM. Alzheimer’s disease, amnestic mild cognitive impairment, and age-associated memory impairment: current understanding and progress toward integrative prevention. Altern Med Rev. 2008;13(2):85–115.
Reger MA, Watson GS, Green PS, Wilkinson CW, Baker LD, Cholerton B, et al. Intranasal insulin improves cognition and modulates beta-amyloid in early AD. Neurology. 2008;70(6):440–8.
de la Monte SM, Wands JR. Alzheimer’s disease is type 3 diabetes: evidence reviewed. J Diabetes Sci Technol. 2008;2(6):1101–13.
Steen E, Terry BM, Rivera EJ, Cannon JL, Neely TR, Tavares R, et al. Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease—is this type 3 diabetes? J Alzheimers Dis. 2005;7(1):63–80.
Moloney AM, Griffin RJ, Timmons S, O’Connor R, Ravid R, O’Neill C. Defects in IGF-1 receptor, insulin receptor and IRS-1/2 in Alzheimer’s disease indicate possible resistance to IGF-1 and insulin signalling. Neurobiol Aging. 2010;31(2):224–43.
Zeki Al Hazzouri A, Stone KL, Haan MN, Yaffe K. Leptin, mild cognitive impairment, and dementia among elderly women. J Gerontol A Biol Sci Med Sci. 2013;68(2):175–80.
Cole GM, Frautschy SA. The role of insulin and neurotrophic factor signaling in brain aging and Alzheimer’s Disease. Exp Gerontol. 2007;42(1–2):10–21.
Calsolaro V, Edison P. Novel GLP-1 (Glucagon-Like Peptide-1) analogues and insulin in the treatment for Alzheimer’s disease and other neurodegenerative diseases. CNS Drugs. 2015;29(12):1023–39.
Holscher C. The incretin hormones glucagonlike peptide 1 and glucose-dependent insulinotropic polypeptide are neuroprotective in mouse models of Alzheimer’s disease. Alzheimers Dement. 2014;10(1 Suppl):S47–54.
Ji C, Xue GF, Li G, Li D, Holscher C. Neuroprotective effects of glucose-dependent insulinotropic polypeptide in Alzheimer’s disease. Rev Neurosci. 2016;27(1):61–70.
Freeman JS. Role of the incretin pathway in the pathogenesis of T2DM. Cleve Clin J Med. 2009;76(Suppl 5):S12–9.
Farr OM, Gavrieli A, Mantzoros CS. Leptin applications in 2015: what have we learned about leptin and obesity? Curr Opin Endocrinol Diabetes Obes. 2015;22(5):353–9.
Yang XN, Zhang CY, Wang B-W, Zhu SG, Zheng RM. Leptin Signalings and Leptin Resistance. Sheng Li Ke Xue Jin Zhan. 2015;46(5):327–33.
Civelek S, Konukoglu D, Erdenen F, Uzun H. Serum neurotrophic factor levels in patients with T2DM: relationship to metabolic syndrome components. Clin Lab. 2013;59(3–4):369–74.
Hristova M, Aloe L. Metabolic syndrome–neurotrophic hypothesis. Med Hypotheses. 2006;66(3):545–9.
Aguirre GA, De Ita JR, de la Garza RG, Castilla-Cortazar I. Insulin-like growth factor-1 deficiency and metabolic syndrome. J Transl Med. 2016;06(14):3.
Cubbon RM, Kearney MT, Wheatcroft SB. Endothelial IGF-1 receptor signalling in diabetes and insulin resistance. Trends Endocrinol Metab. 2016;27(2):96–104.
Ono M, Ichihara J, Nonomura T, Itakura Y, Taiji M, Nakayama C, et al. Brain-derived neurotrophic factor reduces blood glucose level in obese diabetic mice but not in normal mice. Biochem Biophys Res Commun. 1997;238(2):633–7.
Baeza-Raja B, Li P, Le Moan N, Sachs BD, Schachtrup C, Davalos D, et al. p75 neurotrophin receptor regulates glucose homeostasis and insulin sensitivity. Proc Natl Acad Sci USA. 2015;109(15):5838–43.
Ramirez S, Claret M. Hypothalamic ER stress: a bridge between leptin resistance and obesity. FEBS Lett. 2015;589(14):1678–87.
Thon M, Hosoi T, Ozawa K. Possible integrative actions of leptin and insulin signaling in the hypothalamus targeting energy homeostasis. Front Endocrinol (Lausanne). 2016;7:138.
Meier JJ, Nauck MA. Is the diminished incretin effect in type 2 diabetes just an epi-phenomenon of impaired beta-cell function? Diabetes. 2010;59(5):1117–25.
Joao AL, Reis F, Fernandes R. The incretin system ABCs in obesity and diabetes—novel therapeutic strategies for weight loss and beyond. Obes Rev. 2016;17(7):553–72.
de la Monte SM, Neusner A, Chu J, Lawton M. Epidemiological trends strongly suggest exposures as etiologic agents in the pathogenesis of sporadic Alzheimer’s disease, diabetes mellitus, and non-alcoholic steatohepatitis. J Alzheimers Dis. 2009;17(3):519–29.
Pedditizi E, Peters R, Beckett N. The risk of overweight/obesity in mid-life and late life for the development of dementia: a systematic review and meta-analysis of longitudinal studies. Age Ageing. 2016;45(1):14–21.
Alosco ML, Gunstad J. The negative effects of obesity and poor glycemic control on cognitive function: a proposed model for possible mechanisms. Curr Diab Rep. 2014;14(6):495.
Luchsinger JA, Reitz C, Patel B, Tang MX, Manly JJ, Mayeux R. Relation of diabetes to mild cognitive impairment. Arch Neurol. 2007;64(4):570–5.
Noble JM, Manly JJ, Schupf N, Tang MX, Luchsinger JA. Type 2 diabetes and ethnic disparities in cognitive impairment. Ethn Dis. 2012;22(1):38–44.
Naderali EK, Ratcliffe SH, Dale MC. Obesity and Alzheimer’s disease: a link between body weight and cognitive function in old age. Am J Alzheimers Dis Other Demen. 2009;24(6):445–9.
Drab SR. Recognizing the rising impact of diabetes in seniors and implications for its management. Consult Pharm. 2009;24 Suppl B:5–10.
Roriz-Filho JS, Sa-Roriz TM, Rosset I, Camozzato AL, Santos AC, Chaves ML, et al. (Pre)diabetes, brain aging, and cognition. Biochim Biophys Acta. 2009;1792(5):432–43.
de la Monte SM. Metabolic derangements mediate cognitive impairment and Alzheimer’s disease: role of peripheral insulin-resistance diseases. Panminerva Med. 2012;54(3):171–8.
Fotuhi M, Do D, Jack C. Modifiable factors that alter the size of the hippocampus with ageing. Nat Rev Neurol. 2012;8(4):189–202.
Sridhar GR, Lakshmi G, Nagamani G. Emerging links between type 2 diabetes and Alzheimer’s disease. World J Diabetes. 2015;6(5):744–51.
de la Monte SM. Relationships between diabetes and cognitive impairment. Endocrinol Metab Clin North Am. 2014;43(1):245–67.
Li X, Song D, Leng SX. Link between type 2 diabetes and Alzheimer’s disease: from epidemiology to mechanism and treatment. Clin Interv Aging. 2015;10:549–60.
Kim B, Feldman EL. Insulin resistance as a key link for the increased risk of cognitive impairment in the metabolic syndrome. Exp Mol Med. 2015;47:e149.
Cholerton B, Baker LD, Craft S. Insulin resistance and pathological brain ageing. Diabet Med. 2011;28(12):1463–75.
Frisardi V, Solfrizzi V, Seripa D, Capurso C, Santamato A, Sancarlo D, et al. Metabolic-cognitive syndrome: a cross-talk between metabolic syndrome and Alzheimer’s disease. Ageing Res Rev. 2010;9(4):399–417.
Ho L, Qin W, Pompl PN, Xiang Z, Wang J, Zhao Z, et al. Diet-induced insulin resistance promotes amyloidosis in a transgenic mouse model of Alzheimer’s disease. FASEB J. 2004;18(7):902–4.
Moroz N, Tong M, Longato L, Xu H, de la Monte SM. Limited Alzheimer-type neurodegeneration in experimental obesity and T2DM. J Alzheimers Dis. 2008;15(1):29–44.
de la Monte SM, Longato L, Tong M, Wands JR. Insulin resistance and neurodegeneration: roles of obesity, T2DM and non-alcoholic steatohepatitis. Curr Opin Investig Drugs. 2009;10(10):1049–60.
Nagoshi S. Liver diseases. Nihon Rinsho. 2014;72(4):726–9.
Lyn-Cook LE Jr, Lawton M, Tong M, Silbermann E, Longato L, Jiao P, et al. Hepatic ceramide may mediate brain insulin resistance and neurodegeneration in type 2 diabetes and non-alcoholic steatohepatitis. J Alzheimers Dis. 2009;16(4):715–29.
Meda SA, Narayanan B, Liu J, Perrone-Bizzozero NI, Stevens MC, Calhoun VD, et al. A large scale multivariate parallel ICA method reveals novel imaging-genetic relationships for Alzheimer’s disease in the ADNI cohort. Neuroimage. 2012;60(3):1608–21.
Demarin V, Lisak M, Morovic S. Mediterranean diet in healthy lifestyle and prevention of stroke. Acta Clin Croat. 2011;50(1):67–77.
Polidori MC. Preventive benefits of natural nutrition and lifestyle counseling against Alzheimer’s disease onset. J Alzheimers Dis. 2014;42(Suppl 4):S475–82.
Beydoun MA, Beydoun HA, Gamaldo AA, Teel A, Zonderman AB, Wang Y. Epidemiologic studies of modifiable factors associated with cognition and dementia: systematic review and meta-analysis. BMC Public Health. 2014;14:643.
Kalaria RN. Vascular basis for brain degeneration: faltering controls and risk factors for dementia. Nutr Rev. 2010;68(Suppl 2):S74–87.
Stranahan AM, Mattson MP. Bidirectional metabolic regulation of neurocognitive function. Neurobiol Learn Mem. 2011;96(4):507–16.
Rigacci S. Olive oil phenols as promising multi-targeting agents against Alzheimer’s disease. Adv Exp Med Biol. 2015;863:1–20.
Lehtisalo J, Lindstrom J, Ngandu T, Kivipelto M, Ahtiluoto S, Ilanne-Parikka P, et al. Diabetes, glycaemia, and cognition-a secondary analysis of the Finnish Diabetes Prevention Study. Diabetes Metab Res Rev. 2016;32(1):102–10.
Moreira PI. High-sugar diets, type 2 diabetes and Alzheimer’s disease. Curr Opin Clin Nutr Metab Care. 2013;16(4):440–5.
Moreno-Gonzalez I, Estrada LD, Sanchez-Mejias E, Soto C. Smoking exacerbates amyloid pathology in a mouse model of Alzheimer’s disease. Nat Commun. 2013;4:1495.
Daulatzai MA. Neurotoxic saboteurs: straws that break the hippo’s (hippocampus) back drive cognitive impairment and Alzheimer’s Disease. Neurotox Res. 2013;24(3):407–59.
Calderon-Garciduenas L, Maronpot RR, Torres-Jardon R, Henriquez-Roldan C, Schoonhoven R, Acuna-Ayala H, et al. DNA damage in nasal and brain tissues of canines exposed to air pollutants is associated with evidence of chronic brain inflammation and neurodegeneration. Toxicol Pathol. 2003;31(5):524–38.
Calderon-Garciduenas L, Reed W, Maronpot RR, Henriquez-Roldan C, Delgado-Chavez R, Calderon-Garciduenas A, et al. Brain inflammation and Alzheimer’s-like pathology in individuals exposed to severe air pollution. Toxicol Pathol. 2004;32(6):650–8.
de la Monte SM. Type 3 diabetes is sporadic Alzheimers disease: mini-review. Eur Neuropsychopharmacol. 2014;24(12):1954–60.
de la Monte SM, Tong M. Mechanisms of nitrosamine-mediated neurodegeneration: potential relevance to sporadic Alzheimer’s disease. J Alzheimers Dis. 2009;17(4):817–25.
de la Monte SM, Tong M, Lawton M, Longato L. Nitrosamine exposure exacerbates high fat diet-mediated T2DM, non-alcoholic steatohepatitis, and neurodegeneration with cognitive impairment. Mol Neurodegener. 2009;4:54.
Tong M, Neusner A, Longato L, Lawton M, Wands JR, de la Monte SM. Nitrosamine exposure causes insulin resistance diseases: relevance to T2DM, non-alcoholic steatohepatitis, and Alzheimer’s disease. J Alzheimers Dis. 2009;17(4):827–44.
Yalcin E, de la Monte S. Tobacco nitrosamines as culprits in disease: mechanisms reviewed. J Physiol Biochem. 2016;72(1):107–20.
Middleton LE, Yaffe K. Promising strategies for the prevention of dementia. Arch Neurol. 2009;66(10):1210–5.
Standridge JB. Pharmacotherapeutic approaches to the prevention of Alzheimer’s disease. Am J Geriatr Pharmacother. 2004;2(2):119–32.
Boimel M, Grigoriadis N, Lourbopoulos A, Touloumi O, Rosenmann D, Abramsky O, et al. Statins reduce the neurofibrillary tangle burden in a mouse model of tauopathy. J Neuropathol Exp Neurol. 2009;68(3):314–25.
Piedrahita D, Hernandez I, Lopez-Tobon A, Fedorov D, Obara B, Manjunath BS, et al. Silencing of CDK5 reduces neurofibrillary tangles in transgenic alzheimer’s mice. J Neurosci. 2010;30(42):13966–76.
Leroy K, Ando K, Heraud C, Yilmaz Z, Authelet M, Boeynaems JM, et al. Lithium treatment arrests the development of neurofibrillary tangles in mutant tau transgenic mice with advanced neurofibrillary pathology. J Alzheimers Dis. 2010;19(2):705–19.
Villaflores OB, Chen YJ, Chen CP, Yeh JM, Wu TY. Curcuminoids and resveratrol as anti-Alzheimer agents. Taiwan J Obstet Gynecol. 2012;51(4):515–25.
Lazar AN, Mourtas S, Youssef I, Parizot C, Dauphin A, Delatour B, et al. Curcumin-conjugated nanoliposomes with high affinity for Abeta deposits: possible applications to Alzheimer disease. Nanomedicine. 2013;9(5):712–21.
Hamaguchi T, Ono K, Yamada M. REVIEW: curcumin and Alzheimer’s disease. CNS Neurosci Ther. 2010;16(5):285–97.
Olmos-Alonso A, Schetters ST, Sri S, Askew K, Mancuso R, Vargas-Caballero M, et al. Pharmacological targeting of CSF1R inhibits microglial proliferation and prevents the progression of Alzheimer’s-like pathology. Brain. 2016;139(Pt 3):891–907.
Goozee KG, Shah TM, Sohrabi HR, Rainey-Smith SR, Brown B, Verdile G, et al. Examining the potential clinical value of curcumin in the prevention and diagnosis of Alzheimer’s disease. Br J Nutr. 2016;115(3):449–65.
Prakash A, Kumar A. Implicating the role of lycopene in restoration of mitochondrial enzymes and BDNF levels in beta-amyloid induced Alzheimers disease. Eur J Pharmacol. 2014;15(741):104–11.
Mancuso C, Bates TE, Butterfield DA, Calafato S, Cornelius C, De Lorenzo A, et al. Natural antioxidants in Alzheimer’s disease. Expert Opin Investig Drugs. 2007;16(12):1921–31.
Grundman M, Grundman M, Delaney P. Antioxidant strategies for Alzheimer’s disease. Proc Nutr Soc. 2002;61(2):191–202.
Kishi Y, Schmelzer JD, Yao JK, Zollman PJ, Nickander KK, Tritschler HJ, et al. Alpha-lipoic acid: effect on glucose uptake, sorbitol pathway, and energy metabolism in experimental diabetic neuropathy. Diabetes. 1999;48(10):2045–51.
Mitsui Y, Schmelzer JD, Zollman PJ, Mitsui M, Tritschler HJ, Low PA. Alpha-lipoic acid provides neuroprotection from ischemia-reperfusion injury of peripheral nerve. J Neurol Sci. 1999;163(1):11–6.
Hardas SS, Sultana R, Clark AM, Beckett TL, Szweda LI, Murphy MP, et al. Oxidative modification of lipoic acid by HNE in Alzheimer disease brain. Redox Biol. 2013;1:80–5.
Grundman M. Vitamin E and Alzheimer disease: the basis for additional clinical trials. Am J Clin Nutr. 2000;71(2):630S–6S.
Isaac MG, Quinn R, Tabet N. Vitamin E for Alzheimer’s disease and mild cognitive impairment. Cochrane Database Syst Rev. 2008(3):CD002854.
Wattanapenpaiboon N, Wahlqvist MW. Phytonutrient deficiency: the place of palm fruit. Asia Pac J Clin Nutr. 2003;12(3):363–8.
Scali C, Giovannini MG, Prosperi C, Bellucci A, Pepeu G, Casamenti F. The selective cyclooxygenase-2 inhibitor rofecoxib suppresses brain inflammation and protects cholinergic neurons from excitotoxic degeneration in vivo. Neuroscience. 2003;117(4):909–19.
Christen Y. Ginkgo biloba and neurodegenerative disorders. Front Biosci. 2004;1(9):3091–104.
Wang JZ, Wang ZF. Role of melatonin in Alzheimer-like neurodegeneration. Acta Pharmacol Sin. 2006;27(1):41–9.
Ancelin ML, Christen Y, Ritchie K. Is antioxidant therapy a viable alternative for mild cognitive impairment? Examination of the evidence. Dement Geriatr Cogn Disord. 2007;24(1):1–19.
Farina N, Isaac MG, Clark AR, Rusted J, Tabet N. Vitamin E for Alzheimer’s dementia and mild cognitive impairment. Cochrane Database Syst Rev. 2012;11:CD002854.
Polidori MC, Nelles G. Antioxidant clinical trials in mild cognitive impairment and Alzheimer’s disease—challenges and perspectives. Curr Pharm Des. 2014;20(18):3083–92.
Mecocci P, Polidori MC. Antioxidant clinical trials in mild cognitive impairment and Alzheimer’s disease. Biochim Biophys Acta. 2012;1822(5):631–8.
Levey A, Lah J, Goldstein F, Steenland K, Bliwise D. Mild cognitive impairment: an opportunity to identify patients at high risk for progression to Alzheimer’s disease. Clin Ther. 2006;28(7):991–1001.
McGeer PL, McGeer EG. The amyloid cascade-inflammatory hypothesis of Alzheimer disease: implications for therapy. Acta Neuropathol. 2013;126(4):479–97.
Nicoll JA, Wilkinson D, Holmes C, Steart P, Markham H, Weller RO. Neuropathology of human Alzheimer disease after immunization with amyloid-beta peptide: a case report. Nat Med. 2003;9(4):448–52.
Bombois S, Maurage CA, Gompel M, Deramecourt V, Mackowiak-Cordoliani MA, Black RS, et al. Absence of beta-amyloid deposits after immunization in Alzheimer disease with Lewy body dementia. Arch Neurol. 2007;64(4):583–7.
Lemere CA. Developing novel immunogens for a safe and effective Alzheimer’s disease vaccine. Prog Brain Res. 2009;175:83–93.
Kuzuhara S. Treatment strategy of Alzheimer’s disease: pause in clinical trials of Abeta vaccine and next steps. Brain Nerve. 2010;62(7):659–66.
Kobe T, Witte AV, Schnelle A, Lesemann A, Fabian S, Tesky VA, et al. Combined omega-3 fatty acids, aerobic exercise and cognitive stimulation prevents decline in gray matter volume of the frontal, parietal and cingulate cortex in patients with mild cognitive impairment. Neuroimage. 2016;1(131):226–38.
Xu J, Yeon JE, Chang H, Tison G, Chen GJ, Wands J, et al. Ethanol impairs insulin-stimulated neuronal survival in the developing brain: role of PTEN phosphatase. J Biol Chem. 2003;278(29):26929–37.
Chiu SL, Chen CM, Cline HT. Insulin receptor signaling regulates synapse number, dendritic plasticity, and circuit function in vivo. Neuron. 2008;58(5):708–19.
Sun Q, Li J, Gao F. New insights into insulin: the anti-inflammatory effect and its clinical relevance. World J Diabetes. 2014;5(2):89–96.
Vandal M, White PJ, Tremblay C, St-Amour I, Chevrier G, Emond V, et al. Insulin reverses the high-fat diet-induced increase in brain Abeta and improves memory in an animal model of Alzheimer disease. Diabetes. 2014;63(12):4291–301.
Soscia SJ, Tong M, Xu XJ, Cohen AC, Chu J, Wands JR, et al. Chronic gestational exposure to ethanol causes insulin and IGF resistance and impairs acetylcholine homeostasis in the brain. Cell Mol Life Sci. 2006;63(17):2039–56.
Jauch-Chara K, Friedrich A, Rezmer M, Melchert UH, Scholand-Engler HG, Hallschmid M, et al. Intranasal insulin suppresses food intake via enhancement of brain energy levels in humans. Diabetes. 2012;61(9):2261–8.
Schilling TM, Ferreira de Sa DS, Westerhausen R, Strelzyk F, Larra MF, Hallschmid M, et al. Intranasal insulin increases regional cerebral blood flow in the insular cortex in men independently of cortisol manipulation. Hum Brain Mapp. 2014;35(5):1944–56.
Zhao L, Teter B, Morihara T, Lim GP, Ambegaokar SS, Ubeda OJ, et al. Insulin-degrading enzyme as a downstream target of insulin receptor signaling cascade: implications for Alzheimer’s disease intervention. J Neurosci. 2004;24(49):11120–6.
Hanson LR, Frey WH 2nd. Intranasal delivery bypasses the blood-brain barrier to target therapeutic agents to the central nervous system and treat neurodegenerative disease. BMC Neurosci. 2008;9(Suppl 3):S5.
Williams GS. Intranasal drug delivery bypasses the blood-brain barrier. Neurol Rev. 2016;24(4):40–1.
Born J, Lange T, Kern W, McGregor GP, Bickel U, Fehm HL. Sniffing neuropeptides: a transnasal approach to the human brain. Nat Neurosci. 2002;5(6):514–6.
Freiherr J, Hallschmid M, Frey WH 2nd, Brunner YF, Chapman CD, Holscher C, et al. Intranasal insulin as a treatment for Alzheimer’s disease: a review of basic research and clinical evidence. CNS Drugs. 2013;27(7):505–14.
Alagiakrishnan K, Sankaralingam S, Ghosh M, Mereu L, Senior P. Antidiabetic drugs and their potential role in treating mild cognitive impairment and Alzheimer’s disease. Discov Med. 2013;16(90):277–86.
Chen Y, Zhang J, Zhang B, Gong CX. Targeting insulin signaling for the treatment of Alzheimer’s disease. Curr Top Med Chem. 2016;16(5):485–92.
Reger MA, Watson GS, Frey WH 2nd, Baker LD, Cholerton B, Keeling ML, et al. Effects of intranasal insulin on cognition in memory-impaired older adults: modulation by APOE genotype. Neurobiol Aging. 2006;27(3):451–8.
Reger MA, Watson GS, Green PS, Baker LD, Cholerton B, Fishel MA, et al. Intranasal insulin administration dose-dependently modulates verbal memory and plasma amyloid-beta in memory-impaired older adults. J Alzheimers Dis. 2008;13(3):323–31.
Claxton A, Baker LD, Hanson A, Trittschuh EH, Cholerton B, Morgan A, et al. Long-acting intranasal insulin detemir improves cognition for adults with mild cognitive impairment or early-stage Alzheimer’s disease dementia. J Alzheimers Dis. 2015;44(3):897–906.
Hirsch IB, Franek E, Mersebach H, Bardtrum L, Hermansen K. Safety and efficacy of insulin degludec/insulin aspart with bolus mealtime insulin aspart compared with standard basal-bolus treatment in people with Type 1 diabetes: 1-year results from a randomized clinical trial (BOOST(R) T1). Diabet Med. 2016. doi:10.1111/dme.13068.
Christiansen JS, Niskanen L, Rasmussen S, Johansen T, Fulcher G. Lower rates of hypoglycemia during maintenance treatment with insulin degludec/insulin aspart versus biphasic insulin aspart 30: a combined analysis of two Phase 3a studies in type 2 diabetes. J Diabetes. 2016;8(5):720–8.
Kalra S, Gupta Y. Clinical use of insulin degludec: practical experience and pragmatic suggestions. N Am J Med Sci. 2015;7(3):81–5.
Doyle ME, Egan JM. Mechanisms of action of glucagon-like peptide 1 in the pancreas. Pharmacol Ther. 2007;113(3):546–93.
Long-Smith CM, Manning S, McClean PL, Coakley MF, O’Halloran DJ, Holscher C, et al. The diabetes drug liraglutide ameliorates aberrant insulin receptor localisation and signalling in parallel with decreasing both amyloid-beta plaque and glial pathology in a mouse model of Alzheimer’s disease. Neuromolecular Med. 2013;15(1):102–14.
Irwin N, Gault V, Flatt PR. Therapeutic potential of the original incretin hormone glucose-dependent insulinotropic polypeptide: diabetes, obesity, osteoporosis and Alzheimer’s disease? Expert Opin Investig Drugs. 2010;19(9):1039–48.
Yabe D, Seino Y. Incretin actions beyond the pancreas: lessons from knockout mice. Curr Opin Pharmacol. 2013;13(6):946–53.
Goke R, Larsen PJ, Mikkelsen JD, Sheikh SP. Distribution of GLP-1 binding sites in the rat brain: evidence that exendin-4 is a ligand of brain GLP-1 binding sites. Eur J Neurosci. 1995;7(11):2294–300.
Merchenthaler I, Lane M, Shughrue P. Distribution of pre-pro-glucagon and glucagon-like peptide-1 receptor messenger RNAs in the rat central nervous system. J Comp Neurol. 1999;403(2):261–80.
Richards P, Parker HE, Adriaenssens AE, Hodgson JM, Cork SC, Trapp S, et al. Identification and characterization of GLP-1 receptor-expressing cells using a new transgenic mouse model. Diabetes. 2014;63(4):1224–33.
Alhadeff AL, Rupprecht LE, Hayes MR. GLP-1 neurons in the nucleus of the solitary tract project directly to the ventral tegmental area and nucleus accumbens to control for food intake. Endocrinology. 2012;153(2):647–58.
Tang-Christensen M, Larsen PJ, Goke R, Fink-Jensen A, Jessop DS, Moller M, et al. Central administration of GLP-1-(7-36) amide inhibits food and water intake in rats. Am J Physiol. 1996;271(4 Pt 2):R848–56.
Turton MD, O’Shea D, Gunn I, Beak SA, Edwards CM, Meeran K, et al. A role for glucagon-like peptide-1 in the central regulation of feeding. Nature. 1996;379(6560):69–72.
Kastin AJ, Akerstrom V. Entry of exendin-4 into brain is rapid but may be limited at high doses. Int J Obes Relat Metab Disord. 2003;27(3):313–8.
Kanoski SE, Fortin SM, Arnold M, Grill HJ, Hayes MR. Peripheral and central GLP-1 receptor populations mediate the anorectic effects of peripherally administered GLP-1 receptor agonists, liraglutide and exendin-4. Endocrinology. 2011;152(8):3103–12.
Chan SW, Lin G, Yew DT, Rudd JA. A physiological role of glucagon-like peptide-1 receptors in the central nervous system of Suncus murinus (house musk shrew). Eur J Pharmacol. 2011;668(1–2):340–6.
Farr S, Baker C, Naples M, Taher J, Iqbal J, Hussain M, et al. Central nervous system regulation of intestinal lipoprotein metabolism by glucagon-like peptide-1 via a brain-gut axis. Arterioscler Thromb Vasc Biol. 2015;35(5):1092–100.
Griffioen KJ, Wan R, Okun E, Wang X, Lovett-Barr MR, Li Y, et al. GLP-1 receptor stimulation depresses heart rate variability and inhibits neurotransmission to cardiac vagal neurons. Cardiovasc Res. 2011;89(1):72–8.
Rachmany L, Tweedie D, Li Y, Rubovitch V, Holloway HW, Miller J, et al. Exendin-4 induced glucagon-like peptide-1 receptor activation reverses behavioral impairments of mild traumatic brain injury in mice. Age (Dordr). 2013;35(5):1621–36.
Duffy AM, Holscher C. The incretin analogue D-Ala2GIP reduces plaque load, astrogliosis and oxidative stress in an APP/PS1 mouse model of Alzheimer’s disease. Neuroscience. 2013;3(228):294–300.
Garcia-Casares N, Garcia-Arnes JA, Gomez-Huelgas R, Valdivielso-Felices P, Garcia-Arias C, Gonzalez-Santos P. Glucagon-like peptide-1 (GLP-1) mimetics: a new treatment for Alzheimer’s disease? Rev Neurol. 2014;59(11):517–24.
Holscher C. Drugs developed for treatment of diabetes show protective effects in Alzheimer’s and Parkinson’s diseases. Sheng Li Xue Bao. 2014;66(5):497–510.
Holscher C. Insulin, incretins and other growth factors as potential novel treatments for Alzheimer’s and Parkinson’s diseases. Biochem Soc Trans. 2014;42(2):593–9.
Irwin N, Flatt PR. New perspectives on exploitation of incretin peptides for the treatment of diabetes and related disorders. World J Diabetes. 2015;6(15):1285–95.
Kornelius E, Lin CL, Chang HH, Li HH, Huang WN, Yang YS, et al. DPP-4 inhibitor linagliptin attenuates abeta-induced cytotoxicity through activation of AMPK in neuronal cells. CNS Neurosci Ther. 2015;21(7):549–57.
Gejl M, Gjedde A, Egefjord L, Moller A, Hansen SB, Vang K, et al. In Alzheimer’s disease, 6-month treatment with GLP-1 analog prevents decline of brain glucose metabolism: randomized, placebo-controlled. Double-blind clinical trial. Front Aging Neurosci. 2016;8:108.
Liu X, Foo G, Lim WP, Ravikumar S, Sim SH, Win MS, et al. Sulphonylurea usage in melioidosis is associated with severe disease and suppressed immune response. PLoS Negl Trop Dis. 2014;8(4):e2795.
Moore EM, Mander AG, Ames D, Kotowicz MA, Carne RP, Brodaty H, et al. Increased risk of cognitive impairment in patients with diabetes is associated with metformin. Diabetes Care. 2013;36(10):2981–7.
Wheeler S, Moore K, Forsberg CW, Riley K, Floyd JS, Smith NL, et al. Mortality among veterans with type 2 diabetes initiating metformin, sulfonylurea or rosiglitazone monotherapy. Diabetologia. 2013;56(9):1934–43.
Ng TP, Feng L, Yap KB, Lee TS, Tan CH, Winblad B. Long-term metformin usage and cognitive function among older adults with diabetes. J Alzheimers Dis. 2014;41(1):61–8.
Luchsinger JA, Perez T, Chang H, Mehta P, Steffener J, Pradabhan G, et al. Metformin in amnestic mild cognitive impairment: results of a pilot randomized placebo controlled clinical trial. J Alzheimers Dis. 2016;51(2):501–14.
Kliewer SA, Lehmann JM, Milburn MV, Willson TM. The PPARs and PXRs: nuclear xenobiotic receptors that define novel hormone signaling pathways. Recent Prog Horm Res. 1999;54:345–67 (discussion 67–8).
Bright JJ, Kanakasabai S, Chearwae W, Chakraborty S. PPAR regulation of inflammatory signaling in CNS diseases. PPAR Res. 2008;2008:658520.
Fuentes L, Roszer T, Ricote M. Inflammatory mediators and insulin resistance in obesity: role of nuclear receptor signaling in macrophages. Mediat Inflamm. 2010;2010:219583.
Gilde AJ, Van Bilsen M. Peroxisome proliferator-activated receptors (PPARS): regulators of gene expression in heart and skeletal muscle. Acta Physiol Scand. 2003;178(4):425–34.
Lee CH, Olson P, Evans RM. Minireview: lipid metabolism, metabolic diseases, and peroxisome proliferator-activated receptors. Endocrinology. 2003;144(6):2201–7.
Schoonjans K, Staels B, Auwerx J. The peroxisome proliferator activated receptors (PPARS) and their effects on lipid metabolism and adipocyte differentiation. Biochim Biophys Acta. 1996;1302(2):93–109.
Watson GS, Cholerton BA, Reger MA, Baker LD, Plymate SR, Asthana S, et al. Preserved cognition in patients with early Alzheimer disease and amnestic mild cognitive impairment during treatment with rosiglitazone: a preliminary study. Am J Geriatr Psychiatry. 2005;13(11):950–8.
Abbatecola AM, Lattanzio F, Molinari AM, Cioffi M, Mansi L, Rambaldi P, et al. Rosiglitazone and cognitive stability in older individuals with type 2 diabetes and mild cognitive impairment. Diabetes Care. 2010;33(8):1706–11.
Chen J, Li S, Sun W, Li J. Anti-diabetes drug pioglitazone ameliorates synaptic defects in AD transgenic mice by inhibiting cyclin-dependent kinase5 activity. PLoS One. 2015;10(4):e0123864.
Toba J, Nikkuni M, Ishizeki M, Yoshii A, Watamura N, Inoue T, et al. PPARgamma agonist pioglitazone improves cerebellar dysfunction at pre-Abeta deposition stage in APPswe/PS1dE9 Alzheimer’s disease model mice. Biochem Biophys Res Commun. 2016;473(4):1039–44.
Zou C, Shi Y, Ohli J, Schuller U, Dorostkar MM, Herms J. Neuroinflammation impairs adaptive structural plasticity of dendritic spines in a preclinical model of Alzheimer’s disease. Acta Neuropathol. 2016;131(2):235–46.
Roses AD, Saunders AM, Lutz MW, Zhang N, Hariri AR, Asin KE, et al. New applications of disease genetics and pharmacogenetics to drug development. Curr Opin Pharmacol. 2014;14:81–9.
Crenshaw DG, Asin K, Gottschalk WK, Liang Z, Zhang N, Roses AD. Effects of low doses of pioglitazone on resting-state functional connectivity in conscious rat brain. PLoS One. 2015;10(2):e0117973.
Iwashita A, Muramatsu Y, Yamazaki T, Muramoto M, Kita Y, Yamazaki S, et al. Neuroprotective efficacy of the peroxisome proliferator-activated receptor delta-selective agonists in vitro and in vivo. J Pharmacol Exp Ther. 2007;320(3):1087–96.
Aleshin S, Reiser G. Peroxisome proliferator-activated receptor beta/delta (PPARbeta/delta) protects against ceramide-induced cellular toxicity in rat brain astrocytes and neurons by activation of ceramide kinase. Mol Cell Neurosci. 2014;59:127–34.
Aleshin S, Grabeklis S, Hanck T, Sergeeva M, Reiser G. Peroxisome proliferator-activated receptor (PPAR)-gamma positively controls and PPARalpha negatively controls cyclooxygenase-2 expression in rat brain astrocytes through a convergence on PPARbeta/delta via mutual control of PPAR expression levels. Mol Pharmacol. 2009;76(2):414–24.
Santhanam AV, d’Uscio LV, He T, Katusic ZS. PPARdelta agonist GW501516 prevents uncoupling of endothelial nitric oxide synthase in cerebral microvessels of hph-1 mice. Brain Res. 2012;5(1483):89–95.
Yin KJ, Deng Z, Hamblin M, Zhang J, Chen YE. Vascular PPARdelta protects against stroke-induced brain injury. Arterioscler Thromb Vasc Biol. 2011;31(3):574–81.
Saluja I, Granneman JG, Skoff RP. PPAR delta agonists stimulate oligodendrocyte differentiation in tissue culture. Glia. 2001;33(3):191–204.
Kocalis HE, Turney MK, Printz RL, Laryea GN, Muglia LJ, Davies SS, et al. Neuron-specific deletion of peroxisome proliferator-activated receptor delta (PPARdelta) in mice leads to increased susceptibility to diet-induced obesity. PLoS One. 2012;7(8):e42981.
Poon K, Alam M, Karatayev O, Barson JR, Leibowitz SF. Regulation of the orexigenic neuropeptide, enkephalin, by PPARdelta and fatty acids in neurons of the hypothalamus and forebrain. J Neurochem. 2015;135(5):918–31.
Yu S, Levi L, Casadesus G, Kunos G, Noy N. Fatty acid-binding protein 5 (FABP5) regulates cognitive function both by decreasing anandamide levels and by activating the nuclear receptor peroxisome proliferator-activated receptor beta/delta (PPARbeta/delta) in the brain. J Biol Chem. 2014;289(18):12748–58.
Mounsey RB, Martin HL, Nelson MC, Evans RM, Teismann P. The effect of neuronal conditional knock-out of peroxisome proliferator-activated receptors in the MPTP mouse model of Parkinson’s disease. Neuroscience. 2015;6(300):576–84.
Delmedico MK, Severynse-Stevens D, Oliver WR. DB959 is a novel, dual PPAR δ/γ agonist which controls glucose and regulates triglycerides and HDLc in animal models of T2D and dyslipidemia. In: 69th Annual Scientific Sessions of the American Diabetes Association; 2009. p. 365-OR.
Didsbury J. T3D Therapeutics, Inc. Receives FDA IND Approval to Begin Phase 2 Clinical Study of T3D-959 in Alzheimer’s Patients. Research Triangle Park, NC: PRWEB; 2015.
Didsbury J, de la Monte SM. T3D-959: a multi-faceted disease remedial drug candidate for the treatment of Alzheimer’s disease. Alzheimer Dement. 2015;11(7):906.
Tong M, Deochand C, Didsbury J, de la Monte SM. T3D-959: a multi-faceted disease remedial drug candidate for the treatment of Alzheimer’s disease. J Alzheimers Dis. 2016;51(1):123–38.
Acknowledgements
Supported by AA-11431 from the National Institutes of Health.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
No funding was received for the publication of this review.
Conflict of interest
The author has consulted and delivered a paid lecture for Novo Nordisk in 2016, and is on the Scientific Advisory Board for T3D Pharmaceuticals, Inc.
Rights and permissions
About this article

Cite this article
de la Monte, S.M. Insulin Resistance and Neurodegeneration: Progress Towards the Development of New Therapeutics for Alzheimer’s Disease. Drugs 77, 47–65 (2017). https://doi.org/10.1007/s40265-016-0674-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-016-0674-0