
Abstract
An age-associated increase in oxidative damage to nucleic acids, predominantly to RNA, has been recently demonstrated in neurons of human and rodent brains, which may play a fundamental role in the development of age-associated neurodegeneration. Indeed, more prominent levels of neuronal RNA oxidation compared to normal aging have been described in neurodegenerative disorders including Alzheimer disease, Parkinson disease, dementia with Lewy bodies, and amyotrophic lateral sclerosis. Moreover, oxidative damage to RNA has been found also in cellular and animal model of neurodegeneration. Oxidative RNA modification can occur not only in protein-coding RNAs but also in non-coding RNAs that are recently revealed to contribute towards the complexity of the mammalian brain. It has been hypothesized that RNA oxidation causes aberrant expression of microRNAs and proteins and subsequently initiates inappropriate cell fate pathways. While less lethal than mutations in the genome and not inheritable, such sublethal damage to cells might be associated with underlying mechanisms of degeneration, especially age-associated neurodegeneration. Of particular interest, the accumulating evidence obtained from studies on either human samples or experimental models coincidentally suggests that RNA oxidation is a feature in neurons of aging brain and more prominently observed in vulnerable neurons at early-stage of age-associated neurodegenerative disorders, indicating that RNA oxidation actively contributes to the background, the onset, and the development of the disorders. Further investigations aimed at understanding of the processing mechanisms related to oxidative RNA damage and its consequences may provide significant insights into the pathogenesis of neurodegenerative disorders and lead to better therapeutic strategies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Neurodegenerative disorders are common in elderly population; for example, the prevalence in the United States per 1,000 elderly is 67 for Alzheimer disease (AD) and 9.5 for Parkinson disease (PD) (Hirtz et al. 2007), the annual incidence in Italy per 100,000 of general population is 1.7 for amyotrophic lateral sclerosis (ALS) with a peak between 65 and 75 years of age (Logroscino et al. 2005); the prevalence in Finland per 1,000 general population aged 75 years and older is 106 for AD and 50 for dementia with Lewy bodies (DLB) (Rahkonen et al. 2003). Many lines of evidence have indicated that oxidative damage is involved not only in the process of brain aging (Gemma et al. 2007) but also in the pathogenesis of neurodegenerative disorders including AD, PD, and ALS (Sayre et al. 2001; Ischiropoulos and Beckman 2003; Jenner 2003; Andersen 2004; Barnham et al. 2004; Barber et al. 2006; Lin and Beal 2006; Nunomura et al. 2006). Indeed, oxidatively modified products of nucleic acids and proteins, as well as products by lipid peroxidation and glycoxidation, all known markers of oxidative damage, have been demonstrated in the affected lesions in the central nervous system (CNS), cerebrospinal fluid (CSF), plasma or serum, and urine from the patients with these disorders. The increased levels of oxidative damage in such neurodegenerative disorders are often accompanied by the reduced levels of anti-oxidative defense mechanisms in the same subjects (Ischiropoulos and Beckman 2003; Andersen 2004). Remarkably, a number of known genetic and environmental factors of the neurodegenerative disorders, namely disease-specific gene mutations, risk-modifying gene polymorphisms, and risk-modifying life-style factors are closely associated with oxidative damage (Barnham et al. 2004; Ischiropoulos and Beckman 2003; Nunomura et al. 2006, 2007), which implicates a pathogenic role of oxidative damage in the process of neurodegeneration.
Despite the abundant evidence for an involvement of oxidative insults at an early-stage of the neurodegenerative process, interventions such as the administration of one or a few antioxidants have been, at best, modestly successful in clinical trials. The complexity of the metabolism of reactive oxygen species (ROS) suggests that such interventions may be too simplistic and requires more integrated approaches not only to enrich the exogenous antioxidants but also to up-regulate the multilayered endogenous anti-oxidative defense systems (Lin and Beal 2006; Nunomura et al. 2006, 2007). Indeed, the pathomechanisms of neurodegeneration may be better explained by an “age-based” theory rather than a simple proteinopathy hypothesis (Herrup 2010). Therefore, it is clear that there is a considerable need for a better understanding of the association among ROS metabolism, aging, and neurodegeneration, which may constitute a breakthrough in the treatment of neurodegenerative disorders (Mangialasche et al. 2010).
DNA and RNA Oxidation
Oxidative damage to DNA has been well studied and several classes of products such as base oxidation and fragmentation products (e.g., single- and double-strand breaks), inter/intra-strand cross-links, DNA–protein cross-links, and sugar fragmentation products are identified (Evans et al. 2004; Cooke et al. 2006). Far fewer studies have focused on oxidative damage to RNA and only limited kinds of oxidatively modified bases in RNA have been reported previously (Kasai et al. 1986; Ames and Gold 1991; Yanagawa et al. 1992; Schneider et al. 1993; Rhee et al. 1995; Barciszewski et al. 1999). Among multiple adducts of nucleoside oxidation, adducts of deoxyguanosine and guanosine, i.e., 8-hydroxydeoxyguanosine (8-OHdG) and 8-hydroxyguanosine (8-OHG) are two of the best characterized and studied forms of DNA and RNA oxidation, respectively (Fiala et al. 1989; Wamer and Wei 1997; Shen et al. 2000; Hofer et al. 2005, 2006, 2008b). Recently, detection and quantification of oxidation-induced abasic (depurinated/depyrimidinated) RNA have been reported by using an aldehyde reactive probe (Tanaka et al. 2011a, b).
Although RNA should be subject to the same oxidative insults as DNA and other cellular macromolecules, oxidative damage to RNA has not been a major focus in investigating the magnitude and the biological consequences of ROS. Because RNA is largely single-stranded and its bases are not protected by hydrogen bonding and is probably less protected by specific proteins, RNA may be more susceptible to oxidative insults than DNA (Nunomura et al. 1999; Brégeon and Sarasin 2005; Li et al. 2006; Moreira et al. 2008). That RNA is a vulnerable target is also a reasonable proposition given the relative cellular abundance of RNA and the subcellular distribution of RNA that locates in the vicinity of mitochondria, the primary source of ROS (Nunomura et al. 1999). Given these factors, it is not surprising that, using high-performance liquid chromatography coupled with electrochemical detector (HPLC-ECD) or with electrospray tandem mass spectrometry (HPLC–MS/MS), greater oxidation to RNA than to DNA has been shown in an experiment with isolated DNA and RNA (Hofer et al. 2006) as well as in cell lines and tissue, namely, human leukocytes (Shen et al. 2000), human skin fibroblasts (Wamer and Wei 1997), human lung epithelial cells (Hofer et al. 2005), rat skeletal muscle (Hofer et al. 2008b), rat liver (Fiala et al. 1989; Hofer et al. 2006). Urinary excretion of an oxidized form of ribonucleoside in healthy humans and rats (Weimann et al. 2002) not only suggests substantial RNA oxidation in normal metabolism but also the existence of a repair mechanism for the damaged RNA. Greater oxidation to RNA than to DNA has been found also in human urine samples from patients with hereditary hemochromatosis (Broedbaek et al. 2009) as well as workers exposed to styrene or benzene (Manini et al. 2009, 2010). Furthermore, it has been shown recently that a urinary marker of oxidative damage to RNA but not DNA predicts long-term mortality of type 2 diabetic patients (Broedbaek et al. 2011). Indeed, higher urinary levels of oxidized RNA in the patients are associated with higher all-cause mortality and higher diabetes-related mortality, which is a good example showing an involvement of RNA oxidation in the pathophysiology of a chronic disease.
It is now becoming evident that RNA molecules are not only intermediates for the transfer of genetic information from DNA to proteins but also key players in many mechanisms controlling expression of genetic information (Costa 2005; Szymanski et al. 2005; Cao et al. 2006; Mehler and Mattick 2006, 2007). Given the view that RNAs are undergoing a “renaissance”, we and others have developed a hypothesis that RNA damage is involved in the pathomechanisms of neurodegeneration (Brégeon and Sarasin, 2005; Costa 2005; Cao et al. 2006; Li et al. 2006; Mehler and Mattick 2006, 2007; Moreira et al. 2008).
RNA Oxidation and the Biological Consequence
Formation of Oxidized RNA Nucleosides and Strand Scission from RNA Nucleobase Radical
More than 20 different types of oxidatively altered purine and pyrimidine bases have been detected in nucleic acids (Barciszewski et al. 1999; Evans et al. 2004; Ishibashi et al. 2005; Cooke et al. 2006). However, since guanine is the most reactive of the nucleic acid bases (Yanagawa et al. 1992), it is not surprising that the oxidized base, 8-hydroxyguanine is the most abundant among the oxidized bases (Brégeon and Sarasin 2005). The 8-hydroxyguanine-containing nucleoside, 8-OHG, can be formed in RNA by direct oxidation of the base and also by the incorporation of the oxidized base from the cytosolic pool into RNA through the normal action of RNA polymerase (Fig. 1) (Yanagawa et al. 1992; Ishibashi et al. 2005). Not only 8-OHG but also 8-hydroxyadenosine, 5-hydroxycytidine, and 5-hydroxyuridine have been identified in oxidized RNA (Yanagawa et al. 1992), which may have altered pairing capacity and thus be at the origin of erroneous protein production. Indeed, the 8-hydroxyguanine can pair with both adenine and cytosine, and thus oxidized RNA compromises the accuracy of translation (Taddei et al. 1997; Ishibashi et al. 2005).

Mode of formation of 8-OHG-containing RNA and surveillance mechanism for oxidized RNA in mammalian cells. 8-OHG-containing RNA can be generated by direct oxidation to RNA as well as by incorporation of oxidized ribonucleotide, 8-hydroxy-guanosine-triphosphate (8-OH-GTP), which can be generated by direct oxidation of GTP as well as by phosphorylation of 8-hydroxy-guanosine-diphosphate (8-OH-GDP) by NDK. Remarkably, several enzymes involved in nucleotides metabolism show discriminating function against incorporation of oxidized nucleotide into nucleic acids. GK, an enzyme converts GMP to GDP, is inactive on 8-hydroxy-guanosine-monophosphate (8-OH-GMP). Similarly, RNR, an enzyme converts GDP to dGDP, is inactive on 8-OH-GDP, which avoid incorporation of the oxidized nucleotide into DNA synthesis. Additionally, RNA polymerase incorporates 8-OH-GTP into RNA at much lower rate compared with the incorporation of GTP. Furthermore, MutT homologue 1 (MTH1) has the potential to hydrolyze 8-OH-GTP or 8-OH-GDP to 8-oxoGMP and Nudix type 5 (NUDT5) can hydrolyze 8-OH-GDP to 8-oxoGMP, which drastically reduces the possibilities that the oxidized nucleotides are incorporated into RNA. When 8-OHG is misincorporated into RNA, it may cause direct errors in translation or inadequate regulation of protein synthesis and cells may switch from translation to degradation by sequestrating the oxidized RNA with specific binding proteins such as polynucleotide phosphorylase (PNPase), Y-box binding protein 1 (YB-1), and heterogeneous nuclear ribonucleoprotein D0 (HNRNPD). The existence of repair mechanism for oxidized RNA remains to be elucidated
Recently, the chemical characterization of the pathway for direct strand scission from RNA nucleobase radical has been provided (Jacobs et al. 2010). One mechanism involves initial formation of a nucleobase radical and/or the respective peroxyl radical which subsequently abstracts a hydrogen atom from the ribose ring, ultimately resulting in strand scission. Indeed, direct strand scission is proposed to result from as many as 40 % of reactions of •OH with RNA (Jacobs et al. 2010).
Defective Protein Synthesis Due to Oxidized RNA
The biological consequence of oxidatively damaged mRNA species has been investigated in vitro by expressing them in cell lines. Oxidized mRNAs lead to loss of normal protein level and protein function, and potentially produce defective proteins leading to protein aggregation, a common feature of neurodegenerative disorders (Shan et al. 2003). In a recent study, polyribosome analysis indicates that oxidized bases in mRNAs cause ribosome stalling on the transcripts or slow the translation process, which leads to a decrease of protein expression (Shan et al. 2007). When oxidized and non-oxidized luciferase RNAs were subjected to translation in rabbit reticulocyte lysates and the fractions were analyzed by northern blot, the oxidized RNA samples showed a decreased amount of free monosomes and an increased amount of RNA-associated polyribosomes compared to the non-oxidized RNA samples (Shan et al. 2007). In another recent study, the translation of oxidized mRNA in cell lines causes the accumulation of short polypeptides, resulting from the premature termination of the translation process of the oxidized mRNA and/or the proteolytic degradation of the modified protein containing the translation errors due to the oxidized mRNA (Tanaka et al. 2007). Coincidently, oxidative damage to Escherichia coli 16S rRNA results in the formation of short cDNA by the RT-PCR (Gong et al. 2006). The biological consequence of ribosomal oxidation have also been investigated in vitro using translation assays with oxidized ribosomes from rabbit reticulocytes and show a significant reduction of protein synthesis (Honda et al. 2005). Notably, studies on brains of subjects with AD and MCI have demonstrated ribosomal dysfunction associated with oxidative RNA damage (Ding et al. 2005, 2006). Isolated polyribosome complexes from AD and MCI brains show a decreased rate and capability for protein synthesis without alteration in the polyribosome content. Decreased rRNA and tRNA levels and increased 8-OHG in total RNA pool, especially in rRNA, are accompanied by the ribosomal dysfunction, while there is no alteration in the level of initiation factors (Ding et al. 2005).
These findings have indicated that RNA oxidation has detrimental effects on cellular function whether the damaged RNA species are coding for proteins (mRNA) or performing translation (rRNA and tRNA). It is noteworthy in this respect that studies on some anti-cancer agents have shown that RNA damage can lead to cell-cycle arrest and cell death, much as DNA damage does. RNA damage may cause cell death via either pathway involving p53-dependent mechanism associated with inhibition of protein synthesis or p53-independent mechanism different from inhibition of protein synthesis (Bellacosa and Moss 2003).
Coping with RNA Damage
Ribonucleases
Degradation of RNA plays a central role in RNA metabolism and damaged RNA can be removed through degradation by ribonucleases (RNase), but selective degradation activity for oxidized RNA has not been established for known RNases (Deutscher 2006; Li et al. 2006). Oxidative stress induces cytoplasmic mRNA processing bodies (P-bodies), the site of active degradation of mRNA (Sheth and Parker 2003), which is linking with an induction of another cytoplasmic structure called “stress granules” (Kedersha et al. 2005). In contrast to mRNAs with rapid turnover, stable RNAs, consisting primarily of rRNAs and tRNAs and encompassing 98 % of total cellular RNA, may be protected against RNase action by tertiary structure, assembly into ribonucleoprotein complex, or even blocking the RNA’s 3′ terminus (Deutscher 2006).
Repair Mechanisms
Until recently, it has been considered that damaged RNA may be only degraded rather than repaired. However, Aas et al. (2003) has suggested that the cells have at least one specific mechanism to repair RNA damage, indicating that cells may have a greater investment in the protection of RNA than previously suspected (Bellacosa and Moss 2003; Krokan et al. 2004; Brégeon and Sarasin 2005). Indeed, alkylation damage in RNA is repaired by the same mechanism as a DNA-repair, catalyzed in the bacterium Escherichia coli by the enzyme AlkB, and in humans by the related protein (Aas et al. 2003). Alk B and its homologue hABH3 cause hydroxylation of the methyl group on damaged DNA and RNA bases, and thus directly reverse alkylation damage. Alk B and hABH3, but not hABH2, repair RNA, since Alk B and hABH3 prefer single-stranded nucleic acids while hABH2 acts more efficiently on double-stranded DNA (Aas et al. 2003).
DNA damage can be repaired not only by the mechanism of direct reversal of the modified bases but also by a base excision repair mechanism. Specific DNA glycosylases excise the damaged base and DNA polymerases replace the nucleotides (Bellacosa and Moss 2003; Nakabeppu et al. 2004). Defective DNA base excision repair is demonstrated in brain from subjects with AD and MCI (Weissman et al. 2007; Shao et al. 2008). While it remains unknown whether RNA damage can be repaired by base excision, it has been considered that the mechanism is not likely efficient at the RNA level, for the excision repair generally requires a complementary strand (Krokan et al. 2004). However, it has recently been suggested that a DNA-repair enzyme APE/Ref-1, acting as the major apurinic/apyrimidinic endonuclease, is endowed with rRNA quality control (Vascotto et al. 2009). Indeed, inability to remove 8-hydroxyguanine-containing rRNA upon oxidative stress is observed in APE/Ref-1-knocked-down cells, in which impaired translation, lower intracellular protein content, and decreased cell growth rate are also observed.
Avoidance of Oxidized Ribonucleotides Incorporation into Translational Machinery
Cells have mechanisms of dealing with nucleotide damage other than direct repair and excision repair, which seems to be useful for defense against oxidative damage to both DNA and RNA. Because oxidation of nucleotides can occur in the cellular nucleotide pool and the oxidized nucleotide can be incorporated into DNA and RNA, the mechanism avoiding such incorporation of the oxidized nucleotide is involved in coping with nucleic acid damage (Fig. 1) (Bellacosa and Moss 2003; Brégeon and Sarasin 2005; Li et al. 2006). MutT protein in Escherichia coli and its mammalian homologues MutT homologue 1 (MTH1) and Nudix type 5 (NUDT5) proteins participate in this error-avoiding mechanism by hydrolyzing the oxidized nucleoside diphosphates and/or triphosphates to the monophosphates (Taddei et al. 1997; Hayakawa et al. 1999; Nakabeppu et al. 2004, 2006; Ishibashi et al. 2005; Ito et al. 2005). Indeed, the increase in the production of erroneous proteins by oxidative damage is 28-fold over the wild type cells in Escherichia coli mutT deficient cells, which is reduced to 1.2- or 1.4-fold by the expression of MTH1 or NUDT5, respectively (Ishibashi et al. 2005). Correspondingly, MTH1 deficiency augments the RNA oxidation induced by kainic acid treatment in MTH1-null mouse (Kajitani et al. 2006). Indeed, an increase in neuronal RNA oxidation is correlated with decreased expression of MTH1 in the hippocampus of senescence-accelerated SAMP8 mice as well as patients with AD (Song et al. 2011). However, in other studies, an increased expression of MTH1 in the vulnerable neuronal populations has been reported in the brains of AD (Furuta et al. 2001) and PD (Shimura-Miura et al. 1999), which may indicate a compensatory up-regulation of the MTH1 against oxidative stress (Nakabeppu et al. 2006).
In addition to the degradative activity of MTH1 and NUDT5, several enzymes involved in nucleotide metabolism show a discriminator activity against the oxidized nucleotides (Fig. 1). Guanylate kinase (GK), an enzyme that converts GMP to GDP, is inactive on 8-OH-GMP, while nucleotide diphosphate kinase (NDK), an enzyme that converts GDP to GTP, fails to show such discriminating function (Hayakawa et al. 1999). Similarly, ribonucleotide reductase (RNR), an enzyme that catalyzes reduction of four naturally occurring ribonucleoside diphosphates, is inactive on conversion of 8-OH-GDP to 8-OH-dGDP, which avoids incorporation of the oxidized nucleotide into DNA synthesis (Hayakawa et al. 1999). The final “gatekeeper” discriminating the oxidized nucleotide from normal nucleotide is RNA polymerase that incorporates 8-OH-GTP into RNA at a much lower rate compared to the normal GTP incorporation (Taddei et al. 1997; Li et al. 2006). However, a recent study on MCF-7 cells exposed to carbon-labeled 8-OHdG has revealed an incorporation of extracellular 8-OHdG into RNA with 5–6-fold higher levels compared to the incorporation into DNA (Mundt et al. 2008). The proposed mechanism of 8-OHdG metabolism involves phosphorolysis of 8-OHdG to free 8-hydroxyguanine and incorporation of the free 8-hydroxyguanine into RNA synthesis. Therefore, some of the above-mentioned discriminating mechanisms against the oxidized nucleotides may be inefficient.
Proteins Binding Specifically to Oxidized RNA
Then, one important question is whether cells have machinery to deal with oxidatively damaged nucleotides that are contained in RNA, because RNA can be directly oxidized even if the incorporation of oxidized nucleotides into RNA is blocked. Recently, proteins that bind specifically to 8-OHG-containing RNA have been reported, namely, Escherichia coli polynucleotide phosphorylase (PNPase) protein and human PNPase (Hayakawa et al. 2001; Hayakawa and Sekiguchi 2006), human Y-box binding protein 1 (YB-1) (Hayakawa et al. 2002), and heterogeneous nuclear ribonucleoprotein D0 (HNRNPD) (Hayakawa et al. 2010). The binding of the specific protein likely makes the 8-OHG-containing RNA resistant to nuclease degradation (Hayakawa et al. 2001). However, it has been proposed that these proteins may recognize and discriminate the oxidized RNA molecule from normal ones, thus contributing to the fidelity of translation in cells by sequestrating the damaged RNA from the translational machinery (Hayakawa et al. 2001, 2002, 2010; Hayakawa and Sekiguchi 2006). The human PNPase binds to 8-OHG-containing RNA preferentially and cellular amounts of human PNPase decrease rapidly by exposure to agents inducing oxidative stress, while amounts of other proteins in the cells do not change after the treatments (Hayakawa and Sekiguchi 2006). A recent study on HeLa cell exposed to H2O2 has demonstrated that overexpression of human PNPase reduces RNA oxidation and increases cell viability against the oxidative insult, while human PNPase knockdown increases RNA oxidation and decreases cell viability (Wu and Li 2008). Responses of HNRNPD protein to oxidative stress also have been demonstrated by using HeLa cell. The amount of HNRNPD protein rapidly decreases when cells are exposed to H2O2, and suppression of HNRNPD expression by small interfering RNA causes cells to exhibit an increased sensitivity to H2O2 (Hayakawa et al. 2010). Furthermore, human YB-1 has been elucidated to be a component of P-bodies where active degradation of mRNA occurs. YB-1 is translocated from P-bodies to stress granules during oxidative stress, which suggests a dynamic link between P-bodies and stress granules under oxidative stress (Yang and Bloch 2007).
It is possible that the RNA quality control mechanisms are defective or inefficient in cancer cells as well as cells of neurodegenerative diseases. Further elucidation of the mechanisms of repair or avoidance of RNA damage and their potential role in preventing human diseases might provide new approaches to a number issues of life science that have evaded resolution, while it has not been the major focus in investigation for a long period (Brégeon and Sarasin 2005; Li et al. 2006; Yang and Bloch 2007).
Sublethal Damage to RNA and Its Possible Relevance to Neurodegeneration
Recent progress in genetics has revealed an expanding universe of RNA beyond its classical function as intermediates in protein synthesis according to “Central Dogma” that describes the transcription from DNA to messenger RNA (mRNA) and translation from mRNA to proteins. Indeed, it is now evident that only a minority of genetic transcripts (2–3 % in the human) code for proteins. Non-coding RNA (ncRNA), rather than being “junk”, can function directly in structural and catalytic activities and also appears to play a critical role in regulating the timing and rate of gene expression (Costa 2005; Szymanski et al. 2005; Cao et al. 2006; Mehler and Mattick 2006, 2007). Of particular note, the complexity of an organism correlates poorly with the number of protein-coding genes, however, complexity is highly correlated with the number of ncRNAs (Taft et al. 2007). Furthermore, the increasing variety of ncRNAs being identified in the CNS suggests a strong connection between the biogenesis, dynamics of action, and combinational regulatory potential of ncRNAs and the complexity of the CNS (Cao et al. 2006; Mehler and Mattick 2006, 2007). Indeed, specific ncRNAs have been shown to regulate dendritic spine development, neuronal fate specification and differentiation, and synaptic protein synthesis (Satterlee et al. 2007). Therefore, further advances in studies on the mechanisms and consequences of RNA damage and its surveillance may have a significant impact on understanding of the pathophysiology of currently unresolved complex disorders including neurological and psychiatric disorders (Perkins et al. 2005; Cao et al. 2006; Mehler and Mattick 2006, 2007; Satterlee et al. 2007; Taft et al. 2007).
MicroRNAs are small (~22 nucleotides) ncRNAs that participate in mRNA translational regulation. Despite their relatively recent discovery, it is already known that microRNAs play important roles in neural development (Mehler and Mattick 2006). They are also expressed at high levels within mature and even the senescent brain and function to orchestrate the maintenance of adult neural traits, to promote cellular homeostasis against endogenous and exogenous stress, and to modulate multiple parameters associated with synaptic plasticity (Mehler and Mattick 2006). Nelson et al. (2008) have reviewed rapidly accumulating evidence of the roles of microRNAs in neurodegenerative disorders including AD, PD, and triplet repeat disorders. They have discussed a possible involvement of microRNAs oxidation in the pathogenesis of the neurodegenerative disorders. It has been hypothesized that RNA oxidation causes aberrant expression of microRNAs and proteins and subsequently initiates inappropriate cell fate pathways. Recently, Shi and Gibson (2011) have reported that post-transcriptional up-regulation of a mitochondrial enzyme, malate dehydrogenase, by oxidative stress in a neuronal cell line is mediated by microRNA-743a, which provides insights into possible roles of microRNAs in oxidative stress and neurodegeneration. Specifically, several questions remain to be elucidated: (1) whether oxidative modification occurs exactly in microRNAs in neurodegenerative disorders; (2) whether oxidative modification alters function of microRNAs; and (3) whether oxidized microRNAs participate in the induction/promotion of neurodegenerative pathway.
In two of the most frequent neurodegenerative disorders, AD and PD (Hirtz et al. 2007), gene mutations cause hereditary-forms of the disorders and the mutations are associated with protein aggregates (e.g., amyloid-β aggregation in senile plaques in AD and α-synuclein aggregation in Lewy bodies in PD) that form hallmark pathologies in the affected brains (Taylor et al. 2002; Lin and Beal 2006). However, the majority of the patients with AD and PD have sporadic disorder and the etiology of the disorders is largely unknown. Indeed, there is recent evidence suggesting that several known gene mutations in causing familial AD (amyloid-β protein precursor, presenilin-1, or presenilin-2 gene) and familial PD (Parkin, PINK-1, or DJ-1 gene) are associated with increased oxidative stress. Also, several known genetic (e.g., apolipoprotein Eε4 variant) and environmental (e.g., metals or pesticides exposure) risk factors of sporadic AD and/or PD are associated with increased oxidative stress (Nunomura et al. 2007). As we have reviewed here, oxidative damage to neuronal RNA is not only a common feature of AD, PD and associated neurodegenerative disorders but also an early-stage event in the pathological cascade of the disorders, which suggests an involvement of RNA damage in the pathogenic mechanisms of these neurodegenerative disorders. While RNA damage would be less lethal for cells than mutations in the genome, such moderate, sublethal insults to cells might be associated with underlying mechanisms of several human disorders, especially chronic degeneration.
Age-Associated Increase in Oxidative Damage to RNA (Table 1)
Recently, an age-associated increase in neuronal RNA oxidation (8-OHG) has been demonstrated by immunocytochemical detection accompanied with densitometric semi-quantitative analysis on post-mortem human brain samples (Nunomura et al. 2012). Experimental studies on young and old rodents (Liu et al. 2002; Cui et al. 2009) and a strain of senescence-accelerated mice (Song et al. 2011; Shi et al. 2012) also have shown an age-associated increase in neuronal 8-OHG in brain. Of note, antioxidants or mitochondrial metabolites can reduce both oxidative damage and the spatial memory deficit in old animals (Liu et al. 2002). In the C57BL/6J mice, even young animals (10–12 weeks old) show substantial levels of spontaneously oxidized RNA (8-OHG) in neurons of the hippocampus and the substantia nigra (Yamaguchi et al. 2006), which contrasts the observations in human control brains with no apparent level of RNA oxidation at younger ages (Nunomura et al. 2012). It may be interesting to see if the levels of RNA oxidation are different among diverse mammalian species particularly with reference to the maximum life spans of the species which negatively correlates with the levels of DNA oxidation (Foksinski et al. 2004).
An age-associated increase in 8-OHG has been demonstrated also by an analysis of urine from healthy human individuals as a marker of systemic RNA oxidation (Andreoli et al. 2011; Broedbaek et al. 2011). In experimental animals, a recent study has demonstrated that RNA, but not DNA, oxidation in skeletal muscle increases with aging and muscle atrophy, which is related to increased levels of non-heme iron (Hofer et al. 2008b). Also, liver of rodents shows age-associated RNA oxidation (Seo et al. 2006). Specifically, levels of mitochondrial RNA oxidation in skeletal muscle and liver increase with age and correlate with levels of mitochondrial iron (Seo et al. 2008). These findings further support the concept that RNA oxidation is involved in chronic age-associated degeneration.
RNA Oxidation in Various Neuropsychiatric Disorders (Table 2)
RNA Oxidation in AD and PD
The disruption of transcriptional or translational fidelity in neurons leads to the accumulation of aberrant or misfolded proteins and neuronal death (van Leeuwen et al. 1998; Lee et al. 2006), which suggests a role of RNA damage in the underlying mechanisms of neurological disorders. The availability of highly specific antibodies with 8-OHdG/8-OHG has enabled us to perform in situ approaches to examine nucleoside oxidation in post-mortem brain samples taken from patients with neurological disorders (Park et al. 1992; Yin et al. 1995). In 1999, increased levels of 8-OHdG/8-OHG were demonstrated in the vulnerable neuronal populations in post-mortem brains of patients with AD and PD (Nunomura et al. 1999; Zhang et al. 1999). In AD and PD, the neuronal 8-OHdG/8-OHG showed cytoplasmic predominance, which suggested that mitochondrial DNA and cytoplasmic RNA in neurons were major targets of oxidative damage. However, because the neuronal 8-OHdG/8-OHG immunoreactivity in AD brain was diminished greatly by RNase pretreatment but not by DNase pretreatment, we concluded that the oxidized nucleoside was predominantly associated with RNA rather than DNA (Nunomura et al. 1999). This notion was further supported by the immunoelectron microscopic observation that most of the oxidized nucleoside was localized to ribosomes (Nunomura et al. 2001).
RNA Oxidation in Other Neuropsychiatric Disorders
RNA oxidation was also found biochemically in tissue samples of the motor cortex and spinal cord from patients with ALS (Chang et al. 2008). Similar RNA oxidation in neuronal cytoplasm was observed in brain samples of patients with Down syndrome (Nunomura et al. 2000), DLB (Nunomura et al. 2002), dentaterubral-pallidoluisian atrophy (DRPLA) (Miyata et al. 2008), Creutzfeldt–Jakob disease (CJD) (Guentchev et al. 2002), and subacute sclerosing panencephalitis (SSPE) (Hayashi et al. 2002), as summarized in Table 2. The oxidative damage to RNA was demonstrated not only in sporadic-forms of the disorders but also in familial-forms of AD (Nunomura et al. 2004), ALS (Chang et al. 2008), and prion diseases, i.e., familial CJD and Gerstmann–Straussler–Scheinker disease (GSS) (Guentchev et al. 2002; Petersen et al. 2005). Furthermore, nuclear DNA oxidation and cytoplasmic RNA oxidation were observed in brains of patients with a genetic defect of nucleotide excision repair mechanism, xeroderma pigmentosum, showing cutaneous hypersensitivity to sunlight and progressive neurological disturbances (Hayashi et al. 2005). Recently, neuronal RNA oxidation has been demonstrated also in psychiatric diseases and brain dysfunction due to severe somatic diseases; namely, schizophrenia and mood disorders (Che et al. 2010), cirrhosis with hepatic encephalopathy (Görg et al. 2010) and, fatal brain edema of diabetic ketoacidosis (Hoffman et al. 2011). These findings may indicate that RNA oxidation is involved in the pathogenesis of a wide variety of neuropsychiatric disorders including not only progressive neurodegeneration but also potentially reversible neuronal dysfunction.
RNA Oxidation in Extra-Neuronal Tissue in Human Disorders
Additionally, we should pay attention to the fact that RNA oxidation has been demonstrated also in extra-neuronal tissue in human disorders; e.g., muscle cells of patients with rimmed vacuole myopathy (Tateyama et al. 2003) as well as in the smooth muscle cells and endothelial cells of human atherosclerotic plaques (Martinet et al. 2004). It remains to be elucidated whether such extra-neuronal RNA oxidation in these disorders merely represents age-associated or metabolic alterations in each tissue or it specifically represents pathogenic similarities or relevance of these disorders with AD-type neurodegeneration as suggested by previous studies (Tateyama et al. 2003; Honig et al. 2005; Moreira et al. 2006a).
Biochemical Detection of RNA Oxidation
The immunocytochemical studies of neuronal RNA oxidation were followed by biochemical detection of the oxidized nucleoside in AD brain with immunoblot analyses (Shan et al. 2003; Ding et al. 2005, 2006; Honda et al. 2005; Shan and Lin 2006). Shan et al. (2003) used northwestern blotting with a monoclonal anti-8-OHG antibody, to isolate and identify oxidized RNA species and showed that a significant amount of brain poly (A)+ mRNA species were oxidized in AD. The oxidation to mRNA was further confirmed by cDNA synthesis and Southern blotting of the immunoprecipitated mRNA species. Densitometric analysis of the Southern blot results revealed that 30–70 % of the mRNAs from AD frontal cortices were oxidized, while only 2 % of the mRNAs were oxidized in age-matched normal controls (Shan and Lin 2006). Interestingly, reverse transcription polymerase chain reaction (RT-PCR) and filter array analyses of the identified oxidized mRNAs revealed that some species were more susceptible to oxidative damage in AD. While it has been shown in vitro that runs of guanines in RNA are relatively susceptible to oxidation (Holcomb et al. 2010), no common motifs or structures were found in the oxidatively susceptible mRNA species in AD brain (Shan et al. 2003). Some of the identified known oxidized transcripts were related to AD, which included p21ras, mitogen-activated protein kinase (MAPK) kinase 1, carbonyl reductase, copper/zinc superoxide dismutase (SOD1), apolipoprotein D, calpains, but not amyloid-β protein precursor or tau (Shan et al. 2003). The same research group reported that 3–10 % of the mRNAs were oxidized in tissues of the motor cortex and the spinal cord from subjects with ALS (Chang et al. 2008). Although the above-mentioned studies (Shan et al. 2003; Shan and Lin 2006; Chang et al. 2008) focused on mRNA species that account for only a few percent of total cellular RNA, Honda et al. (2005) and Ding et al. (2005, 2006) reported that ribosomal RNA (rRNA), extremely abundant in neurons, contained 8-OHG in AD brain. Remarkably, rRNA showed higher binding capacity to redox-active iron than transfer RNA (tRNA), and consequently the oxidation of rRNA by the Fenton reaction formed 13 times more 8-OHG than tRNA (Honda et al. 2005).
Recently, a direct detection of RNA oxidation has been reported in brains affected with late-stage AD (Weidner et al. 2011). Among five RNA adducts quantified by gas chromatography/mass spectroscopy (GC/MS), only 8-hydroxyadenine was significantly increased in vulnerable brain regions from AD cases compared to age-matched controls.
Selective Neuronal Vulnerability and RNA Oxidation
Of particular interest, both immunocytochemical (Nunomura et al. 1999, 2002, 2012; Zhang et al. 1999) and biochemical (Shan et al. 2003; Ding et al. 2005) studies revealed that the regional distribution of RNA oxidation in the brain was consistent with the selective neuronal vulnerability in each neurological disorder. There were increased levels of 8-OHG in the hippocampus and cerebral neocortex in AD, in the substantia nigra in PD, as well as in the motor cortex and spinal cord in ALS; while no alteration in 8-OHG levels was found in the cerebellum in AD, PD, and ALS compared with controls (Nunomura et al. 1999, 2012; Zhang et al. 1999; Shan et al. 2003; Ding et al. 2005; Chang et al. 2008). Immunocytochemical approaches further enabled confirmation that the oxidized RNAs were localized predominantly in neuronal cells compared with glial cells (Nunomura et al. 1999, 2000, 2001, 2002, 2004; Zhang et al. 1999).
RNA Oxidation as a Possible Biomarker for Neurodegenerative Disorders
In addition to the brain tissue, significantly increased levels of the oxidized RNA nucleoside, 8-OHG, have been identified in CSF collected from patients with AD and PD (Abe et al. 2002, 2003; Kikuchi et al. 2002) as well as in serum of PD patients (Kikuchi et al. 2002). Certainly, further investigations with large sample size are required to test whether 8-OHG detected in the peripheral materials is a suitable biomarker of these disorders. As we describe in the following section, 8-OHG may have diagnostic utility as an early-stage marker of the disorders or a marker predicting conversion from the prodromal stage into early-stage of the disorders. In this context, it is noteworthy that levels of RNA oxidation in white blood cells of 50–60 years old subjects with body mass index between 23.5 and 29.9 are reduced by 12 months intervention of caloric restriction or exercise (Hofer et al. 2008a). It has been suggested that caloric restriction and exercise may be efficacious strategies for the prevention of AD and related neurodegenerative disorders through an enhancement of endogenous antioxidant defenses (Nunomura et al. 2006, 2007). Indeed, activation of endogenous cellular defense pathways including sirtuin and nuclear factor erythroid-2 related factor 2 (Nrf2)/antioxidant response element (ARE) signaling potentially plays a preventive role against neurodegeneration (Calabrese et al. 2010, 2012). An oxidized RNA nucleoside is considered as a biomarker candidate for the assessment of preventive interventions for neurodegenerative disorders.
Neuronal RNA Oxidation in Experimental Conditions (Table 3)
RNA Oxidation in AD and ALS Human Mutant Animal Models
A strong genetic link between oxidative damage and neurodegeneration has been suggested by the finding that about 20 % of patients with familial ALS present a mutation in SOD1, a metalloenzyme that catalyzes the dismutation of the toxic superoxide (O •−2 ) to hydrogen peroxide (H2O2) (Rosen et al. 1993). Although several lines of evidence indicates that the involvement of the SOD1 mutation in neurodegeneration seems to be a toxic gain of function rather than a loss in the SOD1 activity (Andersen 2004), a transgenic mice model of ALS expressing Gly93Ala-SOD1 mutation that develops an ALS phenotype (Gurney et al. 1994) shows increased mRNA oxidation in the motor neurons of the spinal cord (Chang et al. 2008). Identification of oxidized mRNA species in this animal model has revealed that some species are more vulnerable to oxidative damage. Importantly, many oxidized mRNA species have been implicated specifically in the pathogenesis of the disease (Chang et al. 2008), which is demonstrated also in the study on oxidized mRNA species of human AD brain (Shan et al. 2003). Of note, in the transgenic mouse model of ALS, vitamin E reduces the level of mRNA oxidation and delays the progressive loss of motor neurons (Chang et al. 2008).
In double knock-in mice expressing familial AD-linked mutations in amyloid precursor protein (APP) (Swedish KM670/671NL mutation) and presenilin-1 (PS1) (P264L mutation), remarkable RNA oxidation has been observed immunocytochemically in neurons of the cerebral cortex (Lovell et al. 2009). In this animal model, selenium supplementation reduces the level of neuronal RNA oxidation and amyloid-β plaque deposition, which is concomitant with an increase in activity of an antioxidant enzyme, glutathione peroxidase.
RNA Oxidation Induced by Hypoxia, Neurotoxin, and Traumatic CNS Injury
Experimental studies with rodents have shown that neuronal RNA oxidation and spatial memory deficit are observed in animals with intermittent hypoxia, where antioxidants can reduce both oxidative damage and the spatial memory deficit (Row et al. 2003). Animal models of neurodegeneration exposed to neurotoxins demonstrate an involvement of RNA oxidation in the degenerative pathway. Animals treated with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) showing degeneration in nigrostriatal dopaminergic neurons are widely used as an experimental model of PD (Javitch et al. 1985). In MPTP-treated mice, there is a significant increase in neuronal 8-OHG in the substantia nigra (Yamaguchi et al. 2006). Also, kainic acid-mediated excitotoxicity, a model for neurodegeneration (Wang et al. 2005) is also associated with increased levels of 8-OHG in neurons and glial cells of the hippocampus (Kajitani et al. 2006). Recently, an experimental model of cerebral ammonia toxicity reveals that acute ammonia intoxication causes a transient coma in rats and reversible RNA oxidation in neurons and perivascular astrocytes of the brain (Görg et al. 2008). This study may indicate that RNA oxidation is involved in the pathogenesis of a wide variety of neurological disorders including progressive neurodegeneration and reversible neuronal dysfunction. The devastating effects of traumatic CNS injury are a result not only of the initial injury but also a process of secondary degeneration associated with increased ROS formation (King et al. 2006; Huang et al. 2007). Indeed, traumatic spinal cord injury by hemisection (King et al. 2006) or compression (Huang et al. 2007) also causes RNA oxidation in ventral horn neurons of rats, which is significantly reduced by treatment with docosahexaenoic acid.
RNA Oxidation in Cell Culture Models of Neurodegeneration
Cell culture experiments further suggest an association between increased RNA oxidation and neurodegeneration (Ding et al. 2004; Shan et al. 2007). In a mixed astrocyte and neuron culture model (Ding et al. 2004), DNA and RNA oxidation have been observed following proteasome inhibition that is associated with several neurodegenerative features such as protein aggregation, activated apoptotic pathways, and induction of mitochondrial disturbances. Interestingly, in this proteasome inhibition model, neurons underwent larger increases in nucleic acid oxidation compared to astrocyte cultures, and RNA appeared to undergo a greater degree of oxidation than DNA, which was identically observed in AD brain (Nunomura et al. 1999). Various neurodegenerative disorders including AD, PD, and ALS are associated with compromises in the ubiquitin–proteasome system that affect multiple aspects of RNA metabolism and promote RNA pathology (Ding et al. 2007). Another recent study using primary rat cortical cultures has shown that exposures to H2O2 and other oxidative insults cause neuronal RNA oxidation and subsequent neuronal death (Shan et al. 2007). This model has clearly demonstrated a chronological primacy of neuronal RNA oxidation in the process of neurodegeneration.
RNA Oxidation: An Early-Stage Event in Neurodegeneration
Chronological Primacy of RNA Oxidation in AD and PD
Because RNA oxidation is involved in a wide variety of neuropsychiatric disorders (Table 2), it may be considered that RNA oxidation is a common feature in neuronal dysfunction that occurs in late stages of the disorders and is simply epiphenomenal. However, that is not the case in AD and PD, and probably in ALS. In fact, there is considerable evidence supporting an early involvement of RNA oxidation in the pathological cascade of neurodegeneration, especially in AD. For instance, RNA oxidation has been observed in post-mortem brains of cases with early-stage AD (Nunomura et al. 2001, 2006), a presymptomatic case with familial AD mutation (Nunomura et al. 2004), Down syndrome cases with early-stage AD pathology (Nunomura et al. 2000), and subjects with mild cognitive impairment (MCI) who, at least in part, represent prodromal stage of AD (Ding et al. 2005, 2006; Lovell and Markesbery 2008; Nunomura et al. 2012). Indeed, intraneuronal amyloid-β accumulation, an initial change of the AD pathology, and RNA oxidation are observed in the same neuronal population, while there is an inverse relationship between the levels of them (Nunomura et al. 2010). Furthermore, the increased level of RNA oxidation in CSF is more prominent in cases with shorter duration of AD and PD (Abe et al. 2002, 2003) as well as in AD cases with higher scores on a cognitive scale (Abe et al. 2002). In addition, recent studies on MCI subjects have also demonstrated increased oxidation/nitration to protein, lipid peroxidation and DNA oxidation in post-mortem brain (Keller et al. 2005; Wang et al. 2006; Butterfield et al. 2007), increased lipid peroxidation in CSF, plasma, and urine (Praticò et al. 2002), as well as increased DNA oxidation in peripheral leukocytes (Migliore et al. 2005). Decreased levels of defense mechanism against oxidative damage have been also reported in MCI subjects; namely, decreased plasma antioxidant vitamins and enzymes (Rinaldi et al. 2003) and decreased plasma total antioxidant capacity (Guidi et al. 2006).
Recently, preclinical stage of AD has been described in which subjects show no overt clinical manifestations but demonstrate significant AD pathology (Sperling et al. 2011). Two recent studies on RNA oxidation in vulnerable neurons of preclinical AD provide inconsistent results. There is a significant increase in the level of 8-OHG immunoreactions in subjects with preclinical AD compared to controls in one study (Lovell et al. 2011), but not in the other (Nunomura et al. 2012). Although the exact reason for the discrepancy between these studies is not clear, it may come from a difference in brain region studied. Namely, there is a significantly increased level of RNA oxidation in the hippocampus (Lovell et al. 2011) but not in the cerebral neocortex (Nunomura et al. 2012). Indeed, recent studies on preclinical AD brains have reported significantly increased levels of lipid peroxidation products in the hippocampus, but no significant changes in levels of lipid peroxidation and protein oxidation in the cerebral neocortex (Aluise et al. 2010, 2011; Bradley et al. 2010). Clearly, further investigations are required to determine whether a significant level of RNA oxidation is involved at preclinical stage of AD, the earliest stage in transition from normal aging to AD.
Chronological Primacy of RNA Oxidation in SSPE
An early-stage involvement of neuronal RNA oxidation is not only evident in age-associated neurodegenerative disorders, but also in cases with SSPE that is caused by persistent measles virus infection in the CNS (Hayashi et al. 2002). Neuronal RNA oxidation is observed in post-mortem brains of subjects with shorter disease duration. Interestingly, SSPE is pathologically accompanied with brain atrophy and the formation of neurofibrillary tangles which are features of neurodegenerative disorders.
Chronological Primacy of RNA Oxidation in a Transgenic Mouse Model of ALS
As for the transgenic mouse model of ALS, significantly increased RNA oxidation has been observed in spinal cord motor neurons of the familial ALS-linked SOD1 mutant mice at an early, presymptomatic stage before motor neuron death (Chang et al. 2008). In the animals expressing Gly93Ala-SOD1 mutation, the increased neuronal RNA oxidation is observed in the spinal cord motor neurons at age of 60 days, when the neurons still appear to be healthy; i.e., the neurons have normal nuclear and chromatin morphology and have no ubiquitinated protein aggregation and have only minor ultrastructural changes in mitochondria. At the symptomatic stage (90–120 days of age), the dying motor neurons of the mutant mice show abnormal nuclear and chromatin morphology, ubiquitinated protein aggregation, as well as mitochondrial vacuolation, but less neuronal RNA oxidation (Chang et al. 2008). This in vivo model clearly indicates that RNA oxidation is an early-stage event in the process of neurodegeneration.
Chronological Primacy of RNA Oxidation in Cell Culture Models of Neurodegeneration
The chronological primacy of neuronal RNA oxidation in the process of neurodegeneration has been clearly demonstrated also in a model of primary rat cortical cultures (Shan et al. 2007). In the time course after various oxidative insults to the cultures, RNA oxidation occurs primarily in a distinct group of neurons that die later. Together with an observation of decreased protein synthesis due to the oxidized RNA, this study suggests that neuronal RNA oxidation is neither a consequence of dying neurons nor a harmless epiphenomenon but is an early event that precedes neuronal death and contributes to it (Shan et al. 2007). Beyond the chronological issue, the cell culture experiment may indicate that substantial levels of oxidative RNA damage are sufficient to cause cell death via dysfunction of protein synthesis. From a clinical perspective, the notion of an early involvement of oxidative damage in the pathogenesis of these degenerative disorders should have a great importance to the establishment of diagnostic tools and therapeutic targets, as we have reviewed recently (Nunomura et al. 2006, 2007; Liu et al. 2007; Moreira et al. 2006b).
RNA Oxidation as a “Steady-State” Marker of Oxidative Damage Rather than History
While protein carbonyls, lipid peroxidation products, and glycoxidation products are relatively stable at the site of generation due to the formation of cross-links and consequent resistant to degradation, oxidized RNAs are likely turned over more rapidly compared to the other oxidatively modified macromolecules. Therefore, the detection of most oxidative markers using in situ approaches in tissues affected by disease (protein carbonyls, lipid peroxidation products or glycoxidation products) indicates the “history” of oxidative damage. On the other hand, RNA oxidation reflects the “steady-state balance” of oxidative damage at a “snapshot” point (Nunomura et al. 1999; Sayre et al. 1999). In accordance with this concept, protein carbonyls, lipid peroxidation products such as 4-hydroxynonenal and F2-isoprostane, and a glycoxidation product carboxymethyl-lysine have been demonstrated in neurons with and without associated pathology in AD brains (Smith et al. 1996; Sayre et al. 1997; Castellani et al. 2001; Casadesus et al. 2007). These data likely reflect the occurrence of damage throughout the early and advanced stages of neurodegeneration (“history” of the damage). These observations contrast remarkably with the pattern of the RNA oxidation, a “steady-state” marker, which is prominent in neurons without pathology and is present in lesser amounts in neurons containing pathology (Nunomura et al. 2000, 2001). Among the markers for oxidative damage, 3-nitrotyrosine may be another steady-state marker of oxidative damage. 3-Nitrotyrosine is formed by a modification of tyrosine residue of proteins by an attack of peroxynitrite (ONOO−), a powerful oxidant produced from the reaction of O •−2 with nitric oxide (NO•), and is not known to accumulate in cells. As such, it is not surprising that intracellular level of 3-nitrotyrosine parallels the level of 8-OHG in AD and Down syndrome brains (Nunomura et al. 2000, 2001).
Sources of Reactive Oxygen Species (ROS) Responsible for RNA Oxidation
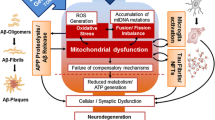
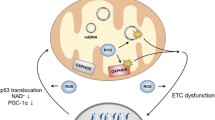
The brain is especially vulnerable to oxidative damage because of its high content of easily peroxidizable unsaturated fatty acids, high oxygen consumption rate (accounting for 20–25 % of total body oxygen consumption but less than 2 % of total body weight), and relative paucity of antioxidant enzymes compared with other organs (e.g., the content of catalase in brain is only 10–20 % of liver and heart) (Coyle and Puttfarcken 1993; Mattson et al. 2002). Given this environment, neurons are continuously exposed to ROS such as O •−2 , H2O2, and hydroxyl radical (•OH) that are produced from the mitochondrial electron transport chain through normal cellular metabolism (Halliwell 1992; Coyle and Puttfarcken 1993; Mattson et al. 2002). •OH can diffuse through tissue only in the order of several nanometers (Joenje 1989) and O •−2 is hardly permeable through cell membranes (Takahashi and Asada 1983). In consideration of the widespread damage to cytoplasmic RNA in neurodegenerative diseases, RNA species are likely attacked by •OH, which is formed from the reaction of highly diffusible H2O2 (Schubert and Wilmer 1991) with redox-active metals through the Fenton reaction (Honda et al. 2005). In the AD brain, disrupted mitochondria likely play a central role in producing abundant ROS as well as supplying redox-active iron into the cytosol (Fig. 2) (Hirai et al. 2001; Perry et al. 2002a, b; Lin and Beal 2006). Indeed, ribosomes purified from AD hippocampus contain significantly higher levels of redox-active iron compared to controls, and the iron is bound to rRNA (Honda et al. 2005). Therefore, mitochondrial abnormalities coupled with metal dysregulation of metal homeostasis are key features closely associated with ROS formation responsible for the RNA oxidation in AD (Smith et al. 2000). Interestingly, mitochondrial abnormalities (Schapira et al. 1990; Gu et al. 2002) and metal ion dysregulation (Sofic et al. 1988; Berg et al. 2002) are also found in the substantia nigra of PD, making this mechanism a common theme in neurodegenerative cascades.

Hypothetical schema of upstream mediators and downstream consequences of cellular RNA oxidation. Disrupted mitochondria (Hirai et al. 2001) likely play a central role in producing ROS as well as supplying redox-active metals (iron and copper) in the cytosol to catalyze •OH production through the Fenton/Harber–Weiss reactions. Oxidation of coding RNAs leads to reduced level of functional proteins, and to formation of non-functional, truncated proteins or mutated proteins. Non-coding RNAs could also be oxidized and their complex regulatory functions in protein synthesis should be impeded, while this interesting aspect still remains largely unexplored
Conclusion
Involvement of RNA oxidation in the process of aging and neurodegeneration has been demonstrated in human brain tissue and experimental models. Indeed, remarkable RNA oxidation has been observed in vulnerable neuronal population of AD, PD, DLB, ALS, DRPLA (a CAG-repeat disease), and prion diseases (CJD and GSS). Particular emphasis should be placed on the early-stage involvement of RNA oxidation in the process of neurodegeneration, which suggests a primary role of RNA oxidation in the pathomechanisms. Indeed, oxidized RNA is associated with a disturbance in protein synthesis in vitro and in vivo. There are only a small number of studies suggesting the existence of coping mechanisms for RNA damage at present. Understanding of the consequences and cellular handling mechanisms of the oxidative RNA damage may provide clues to the underlying mechanisms of aging as well as pathophysiology of chronic degenerative disorders and lead to better anti-aging and therapeutic strategies.
References
Aas PA, Otterlei M, Falnes PO et al (2003) Human and bacterial oxidative demethylases repair alkylation damage in both RNA and DNA. Nature 421:859–863
Abe T, Tohgi H, Isobe C, Murata T, Sato C (2002) Remarkable increase in the concentration of 8-hydroxyguanosine in cerebrospinal fluid from patients with Alzheimer’s disease. J Neurosci Res 70:447–450
Abe T, Isobe C, Murata T, Sato C, Tohgi H (2003) Alteration of 8-hydroxyguanosine concentrations in the cerebrospinal fluid and serum from patients with Parkinson’s disease. Neurosci Lett 336:105–108
Aluise CD, Robinson RA, Beckett TL et al (2010) Preclinical Alzheimer disease; brain oxidative stress, Aβ peptide and proteomics. Neurobiol Dis 39:221–228
Aluise CD, Robinson RA, Cai J, Pierce WM, Markesbery WR, Butterfield DA (2011) Redox proteomics analysis of brains from subjects with amnestic mild cognitive impairment compared to brains from subjects with preclinical Alzheimer’s disease; insights into memory loss in MCI. J Alzheimers Dis 23:257–269
Ames BN, Gold LS (1991) Endogenous mutagens and the causes of aging and cancer. Mutat Res 250:3–16
Andersen JK (2004) Oxidative stress in neurodegeneration: cause or consequence? Nat Med 10(Suppl):S18–S25
Andreoli R, Mutti A, Goldoni M, Manini P, Apostoli P, De Palma G (2011) Reference ranges of urinary biomarkers of oxidized guanine in (2′-deoxy)ribonucleotides and nucleic acids. Free Radic Biol Med 50:254–261
Barber SC, Mead RJ, Shaw PJ (2006) Oxidative stress in ALS: a mechanism of neurodegeneration and a therapeutic target. Biochim Biophys Acta 1762:1051–1067
Barciszewski J, Barciszewska MZ, Siboska G, Rattan SI, Clark BF (1999) Some unusual nucleic acid bases are products of hydroxyl radical oxidation of DNA and RNA. Mol Biol Rep 26:231–238
Barnham KJ, Masters CL, Bush AI (2004) Neurodegenerative diseases and oxidative stress. Nat Rev Drug Discov 3:205–214
Bellacosa A, Moss EG (2003) RNA repair: damage control. Curr Biol 13:R482–R484
Berg D, Roggendorf W, Schroder U et al (2002) Echogenicity of the substantia nigra: association with increased iron content and marker for susceptibility to nigrostriatal injury. Arch Neurol 59:999–1005
Bradley MA, Markesbery WR, Lovell MA (2010) Increased levels of 4-hydroxynonenal and acrolein in the brain in preclinical Alzheimer disease. Free Radic Biol Med 48:1570–1576
Brégeon D, Sarasin A (2005) Hypothetical role of RNA damage avoidance in preventing human disease. Mutat Res 577:293–302
Broedbaek K, Poulsen HE, Weimann A et al (2009) Urinary excretion of biomarkers of oxidatively damaged DNA and RNA in hereditary hemochromatosis. Free Radic Biol Med 47:1230–1233
Broedbaek K, Siersma V, Henriksen T et al (2011) Urinary markers of nucleic acid oxidation and long-term mortality of newly diagnosed type 2 diabetic patients. Diabetes Care 34:2594–2596
Butterfield DA, Reed TT, Perluigi M et al (2007) Elevated levels of 3-nitrotyrosine in brain from subjects with amnestic mild cognitive impairment: implications for the role of nitration in the progression of Alzheimer’s disease. Brain Res 1148:243–248
Calabrese V, Cornelius C, Dinkova-Kostova AT, Calabrese EJ, Mattson MP (2010) Cellular stress responses, the hormesis paradigm, and vitagenes: novel targets for therapeutic intervention in neurodegenerative disorders. Antioxid Redox Signal 13:1763–1811
Calabrese V, Cornelius C, Dinkova-Kostova AT et al (2012) Cellular stress responses, hormetic phytochemicals and vitagenes in aging and longevity. Biochim Biophys Acta 1822:753–783
Cao X, Yeo G, Muotri AR, Kuwabara T, Gage FH (2006) Noncoding RNAs in the mammalian central nervous system. Annu Rev Neurosci 29:77–103
Casadesus G, Smith MA, Basu S et al (2007) Increased isoprostane and prostaglandin are prominent in neurons in Alzheimer disease. Mol Neurodegener 2:2
Castellani RJ, Harris PL, Sayre LM et al (2001) Active glycation in neurofibrillary pathology of Alzheimer disease: N(epsilon)-(carboxymethyl) lysine and hexitol-lysine. Free Radic Biol Med 31:175–180
Chang Y, Kong Q, Shan X et al (2008) Messenger RNA oxidation occurs early in disease pathogenesis and promotes motor neuron degeneration in ALS. PLoS ONE 3:e2849
Che Y, Wang JF, Shao L, Young T (2010) Oxidative damage to RNA but not DNA in the hippocampus of patients with major mental illness. J Psychiatry Neurosci 35:296–302
Cooke MS, Olinski R, Evans MD (2006) Does measurement of oxidative damage to DNA have clinical significance? Clin Chim Acta 365:30–49
Costa FF (2005) Non-coding RNAs: new players in eukaryotic biology. Gene 357:83–94
Coyle JT, Puttfarcken P (1993) Oxidative stress, glutamate, and neurodegenerative disorders. Science 262:689–695
Cui L, Hofer T, Rani A, Leeuwenburgh C, Foster TC (2009) Comparison of lifelong and late life exercise on oxidative stress in the cerebellum. Neurobiol Aging 30:903–909
Deutscher MP (2006) Degradation of RNA in bacteria: comparison of mRNA and stable RNA. Nucleic Acids Res 34:659–666
Ding Q, Dimayuga E, Markesbery WR, Keller JN (2004) Proteasome inhibition increases DNA and RNA oxidation in astrocyte and neuron cultures. J Neurochem 91:1211–1218
Ding Q, Markesbery WR, Chen Q, Li F, Keller JN (2005) Ribosome dysfunction is an early event in Alzheimer’s disease. J Neurosci 25:9171–9175
Ding Q, Markesbery WR, Cecarini V, Keller JN (2006) Decreased RNA, and increased RNA oxidation, in ribosomes from early Alzheimer’s disease. Neurochem Res 31:705–710
Ding Q, Cecarini V, Keller JN (2007) Interplay between protein synthesis and degradation in the CNS: physiological and pathological implications. Trends Neurosci 30:31–36
Evans MD, Dizdaroglu M, Cooke MS (2004) Oxidative DNA damage and disease: induction, repair and significance. Mutat Res 567:1–61
Fiala ES, Conaway CC, Mathis JE (1989) Oxidative DNA and RNA damage in the livers of Sprague-Dawley rats treated with the hepatocarcinogen 2-nitropropane. Cancer Res 49:5518–5522
Foksinski M, Rozalski R, Guz J et al (2004) Urinary excretion of DNA repair products correlates with metabolic rates as well as with maximum life spans of different mammalian species. Free Radic Biol Med 37:1449–1454
Furuta A, Iida T, Nakabeppu Y, Iwaki T (2001) Expression of hMTH1 in the hippocampi of control and Alzheimer’s disease. NeuroReport 12:2895–2899
Gemma C, Vila J, Bachstetter A, Bickford PC (2007) Oxidative stress and the aging brain: from theory to prevention, Chapter 15. In: Riddle DR (ed) Brain aging: models, methods, and mechanisms. CRC Press, Boca Raton, FL
Gong X, Tao R, Li Z (2006) Quantification of RNA damage by reverse transcription polymerase chain reactions. Anal Biochem 357:58–67
Görg B, Qvartskhava N, Keitel V et al (2008) Ammonia induces RNA oxidation in cultured astrocytes and brain in vivo. Hepatology 48:567–579
Görg B, Qvartskhava N, Bidmon HJ et al (2010) Oxidative stress markers in the brain of patients with cirrhosis and hepatic encephalopathy. Hepatology 52:256–265
Gu G, Reyes PE, Golden GT et al (2002) Mitochondrial DNA deletions/rearrangements in Parkinson disease and related neurodegenerative disorders. J Neuropathol Exp Neurol 61:634–639
Guentchev M, Siedlak SL, Jarius C et al (2002) Oxidative damage to nucleic acids in human prion disease. Neurobiol Dis 9:275–281
Guidi I, Galimberti D, Lonati S et al (2006) Oxidative imbalance in patients with mild cognitive impairment and Alzheimer’s disease. Neurobiol Aging 27:262–269
Gurney ME, Pu H, Chiu AY et al (1994) Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science 264:1772–1775
Halliwell B (1992) Reactive oxygen species and the central nervous system. J Neurochem 59:1609–1623
Hayakawa H, Sekiguchi M (2006) Human polynucleotide phosphorylase protein in response to oxidative stress. Biochemistry (Mosc) 45:6749–6755
Hayakawa H, Hofer A, Thelander L et al (1999) Metabolic fate of oxidized guanine ribonucleotides in mammalian cells. Biochemistry (Mosc) 38:3610–3614
Hayakawa H, Kuwano M, Sekiguchi M (2001) Specific binding of 8-oxoguanine-containing RNA to polynucleotide phosphorylase protein. Biochemistry (Mosc) 40:9977–9982
Hayakawa H, Uchiumi T, Fukuda T et al (2002) Binding capacity of human YB-1 protein for RNA containing 8-oxoguanine. Biochemistry (Mosc) 41:12739–12744
Hayakawa H, Fujikane A, Ito R, Matsumoto M, Nakayama KI, Sekiguchi M (2010) Human proteins that specifically bind to 8-oxoguanine-containing RNA and their responses to oxidative stress. Biochem Biophys Res Commun 403:220–224
Hayashi M, Arai N, Satoh J et al (2002) Neurodegenerative mechanisms in subacute sclerosing panencephalitis. J Child Neurol 17:725–730
Hayashi M, Araki S, Kohyama J, Shioda K, Fukatsu R (2005) Oxidative nucleotide damage and superoxide dismutase expression in the brains of xeroderma pigmentosum group A and Cockayne syndrome. Brain Dev 27:34–38
Herrup K (2010) Reimagining Alzheimer’s disease—an age-based hypothesis. J Neurosci 30:16755–16762
Hirai K, Aliev G, Nunomura A et al (2001) Mitochondrial abnormalities in Alzheimer’s disease. J Neurosci 21:3017–3023
Hirtz D, Thurman DJ, Gwinn-Hardy K, Mohamed M, Chaudhuri AR, Zalutsky R (2007) How common are the “common” neurologic disorders? Neurology 68:326–337
Hofer T, Badouard C, Bajak E, Ravanat JL, Mattsson A, Cotgreave IA (2005) Hydrogen peroxide causes greater oxidation in cellular RNA than in DNA. Biol Chem 386:333–337
Hofer T, Seo AY, Prudencio M, Leeuwenburgh C (2006) A method to determine RNA and DNA oxidation simultaneously by HPLC-ECD: greater RNA than DNA oxidation in rat liver after doxorubicin administration. Biol Chem 387:103–111
Hofer T, Fontana L, Anton SD et al (2008a) Long-term effects of caloric restriction or exercise on DNA and RNA oxidation levels in white blood cells and urine in humans. Rejuvenation Res 11:793–799
Hofer T, Marzetti E, Xu J et al (2008b) Increased iron content and RNA oxidative damage in skeletal muscle with aging and disuse atrophy. Exp Gerontol 43:563–570
Hoffman WH, Siedlak SL, Wang Y, Castellani RJ, Smith MA (2011) Oxidative damage is present in the fatal brain edema of diabetic ketoacidosis. Brain Res 1369:194–202
Holcomb DR, Ropp PA, Theil EC, Thorp HH (2010) Nature of guanine oxidation in RNA via the flash-quench technique versus direct oxidation by a metal oxo complex. Inorg Chem 49:786–795
Honda K, Smith MA, Zhu X et al (2005) Ribosomal RNA in Alzheimer disease is oxidized by bound redox-active iron. J Biol Chem 280:20978–20986
Honig LS, Kukull W, Mayeux R (2005) Atherosclerosis and AD: analysis of data from the US National Alzheimer’s Coordinating Center. Neurology 64:494–500
Huang WL, King VR, Curran OE et al (2007) A combination of intravenous and dietary docosahexaenoic acid significantly improves outcome after spinal cord injury. Brain 130:3004–3019
Ischiropoulos H, Beckman JS (2003) Oxidative stress and nitration in neurodegeneration: cause, effect, or association? J Clin Invest 111:163–169
Ishibashi T, Hayakawa H, Ito R, Miyazawa M, Yamagata Y, Sekiguchi M (2005) Mammalian enzymes for preventing transcriptional errors caused by oxidative damage. Nucleic Acids Res 33:3779–3784
Ito R, Hayakawa H, Sekiguchi M, Ishibashi T (2005) Multiple enzyme activities of Escherichia coli MutT protein for sanitization of DNA and RNA precursor pools. Biochemistry (Mosc) 44:6670–6674
Jacobs AC, Resendiz MJ, Greenberg MM (2010) Direct strand scission from a nucleobase radical in RNA. J Am Chem Soc 132:3668–3669
Javitch JA, D’Amato RJ, Strittmatter SM, Snyder SH (1985) Parkinsonism-inducing neurotoxin, N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine: uptake of the metabolite N-methyl-4-phenylpyridine by dopamine neurons explains selective toxicity. Proc Natl Acad Sci USA 82:2173–2177
Jenner P (2003) Oxidative stress in Parkinson’s disease. Ann Neurol 53(Suppl 3):S26–S36
Joenje H (1989) Genetic toxicology of oxygen. Mutat Res 219:193–208
Kajitani K, Yamaguchi H, Dan Y, Furuichi M, Kang D, Nakabeppu Y (2006) MTH1, an oxidized purine nucleoside triphosphatase, suppresses the accumulation of oxidative damage of nucleic acids in the hippocampal microglia during kainate-induced excitotoxicity. J Neurosci 26:1688–1698
Kasai H, Crain PF, Kuchino Y, Nishimura S, Ootsuyama A, Tanooka H (1986) Formation of 8-hydroxyguanine moiety in cellular DNA by agents producing oxygen radicals and evidence for its repair. Carcinogenesis 7:1849–1851
Kedersha N, Stoecklin G, Ayodele M et al (2005) Stress granules and processing bodies are dynamically linked sites of mRNP remodeling. J Cell Biol 169:871–884
Keller JN, Schmitt FA, Scheff SW et al (2005) Evidence of increased oxidative damage in subjects with mild cognitive impairment. Neurology 64:1152–1156
Kikuchi A, Takeda A, Onodera H et al (2002) Systemic increase of oxidative nucleic acid damage in Parkinson’s disease and multiple system atrophy. Neurobiol Dis 9:244–248
King VR, Huang WL, Dyall SC, Curran OE, Priestley JV, Michael-Titus AT (2006) Omega-3 fatty acids improve recovery, whereas omega-6 fatty acids worsen outcome, after spinal cord injury in the adult rat. J Neurosci 26:4672–4680
Krokan HE, Kavli B, Slupphaug G (2004) Novel aspects of macromolecular repair and relationship to human disease. J Mol Med 82:280–297
Lee JW, Beebe K, Nangle LA et al (2006) Editing-defective tRNA synthetase causes protein misfolding and neurodegeneration. Nature 443:50–55
Li Z, Wu J, Deleo CJ (2006) RNA damage and surveillance under oxidative stress. IUBMB Life 58:581–588
Lin MT, Beal MF (2006) Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443:787–795
Liu J, Head E, Gharib AM et al (2002) Memory loss in old rats is associated with brain mitochondrial decay and RNA/DNA oxidation: partial reversal by feeding acetyl-L-carnitine and/or R-alpha -lipoic acid. Proc Natl Acad Sci USA 99:2356–2361
Liu Q, Xie F, Rolston R et al (2007) Prevention and treatment of Alzheimer disease and aging: antioxidants. Mini Rev Med Chem 7:171–180
Logroscino G, Beghi E, Zoccolella S et al (2005) Incidence of amyotrophic lateral sclerosis in southern Italy: a population based study. J Neurol Neurosurg Psychiatry 76:1094–1098
Lovell MA, Markesbery WR (2008) Oxidatively modified RNA in mild cognitive impairment. Neurobiol Dis 29:169–175
Lovell MA, Xiong S, Lyubartseva G, Markesbery WR (2009) Organoselenium (Sel-Plex diet) decreases amyloid burden and RNA and DNA oxidative damage in APP/PS1 mice. Free Radic Biol Med 46:1527–1533
Lovell MA, Soman S, Bradley MA (2011) Oxidatively modified nucleic acids in preclinical Alzheimer’s disease (PCAD) brain. Mech Ageing Dev 132:443–448
Mangialasche F, Solomon A, Winblad B, Mecocci P, Kivipelto M (2010) Alzheimer’s disease: clinical trials and drug development. Lancet Neurol 9:702–716
Manini P, De Palma G, Andreoli R et al (2009) Biomarkers of nucleic acid oxidation, polymorphism in, and expression of, hOGG1 gene in styrene-exposed workers. Toxicol Lett 190:41–47
Manini P, De Palma G, Andreoli R et al (2010) Occupational exposure to low levels of benzene: biomarkers of exposure and nucleic acid oxidation and their modulation by polymorphic xenobiotic metabolizing enzymes. Toxicol Lett 193:229–235
Martinet W, de Meyer GR, Herman AG, Kockx MM (2004) Reactive oxygen species induce RNA damage in human atherosclerosis. Eur J Clin Invest 34:323–327
Mattson MP, Chan SL, Duan W (2002) Modification of brain aging and neurodegenerative disorders by genes, diet, and behavior. Physiol Rev 82:637–672
Mehler MF, Mattick JS (2006) Non-coding RNAs in the nervous system. J Physiol 575:333–341
Mehler MF, Mattick JS (2007) Noncoding RNAs and RNA editing in brain development, functional diversification, and neurological disease. Physiol Rev 87:799–823
Migliore L, Fontana I, Trippi F et al (2005) Oxidative DNA damage in peripheral leukocytes of mild cognitive impairment and AD patients. Neurobiol Aging 26:567–573
Miyata R, Hayashi M, Tanuma N, Shioda K, Fukatsu R, Mizutani S (2008) Oxidative stress in neurodegeneration in dentatorubral-pallidoluysian atrophy. J Neurol Sci 264:133–139
Moreira PI, Honda K, Zhu X et al (2006a) Brain and brawn: parallels in oxidative strength. Neurology 66(Suppl 1):S97–S101
Moreira PI, Zhu X, Nunomura A, Smith MA, Perry G (2006b) Therapeutic options in Alzheimer’s disease. Expert Rev Neurother 6:897–910
Moreira PI, Nunomura A, Nakamura M et al (2008) Nucleic acid oxidation in Alzheimer disease. Free Radic Biol Med 44:1493–1505
Mundt JM, Hah SS, Sumbad RA, Schramm V, Henderson PT (2008) Incorporation of extracellular 8-oxodG into DNA and RNA requires purine nucleoside phosphorylase in MCF-7 cells. Nucleic Acids Res 36:228–236
Nakabeppu Y, Tsuchimoto D, Ichinoe A et al (2004) Biological significance of the defense mechanisms against oxidative damage in nucleic acids caused by reactive oxygen species: from mitochondria to nuclei. Ann N Y Acad Sci 1011:101–111
Nakabeppu Y, Kajitani K, Sakamoto K, Yamaguchi H, Tsuchimoto D (2006) MTH1, an oxidized purine nucleoside triphosphatase, prevents the cytotoxicity and neurotoxicity of oxidized purine nucleotides. DNA Repair (Amst) 5:761–772
Nelson PT, Wang WX, Rajeev BW (2008) MicroRNAs (miRNAs) in neurodegenerative diseases. Brain Pathol 18:130–138
Nunomura A, Perry G, Pappolla MA et al (1999) RNA oxidation is a prominent feature of vulnerable neurons in Alzheimer’s disease. J Neurosci 19:1959–1964
Nunomura A, Perry G, Pappolla MA et al (2000) Neuronal oxidative stress precedes amyloid-β deposition in Down syndrome. J Neuropathol Exp Neurol 59:1011–1017
Nunomura A, Perry G, Aliev G et al (2001) Oxidative damage is the earliest event in Alzheimer disease. J Neuropathol Exp Neurol 60:759–767
Nunomura A, Chiba S, Kosaka K et al (2002) Neuronal RNA oxidation is a prominent feature of dementia with Lewy bodies. NeuroReport 13:2035–2039
Nunomura A, Chiba S, Lippa CF et al (2004) Neuronal RNA oxidation is a prominent feature of familial Alzheimer’s disease. Neurobiol Dis 17:108–113
Nunomura A, Castellani RJ, Zhu X, Moreira PI, Perry G, Smith MA (2006) Involvement of oxidative stress in Alzheimer disease. J Neuropathol Exp Neurol 65:631–641
Nunomura A, Moreira PI, Lee HG et al (2007) Neuronal death and survival under oxidative stress in Alzheimer and Parkinson diseases. CNS Neurol Disord: Drug Targets 6:411–423
Nunomura A, Tamaoki T, Tanaka K et al (2010) Intraneuronal amyloid β accumulation and oxidative damage to nucleic acids in Alzheimer disease. Neurobiol Dis 37:731–737
Nunomura A, Tamaoki T, Motohashi N et al (2012) The earliest stage of cognitive impairment in transition from normal aging to Alzheimer disease is marked by prominent RNA oxidation in vulnerable neurons. J Neuropathol Exp Neurol 71:233–241
Park EM, Shigenaga MK, Degan P et al (1992) Assay of excised oxidative DNA lesions: isolation of 8-oxoguanine and its nucleoside derivatives from biological fluids with a monoclonal antibody column. Proc Natl Acad Sci USA 89:3375–3379
Perkins DO, Jeffries C, Sullivan P (2005) Expanding the ‘central dogma’: the regulatory role of nonprotein coding genes and implications for the genetic liability to schizophrenia. Mol Psychiatry 10:69–78
Perry G, Nunomura A, Cash AD et al (2002a) Reactive oxygen: its sources and significance in Alzheimer disease. J Neural Transm Suppl 62:69–75
Perry G, Nunomura A, Hirai K et al (2002b) Is oxidative damage the fundamental pathogenic mechanism of Alzheimer’s and other neurodegenerative diseases? Free Radic Biol Med 33:1475–1479
Petersen RB, Siedlak SL, Lee HG et al (2005) Redox metals and oxidative abnormalities in human prion diseases. Acta Neuropathol 110:232–238
Praticò D, Clark CM, Liun F, Rokach J, Lee VY, Trojanowski JQ (2002) Increase of brain oxidative stress in mild cognitive impairment: a possible predictor of Alzheimer disease. Arch Neurol 59:972–976
Rahkonen T, Eloniemi-Sulkava U, Rissanen S, Vatanen A, Viramo P, Sulkava R (2003) Dementia with Lewy bodies according to the consensus criteria in a general population aged 75 years or older. J Neurol Neurosurg Psychiatry 74:720–724
Rhee Y, Valentine MR, Termini J (1995) Oxidative base damage in RNA detected by reverse transcriptase. Nucleic Acids Res 23:3275–3282
Rinaldi P, Polidori MC, Metastasio A et al (2003) Plasma antioxidants are similarly depleted in mild cognitive impairment and in Alzheimer’s disease. Neurobiol Aging 24:915–919
Rosen DR, Siddique T, Patterson D et al (1993) Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362:59–62
Row BW, Liu R, Xu W, Kheirandish L, Gozal D (2003) Intermittent hypoxia is associated with oxidative stress and spatial learning deficits in the rat. Am J Respir Crit Care Med 167:1548–1553
Satterlee JS, Barbee S, Jin P et al (2007) Noncoding RNAs in the Brain. J Neurosci 27:11856–11859
Sayre LM, Zelasko DA, Harris PL, Perry G, Salomon RG, Smith MA (1997) 4-Hydroxynonenal-derived advanced lipid peroxidation end products are increased in Alzheimer’s disease. J Neurochem 68:2092–2097
Sayre LM, Perry G, Smith MA (1999) In situ methods for detection and localization of markers of oxidative stress: application in neurodegenerative disorders. Methods Enzymol 309:133–152
Sayre LM, Smith MA, Perry G (2001) Chemistry and biochemistry of oxidative stress in neurodegenerative disease. Curr Med Chem 8:721–738
Schapira AH, Cooper JM, Dexter D, Clark JB, Jenner P, Marsden CD (1990) Mitochondrial complex I deficiency in Parkinson’s disease. J Neurochem 54:823–827
Schneider JE Jr, Phillips JR, Pye Q, Maidt ML, Price S, Floyd RA (1993) Methylene blue and rose bengal photoinactivation of RNA bacteriophages: comparative studies of 8-oxoguanine formation in isolated RNA. Arch Biochem Biophys 301:91–97
Schubert J, Wilmer JW (1991) Does hydrogen peroxide exist “free” in biological systems? Free Radic Biol Med 11:545–555
Seo AY, Hofer T, Sung B, Judge S, Chung HY, Leeuwenburgh C (2006) Hepatic oxidative stress during aging: effects of 8% long-term calorie restriction and lifelong exercise. Antioxid Redox Signal 8:529–538
Seo AY, Xu J, Servais S et al (2008) Mitochondrial iron accumulation with age and functional consequences. Aging Cell 7:706–716
Shan X, Lin CL (2006) Quantification of oxidized RNAs in Alzheimer’s disease. Neurobiol Aging 27:657–662
Shan X, Tashiro H, Lin CL (2003) The identification and characterization of oxidized RNAs in Alzheimer’s disease. J Neurosci 23:4913–4921
Shan X, Chang Y, Lin CL (2007) Messenger RNA oxidation is an early event preceding cell death and causes reduced protein expression. FASEB J 21:2753–2764
Shao C, Xiong S, Li GM et al (2008) Altered 8-oxoguanine glycosylase in mild cognitive impairment and late-stage Alzheimer’s disease brain. Free Radic Biol Med 45:813–819
Shen Z, Wu W, Hazen SL (2000) Activated leukocytes oxidatively damage DNA, RNA, and the nucleotide pool through halide-dependent formation of hydroxyl radical. Biochemistry (Mosc) 39:5474–5482
Sheth U, Parker R (2003) Decapping and decay of messenger RNA occur in cytoplasmic processing bodies. Science 300:805–808
Shi Q, Gibson GE (2011) Up-regulation of the mitochondrial malate dehydrogenase by oxidative stress is mediated by miR-743a. J Neurochem 118:440–448
Shi F, Gan W, Nie B et al (2012) Greater nucleic acids oxidation in the temporal lobe than the frontal lobe in SAMP8. NeuroReport 23:508–512
Shimura-Miura H, Hattori N, Kang D, Miyako K, Nakabeppu Y, Mizuno Y (1999) Increased 8-oxo-dGTPase in the mitochondria of substantia nigral neurons in Parkinson’s disease. Ann Neurol 46:920–924
Smith MA, Perry G, Richey PL et al (1996) Oxidative damage in Alzheimer’s. Nature 382:120–121
Smith MA, Nunomura A, Zhu X, Takeda A, Perry G (2000) Metabolic, metallic, and mitotic sources of oxidative stress in Alzheimer disease. Antioxid Redox Signal 2:413–420
Sofic E, Riederer P, Heinsen H et al (1988) Increased iron (III) and total iron content in post mortem substantia nigra of parkinsonian brain. J Neural Transm 74:199–205
Song XN, Zhang LQ, Liu DG et al (2011) Oxidative damage to RNA and expression patterns of MTH1 in the hippocampi of senescence-accelerated SAMP8 mice and Alzheimer’s disease patients. Neurochem Res 36:1558–1565
Sperling RA, Aisen PS, Beckett LA et al (2011) Toward defining the preclinical stages of Alzheimer’s disease; recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 7:280–292
Szymanski M, Barciszewska MZ, Erdmann VA, Barciszewski J (2005) A new frontier for molecular medicine: noncoding RNAs. Biochim Biophys Acta 1756:65–75
Taddei F, Hayakawa H, Bouton M et al (1997) Counteraction by MutT protein of transcriptional errors caused by oxidative damage. Science 278:128–130
Taft RJ, Pheasant M, Mattick JS (2007) The relationship between non-protein-coding DNA and eukaryotic complexity. BioEssays 29:288–299
Takahashi MA, Asada K (1983) Superoxide anion permeability of phospholipid membranes and chloroplast thylakoids. Arch Biochem Biophys 226:558–566
Tanaka M, Chock PB, Stadtman ER (2007) Oxidized messenger RNA induces translation errors. Proc Natl Acad Sci USA 104:66–71
Tanaka M, Han S, Küpfer PA, Leumann CJ, Sonntag WE (2011a) Quantification of oxidized levels of specific RNA species using an aldehyde reactive probe. Anal Biochem 417:142–148
Tanaka M, Han S, Küpfer PA, Leumann CJ, Sonntag WE (2011b) An assay for RNA oxidation induced abasic sites using the Aldehyde Reactive Probe. Free Radic Res 45:237–247
Tateyama M, Takeda A, Onodera Y et al (2003) Oxidative stress and predominant Aβ42(43) deposition in myopathies with rimmed vacuoles. Acta Neuropathol 105:581–585
Taylor JP, Hardy J, Fischbeck KH (2002) Toxic proteins in neurodegenerative disease. Science 296:1991–1995
van Leeuwen FW, de Kleijn DP, van den Hurk HH et al (1998) Frameshift mutants of β amyloid precursor protein and ubiquitin-B in Alzheimer’s and Down patients. Science 279:242–247
Vascotto C, Fantini D, Romanello M et al (2009) APE1/Ref-1 interacts with NPM1 within nucleoli and plays a role in the rRNA quality control process. Mol Cell Biol 29:1834–1854
Wamer WG, Wei RR (1997) In vitro photooxidation of nucleic acids by ultraviolet A radiation. Photochem Photobiol 65:560–563
Wang Q, Yu S, Simonyi A, Sun GY, Sun AY (2005) Kainic acid-mediated excitotoxicity as a model for neurodegeneration. Mol Neurobiol 31:3–16
Wang J, Markesbery WR, Lovell MA (2006) Increased oxidative damage in nuclear and mitochondrial DNA in mild cognitive impairment. J Neurochem 96:825–832
Weidner AM, Bradley MA, Beckett TL et al (2011) RNA oxidation adducts 8-OHG and 8-OHA change with Aβ42 levels in late-stage Alzheimer’s disease. PLoS ONE 6(9):e24930
Weimann A, Belling D, Poulsen HE (2002) Quantification of 8-oxo-guanine and guanine as the nucleobase, nucleoside and deoxynucleoside forms in human urine by high-performance liquid chromatography-electrospray tandem mass spectrometry. Nucleic Acids Res 30:e7
Weissman L, Jo DG, Sørensen MM et al (2007) Defective DNA base excision repair in brain from individuals with Alzheimer’s disease and amnestic mild cognitive impairment. Nucleic Acids Res 35:5545–5555
Wu J, Li Z (2008) Human polynucleotide phosphorylase reduces oxidative RNA damage and protects HeLa cell against oxidative stress. Biochem Biophys Res Commun 372:288–292
Yamaguchi H, Kajitani K, Dan Y et al (2006) MTH1, an oxidized purine nucleoside triphosphatase, protects the dopamine neurons from oxidative damage in nucleic acids caused by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Cell Death Differ 13:551–563
Yanagawa H, Ogawa Y, Ueno M (1992) Redox ribonucleosides. Isolation and characterization of 5-hydroxyuridine, 8-hydroxyguanosine, and 8-hydroxyadenosine from Torula yeast RNA. J Biol Chem 267:13320–13326
Yang WH, Bloch DB (2007) Probing the mRNA processing body using protein macroarrays and “autoantigenomics”. RNA 13:704–712
Yin B, Whyatt RM, Perera FP, Randall MC, Cooper TB, Santella RM (1995) Determination of 8-hydroxydeoxyguanosine by an immunoaffinity chromatography-monoclonal antibody-based ELISA. Free Radic Biol Med 18:1023–1032
Zhang J, Perry G, Smith MA et al (1999) Parkinson’s disease is associated with oxidative damage to cytoplasmic DNA and RNA in substantia nigra neurons. Am J Pathol 154:1423–1429
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Nunomura, A., Moreira, P.I., Castellani, R.J. et al. Oxidative Damage to RNA in Aging and Neurodegenerative Disorders. Neurotox Res 22, 231–248 (2012). https://doi.org/10.1007/s12640-012-9331-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12640-012-9331-x