
Abstract
Deferoxamine (DFO), a metal chelator, has been previously reported to slow the loss of spatial memory in a mouse model of amyloid accumulation when delivered intranasally (IN). In this study, we determined whether IN DFO also has beneficial effects in the P301L mouse, which accumulates hyperphosphorylated tau. Mice were intranasally treated three times per week with either 10 % DFO (2.4 mg) or saline for 5 months, and a battery of behavioral tests were conducted before tissue collection and biochemical analyses of brain tissue with Western blot and ELISA. Wild-type (WT) mice statistically outperformed transgenic (TG) saline mice in the radial arm water maze, while performance of TG-DFO mice was not different than WT mice, suggesting improved performance in the radial arm water maze. Other behavioral changes were not evident. Beneficial changes in brain biochemistry were evident in DFO-treated mice for several proteins. The TG mice had significantly less pGSK3β and HIF-1α, with more interleukin-1β and total protein oxidation than wild-type controls, and for each protein, DFO treatment significantly reduced these differences. There was not a significant decrease in phosphorylated tau in brain tissue of DFO-treated mice at the sites we measured. These data suggest that IN DFO is a potential treatment not only for Alzheimer’s disease, but also for other neurodegenerative diseases and psychiatric disorders in which GSK3β and HIF-1α play a prominent role.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Dysregulation of metals in neurodegenerative disease is well established, and chelation therapy has been suggested as a possible treatment (Bandyopadhyay et al. 2010). Deferoxamine (DFO), a metal chelator commonly used for the treatment of iron-overload, is a good option because it not only chelates metals, but it has also been shown to influence some of the other pathways involved in the pathogenesis of Alzheimer’s disease (AD). For example, DFO stabilizes hypoxia inducible factor-1α (HIF-1α), which leads to increased utilization of insulin in rat liver cells (Schubert et al. 2009; Soucek et al. 2003). This is relevant because insulin utilization has been shown to be decreased in the brains of patients with AD (Hallschmid and Schultes 2009). HIF-1α also increases glycolysis and the associated hexose monophosphate shunt pathway, which increases levels of glutathione and other reducing agents that diminish oxidative damage that is increased with AD (Dongiovanni et al. 2008; Semenza et al. 1994). Further, DFO leads to the phosphorylation and subsequent inactivation of GSK3β in bone tissue (Qu et al. 2008), which is important because dysregulation of GSK3β has been suggested as one of the main culprits in the development of AD (Hooper et al. 2008; Qu et al. 2008). Specifically, GSK3β has been shown to phosphorylate microtubule associated protein tau, which forms neurofibrillary tangles and to augment production of Aβ (Hooper et al. 2008). Finally, DFO has been shown in postnatal rat dorsal root ganglion cells to increase neurite outgrowth and synapse formation, which may also be beneficial in the AD brain (Nowicki et al. 2009).
DFO was tested in a single clinical trial in patients with AD in the early 1990s (Crapper McLachlan et al. 1991). Twice daily intramuscular (IM) injections significantly slowed decline of daily living skills by 50 % over 2 years. However, the IM injections were painful and side effects included loss of appetite and weight. Intranasal (IN) administration is an alternative to IM injections that targets DFO, which has a short half-life in blood, directly to the central nervous system (CNS) (Hanson et al. 2009; Dhuria et al. 2010; Peters et al. 1966). Drugs administered IN travel extracellularly along the olfactory and trigeminal nerves to gain direct access to the CNS, bypassing the blood–brain barrier and minimizing systemic exposure (Dhuria et al. 2010; Jogani et al. 2008). Delivery to the CNS following intranasal administration of a variety of drugs and biopharmaceuticals of differing size and structure has been demonstrated in rodents, primates, and humans (Dhuria et al. 2009; Craft et al. 2011; Thorne et al. 2008). Recently, IN DFO treatment of APP/PS1 mice was shown to improve performance in the Morris water maze test of spatial memory, although the mechanism of action is unclear (Hanson et al. 2012).
In the current study, we determined whether IN DFO would decrease loss of memory in the P301L mouse, which accumulates hyperphosphorylated tau, and how the treatment might affect AD neuropathology. DFO is a good candidate to decrease memory loss in these mice both because of the ability of DFO to inactivate GSK3β, which hyperphosphorylates tau (Deng et al. 2009), and because IM DFO was shown to decrease neurofibrillary aggregation in a rabbit model of tauopathy (Savory et al. 1998). We found that IN DFO decreased loss of working memory as measured in the radial arm water maze, and lead to increases in HIF-1α and pGSK3β. These results further demonstrate the potential of DFO as a treatment not only for Alzheimer’s disease, but other neurodegenerative diseases and psychiatric disorders for which GSK3β and HIF-1α play a prominent role.
Methods
Animal care and treatment
A total of 42 mice were purchased from Taconic Farms, Inc. (Germantown, NY). For chronic dosing and behavior tests, 28 transgenic (TG) tau mice at 7–8 weeks of age were used (Stock Tg(Prnp-MAPT*P301L)JNPL3Hlmc; model 002508-M–M), which were derived from a C57BL/6, DBA/2, SW mixed background. Fourteen wild-type (WT) controls at 8 weeks of age were also used for behavior (model 001638-W-M). Mice were individually housed and had continuous access to rodent chow and water during a 12-hour light/dark cycle. Behavioral testing and dosing was performed only during the day portion of the circadian cycle. All procedures were approved by the Animal Care and Use Committee of HealthPartners Research Foundation at Regions Hospital under protocol #05-097.
Experimental design
Mice were divided into three treatment groups of 14 each: 1) TG mice treated with IN DFO (TG-DFO), 2) TG mice treated with IN phosphate-buffered saline (TG-PBS), and 3) WT mice treated with IN PBS (WT-PBS). Mice were acclimated to handling over a period of 3 weeks and then treated intranasally every Monday, Wednesday, and Friday starting at 11 weeks of age. Behavior tests started at 32 weeks of age. Dosing continued during the 5 weeks of behavior tests. After behavior tests, mice were dosed a final time, and 24 h later, euthanized and tissues collected for biochemical analyses.
Drug treatment and dosing
Deferoxamine mesylate salt was purchased from Sigma (D9533; St. Louis, MO) and was delivered as a 10 % solution in 0.2× PBS (Sigma; P5493) at a pH of 6.0. The vehicle consisted of 0.2× PBS without DFO. For intranasal delivery, after 3 weeks of acclimation to handling, unanesthetized mice were gripped by the skin of their necks and held gently, but firmly, upside down in the palm of the hand. A 20-μl pipettor was used to administer four 6-μl nose drops (alternating nares; 24 μl total; 2.4 mg DFO) to each mouse. Each mouse was released for 2-min. intervals between each pair of nasal drops as described in Hanson et al. (2004).
Behavioral assessment
Over a 5-week period, mice were subjected to a battery of behavior tests to determine their cognitive, anxiolytic, and sensorimotor functions in the order described. All dry mazes were cleaned with a vinegar/water mixture between each mouse.
Morris water maze (MWM; hidden platform, probe, visual platform)
The MWM was performed in a round, flat-bottomed, black plastic tub (Polytank Inc., Litchfield, MN). The diameter at the surface of the water was 108 cm. Water was maintained at room temperature. Nontoxic white paint was added to obscure the hidden platform. Drop points 1, 2, 3, and 4 were located starting at the observer’s left and were distributed clockwise every quarter of the tank, respectively. The platform was a clear, plastic square measuring 9 × 9 cm placed 1 cm below the surface of the water. Tests with hidden platform were performed in eight successive blocks at two blocks/day (M-T-W–Th). Each block consisted of four trials of up to 60 s (with 20 s between trials to sit on the platform). Each trial consisted of a drop at one of the four drop points. Mice that did not reach the platform after 60 s were guided to the platform, which remained at the same location. For the probe test Friday, the hidden platform was removed and mice were allowed to swim in the maze for 1 min. The percent time in each quadrant was measured. After the probe, there was a visual platform test to determine visual ability that was the same as the hidden platform test, except the platform was striped, had a small flag, and remained visible above the surface. Data was digitally acquired with an overhead camera connected to a DVD recorder. Escape latency was recorded with a timer. For probe tests, the percent time spent in the quadrant with the platform was analyzed. For hidden platform tests, the four trials were averaged for each block.
Open field test
Open field test was used to assess levels of activity and exploration. The open field was a white square open-top box measuring 85 cm (length) × 77 cm (width) × 28 cm (height). Each mouse was placed in the center of the box and allowed to explore for 5 min. Data were digitally acquired with an overhead camera connected to a DVD recorder. Using EthoVision tracking system (Version 3.1; Noldus, Leesburg, VA, USA), the area of the box floor was divided into 16 boxes of equal size and the total number of line crossings was recorded.
Elevated plus maze
To assess anxiety, this maze was white and had four arms (each measuring 30 × 5 cm) in a plus (+) shape, surrounding a central area of 5 × 5 cm, elevated 76 cm. Two opposing arms were open, while the other arms had walls at a height of 16 cm. One trial per mouse was performed in which the mouse was placed in the center of the maze and allowed to explore for 5 min. Trials were recorded with an overhead camera and analyzed with the Ethovision software. The amount of time in open and closed arms was measured.
Y-maze
Y-maze was used to assess basic activity (total arm entries) and mnemonic function (percent alternation). Mice were tested in a white maze with three arms that measured 29 cm (length) × 5 cm (width) × 15 cm (height). Each mouse was placed in the center of the maze and allowed to explore for 5 min. Trials were recorded with an overhead camera and analyzed manually by counting arm entries and their sequence. Percent alternation was expressed as the ratio of arm entries differing from the previous two choices divided by the number of opportunities to make an alternate arm entry from the previous two.
Rotarod
Motor coordination was measured using a rotarod (IITC Life Systems, Woodland Hills, CA). Mice were given three trials of up to 120 s on a rotating rod that started at 10 rpm and accelerated to 22 rpm over 30 s. The inter-trial interval was 3 min. Time to fall from the rod was measured, and the highest score for each mouse was recorded and averaged for each group.
Radial arm water maze (RAWM)
To assess working memory, the same tank and setup were used as for MWM except that inserts were included to divide the maze into six radially arranged arms. Arms were 41 × 15 cm, and walls extended 5.5 cm above the water surface. For 12 successive days of testing, each mouse was assessed in four successive acquisition trials and a fifth retention trial 30 min later. The last acquisition trial (T4) and the retention trial (T5) are considered the index of working memory. A clear platform (6.4 × 6.4 cm) was submerged 1 cm at the end of one of the arms. Platform location was changed for each of the 12 days. The start arms for each of the acquisition trials (T1–T4), and the retention trial (T5) were selected in a semi-random sequence from swim arms lacking the platform. For each trial, the mouse was placed at the end of the selected swim arm, facing the wall of the pool. Each trial was continued for up to 60 s or until the mouse located the platform. During the trial, each mouse was allowed to leave the drop arm to search for the platform. An error was recorded when the mouse entered any of the five arms not containing the platform, or failed to make an arm choice within 20 s. Following each error, the mouse was returned to the start arm and allowed to continue. Once the mouse located the platform, it was allowed to stay on for 20 s. If the mouse failed to locate the platform after 60 s, it was placed on the platform and allowed to stay for 20 s. Along with the number of errors made per trial, the escape latency of finding the hidden platform was recorded. After completion of the acquisition trials (T1–T4), the mouse was returned to its cage for 30 min, after which it was returned to the pool for the retention trial (T5).
Optomotor visual test
Optomotor was used as a second, more sensitive test to assess vision. The optomotor device consisted of a free-rotating cylinder (height 43 cm, diameter 23 cm) with black and white vertical stripes of equal width (3/4 cm) lining the inner wall. The mouse was placed on a stationary platform (6 × 6 cm, elevated 10 cm) in the center and monitored by an overhead video camera as the cylinder was rotated clockwise at 2 rpm for 2 min. Visual acuity was measured by the frequency of “head turns” as an indicator of the mouse recognizing contrast and following it in motion. A head turn was defined as a sweeping head motion of at least 15-degrees at pace with the rotating cylinder, as in (Thaung et al. 2002).
Euthanasia and tissue collection
Mice were euthanized by CO2 asphyxiation followed by immediate decapitation. Blood was processed with a serum separator tube (BD Microtainer, Franklin Lakes, NJ), and serum was snap-frozen in liquid nitrogen. The brain was removed from the skull. Olfactory bulbs and cerebellum were discarded, the brain was hemisected sagitally, each half was snap-frozen in liquid nitrogen, and the tubes were stored at −70 °C until biochemical analysis.
Protein extraction
Frozen brain tissues were homogenized in 5 volumes of ice-cold RIPA buffer (50 mM Tris–HCl, pH 7.4, 150 mM NaCl, 2 mM EDTA, 1 % Sodium Deoxycholate, 1 % NP-40, 0.1 % SDS) supplemented with protease inhibitor cocktail (Roche, Boulder, CO) and phosphatase inhibitor cocktail (Sigma, St. Louis, MO). Homogenates were centrifuged at 20,000×g for 20 min at 4 °C. Supernatant was stored at −70 °C until analysis by western blot and ELISA.
Western blot analysis
Protein concentrations were determined using the BCA (bicinchoninic acid) method. Equal amounts of total protein (50ug for HIF-1αlpha and 25ug for all other protein targets) were diluted in Laemmli buffer, separated by SDS-PAGE, and transferred to PVDF membranes (Millipore, Billerica, MA, USA). The membranes were blocked with 5 % dry milk in Tris-buffered saline/0.1 % Tween (TBS-T) overnight at 4 °C on a shaking platform. Membranes were then incubated for 1 h with one of the following antibodies in TBS-T: phospho-GSK-3β (Ser9) rabbit antibody (#9336, Cell Signaling Technology, Beverly, MA, USA), GSK-3β (27C10) rabbit monoclonal antibody (#9315, Cell Signaling Tech.), HIF-1α rabbit polyclonal antibody (NB100-479, Novus Biologicals, Littleton, CO), GLUT1 rabbit polyclonal antibody (ab652, Abcam, Cambridge, MA, USA), β-catenin E-5 (sc-7963, Santa Cruz Biotechnology, Santa Cruz, CA, USA), p-Tau (Ser 262) (sc-101813, Santa Cruz Biotech.), p-Tau AT8 (Ser202/Thr205) (MN1020), p-Tau AT100 (Ser212/Thr214) (MN1060), or human Tau HT7 (MN1000, Thermo Scientific, Rockford, IL, USA). Rabbit anti-actin polyclonal antibody was detected on all blots as a loading control (NB600-532, Novus Biologicals). Membranes were rinsed in TBS-T and then incubated in either anti-rabbit or anti-mouse IgG conjugated to horseradish peroxidase (#7074, #7076, Cell Signaling Tech.) in TBS-T with 5 % dry milk for 1 h. Enhanced Chemiluminescence Plus Western blotting detection reagent (GE Healthcare, Buckinghamshire, UK) was used to visualize peroxidase enzymatic activity on X-ray film. Exposed films were quantified using Image J software provided by the National Institutes of Health (Bethesda, MD, USA). Protein oxidation was measured using an Oxyblot Protein Oxidation detection kit (S1750; Millipore, Billerica, MA, USA).
ELISA
Interleukin-1β (IL-1β) in the brain was measured using a commercially available ELISA kit (MLB00B; R&D Systems, Minneapolis, MN) according to the manufacturer’s instructions.
Statistical analyses
Analyses for behavioral tests included parametric and non-parametric analyses. Non-parametric analysis was used for tests that included a truncation effect, which violated assumptions of normality; these included MWM visual platform and probe, rotarod, and RAWM escape latency. For comparisons of all three treatment groups, ANOVA with Fisher’s LSD post-test (parametric) or Kruskal–Wallis with Dunn’s post-test (non-parametric) was used. Repeated measures ANOVA was used for hidden platform tests in MWM. For all biochemical tests, t tests or ANOVAs were used to compare treatment groups.
Results
General health
During the course of the study, two mice in the TG-PBS group died of natural causes before the appointed sacrifice time at 8.5 months of age. There were no significant differences in average weight for each treatment group at the time of sacrifice. Average weight (±SEM) was 42.4 ± 1.4 g for WT-PBS mice, 42.7 ± 1.4 g for TG-DFO mice, and 41.5 ± 1.4 g for TG-PBS mice. No obvious physical differences were observed between groups.
Memory tests
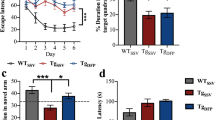
In the RAWM, WT-PBS mice had significantly shorter escape latencies than TG-PBS mice in trials four and five of blocks three and four (p < 0.05; Fig. 1a), but not significantly shorter than TG-DFO. This result was the same in trials four and five of block four for number of errors (p < 0.05; Fig. 1b). There were no significant differences in blocks one and two (data not shown).

Radial arm water maze data from blocks 3 and 4 for a escape latency and b errors. TG mice are P301L dosed with IN DFO or PBS for 4 mo. (n = 12–14 mice/group). Blocks 3 and 4 are an average of days 7–9 and 10–12, respectively. Data for trials 1, 4, and 5 are shown. Asterisks on trials 4 and 5 represent significant differences among the three groups (*p < 0.05). In each case, the difference was between WT-PBS and TG-PBS but not between WT-PBS and TG-DFO, indicating that DFO-treated mice performed as well as WT mice. TG transgenic, IN intranasal, DFO deferoxamine, WT wild type, PBS phosphate-buffered saline
For hidden platform tests in MWM, WT-PBS mice had shorter escape latencies than both groups of TG mice when analyzed with a repeated measures ANOVA (p < 0.05) (Fig. 2). There were no differences between the two groups of TG mice. When compared with a Chi-square test, WT-PBS mice had significantly fewer trials that took 60 s to find the platform (7) than TG-DFO (32) and TG-PBS (41) (p < 0.05). For probe tests in MWM, there were no statistical differences between groups. The average percent time (±SEM) in the target quadrant was 38.5 ± 4.2 for WT-PBS, 37.5 ± 4.1 for TG-PBS, and 30.6 ± 3.8 for TG-DFO. There were no significant differences between groups for velocity in the MWM.

Hidden platform trials in Morris water maze. TG mice are P301L dosed with IN DFO or PBS for 4 mo. (n = 12–14 mice/group). WT-PBS mice had shorter escape latencies than both drug- and vehicle-treated TG mice with repeated measures ANOVA (*p < 0.05), indicating a strong transgenic effect. Error bars are SEM. TG transgenic, IN intranasal, DFO deferoxamine, WT wild type, PBS phosphate-buffered saline, SEM standard error of the mean
There were no statistically significant differences between groups for arm entries or percent arm alternations in the Y-maze. Average arm entries (±SEM) were 16.8 ± 1.2 for WT-PBS, 14.4 ± 1.6 for TG-DFO, and 17 ± 1.1 for TG-PBS. Percent alternations (±SEM) were 55.1 ± 2.9 for WT-PBS, 53.1 ± 5.6 for TG-DFO, and 52.6 ± 4.7 for TG-PBS.
Visual tests
For visual platform tests in the MWM, WT-PBS mice had significantly shorter escape latencies than TG-DFO (20.3 ± 2.6) and TG-PBS (20.2 ± 2.6) (p < 0.05) (Fig. 3a). For optomotor test, WT-PBS mice had significantly more head turns than both TG groups (p < 0.05) (Fig. 3b). Average head turns (±SEM) were 8.8 ± 1.9 for WT-PBS, 3.3 ± 1.2 for TG-PBS, and 3.8 ± 1.0 for TG-DFO.

Visual ability of mice in this study as measured by visual platform test in Morris water maze (a) and optomotor task (b). TG mice are P301L dosed with IN DFO or PBS for 4 mo. (n = 12–14 mice/group). For visual platform test, escape latencies were significantly shorter for WT-PBS than for both groups of TG mice (*p < 0.05), indicating impaired vision. For optomotor task, sweeping head turns of 15° were measured for 2 min WT-PBS mice had significantly more head turns than both groups of TG mice (*p < 0.05), indicating impaired visual ability for TG mice. Error bars are SEM. TG transgenic, IN intranasal, DFO deferoxamine, WT wild type, PBS phosphate-buffered saline, SEM standard error of the mean
Exploratory and anxiolytic tests
In the open field maze, there were no significant differences in line crossings between the three treatment groups (p = 0.30). Total line crossings (±SEM) were 80.9 ± 6.5 for WT-PBS, 98.0 ± 9.2 for TG-DFO, and 83.6 ± 9.4 for TG-PBS. In the elevated plus maze, there were no statistically significant differences in any groups for time spent in open arms (p = 0.87). Seconds spent in open arms for WT-PBS was 19.5 ± 11.7 s, for TG-DFO it was 15.3 ± 3.3 s, and for TG-PBS it was 16.2 ± 5.0 s.
Motor function
On the rotarod, there was no statistical difference between treatment groups (p = 0.31). Average time (sec) spent on rod (±SEM) was 30.6 ± 6.9 for WT-PBS, 32.6 ± 5.4 for TG-DFO, and 50.6 ± 10.8 for TG-PBS.
Tau
Phosphorylated tau was easily measured in both treatment groups of P301L mice, but was not detectable in the WT-PBS mice. Within the TG groups, there was no statistical difference between the TG treatment groups for either total tau or the phosphorylated isoforms of tau recognized by p-Tau (ser262; ptau/total tau for TG-PBS = 0.2, TG-DFO = 0.18), AT8 (Ser202/Thr205; TG-PBS = 6,566 ± 3,934, TG-DFO = 6,363 ± 4,018), or AT100 (Ser212/Thr214; TG-PBS = 6,481 ± 2,854, TG-DFO = 6,973 ± 4,833), (p > 0.05, t tests).
GSK3β and β-catenin
The P301L mice had lower levels of pGSK3β and IN DFO treatment restored normal levels. TG-PBS mice had significantly less pGSK3β than both WT-PBS (45 % less) and TG-DFO mice (49 % less) (p < 0.05; Fig. 4a). There was no difference between the WT-PBS and TG-DFO groups. The same relationships were seen when analyzing the ratio of pGSK3β/total GSK3β (Fig. 4b). There were no significant differences among groups for total GSK (data not shown) or β-catenin (p > 0.05; Fig. 4c).

Representative blots and histograms for proteins analzyed by Western blot. TG mice are P301L dosed with IN DFO or PBS for 4 mo. (n = 12–14 mice/group). a pGSK3β, b pGSK3β/GSK3β, c β-catenin d HIF-1α, e GLUT1, and f Oxyblot. For oxyblot, carbonyl groups introduced into proteins by oxidative reactions were derivatized to 2,4-Dinitropheylhydrazone (DNP), and the DNP was quantified. Blots are representative of each treatment group, while histograms are percent of change from WT-PBS mice. Error bars are SEM converted from the raw data to percentages and are only shown to help visualize differences. Statistically significant differences between groups as measured by ANOVA with Fisher’s LSD post-test are represented with an asterisk (*p < 0.05) and were performed on the raw data as optical density, not from percent changes. TG transgenic, IN intranasal, DFO deferoxamine, WT wild type, PBS phosphate-buffered saline, SEM standard error of the mean
HIF-1α and GLUT1
The P301L mice had lower levels of HIF-1α and IN DFO treatment restored normal levels. TG-PBS mice had significantly less HIF-1α than both WT-PBS (20 % less) and TG-DFO mice (32 % less) (p < 0.05) (Fig. 4d). There was no difference between WT-PBS and TG-DFO mice. There was also an increase in GLUT1 in both groups of TG mice, and this increase was significant for the TG-PBS mice (Fig. 4e).
Oxidation and inflammation
The P301L mice had higher levels of inflammation and protein oxidation and IN DFO treatment restored normal levels. TG-PBS mice had significantly more protein oxidation than both WT-PBS (43 % more) and TG-DFO mice (33 % more) (p < 0.05) (Fig. 4f). There was no difference in protein oxidation between the WT-PBS and TG-DFO groups. Levels of IL-1β were highest in the TG-PBS model where they measured 43.7 (±3.0 SEM) pg/g brain tissue; levels were lower (though not significantly) in WT-PBS mice at 34.3 (±4.0 SEM) pg/g, and significantly lower in the TG-DFO mice at 28.0 (±3.9 SEM) (p < 0.05).
Discussion
Intranasal DFO treatment of the P301L mouse model of tauopathy improved performance in the radial arm water maze, increased phosphorylated GSK3β, stabilized HIF-1α, and decreased inflammation and protein oxidation. IN DFO was safe and well tolerated as evidenced by no change in body weight and no death in the DFO-treated group. These data demonstrate that the biochemical effects of DFO previously seen in in vitro experiments occurred in brain tissue in vivo when delivered intranasally. While a change in total or hyperphosphorylated tau was not observed, which was expected with the decreased activity of GSK3β, these data suggest that IN DFO has potential as a treatment for AD through the stabilization of HIF-1α and decrease in inflammation and protein oxidation. IN DFO is also a potential treatment of other neurodegenerative and psychiatric disorders with dysregulation of GSK3β, for example, Huntington’s disease or schizophrenia (Aghdam and Barger 2007; Hur and Zhou 2010).
This study yielded several novel observations using the P301L mouse model of tauopathy. First, we were able to observe a behavioral deficit in a single behavioral task, the radial arm water maze. A behavioral deficiency was seen in this same model previously (Arendash et al. 2004), but in that study, the behavioral deficiency was observed as a result of a multi-metric analysis of several behavioral assays with a discriminant function analysis. Also, we performed a behavioral task to specifically measure visual ability, the optomotor task, which is free of confounding cognitive elements (whereas visual platform tests in the Morris water maze are not). This task clearly demonstrated that P301L mice had impaired vision. Several biochemical changes in the brains of the TG model were observed that had not been previously reported. These included decreased levels of pGSK3β and the ratio of pGSK3β/GSK3β, a decrease in HIF1α, increased levels of GLUT1, and increased levels of oxidative damage and inflammation as measured by oxyblot and IL-1B, respectively.
Although there were visual impairments in P301L mice, there was still a significant drug effect of IN DFO when tested in the radial arm water maze. The decreased visual ability was first observed in the Morris water maze visual platform test, which prompted the use of the more rigorous optomotor test. Optomotor data confirmed visual impairment in both treatment groups of TG mice. However, the fact that these mice improved performance in the radial arm water maze from Trial 1 to Trial 4 on a daily basis showed that although vision was compromised, the mice were not completely blind. Similarly, mice routinely found the platform within the 60 s period given for trials. Visual problems with this mouse model were also observed in other attempts to quantify behavioral activity (Arendash et al. 2004). The improvement in performance in the RAWM due to DFO treatment could be considered relatively weak, as the significant difference was between the WT-PBS and TG-PBS groups, but not the TG-PBS and TG-DFO groups. Although drug differences were noted in the radial arm water maze and visual tests, there were no other statistically significant differences with any of the other behavior tests. The strong transgenic effect observed in the Morris water maze is highly likely due to the compromised visual ability in the TG mice. In the future, olfactory-based memory tests such as a novel odor recognition task might be more appropriate for this mouse model, or having the mice tested for the rd1 gene, which is now available for commercially purchased transgenic mice (Garcia et al. 2004).
The transgenic mice had higher levels of GSK3β activity than wild-type controls, and this activity was restored to normal levels by IN DFO treatment. Decreased activity of GSK3β with DFO treatment was also seen previously using in vitro studies with osteoblasts (Qu et al. 2008), and with in vitro studies of another metal chelator, M30 (Avramovich-Tirosh et al. 2010). The demonstration that IN DFO affects GSK3β activity in vivo is important as GSK3β has been implicated in contributing to several of the neuropathologies of AD (Hooper et al. 2008), as well as in playing a role in frontotemporal dementia, other neurodegenerative disorders, and psychiatric conditions such as schizophrenia, autism, and depression (Hur and Zhou 2010; Aghdam and Barger 2007). The decreased activity of GSK3β might also have been expected to lead to a decrease in hyperphosphorylated tau, for which the link is now well established (Lucas et al. 2001; Terwel et al. 2008). However, this did not occur in this study and there are several possibilities why this may be the case. Hyperphosphorylation of tau is a complicated process that depends on the animal model, the order of phosphorylation sites, pre-phosphorylation at certain sites that affect others, and pre-phosphorylation with other kinases such as protein kinase A or CK-1 (Munoz-Montano et al. 1997; Wang et al. 1998; Bertrand et al. 2010). Further, it is possible that the timing of DFO dose and the timing of protein turnover may explain the lack of change in pTau. Although we did not detect changes in ptau in this study, it is still possible that there were changes we did not detect. Much like with tau, the decreased activity of GSK3β might have led to an associated increase in β-catenin, as GSK3β will phosphorylate β-catenin to destabilize it in the absence of the Wnt signaling pathway, but this did not occur in our study (Liu et al. 2005). A similar lack of a change in β-catenin in the presence of a change in GSK3β has been previously observed in cell lines created lacking GSK3β that had normal levels of β-catenin (Doble et al. 2007).
This is the first report that treatment with IN DFO leads to an in vivo increase in brain levels of HIF-1α. It is well established that DFO stabilizes HIF-1α by binding iron, which is a co-factor for prolyl hydroxylase, the enzyme responsible for breakdown of HIF (Dongiovanni et al. 2008; Schubert et al. 2009). The fact the DFO modulates HIF-1α is important because HIF-1α works as a transcription factor that binds to conserved regulatory sequences in the promoter of several target genes, many of which may play a role in neuroprotection. Thus, there are several mechanisms by which HIF-1α may modify the neuropathology associated with Alzheimer’s disease. For one, HIF-1α leads to an increase in glycolysis by up-regulating many of the glycolytic enzymes (Semenza et al. 1994; Mottet et al. 2003), which leads to increased utilization of insulin, and insulin has been shown to be decreased in the brains of patients with AD (Schubert et al. 2009; Soucek et al. 2003). The increase in glycolysis also leads to more glutathione and other reducing agents, which can decrease oxidative damage (which was observed in the mice in this study). Other downstream targets of HIF-1α lead to increases in erythropoeisis (from erythropoietin) and angiogenesis (from VEGF) (Dongiovanni et al. 2008; Semenza et al. 1994). Also, the increase in ATP from glycolysis and the decrease in oxidative damage could decrease mitochondrial dysfunction, another contributing factor to the pathogenesis of AD (Atamna and Frey 2007).
Levels of GLUT1 in the transgenic model were affected, which is important because of the role that impaired glucose utilization as well as insulin signaling play in the neuropathology associated with Alzheimer’s disease. In vivo PET imaging demonstrates a progressive decrease in glucose utilization in AD patients, the extent of which correlates to symptom severity (Mosconi et al. 2008). Further, expression of insulin and insulin receptors in the hippocampus is markedly lower in post-mortem brain of AD patients as compared to normal brains (Steen et al. 2005). The transgenic and drug effects on GLUT1 expression observed in this study were unexpected based on the observed increase in HIF-1α in DFO-treated mice. Levels of GLUT1 were elevated in both groups of TG mice, significantly so in the TG-PBS mice. However, we would have expected a decrease in GLUT1 in the TG-PBS mice, as Chen et al. (2001) showed that GLUT1 mRNA is controlled by HIF-1α, which was decreased in this treatment group. A decrease in insulin availability in the TG mice in this study may explain the increase in GLUT1. It has been found that depletion of insulin, which decreases HIF-1α, can induce hyperglycemic conditions in which blood glucose levels are increased (Ke et al. 2009). Furthermore, conditions of hyperglycemia-induced insulin resistance can lead to enhanced glucose transport through insulin-independent GLUT1 and the hexosamine pathway, instead of through GLUT4 (Ebeling et al. 1998). Although it has been shown that HIF-1α induces GLUT1 mRNA (Chen et al. 2001), it is not the primary inducer in hyperglycemic conditions, which could have been generated in the TG-PBS mice.
Intranasal treatment with DFO decreased both inflammation and oxidation, two phenomena known to occur with AD (Shaftel et al. 2008; Zhu et al. 2007). The TG-PBS mice had higher levels of IL-1β, a measure of neuroinflammation, while the TG-DFO mice had levels similar to the WT mice. IL-1β has been shown to be elevated in brain tissue of both AD patients and some animal models of AD, and local expression of IL-1β has been implicated in impairment of hippocampal dependent memory process (Shaftel et al. 2008; Schneider et al. 1998). IL-1β has also been implicated as a potential regulator of HIF-1α (Thornton et al. 2000), which was also effected by IN DFO in these mice. The transgenic mice also had increased levels of oxidation as measured by oxyblot, and this effect was alleviated by IN DFO. Possible mechanisms by which DFO could decrease oxidative damage are the increase in glycolysis and the associated hexose monophosphate shunt from the stabilization of HIF-1α, which reduces oxidative damage (Dongiovanni et al. 2008; Semenza et al. 1994; Tonin et al. 2010). DFO also directly binds unbound iron, which leads to a decrease in Fenton chemistry and thus less oxidative damage (Domingo 2006).
In conclusion, IN DFO treatment produces a number of beneficial effects in this and other transgenic mouse models of AD pathology in support of its therapeutic potential for AD and other neurodegenerative and psychiatric disorders. It was safe and well tolerated by the mice, and there were no side effects as measured with the non-cognitive behavioral tests like rotarod. Indeed, two of the mice in the TG-PBS group died while none of the TG-DFO mice died, indicating it may have been beneficial for survival. This same phenomenon was noted in Hanson et al. (Hanson et al. 2012). Although the exact mechanism of how this drug produces beneficial effects was not determined, some of the proteins that are likely involved were identified, most notably HIF-1α and GSK3β. There are likely other proteins involved that will be identified in future studies. Because of the many mechanisms in which DFO seems to be beneficial for AD, it may offer a different type of treatment than those currently accepted for the treatment of AD. Toxicology studies are in progress for eventual human clinical trials with IN DFO.
References
Aghdam SY, Barger SW (2007) Glycogen synthase kinase-3 in neurodegeneration and neuroprotection: lessons from lithium. Curr Alzheimer Res 4(1):21–31
Arendash GW, Lewis J, Leighty RE, McGowan E, Cracchiolo JR, Hutton M, Garcia MF (2004) Multi-metric behavioral comparison of APPsw and P301L models for Alzheimer’s disease: linkage of poorer cognitive performance to tau pathology in forebrain. Brain Res 1012(1–2):29–41
Atamna H, Frey WH 2nd (2007) Mechanisms of mitochondrial dysfunction and energy deficiency in Alzheimer’s disease. Mitochondrion 7(5):297–310
Avramovich-Tirosh Y, Bar-Am O, Amit T, Youdim MB, Weinreb O (2010) Up-regulation of hypoxia-inducible factor (HIF) -1alpha and HIF-target genes in cortical neurons by the novel multifunctional iron chelator anti-Alzheimer drug, M30. Curr Alzheimer Res 7(4):300–306
Bandyopadhyay S, Huang X, Lahiri DK, Rogers JT (2010) Novel drug targets based on metallobiology of Alzheimer’s disease. Expert Opin Ther Targets 14(11):1177–1197
Bertrand J, Plouffe V, Senechal P, Leclerc N (2010) The pattern of human tau phosphorylation is the result of priming and feedback events in primary hippocampal neurons. Neuroscience 168(2):323–334
Chen C, Pore N, Behrooz A, Ismail-Beigi F, Maity A (2001) Regulation of glut1 mRNA by hypoxia-inducible factor-1. Interaction between H-ras and hypoxia. J Biol Chem 276(12):9519–9525
Craft S, Baker LD, Montine TJ, Minoshima S, Watson GS, Claxton A, Arbuckle M, Callaghan M, Tsai E, Plymate SR, Green PS, Leverenz J, Cross D, Gerton B (2011) Intranasal insulin therapy for Alzheimer Disease and Amnestic mild cognitive impairment: a pilot clinical trial. Arch Neurol. doi:10.1001/archneurol.2011.233
Crapper McLachlan DR, Dalton AJ, Kruck TP, Bell MY, Smith WL, Kalow W, Andrews DF (1991) Intramuscular desferrioxamine in patients with Alzheimer’s disease. Lancet 337(8753):1304–1308
Deng Y, Li B, Liu Y, Iqbal K, Grundke-Iqbal I, Gong CX (2009) Dysregulation of insulin signaling, glucose transporters, O-GlcNAcylation, and phosphorylation of tau and neurofilaments in the brain: implication for Alzheimer’s disease. Am J Pathol 175(5):2089–2098
Dhuria SV, Hanson LR, Frey WH 2nd (2009) Intranasal drug targeting of hypocretin-1 (orexin-A) to the central nervous system. J Pharm Sci 98(7):2501–2515
Dhuria SV, Hanson LR, Frey WH 2nd (2010) Intranasal delivery to the central nervous system: mechanisms and experimental considerations. J Pharm Sci 99(4):1654–1673
Doble BW, Patel S, Wood GA, Kockeritz LK, Woodgett JR (2007) Functional redundancy of GSK-3alpha and GSK-3beta in Wnt/beta-catenin signaling shown by using an allelic series of embryonic stem cell lines. Dev Cell 12(6):957–971
Domingo JL (2006) Aluminum and other metals in Alzheimer’s disease: a review of potential therapy with chelating agents. J Alzheimers Dis 10(2–3):331–341
Dongiovanni P, Valenti L, Ludovica Fracanzani A, Gatti S, Cairo G, Fargion S (2008) Iron depletion by deferoxamine up-regulates glucose uptake and insulin signaling in hepatoma cells and in rat liver. Am J Pathol 172(3):738–747
Ebeling P, Koistinen HA, Koivisto VA (1998) Insulin-independent glucose transport regulates insulin sensitivity. FEBS Lett 436(3):301–303
Garcia MF, Gordon MN, Hutton M, Lewis J, McGowan E, Dickey CA, Morgan D, Arendash GW (2004) The retinal degeneration (rd) gene seriously impairs spatial cognitive performance in normal and Alzheimer’s transgenic mice. NeuroReport 15(1):73–77
Hallschmid M, Schultes B (2009) Central nervous insulin resistance: a promising target in the treatment of metabolic and cognitive disorders? Diabetologia 52:2264–2269
Hanson LR, Martinez PM, Taheri S, Kamsheh L, Mignot E, Frey WHI (2004) Intranasal administration of hypocretin 1 (Orexin A) bypasses the blood-brain barrier & targets the brain: a new strategy for the treatment of narcolepsy. Drug Deliv Technol 4(4):81–86
Hanson LR, Roeytenberg A, Martinez PM, Coppes VG, Sweet DC, Rao RJ, Marti DL, Hoekman JD, Matthews RB, Frey WH 2nd, Panter SS (2009) Intranasal deferoxamine provides increased brain exposure and significant protection in rat ischemic stroke. J Pharmacol Exp Ther 330(3):679–686
Hanson L, Fine J, Renner D, Svitak A, Burns R, Nguyen T, Tuttle N, Marti D, Panter S, Frey W (2012) Intranasal delivery of deferoxamine reduces spatial memory loss in APP/PS1 mice. Drug Deliv Transl Res:1–9. doi:10.1007/s13346-011-0050-2
Hooper C, Killick R, Lovestone S (2008) The GSK3 hypothesis of Alzheimer’s disease. J Neurochem 104(6):1433–1439
Hur EM, Zhou FQ (2010) GSK3 signalling in neural development. Nat Rev 11(8):539–551
Jogani V, Jinturkar K, Vyas T, Misra A (2008) Recent patents review on intranasal administration for CNS drug delivery. Recent Pat Drug Deliv Formul 2(1):25–40
Ke YD, Delerue F, Gladbach A, Gotz J, Ittner LM (2009) Experimental diabetes mellitus exacerbates tau pathology in a transgenic mouse model of Alzheimer’s disease. PLoS ONE 4(11):e7917
Liu X, Rubin JS, Kimmel AR (2005) Rapid, Wnt-induced changes in GSK3beta associations that regulate beta-catenin stabilization are mediated by Galpha proteins. Curr Biol 15(22):1989–1997
Lucas JJ, Hernandez F, Gomez-Ramos P, Moran MA, Hen R, Avila J (2001) Decreased nuclear beta-catenin, tau hyperphosphorylation and neurodegeneration in GSK-3beta conditional transgenic mice. EMBO J 20(1–2):27–39
Mosconi L, Pupi A, De Leon MJ (2008) Brain glucose hypometabolism and oxidative stress in preclinical Alzheimer’s disease. Ann N Y Acad Sci 1147:180–195
Mottet D, Dumont V, Deccache Y, Demazy C, Ninane N, Raes M, Michiels C (2003) Regulation of hypoxia-inducible factor-1alpha protein level during hypoxic conditions by the phosphatidylinositol 3-kinase/Akt/glycogen synthase kinase 3beta pathway in HepG2 cells. J Biol Chem 278(33):31277–31285
Munoz-Montano JR, Moreno FJ, Avila J, Diaz-Nido J (1997) Lithium inhibits Alzheimer’s disease-like tau protein phosphorylation in neurons. FEBS Lett 411(2–3):183–188
Nowicki M, Kosacka J, Spanel-Borowski K, Borlak J (2009) Deferoxamine-induced neurite outgrowth and synapse formation in postnatal rat dorsal root ganglion (DRG) cell cultures. Eur J Cell Biol 88(10):551–562
Peters G, Keberle H, Schmid K, Brunner H (1966) Distribution and renal excretion of desferrioxamine and ferrioxamine in the dog and in the rat. Biochem Pharmacol 15(1):93–109
Qu ZH, Zhang XL, Tang TT, Dai KR (2008) Promotion of osteogenesis through beta-catenin signaling by desferrioxamine. Biochem Biophys Res Commun 370(2):332–337
Savory J, Huang Y, Wills MR, Herman MM (1998) Reversal by desferrioxamine of tau protein aggregates following two days of treatment in aluminum-induced neurofibrillary degeneration in rabbit: implications for clinical trials in Alzheimer’s disease. Neurotoxicology 19(2):209–214
Schneider H, Pitossi F, Balschun D, Wagner A, del Rey A, Besedovsky HO (1998) A neuromodulatory role of interleukin-1beta in the hippocampus. Proc Natl Acad Sci USA 95(13):7778–7783
Schubert D, Soucek T, Blouw B (2009) The induction of HIF-1 reduces astrocyte activation by amyloid beta peptide. Eur J Neurosci 29(7):1323–1334
Semenza GL, Roth PH, Fang HM, Wang GL (1994) Transcriptional regulation of genes encoding glycolytic enzymes by hypoxia-inducible factor 1. J Biol Chem 269(38):23757–23763
Shaftel SS, Griffin WS, O’Banion MK (2008) The role of interleukin-1 in neuroinflammation and Alzheimer disease: an evolving perspective. J Neuroinflammation 5:7
Soucek T, Cumming R, Dargusch R, Maher P, Schubert D (2003) The regulation of glucose metabolism by HIF-1 mediates a neuroprotective response to amyloid beta peptide. Neuron 39(1):43–56
Steen E, Terry BM, Rivera EJ, Cannon JL, Neely TR, Tavares R, Xu XJ, Wands JR, de la Monte SM (2005) Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease–is this type 3 diabetes? J Alzheimers Dis 7(1):63–80
Terwel D, Muyllaert D, Dewachter I, Borghgraef P, Croes S, Devijver H, Van Leuven F (2008) Amyloid activates GSK-3beta to aggravate neuronal tauopathy in bigenic mice. Am J Pathol 172(3):786–798
Thaung C, Arnold K, Jackson IJ, Coffey PJ (2002) Presence of visual head tracking differentiates normal sighted from retinal degenerate mice. Neurosci Lett 325(1):21–24
Thorne RG, Hanson LR, Ross TM, Tung D, Frey WH 2nd (2008) Delivery of interferon-beta to the monkey nervous system following intranasal administration. Neuroscience 152(3):785–797
Thornton RD, Lane P, Borghaei RC, Pease EA, Caro J, Mochan E (2000) Interleukin 1 induces hypoxia-inducible factor 1 in human gingival and synovial fibroblasts. Biochem J 350(Pt 1):307–312
Tonin AM, Grings M, Busanello EN, Moura AP, Ferreira GC, Viegas CM, Fernandes CG, Schuck PF, Wajner M (2010) Long-chain 3-hydroxy fatty acids accumulating in LCHAD and MTP deficiencies induce oxidative stress in rat brain. Neurochem Int 56(8):930–936
Wang JZ, Wu Q, Smith A, Grundke-Iqbal I, Iqbal K (1998) Tau is phosphorylated by GSK-3 at several sites found in Alzheimer disease and its biological activity markedly inhibited only after it is prephosphorylated by A-kinase. FEBS Lett 436(1):28–34
Zhu X, Su B, Wang X, Smith MA, Perry G (2007) Causes of oxidative stress in Alzheimer disease. Cell Mol Life Sci 64(17):2202–2210
Acknowledgments
The authors would like to thank Katherine Faltesek and Jodi Henthorn for animal maintenance and help with dosing and behavior studies. Funding by private donations to the Alzheimer’s Research Center.
Conflict of interest
WHF and LRH are inventors on a patent owned by HealthPartners Research Foundation related to intranasal deferoxamine.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Fine, J.M., Baillargeon, A.M., Renner, D.B. et al. Intranasal deferoxamine improves performance in radial arm water maze, stabilizes HIF-1α, and phosphorylates GSK3β in P301L tau transgenic mice. Exp Brain Res 219, 381–390 (2012). https://doi.org/10.1007/s00221-012-3101-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00221-012-3101-0