
Abstract
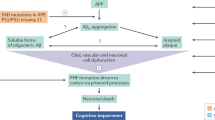
The amyloid cascade hypothesis is widely accepted as the centerpiece of Alzheimer disease (AD) pathogenesis. It proposes that abnormal production of beta amyloid protein (Abeta) is the cause of AD and that the neurotoxicity is due to Abeta itself or its oligomeric forms. We suggest that this, in itself, cannot be the cause of AD because demonstrating such toxicity requires micromolar concentrations of these Abeta forms, while their levels in brain are a million times lower in the picomolar range. AD probably results from the inflammatory response induced by extracellular Abeta deposits, which later become enhanced by aggregates of tau. The inflammatory response, which is driven by activated microglia, increases over time as the disease progresses. Disease-modifying therapeutic attempts to date have failed and may continue to do so as long as the central role of inflammation is not taken into account. Multiple epidemiological and animal model studies show that NSAIDs, the most widely used antiinflammatory agents, have a substantial sparing effect on AD. These studies provide a proof of concept regarding the anti-inflammatory approach to disease modification. Biomarker studies have indicated that early intervention may be necessary. They have established that disease onset occurs more than a decade before it becomes clinically evident. By combining biomarker and pathological data, it is possible to define six phases of disease development, each separated by about 5 years. Phase one can be identified by decreases in Abeta in the CSF, phase 2 by increases of tau in the CSF plus clear evidence of Abeta brain deposits by PET scanning, phase 3 by slight decreases in brain metabolic rate by PET-FDG scanning, phase 4 by slight decreases in brain volume by MRI scanning plus minimal cognitive impairment, phase 5 by increased scanning abnormalities plus clinical diagnosis of AD, and phase 6 by advanced AD requiring institutional care. Utilization of antiinflammatory agents early in the disease process remains an overlooked therapeutic opportunity. Such agents, while not preventative, have the advantage of being able to inhibit the consequences of both Abeta and tau aggregation. Since there is more than a decade between disease onset and cognitive decline, a window of opportunity exists to introduce truly effective disease-modifying regimens. Taking advantage of this opportunity is the challenge for the future.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Historical highlights
It would be difficult to overstate the urgency of finding solutions to the Alzheimer disease (AD) problem. Alzheimer Disease International, in their 2010 World Alzheimer Report, estimates that there are 35 million people suffering from this disorder at an annual cost of $604 billion (http://www.alz.co.uk). The 2013 United States Alzheimer’s Association Report estimates that there are 5.2 million cases in that country alone, with a new case being identified every 68 s. The cost in the US is estimated to be $ 203 billion, not including the unpaid care costs of patients, at $216 billion [156]. More ominously, both reports conclude that unless effective measures of prevention and treatment are discovered, the numbers of cases may increase two- to threefold by 2050.
Such a situation could hardly have been imagined by Alois Alzheimer when, in November 1907, he reported at a meeting in Tübingen “Ueber eine eigenartige Erkrankung der Hirnrinde” (On an unusual illness of the cerebral cortex). His two-page paper is worth reading by every AD researcher today. A translation into English is available [151]. The paper gives a succinct description of the clinical history in a case of dementia with onset at age 51 and the unique pathological findings of plaques and tangles. It concludes with a warning that researchers should not be classifying clinically undetermined cases by forcing them into categories of recognized illnesses. His report ends with this disappointing footnote: “Keine Diskussion”.
The failure to pay much attention to this hallmark paper continued for a further seven decades. It fell to Robert Katzman to ring the alarm bells. In a 1976 editorial he pointed out that AD and senile dementia were indistinguishable and ranked as the fourth or fifth most common cause of death in the US [83].
The decade from 1982 to 1992 was a groundbreaking era for unraveling the mysteries of AD. Up to then, age was the only known risk factor. Below age 65 AD was found to be rare, but the prevalence had been reported to double every 5 years thereafter, reaching about 25 % in the 80–85 year age bracket [79]. There was no explanation then, nor is there now, as to why age is so critical.
The most important development of that decade was Glenner and Wong’s [48] identification of a cerebrovascular amyloid protein which turned out to be Abeta. They then showed the same protein could be found in Down’s syndrome cases, correctly predicting that the genetic defect causing AD would be found on chromosome 21 [49].
The gene for amyloid precursor protein (APP) was soon cloned by three groups [52, 109, 133]. The full structure of Abeta was then apparent. It was concluded that an abundant presence of Abeta immunoreactive plaque lesions throughout the cortex and subcortical gray matter structures was typical of AD [70].
A point mutation in Abeta was then found that produced cerebral hemorrhages and heavy deposits of Abeta in blood vessels and brain [99, 158]. In further screening of chromosome 21, Goate et al. [50] found an AD causing point mutation in Abeta, providing a direct link between Abeta metabolism and AD.
The second pathological hallmark of AD is the presence of intracellular neurofibrillary tangles. The core protein was identified as tau [51, 54, 88] and its incorporation into tangles involved phosphorylation [69]. Since tangles were known to appear in diseases where plaques were absent, tau could be discounted as the fundamental cause [171]. Nevertheless tangled neurons die, so they are an extremely harmful consequence of AD pathogenesis even if they are a secondary phenomenon. The dead neurons leave behind their tau deposits as ghost tangles.
Braak and Braak [16] then published their classic paper on the neuropathological staging of AD in 1991. It was not based on Abeta deposits, but on the hierarchical development of neurofibrillary tangles (NFTs) and neuropil threads (NTs). This was a paradoxical finding, since genetic studies had already established that Abeta abnormalities induced tau abnormalities and not vice versa. In the Braak and Braak series of cases, amyloid deposits, even though variable, frequently appeared prior to tau deposits in pre-dementia stages, so there may not have been an incompatibility between genetic and early pathological findings.
How amyloid deposits spur tau aggregation is still a mystery. However, it is known that extracellular Abeta deposits induce inflammation. That inflammatory stimulation may enhance tau upregulation, which, in turn, may promote intracellular tau aggregation in vulnerable neurons. Tau aggregation can be self-sustaining. There is increasing evidence that such self-propagating tangle formation occurs following concussions in sports such as boxing and football. Dementia developing many years later is the end result [117, 119]. These data have profound implications for therapeutic strategies. If tau aggregation, once initiated, develops a life of its own independently of Abeta aggregation, then therapy based only upon limiting Abeta production or clearance will be of limited value.
Selkoe [143] first integrated the accumulated data into the amyloid cascade hypothesis of AD. This concept was similarly advanced by Hardy and Allsop [57]. The amyloid cascade hypothesis is nevertheless incomplete since it fails to take into proper account the neurotoxicity associated with the resulting neuroinflammation. Eikelenboom and Stam [37] first drew attention to an inflammatory process by reporting that AD senile plaques were positive for the opsonizing components of complement. Griffin et al. [53] noted that the inflammatory cytokine IL-1 was elevated in the brains of both AD and Down syndrome cases. When complement activation was further explored, a fundamental dichotomy was discovered. It was found that, while the opsonizing components decorated senile plaques, the terminal components, which form the membrane attack complex (MAC), were associated with dystrophic neurites [112, 113].
The reason why the complement system is so important in AD came with the discovery by Rogers et al. that Abeta and its N-terminal fragments bind to C1q. Thus Abeta deposits directly activate the complement system, initiating all the inflammatory consequences which occur in AD [134].
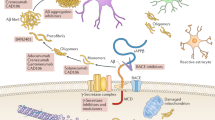
These results, as illustrated in Fig. 1, show the features of neuroinflammation in AD as revealed by immunohistochemistry. They demonstrate that deposits of Abeta initiate activation of the classical complement pathway in AD, causing opsonization of the plaques. Microglia then become activated and aggregate over the opsonized material. While such opsonization promotes their phagocytosis, continuation of the complement cascade to formation of the MAC results in self-damage by the MAC inserting itself into dystrophic neurites (Fig. 1c). These dystrophic neurites are characterized by intracellular tau aggregates (Fig. 1f). The MAC, which is intended to destroy invaders, instead turns on the host, destroying viable cells and their processes. As inflammation intensifies, so does the process of neuronal damage.

Alzheimer disease markers illustrating the inflammatory reaction process. a Alzheimer senile plaques immunostained with the Abeta antibody 6F3D (l0X). b Alzheimer senile plaques immunostained with C4d antibody (10X). Notice the similar staining indicating that the classical complement pathway has been activated on senile plaques, thus opsonizing them. c Alzheimer brain tissue immunostained with an antibody to the membrane attack complex of complement C5b-9 (20X). The immunostaining is completely different. Dystrophic neurites within and around a senile plaque are immunostained, indicating that the autodestructive MAC has inserted itself into viable, but damaged neuronal processes. d Shows Alzheimer disease senile plaques doubly immunostained for Abeta (brown) and microglia (CD 11b, blue, ×10). The microglia are activated and agglomerated over plaques but are not activated in the surround. e Alzheimer brain tissue doubly immunostained for C4d (brown) and microglia (CD 11b, blue, ×10). The immunostaining is similar to D, indicating that it is the opsonization of the plaque that has attracted and activated the microglia. f Alzheimer tissue doubly immunostained for phosphorylated tau (AT8, brown) and C5b-9 (blue, ×10). Phosphorylated tau deposits are noticeable as many brown neuropile threads, while some, colored blue, are under attack from the MAC. g Alzheimer brain tissue doubly immunostained for microglial cells (IbA1, blue) and Abeta (6F3D, brown, ×40). The senile plaque is being attacked by agglomerated and activated microglia in contrast to the surround. h Control brain tissue doubly immunostained for microglia (IbA1, blue) and Abeta (6F3D, brown, ×10). Only inactive microglia appear, as there are no brown Abeta deposits. i Control tissue (10X) doubly immunostained for microglia (IbA1, blue) and phosphorylated tau (AT8, brown). Again, only resting microglia appear as there are no phosphorylated tau deposits
Since self-damage from the MAC is prominent in AD, it is logical to suppose that those taking antiinflammatory agents should be relatively spared from the disease. Rheumatoid arthritis were a good population to study, since disease onset occurs earlier than in AD, and patients are known to be chronic users of antiinflammatory agents. An initial epidemiological study was carried out in which the prevalence of AD amongst rheumatoid arthritics over the age of 65 was compared with the prevalence of AD reported to occur in age-matched general populations. An apparent sixfold sparing was noted [115]. Numerous epidemiological studies have since been carried out with some variation in the results. Nevertheless, the overwhelming consensus is that antiinflammatory agents, particularly NSAIDs, if started early enough, have a sparing effect on AD. These studies are summarized in Table 1.
During the 1982–92 decade, these key discoveries provided insight into how successful treatments for AD could be developed. Yet, 20 years later, no disease-modifying therapies have reached the bedside. Failures have too often been blamed on the amyloid cascade hypothesis being in error, when the evidence indicates that inappropriate attempts to apply it may be at fault. As Schneider and Lahiri [141] observed “It seems—as if both large and small companies keep repeating each others’ mistakes, deriving their development program from an earlier, often unproven one”. A comprehensive list of clinical trials, either completed or underway as of 2011, is summarized by Mangialasche et al. [108].
Mutations causing Alzheimer disease
The central role of Abeta in AD pathology has been further strengthened by the discovery of additional autosomal dominant mutations in APP, either within Abeta itself, or close to the N- and C-terminal regions. These include the regions Abeta minus1-2, Abeta6-7, Abeta22-25, and Abeta43-46 [65]. It is unclear why these mutations cause disease. There are additional polymorphisms and mutations which enhance the risk of AD without directly causing it. These provide clues as to additional factors which may be involved in AD pathogenesis. By far the most important of these is apolipoprotein E (ApoE). One copy of ApoE4 has been shown to enhance age-specific AD prevalence by three- to fourfold and two alleles by about 15-fold [136]. In contrast, ApoE2 is mildly protective. ApoE is involved in cholesterol metabolism, indicating an important, but not fully understood role in Abeta metabolism. Significant, but much less important, polymorphisms in other genes have been identified as risk factors. These include complement receptor 1 (CR1) [30, 60] and the triggering receptor on myeloid cells (TREM 2) [55, 78], both of which are involved in responses to inflammation. The phosphatidylinositol binding clathrin assembly binding protein (PICALM) has also been identified as a risk factor [58].
Neurotoxicity associated with Abeta
Despite the evidence provided by AD causing mutations, there are significant weaknesses associated with the amyloid cascade hypothesis. More than 99 % of all AD cases fall into the wild-type category, also known as sporadic or late onset AD (LOAD), for which no explanation exists. Moreover, the hypothesis fails to explain how Abeta in any of its forms causes neuronal death. That is why an extension to an amyloid cascade-inflammatory hypothesis is appropriate. If neuronal death is to be arrested or prevented, therapeutic strategies based on blocking the resulting neuroinflammation may need to be followed.
The levels of Abeta in brain are very low, being in the pmol/g range. Xia et al. [176] found the in vivo levels for Abeta 1-40 to be 1.1 pmol/g in AD brain compared with 0.7 pmol/g tissue for controls. They found Abeta 1-42 levels in AD brain to be comparable to Abeta 1-40 levels, but Abeta 1-42 levels in controls to be much lower. Steinerman et al. [150] found soluble Abeta 1-40 in AD patients to be 2.7 pmol/g, while control levels were only 0.8 pmol/g. They reported Abeta 1-42 levels to be 14.6 pmol/g, while controls were only 0.8 pmol/g. Brody et al. [20] measured Abeta 1-x levels in vivo by dialysis of interstitial fluid in the white matter of brain injured patients. They found levels of 0.44 pmol/ml in patients with axonal brain injury but in other types of brain injury they were less than 0.22 pmol/g. While there is some variability in these results, they provide convincing evidence that soluble levels of Abeta species in human brain in vivo are in the very low pmol range.
Hypotheses have been put forward that Abeta itself, a toxic fraction such as Abeta 25-35, fibrillar Abeta, or oligomers of Abeta, are directly neurotoxic. In all cases, neurotoxicity has been in the micromolar range, more than a million times too high to lend credibility to the theory that they are neurotoxic in vivo. Yankner and colleagues [178] initially proposed that a fraction of Abeta was directly toxic to primary hippocampal cultured neurons. May et al. [111] then found the required concentration to produce significant toxicity was 10 μm, while Forloni et al. [42] found that repeated additions of 25–100 μm of Abeta 25-35 were required.
Next it was proposed that fibrillized Abeta or soluble oligomers of Abeta were the neurotoxins [104]. Numerous publications have since appeared in which the comparative neurotoxicity of Abeta, fibrillized Abeta, and oligomerized Abeta have been tested against primary cultured neurons [34, 85, 126, 167] with toxicity always in the micromolar range.
The logical conclusion is that it is deposits of Abeta that are the problem. They are neurotoxic due to the inflammatory reaction they stimulate. As Fig. 1 shows, each consolidated Abeta deposit is a focus of inflammation. While the numbers of such deposits in AD have never been counted, Bussiere et al. [24] have estimated their volume to be as high as one-third of the total volume in highly vulnerable brain regions such as the subiculum. Accordingly, therapeutic strategies that ignore the inflammation these deposits provoke are unlikely to be successful.
Enzymatic cleavage of APP
Since inability to handle an Abeta load must be a precipitating factor in AD, it is critical to have a clear understanding of the key enzymes involved. Those responsible for cleaving APP have been named α, β, and γ secretases. α-Secretase is actually a family of disintegrin and metalloprotease domain (ADAM) enzymes which cleave APP at the glutamine–lysine site within Abeta (Abeta 16-17). Such a cleavage prevents Abeta formation. ADAM 10 is considered to be the most important member of this family, but stimulation of its activity is considered a risky therapeutic strategy due to cleavage of other substrates that may stimulate tumorigenesis [129].
After APP is generated, it is transported to the cell surface. The N-terminal region traverses the external membrane and is cleaved by β-secretase or BACE-1 [65, 67, 162]. For Abeta to form, this enzyme must have preferred access to APP compared with α-secretase. Cleavage by BACE-1 occurs within the cellular membrane leaving the C-terminal region containing Abeta 1-29 still within the membrane [65].
Here it can be cleaved by the third enzyme, γ-secretase. It is a trans-membrane protease complex with multiple components embedded in the membrane. Knowledge concerning this complex is still not complete, but it is known to include several proteins. Initially, two genes linked to the catalytic subunits of γ-secretase were identified by genetic screening of familial AD cases. They were named presenilin-1 (PS-1), located on chromosome 14 [146], and presenilin-2 (PS-2), located on chromosome 1 [98]. Multiple mutations in these genes were found to cause autosomal dominant AD. By 2012, their numbers had reached 185 for PS-1 and 13 for PS-2 [26]. For reasons unknown, the cleavage process is variable. The main product is Abeta40, followed by Abeta42, but the cleavage also occurs at Abeta38 and other sites beyond Abeta42. The cleaved products are all released extracellularly.
In a series of experiments Haass and colleagues demonstrated the assembly route of γ-secretase. It commences in the endoplasmic reticulum where nicastrin, anterior pharynx-defective 1 (APH-1), and presenilin enhancer 2 (PEN2) are separately expressed and bind together in the Golgi apparatus. Trafficking then takes the complex to the transmembrane region and lysosomes where it becomes activated [80].
It would logically be presumed that the presenilin mutations would produce a gain of function, thus enhancing the production of Abeta. But at least one-third are nonsense mutations or substitutions which clearly produce a loss of function. If Abeta overproduction is causative for AD, how is it possible that reducing γ-secretase through one incompetent gene can result in autosomal dominant AD? If γ-secretase is a target for Abeta therapy, should its action be enhanced or inhibited? These and other questions regarding the viability of Abeta-degrading enzymes remain unanswered [81, 123, 138].
Abeta deposition can be prevented through cleavage by extracellular enzymes. Neprilysin is the major peptidase carrying out this function [38, 74]. Both the mRNA [179] and the protein [73] are down regulated in vulnerable areas of AD brain, suggesting this may be an initiating factor in the disease. Insulin-degrading enzyme and several minor peptidases can also cleave extracellular Abeta [123]. Up-regulation of Abeta-cleaving enzymes is an attractive strategy, but no practical methods for doing so have so far been developed.
Transgenic animals
A new dimension was added to AD research when Games et al. [45] published the first transgenic mouse (Tg) model that developed amyloid plaque pathology. They transfected mice with a minigene encoding the human disease-causing mutant APPV717F (PDAPP) behind a platelet-derived growth beta promoter. The mice developed plaque deposits at 6–9 months of age. This success was soon confirmed by the creation of new models [68] which involved transfecting mice with single, or multiple AD causing genetic mutations behind various promoters. Within the first decade, eight had been developed incorporating APP mutations, six incorporating PS-1 mutations, and three incorporating PS-2 mutations, plus various combinations [116]. The most aggressive so far is the B6SJL-5X Tg model, developed by Jackson Laboratories, which produces plaques starting at 6–8 weeks of age.
These mouse models have been utilized as the gold standard for developing new AD treatments. But it must be kept in mind that they do not fully replicate the pathology of AD. They are, therefore, incomplete models [87, 142]. The most obvious differences are the failure to develop neurofibrillary tangles and the failure to demonstrate significant neuronal loss. A more subtle difference is the relatively weak inflammatory response in the mouse models.
Abeta deposits in mouse models are readily soluble in SDS, while AD deposits can only be solubilized by such harsh treatment as hot formic acid. Human Abeta deposits undergo such post-translational modifications as N-terminal degradation, pyroglutamyl formation, and cross-linking, while mouse deposits do not [82, 91].
There is also a major difference in the response of the complement system. Mouse C1q poorly recognizes human Abeta deposits in Tg mice, so the overall response is weak compared with human AD. There are differences in human and mouse Abeta at residues 5, 10, and 13 which may play a role in this weak recognition [41, 169, 184].
There is no expression of the MAC in Tg mouse models [82, 142], while it plays a prominent part in AD pathogenesis [112, 113, 168, 181]. While stimulation of an inflammatory response in Tg mice might be helpful by promoting phagocytosis without inducing the self-damaging MAC, comparable stimulation in AD could be anticipated to produce highly damaging results [114].
The discovery by Schenk et al. [140] that Abeta deposits in PDAPP mice could be attenuated by vaccination with full-length Abeta opened up a revolutionary approach to treating AD. The beneficial effect of active immunization in mice, including behavioral improvement, was soon confirmed by others [77, 120, 175]. These findings were then extended to passive immunization, where peripherally administered Abeta antibodies were effective in assisting efflux of Abeta from the brains of mice, although a heavy Abeta load was less effective [32, 33].
Translational experiments
Clinical trials efforts based on immunization
The ease of plaque clearance by active and passive immunization of mice raised hopes that a similar strategy would be successful in treating human AD. They were quickly dampened when the first clinical trial of an Abeta vaccine named AN 1792 was halted because of dangerous side effects. These included death, stroke, and sterile meningoencephalitis in about 6 % of patients [127]. Nicoll et al. [124] then reported widespread encephalitis in an autopsy case although there was plaque clearance. But this clearance could have occurred due to macrophage invasion after remaining dystrophic neurites had been destroyed [5, 6]. Consistent with this interpretation, it was concluded in a follow-up of 80 patients, including post-mortem analysis of eight cases, that there was no slowing of disease progression and no clearance of tau deposits [66].
One explanation offered for the dangerous side effects in the AN 1792 trial was adjuvant-dependent modulation of Th-1 and Th2 responses. Novartis then developed a vaccine designated avagacestat, against gamma secretase. In an initial study involving 46 AD patients, no cases of meningoencephalitis were identified, suggesting that it had an acceptable safety profile [29]. But Bristol-Myers Squib terminated clinical trials in December 2012 due to lack of efficacy in AD. The fundamental problem is that an autoimmune disease is induced by active immunization, and it has not been demonstrated that this can be done without causing self-damage from the MAC. It has even been suggested that vaccination with DNA is a possibility and that immune-suppressive therapy should accompany active vaccination attempts [31].
One possibility for the future is to combat any such self-damage with a specific inhibitor of MAC formation, such as aurin tricarboxylic acid. In a pilot trial using 5X B6SJL-Tg mice it was found to be without toxicity and effective in preserving memory [95].
Passive immunization attempts have also been made, using antibodies against specific regions of Abeta. While such a strategy avoids the complication of inducing an autoimmune disease, it does require the relatively invasive and highly costly procedure of repeated infusions of antibody. Again, the theoretical basis for believing it could be effective in AD is tenuous, since the transgenic mouse models where they have been tested are not a reliable indicator of what will take place in humans. So far all attempts at passive immunization have failed.
Bapineuzumab is an example. It is a humanized monoclonal antibody directed at the N-terminal sequence of Abeta. Four clinical trials have been undertaken. After the failure of two of these trials, development by Johnson and Johnson and Pfizer was discontinued. Altogether 4,500 mild to moderate AD patients, randomized to the antibody or placebo, were tested over 18 months. Side effects included vasogenic edema at higher dose levels. In the CSF, no difference was noted in Abeta levels between test and control patients, although bapineuzumab patients showed a decrease in total tau and p-tau [15].
Yet another antibody is solanezumab, developed by Eli Lilly. This antibody is directed at the mid-region of Abeta and is thought to have an advantage over bapineuzumab by binding with much higher affinity and only to soluble Abeta. Its safety profile is higher [71] but, as with bapineuzumab, it failed in clinical trials [155]. In this case more than 2,000 patients did not meet the primary outcome measures of cognition and function. Nevertheless, Eli Lilly has plans to carry on with clinical trials.
Ponezumab, developed by Pfizer, is a humanized monoclonal antibody which binds specifically to the C-terminus of Abeta40. As with the other humanized antibodies, it showed efficacy in Tg mice [94]. But, after a phase 2 clinical trial, it was halted by Pfizer for unspecified reasons.
The most ambitious trial of all is that being undertaken by Genentech of the humanized monoclonal antibody against Abeta40 and Abeta42 named crenezumab [122]. The trial, projected to cost over $100 million, will test the effects on 300 individuals from Colombia known to have a PSEN-1 mutation.
There are continuing plans to develop a successful treatment of AD with active or passive immunization strategies. These are based on the belief that the science behind the strategy is sound, but failures have also been interpreted as being due to incorrectness of the amyloid cascade hypothesis itself [155]. They overlook the role of inflammation and the fact that immunization, whether active or passive, will stimulate further inflammation rather than compensate for its effects. According to this interpretation, future immunization trials are unlikely to succeed.
Clinical trial efforts based on inhibiting Abeta levels
Weggen et al. [170] found that certain NSAIDS such as flurbiprofen and ibuprofen modified production of Abeta42 in favor of Abeta1-38. These profens exist in S and R enantiomeric forms. Only the S form inhibits COX, but both forms equally affect Abeta production [121]. The authors proposed that the R-form might be an effective treatment for AD and showed that chronic administration was able to attenuate spatial learning deficits in Tg2576 mice [90]. The R form of flurbiprofen, produced as Flurizan, was tested in a trial involving 1,800 AD patients over an 18-month period by Myriad Genetics. It showed no positive effect and was abandoned. A major problem was that the concentrations required to produce the effect on Abeta42 production were more than 100-fold higher than those required to inhibit COX activity [86]. It is not surprising that Szekely et al. [154] found no advantage of Abeta-42-lowering NSAIDs, compared with non-lowering NSAIDs, for prevention of dementia in a data set of six pooled epidemiological studies.
Semagacestat is a γ-secretase inhibitor which was shown to lower plasma, CSF, and Abeta levels in animals [62]. A large international trial of semagacestat was halted before completion because those on the drug fared worse than the placebo patients, and, some, receiving the higher dose of 140 mg/day rather than 100 mg/day, had adverse effects such as skin cancer and infections [35]. The problems have been attributed, at least in part, to inhibition of notch cleavage by semagacestat. Notch is a highly conserved protein with a multitude of essential physiological functions. Inhibitors of γ-secretase cleavage will have dangerous side effects if they also inhibit notch cleavage [36, 80, 173]. Bristol Myers Squib is currently testing their BMS-708163 which is more selective for γ-secretase compared with notch. But, in view of the fact that many of the AD causative PS-1 mutations are nonsense mutations, even specific inhibitors of γ-secretase may have adverse effects.
β-Secretase (BACE 1) inhibitors do not have the systemic drawbacks of γ-secretase inhibitors, but there are already disappointing results. Eli Lilly discontinued development of its β-secretase candidate LY2886721, citing abnormal liver tests as the reason. Merck has initiated a 78-week phase II/III trial of its candidate MK 8931, and others are under development. Mullard [122] has summarized the latest β- and γ-secretase inhibitors, as well as monoclonal antibodies and vaccines that are under trial. There is optimism that β-secretase inhibitors will eventually be successful [47], but, to be clinically effective, an inhibitor will need to be highly selective, very potent, and administered in the early stages of the disease.
Clinical trials based on inhibiting Abeta aggregation
In theory, inhibiting Abeta aggregation is a more attractive strategy than inhibiting its production with enzyme inhibitors. There are no concerns about interfering with any normal functions of Abeta throughout the body, and no concerns about the inevitable side effects of enzyme inhibition. Unfortunately, the aggregation inhibitors that have been tested to date have failed to demonstrate efficacy.
Tramiprosate (homotaurine), an agent which binds to Abeta, thus preventing fibrillization, failed to show efficacy in a phase III trial of 1,052 patients [2]. Scyllo-inositol, a neutralizer of Abeta42 trimers, showed promise in animal experiments [106]. The drug was abandoned, following a trial involving 350 patients, after high doses resulted in infections and death, and low doses failed to show efficacy [137].
Our laboratory has developed a simple in vitro screening assay to replicate as closely as possible the events presumed to be occurring in AD brain [56]. The technique is to take slices of frozen post mortem AD brain tissue and incubate them with a fluorophore (HiLyte488) labeled Abeta42. The Abeta42 derivative is then visualized binding selectively to senile plaques in the AD brain. Candidate inhibitors are next added to determine their relative ability to inhibit the Abeta42 build-up. By this test, tramiprosate and scyllo-inositol were inactive, even at very high concentrations. Agents that showed some promise included 1,2,3,4,6-penta-O-galloyl-d-glucopyranose (PGG), epigallocatechin gallate, and resveratrol. Of potentially greater interest was the inhibitory effect of a number of food products. These included ginger, rhubarb, blueberries, cinnamon, and turmeric. The implication is that each of these products contains some Abeta42 aggregation inhibitors. Of course, such inhibitors would need to reach the brain to be effective in vivo. Many other possible inhibitors have been suggested, with some in various stages of development [172].
Antiinflammatory approaches
Antiinflammatory approaches are based on pathological evidence of neuroinflammation in AD, coupled with epidemiological evidence of AD sparing in individuals consuming NSAIDs. Following the initial report of a sparing of AD in rheumatoid arthritis patients [115], multiple epidemiological studies have been carried out (Table 1). These include incident, population-based, and case–control methodologies.
Seven incidence studies have been carried out (Table 1) [10, 44, 72, 103, 152, 165, 182]. The most comprehensive study to date is that of Vlad et al. [165]. They compared a population from the US Veterans Affairs Health Care System of 49,359 cases of AD with 196,850 controls. The base was large enough to permit analysis of length of any NSAID use and to compare the effects of various NSAIDs.
They found that for more than 5 years of use, the most effective preventative NSAIDs were indomethacin (OR 0.45), piroxicam (OR 0.54), and ibuprofen (0.56). Naproxen (OR 0.78) was considerably less effective. In contrast, COX-2 inhibitors substantially enhanced the risk of developing AD. The odds ratio for rofecoxib climbed from 0.90 for less than 1 year of use to 5.89 for more than 5 years of use. For celecoxib the odds ratio for less than 1 year of use was 0.94, climbing to 1.27 for more than 5 years of use. Such a negative result for preferential COX-2 inhibitors was to be expected given that brain is one of the few organs where COX-2 is constitutively expressed at high levels. It is particularly concentrated in hippocampal neurons, implying a significant role in synaptic transmission [84].
As for the traditional NSAIDs, those with a high COX1/COX 2 inhibitory ratio [43] appeared to give the most protection. Naproxen, utilized in two failed clinical trials, has a relatively unfavorable odds ratio of 0.78.
Int Veld et al. [72] had previously shown in the Rotterdam prospective study of 6,989 subjects that the relative risk of AD amongst NSAID users was 0.95 for 1 month use or less, 0.83 for 1–2 years of use, and 0.20 for longer than 2 years of use. Diclofenac, ibuprofen, and naproxen were the most widely used NSAIDs. Similarly, Zandi et al. [182], in the Cache County Utah study of 3,227 participants, found a risk ratio of 0.75 for less than 2 years of use, decreasing to 0.45 for longer than 2 years of use. Steward et al., in the Baltimore Longitudinal study of 1,686 participants [154], found the odds ratio to be 0.40 for more than 2 years use of NSAIDs but 0.65 for less than 2 years. Ibuprofen was the most commonly used NSAID. Lindsay et al., in the Canadian Study on Health and Aging, found an odds ratio of 0.65 for NSAID use, but the amount and type were not recorded [103].
In contrast to these studies, Arvenitakis et al. [10], in a much smaller population of 1,019 Catholics enrolled in the Religious Orders Study, found no apparent relationship between NSAID use and incident AD. No data were provided on duration of use. Fourier et al. [44] in the French Paquid cohort found no sparing of AD in 53 NSAID users compared with 1,199 non-users, but only for 1 year of use.
The six case–control studies shown in Table 1 are of smaller size [7, 13, 18, 19, 174, 180]. Where the duration of use is recorded, the results are in line with the incidence data in showing a lower odds ratio for longer NSAID use. Duration of use is not given in the three population-based prevalence studies [8, 21, 93].
The overall conclusion from these epidemiological studies is that significant sparing of AD occurs only when NSAIDs have been used for at least 2 years prior to onset of clinical symptoms. Selective COX-2 inhibitors are harmful, and those traditional NSAIDs with the highest COX-1/COX-2 inhibitory ratio are the most beneficial.
Tg mouse studies involving NSAIDs
Table 2 gives a list of 14 Tg mouse studies where the effects of NSAIDs have been tested [27, 61, 63, 76, 89, 101, 102, 118, 131, 153, 159–161, 177]. These studies have the advantage over human epidemiological studies in that the dose, duration, and NSAID involved are known. They suffer the disadvantage of being tested in a heterogeneous collection of six mouse models.
Traditional NSAIDs were reported to have an ameliorating effect on Tg pathology or behavior in 13 of the 14 studies where they were tested. Ibuprofen was the NSAID of choice in ten of these studies. The selective COX-2 inhibitors nimusulide and celecoxib were ineffective, and, in one study, celecoxib tripled the plaque load [89]. Although there are correlations between the results of mouse models and epidemiological studies, they must be regarded with caution.
Clinical trials involving NSAIDs
Table 3 lists the nine clinical trials so far conducted using COX-1 and COX-2 inhibiting agents. Initially, a small pilot clinical trial of indomethacin was carried out which appeared to show some benefit in mild to moderate AD [135], while another with diclofenac/misoprostol showed a non-significant trend towards protection [139]. Gastrointestinal problems led to a high dropout. These trials, which suffered from short duration, low power and high drop-out, could only be interpreted as indicating the need to undertake more definitive trials.
This was followed by four clinical trials with selective COX-2 inhibitors: one with nimesulide [4], one with celecoxib [147], and two with rofecoxib [3, 132]. These trials were undertaken in the absence of any epidemiological evidence of a protective effect of selective COX-2 inhibiting NSAIDs. In contrast, there was evidence of potential harm due to the high constitutive expression of COX-2 in cortical and hippocampal neurons and a presumed role in synaptic signaling [84]. Although the trials were adequately powered and therapeutic doses were utilized, there was no therapeutic effect. The harmful effects of COX-2 inhibition only appeared in later epidemiological [165] and animal model [89] studies. It must be concluded that selective COX-2 inhibitors do not have a future in the treatment of AD.
Naproxen has been utilized in two clinical trials. A multi-center, randomized, double blind, placebo-controlled parallel group trial with 1 year exposure was evaluated [3]. There were 111 placebo patients, 88 of whom completed the study, and 118 naproxen cases, 90 of whom completed the study. Naproxen was administered at a sub-therapeutic dose of 440 mg/day. In this trial, there was not a significant drop-out due to gastrointestinal problems, perhaps because of the relatively low dose of naproxen. No differences in deterioration were noted between the test and control groups.
The National Institute on Aging sponsored the Alzheimer disease Antiinflammatory Prevention Trial (ADAPT) to address the question of NSAID prevention of AD [1]. Cases were drawn from populations at high risk of developing dementia prior to the onset of symptoms. Celecoxib and naproxen were selected as representatives of COX-2 and COX-1 preferential NSAIDs, respectively. It is difficult to understand the selection of these agents, given their poor epidemiological, clinical, and Tg mouse record.
Enrollment commenced in 2001 but was terminated in December 2004 because of perceived cardiovascular side effects [1]. That is also difficult to understand, given that the differences in cardiovascular events between placebo, celecoxib and naproxen were marginal. While naproxen is considered safe enough to be an over-the-counter agent, it has nevertheless been reported that there is a small, but significantly increased risk of myocardial infarction in patients taking naproxen, rofecoxib, celecoxib, diclofenac, and ibuprofen [64]. In the long run, the benefits of preventing as deadly a disease as AD need to be weighed against the low risk of developing heart disease. Follow-ups have been reported on the ADAPT cohort, but they are difficult to interpret, given the termination of treatment followed by a period of non-treatment [96, 97].
The most relevant clinical trial involving COX inhibitors is that of Pasaqualletti et al. [128]. They administered ibuprofen (400 mg bid plus esomeprazole) or placebo to 132 AD cases (MMSE 15-26) over a 1-year period. There were 66 patients in each arm. The only treatment group not showing a decline was the ApoE4 carriers.
These results have raised questions as to whether the theory itself is wrong, or whether NSAIDs can act only as preventative agents if started very early in disease development. The protective effects of NSAIDs revealed in epidemiological studies all involved up to 2 years of use before clinical diagnosis. To be effective, it can be expected that NSAIDs will need to be administered very early in disease development and at doses which are appropriate to the advancing inflammation.
Biomarker studies
Biomarker studies have opened up a new era for Alzheimer research. So far there are three reliable biomarker types: CSF to determine Abeta and tau secretion levels; positron emission tomography (PET) to determine Abeta deposit level (Pittsburgh Compound B), and metabolic rate (FDG); and MRI to determine brain volume. Overall, they show that AD onset occurs a decade or more before clinical symptoms appear.
Sperling et al. [148] proposed a sequential model of the preclinical stages of AD focusing on the 10-year period before the emergence of cognitive decline. Their model proposed a series of steps but no time line was attached to them. Bateman et al. [12], in a landmark study of 128 participants, identified the same steps while including a time line. Concentrations of Abeta42 in the CSF declined 25 years before the expected clinical onset. Abeta deposits in the brain, as revealed by Pittsburgh compound B, were detected 15 years before clinical onset, along with increased tau in the CSF and an increase in brain atrophy. Impaired episodic memory was observed 10 years before the expected clinical diagnosis, and declines in MMSE and the CDRE rating scale were detected 5 years before the expected clinical diagnosis.
Comparable findings were reported by Villemagne et al. [163], who estimated that it took 19.2 years of linear Abeta accumulation, 4.2 years of hippocampal atrophy, and 3.3 years of memory impairment to reach AD clinical diagnostic levels. Seppala et al. [145] correlated CSF findings with cortical biopsy analysis and found that patients with Abeta cortical plaques in biopsy samples had lower Abeta42 CSF levels than those without plaques. Prestia et al. [130], following patients with MCI, found that conversions to dementia increased as patients went from Abeta42 in the CSF, to Abeta42 plus FDG-PET, to Abeta42 plus FDG-PET plus hippocampal atrophy.
Okonko et al. [125] found that abnormal Abeta42, but not tau alterations, were associated with increased risk of AD, and Buchhave et al. [23] reported similar results in a study of patients with MCI followed for a median of 9.2 years. They concluded that 90 % of patients with MCI and pathologic CSF biomarkers develop AD within 9–10 years, and that Abeta42 is fully decreased 5–10 years before appearance of dementia. Shaw et al. measured CSF biomarkers in mild AD and MCI patients compared with controls, as well as autopsy confirmed cases compared with controls. They concluded that Abeta42 plus total tau predicted conversion of MCI to AD and that Abeta42 was the most sensitive marker in the autopsy cases [144]. Similarly, Visser et al. [164], in the DESCRIPA study involving a prospective cohort, found that patients with an AD profile in their CSF were prone to advance from MCI to AD type dementia.
Zetterberg et al. [183] found that CSF biomarkers remained stable over a 2-year period even with cognitive decline. Mattson et al. also found CSF biomarkers to be stable over a 4-year period in MCI patients. They reached a similar conclusion that a CSF profile shifting toward normal would be useful in tracking disease-modifying drugs [110].
As for Abeta deposits in the brain, Vlassenko et al. found that scans with Pittsburgh compound B about 2 years apart in cognitively normal adults showed that those with elevated binding showed enhancement of binding, indicative of increased brain Abeta deposits. They concluded that a major growth in the Abeta burden occurs during a preclinical stage of AD [166]. Bruck et al. [22] compared the prognostic value of Pittsburgh compound B (PIB)-PET, FDG-PET and hippocampal volume MRI for their prognostic value in predicting conversion of MCI to AD. Of the 29 patients, 17 converted to AD after 2 years. They concluded that the PET methods were superior to hippocampal volume methods in predicting the conversion. Hatashita and Yamasaki [59] followed 68 MCI patients by PIB-PET and FDG-PET. Over 19 months, 44 % of patients converted to AD. They found PIB-PET as the most definitive marker of MCI. Jack et al. [75] have proposed a model in which Abeta biomarkers become abnormal first, with neurodegenerative biomarkers becoming abnormal later, correlating with clinical symptom severity.
Despite the biomarker data, Braak et al. [17] have proposed, on the basis that tau aggregations in subcortical and paleocortical nuclei precede Abeta aggregations, that such tau aggregations must induce A-beta aggregations. But, as previously noted, this predicts that at least some tau mutations promoting tau aggregation should cause AD, which they do not. A possible explanation is that the minimal sub-cortical tau aggregations reported by Braak et al. are not decisive in influencing Abeta depositions in the neocortex. It is these much more widespread neocortical Abeta depositions that correlate with biomarker studies indicative of disease onset.
Figure 2 illustrates this principle. It shows the relative development of Abeta and tau deposits in the temporal cortex of three cases from our Kinsmen Laboratory brain bank. They are at different phases of AD progression. Figure 2a, b are immunostained temporal cortex tissue from an 88-year-old lady who died suddenly at home. There are extensive Abeta deposits, but a total absence of tau deposits, indicative of very early disease onset. Figure 2c, d are from the temporal cortex of a 72-year-old male who was admitted to hospital with mild disorientation and a vaguely documented history of some decline in cognitive function. He died in hospital of undetermined causes. They show extensive Abeta deposits as well as some tau deposits, consistent with early cognitive decline. Figure 2e, f are from the temporal cortex of an 87-year-old lady who had suffered from Alzheimer disease for at least 8 years. They show extensive Abeta deposits, and, in addition, very extensive tau deposits, consistent with advanced AD.

Comparative temporal cortex immunostaining for Abeta (6F3D) and tau (tau 0024) at various stages of AD development. a Extensive Abeta deposits in the temporal cortex of an 88-year-old lady who died suddenly at home. b The same area shows a total absence of tau deposits. This corresponds to the expected pathology in phase 1 and early phase 2 of disease development. c Extensive Abeta deposits in the temporal cortex of a 72-year-old male who was admitted to hospital with mild disorientation and a vaguely documented history of some decline in cognitive function. He died in hospital of undetermined causes. d The development of early tangles in the same area of the temporal cortex. This corresponds to the expected pathology in phase 4 of disease development. e Extensive Abeta deposits in the temporal cortex of an 87-year-old lady who had suffered from Alzheimer disease for at least 8 years. f The same area with very extensive tangle development. This corresponds to the expected pathology in phase 6 of disease development
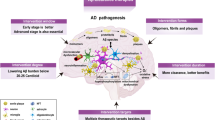
In summary, these data indicate that an extended window of opportunity exists for appropriate AD therapy to ameliorate, or even prevent disease development. They establish that disease onset can be detected by a reduction of Abeta42 in the CSF at least ten, and possibly 20 years prior to clinical diagnosis. This is followed by an increase in tau species in the CSF. Progression can be measured by PET and MRI scanning, all of which become abnormal years before AD can be diagnosed clinically.
Therapeutic implications
Table 4 is a theoretical construct of the phases of AD disease development and the implications for therapeutic intervention. Since the measurable prevalence of clinical AD commences at age 65 [79], the measurable prevalence for disease onset can be hypothesized to occur approximately 10 years earlier.
On this basis, the risk of Phase 1, marking disease onset, can be hypothesized to commence at age 55. Thereafter it will double every 5 years [79]. It is characterized by the commencement of Abeta deposition in brain with consequent decreases in the CSF. Therapeutic opportunities are at their highest. Any strategy which limits Abeta production, enhances its clearance, or prevents its aggregation should be disease modifying. Effectiveness of treatment should be measurable by CSF Abeta levels returning towards normal. The current problem is that there are no simple ways of making the diagnosis of wild-type AD at this very early stage.
Biomarker studies suggest that phase 2 sets in about 5 years later. Abeta deposits in the brain have built up to the level where they can be detected by PET scanning with Pittsburgh compound B. They have continued to show decreases in the CSF while tau levels in the CSF have now increased. Therapeutic opportunities have declined because cortical tau aggregation in brain has been induced. Ideally, a tau aggregation inhibitor should now be added to any therapeutic regimen, but so far none have been produced. Given that tau aggregation is the central pathogenic event in more than two dozen neurodegenerative diseases, developing such an agent is an urgent priority. Numerous proposals have been made [149], and at least two are in clinical trials [122]. The next best method of intervention is to administer antiinflammatory agents. NSAIDs are the most widely utilized, but they are only partial inhibitors and their effectiveness will wane as plaque and tangle levels in the brain increase.
In phase 3, typically in another 5 years, there is a slight metabolic decline that can be demonstrated by a reduction in FDG uptake by PET scanning. Presumably this is due to synaptic loss. Cortical tangle and thread development has increased to Braak stage III. Abeta and tau continue to be expressed at the same levels in the CSF. Pittsburgh compound B scanning intensity has increased. Therapeutic opportunities have further declined. Higher doses of therapeutic agents may be required to arrest disease development or to provide significant disease modification.
Phase 4, in another 5 years, represents disease advance to the level where cognitive deficits are detectable as mild cognitive impairment (MCI). Hippocampal atrophy has become evident by MRI scanning. The disease can no longer be totally prevented. Irreversible brain damage has begun to occur. Abeta and tau continue to be expressed at the same levels in the CSF but Braak stage IV pathology has been reached in brain. Nevertheless, substantial slowing, or perhaps even arrest, could be achieved by anti-Abeta agents combined with antiinflammatory agents such as NSAIDs.
Phase 5, in another 5 years, represents the level where AD can be diagnosed clinically. Neuronal damage and loss is evidenced by further PET-FDG metabolic decline and increasing brain volume loss by MRI. Abeta deposits in brain continue to accumulate, and Braak stage V levels of NFTs and NTs have been reached. Opportunities for therapeutic intervention have seriously declined.
Phase 6 defines the period of progressive clinical decline in all aspects of the disease. Cognitive deficits progress from mild to severe. Full-time care of patients becomes necessary. Costs associated with their care escalate. Therapeutic opportunities are minimal and yet it is in this phase that most clinical trials have been conducted.
To be a highly successful disease-modifying agent, a therapeutic candidate needs to deter Abeta42 aggregation, or intervene in its consequences, prior to the time when tau aggregation takes hold. For example, early application of direct inhibitors of Abeta aggregation, or inhibitors of β-secretase, or γ-secretase might succeed. Active immunization is highly questionable, given the dangers of inducing an autoimmune disorder and stimulating self-attack by complement. Passive immunization is also unlikely to succeed, due to the problems of blood–brain barrier penetration. Several failures have already been demonstrated.
NSAIDs offer the best current opportunity for disease modification since they have the capacity to combat the inflammation caused by both Abeta and tau aggregation. But they will need to be administered at higher doses and earlier in disease development than in previous trials. The effectiveness in each subject of such trials could theoretically be monitored by a shift in CSF Abeta and tau levels towards normal.
The difficulty in starting early treatment is in recognizing when disease onset has commenced. Obviously widespread screening of CSF is impractical. Screening for Abeta levels in blood is more practical but much less certain [157]. Screening saliva for Abeta42 levels is a possibility for the future. It has the advantage of being completely non-invasive. There is one report that Abeta42 saliva levels are increased above normal in AD cases [14]. We have been able to confirm this finding in a very small pilot study (unpublished data). Much more investigation will be required to determine if this is a possibility.
Dietary factors
The value of knowledge concerning dietary factors is that they are self-protective and dietary choices can be made long before the age of AD risk commences. The Mediterranean diet is the most widely studied. In a comprehensive review, Lourida et al. [105] reported that nine out of twelve epidemiological studies reported a lower risk of AD amongst those adhering to this diet. Laitenen et al. [92] found that a moderate intake of polyunsaturates decreased the risk of dementia, whereas a moderate intake of saturated fat may increase the risk.
Caffeine reduced plaque development and improved memory in two AD transgenic mouse studies [9, 28]. In the CAIDE study, Eskilinen and Kivipelto [40] found that drinking 3–5 cups of coffee per day at midlife was associated with a decreased risk of dementia/AD by about 65 % at late life. Maia and de Mendonca [107], in a case–control study, found that high caffeine intake was associated with a significantly lower risk of AD (odds ratio 0.40). However, Gelber et al. [46], in the Honolulu-Asia Aging study, found no overall effect of coffee, except for fewer lesions in men in the highest quartile of caffeine intake compared with the lowest.
Ballon et al. [11] analysed four studies for a specific association between AD and serum vitamin D levels. They concluded that those with levels of 50 nmol or higher were at lower risk. Vitamin D may have a mild protective effect.
As for antioxidants, Butterfield et al. [25] pointed out long ago that oxidative damage is characteristic of AD brain and that antioxidant therapy is a promising area of research. Engelhart et al. [39] reported that in the Rotterdam cohort of 5,395 participants, high intake of vitamin C and vitamin E was associated with a lower risk of AD (OR for each 0.82). Li et al. [100] did a meta-analysis of seven studies on the intake of vitamins E, C, and β-carotene, and the risk of AD. They concluded that each antioxidant had a mild protective effect with vitamin E being the most effective.
Summary
It is more than 20 years since the amyloid cascade hypothesis was first put forward. Yet it has not gained universal support because of the many unsuccessful attempts to develop disease-modifying treatments. One problem has been a failure to recognize that it is the inflammatory response to Abeta deposits, and later cortical tau aggregation, which drives the pathology. NSAIDs in general, and ibuprofen in particular, provide the best practical opportunity for therapeutic exploitation of this principle. Ibuprofen and related NSAIDs, which are widely used for the relatively trivial condition of osteoarthritis, have been shown to reduce the risk of AD in multiple epidemiological studies. They have also been shown to reduce the pathology in even more numerous transgenic mouse models of AD. Early administration may be the key to success. Such agents are relatively safe and highly available.
Recent biomarker studies indicate that disease onset commences as much as 10–15 years before clinical symptoms become manifest. If simple methods of preclinical diagnosis can be achieved, a 10-year window may exist for measures to be instituted that prevent, delay, or ameliorate the disease.
Search strategy and selection criteria. References for this Review were identified by searches of PubMed between 1969 and June, 2013. Among the search terms used were “Alzheimer disease and amyloid beta protein, tau, inflammation, complement, epidemiology, transgenic mice, clinical trials, and biomarkers”. There were no language restrictions. The final reference list, which could not include all possible contributions, was generated on the basis of highest relevance to the topics covered in this Review.
References
ADAPT Research Group (2006) Cardiovascular and cerebrovascular events in the randomized, controlled Alzheimer’s disease anti-inflammatory prevention trial (ADAPT). PLOS. doi:10.1371/journal.pctr.0010033
Aisen PS, Gauthier S, Ferris SH et al (2011) Tramiprosate in mild to moderate Alzheimer’s disease- a randomized, double blind, placebo-controlled, multi-center study (the Alphase study). Arch Med Sci 7:102–104
Aisen PS, Schafer KA, Grundman M et al (2003) Effects of rofecoxib or naproxen vs. placebo on Alzheimer’s disease progression: a randomized controlled trial. JAMA 289:2819–2826
Aisen PS, Schmeidler J, Pasinetti GM (2002) Randomized pilot study of nimesulide treatment in Alzheimer’s disease. Neurology 58:1050–1054
Akiyama H, McGeer PL (2004) Specificity of mechanisms for plaque removal after Aβ immunotherapy for Alzheimer disease. Nature Med 10:117–118
Akiyama H, Schwab C, Kondo H et al (1996) Granules in glial cells of patients with Alzheimer’s disease are immunopositive for C-terminal sequences of beta-amyloid protein. Neurosci Lett 206:169–172
Anon (1994) The Canadian Study of Health and Aging: risk factors for Alzheimer’s disease in Canada. Neurology 44: 2073–2080
Anthony JC, Breitner JC, Zandi PP et al (2000) Reduced prevalence of AD in users of NSAIDs and H2 receptor antagonists: the Cache County study. Neurology 54:2066–2071
Arendish GW, Mori T, Cao C et al (2009) Caffeine reverses cognitive impairment and decreases brain amyloid-beta levels in aged Alzheimer’s disease mice. J Alzheimer’s Dis 17:661–680
Arvenitakis Z, Grodstein F, Schneider JA et al (2008) Relation of NSAIDs to incident AD, change in cognitive function and AD pathology. Neurology 70:2219–2225
Ballon C, Griffith LE, Strifler L et al (2012) Vitamin D, cognition, and dementia: a systemic review and meta-analysis. Neurology 79:1397–1405
Bateman RJ, Xiong C, Benziger TLS et al (2012) Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. New Engl J Med 367(9):793–804
Beard CM, Waring SC, O’Brien PC et al (1988) Nonsteroidal anti-inflammatory drug use and Alzheimer’s disease: a case control study in Rochester, Minnesota, 1980 through 1984. Mayo Clin Proc 73:951–955
Bermajo-Parejo F, Antequera D, Vargas JA et al (2010) Saliva levels of Abeta1–42 as potential biomarker of Alzheimer’s disease: a pilot study. BMC Neurol 10:108
Blenow K, Zetterberg H, Rinne JO et al (2012) Effect of immunotherapy with bapineuzumab on cerebral biomarker levels in patients with mild to moderate Alzheimer disease. Arch Neurol 69:1002–1010
Braak H, Braak E (1991) Neuropathological staging of Alzheimer-related changes. Acta Neuropathol 82:239–259
Braak H, Zetterberg H, Del Tredici K, Blennow K (2013) Intraneuronal tau aggregation precedes diffuse plaque deposition, but amyloid-b changes occur before increases of tau in spinal fluid. Acta Neuropathol PMID:23756600
Breitner JC, Gau BA, Welsh KA et al (1994) Inverse association of anti-inflammatory treatments and Alzheimer’s disease: initial results of a co-twin control study. Neurology 44:227–232
Breitner JC, Welsh KA, Helms MJ et al (1995) Delayed onset of Alzheimer’s disease with nonsteroidal anti-inflammatory and histamine H2 blocking drugs. Neurobiol Aging 16:523–530
Brody DL, Magnoni S, Schwetye KE et al (2008) Amyloid-beta dynamics correlate with neurological status in the injured human brain. Science 321:1221–1224
Broe GA, Grayson DA, Creasy HM et al (2000) Anti-inflammatory drugs protect against Alzheimer disease at low doses. Arch Neurol 57:1586–1591
Bruck A, Virta JR, Koiyunen J et al (2013) [11C]PIB, {18F}FDG and MY imaging in patients with mild cognitive impairment. Eur J Nucl Med Imaging PMID:23801168
Buchhave P, Minthon L, Zetterberg H et al (2012) Cerebrospinal fluid levels of β-amyloid 1-42, but not of tau, are fully changed already 5 to 10 years before the onset of Alzheimer dementia. Arch Gen Psychiatry 69(1):98–106
Bussiere T, Friend N, Sadegh B et al (2002) Stereologic assessment of the total cortical volume occupied by amyloid deposits and its relationship with cognitive status in aging and Alzheimer’s disease. Neuroscience 112:75–81
Butterfield DA, Griffin S, Munch G, Pasinetti GM (2002) Amyloid beta-peptide and amyloid pathology are central to the oxidative stress and inflammatory cascades under which Alzheimer disease brain exists. J Alzheimers Dis 4(3):193–201
Cacquevel M, Aesschbach l, Houacine J, Fraering PC (2012) Alzheimer disease-linked mutations in presenilin-1 result in a drastic loss of activity in purified γ-secretase. PLOS One. doi:10.1371/journal.pone.0035133
Choi SH, Aid S, Caracciolo L et al (2013) Cyclooxygenase-1 inhibition reduces amyloid pathology and improves memory deficits in a mouse model of Alzheimer’s disease. J Neurochem 124:59–68
Chu YE, Chang WH, Black RM et al (2012) Crude caffeine reduces memory impairment and amyloid β(1-42) levels in an Alzheimer’s mouse model. Food Chem 135:2095–2102
Coric V, van Dyck CH, Salloway S et al (2012) Safety and tolerability of the γ-secretase inhibitor avagacestat in a phase 2 study of mild to moderate Alzheimer disease. Arch Neurol 69:1430–1440
Crehan H, Holton P, Wray S et al (2012) Complement receptor 1 (CR1) and Alzheimer’s disease. Immunobiology 217:244–250
Cribbs DH (2010) Abeta DNA vaccination for Alzheimer’s disease: focus on disease prevention. CNS Neurol Disord Drug Targets 9:207–216
Das P, Murphy MP, Younkin LH et al (2001) Reduced effectiveness of Aβ1-42 immunization in APP transgenic mice with significant amyloid deposition. Neurobiol Aging 22:721–727
DeMattos RB, Bales KR, Cummins DJ et al (2001) Peripheral anti-Abeta antibody alters CNS and plasma Abeta clearance and decreases brain Abeta burden in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci USA 98:8550–8555
Deshpande A, Mina E, Glabe C, Busciglio J (2006) Different conformations of amyloid beta induce neurotoxicity by distinct mechanisms in human cortical neurons. J Neurosci 26:6011–6018
Doody RS, Raman R, Farlow M et al (2013) A phase 3 trial of semagacestat for treatment of Alzheimer disease. New Engl J Med 369(4):341–350
D’Onofrio G, Panza F, Frisardi V et al (2012) Advances in the identification of γ-secretase inhibitors for the treatment of Alzheimer’s disease. Expert Opin Drug Discov 7:19–37
Eikelenboom P, Stam FC (1982) Immunoglobulins and complement factors in senile plaques in Alzheimer’s dementia. Acta Neuropathol 57:239–242
El-Amouri SS, Zhu H, Yu J et al (2008) Neprilysin: an enzyme candidate to slow the progression of Alzheimer’ disease. Am J Pathol 172:1342–1354
Engelhart MJ, Geerlings M, Ruitenberg A et al (2002) Dietary intake of antioxidants and risk of Alzheimer disease. JAMA 287:3223–3229
Eskilinen MH, Kivipelto M (2010) Caffeine as a protective factor in dementia and Alzheimer’s disease. J Alzheimer’s Dis 20(Suppl 1): S167–S174
Fonseca MI, Chu SH, Berci AM et al (2011) Contribution of complement activation pathways to neuropathology differs among mouse models of Alzheimer disease. J Neuroinflamm 8(1):4. doi:10:1186/1742-2094-8-4
Forloni G, Chiesa R, Smiroldo S et al (1993) Apoptosis mediated neurotoxicity induced by chronic application of beta amyloid fragment 25-35. NeuroReport 4:523–526
Frohlich JC (1997) A classification of NSAIDs according to the relative inhibition of cyclooxygenase isoenzymes. TIPS 18:3–34
Fourier A, Letenneur I, Begaud B, Dartigues JF (1996) Nonsteroidal anti-inflammatory drug use and cognitive function in the elderly: inconclusive results from a population-based cohort study. J Clin Epidemiol 49:1201
Games D, Adams D, Allesandrini R et al (1995) Alzheimer-type neuropathology in transgenic mice overexpressing V717F beta-amyloid precursor protein. Nature 373:523–527
Gelber RP, Petrovich H, Masaki KH et al (2011) Coffee intake in midlife and risk of dementia and its neuropathological correlates. J Alzheimer’s Dis 23:607–615
Ghosh AK, Brindisi M, Tang J (2012) Developing beta-secretase inhibitors for treatment of Alzheimer’s disease. J Neurochem 120(Suppl 1):71–83
Glenner GG, Wong CW (1984) Alzheimer’s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun 120:885–890
Glenner GG, Wong CW (1984) Alzheimer’s disease and Down’s syndrome: sharing of a unique cerebrovascular amyloid fibril protein. Biochem Biophys Res Commun 122:1131–1135
Goate A, Chartier-Harlin M-C, Mullan M et al (1991) Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature 391:704–706
Goedert M, Wischik RA, Crowther J et al (1988) Cloning and sequencing of the cDNA encoding a core protein of the paired helical filament of Alzheimer disease identification as the microtubule-associated protein tau. Proc Natl Acad Sci USA 85:4051–4055
Goldgaber D, Lerman MI, McBride OW et al (1987) Characterization and chromosomal localization of a cDNA encoding brain amyloid of Alzheimer’s disease. Science 235:877–880
Griffin WS, Stanley LC, Ling C et al (1989) Brain interleukin 1 and S-100 immunoreactivity are elevated in Down syndrome and Alzheimer disease. Proc Natl Acad Sci USA 86(19):7611–7615
Grundke-Iqbal I, Iqbal K, Tung YC et al (1986) Abnormal phosphorylation of the microtubule-associated protein tau in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci USA 83:44913–44917
Guerreiro R, Wojtas A, Bras J et al (2013) TREM 2 variants in Alzheimer’s disease. New Engl J Med 368:117–127
Guo J-P, Yu S, McGeer PL (2010) Simple in vitro assays to identify amyloid-beta aggregation blockers for Alzheimer’s disease therapy. J Alzheimers Dis 19:1359–1370
Hardy J, Allsop D (1991) Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol Sci 12:383–388
Harold D, Abraham R, Hollingworth P et al Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat Genet 41:1088–1093
Hatashita S, Yamasaki H (2009) Diagnosed mild cognitive impairment due to Alzheimer’s disease with PET biomarkers of beta amyloid and neuronal dysfunction. PloS One. doi: 10.1371/journal.pone/0066877
Hazrati LN, Cauwenberghe C, Brooks PL et al (2012) Genetic association of CR1 with Alzheimer’s disease: a tentative disease mechanism. Neurobiol Aging 33:2949
Heneka MT, Sastre M, Dumitrescu-Ozimek L et al (2005) Acute treatment with the PPAR gamma agonist pioglitazone and ibuprofen reduces glial inflammation and Abeta1-42 levels in APPV171 transgenic mice. Brain 128:1442–1453
Henley DB, May PC, Dean RA, Siemers ER (2009) Development of semegacestat (LY450129), a functional gamma-secretase inhibitor, for the treatment of Alzheimer’s disease. Expert Opin Pharmacother 10:1657–1664
Hillman A, Hahn S, Schilling S et al (2012) No improvement after chronic ibuprofen treatment in the 5XFAD mouse model of Alzheimer’s disease. Neurobiol Aging 33:833
Hippisley-Cox J, Coupland C (2006) Risk of myocardial infarction in patients taking cyclo-oxygenase-2 inhibitors or conventional non-steroidal anti-inflammatory drugs. BMJ 330:1–7
Holtzman D, Morris JC, Goate AM (2011) Alzheimer’s disease: the challenge of the second century. Sci Transl Med 3(77):1–17
Holmes C, Boche D, Wilkinson D et al (2008) Long term effects of Ab42 immunization in Alzheimer’s disease: follow-up of a randomized, placebo-controlled phase 1 trial. Lancet 372:216–223
Hong L, Koelsch G, Lin X et al (2000) Structure of the protease domain of memapsin 2 (beta-secretase) complexed with inhibitor. Science 290:150–153
Hsiao K, Chapman P, Nilsen S et al (1996) Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science 274:99–102
Ihara Y, Nukina N, Miura R, Ogawara M (1986) Phosphorylated tau protein is integrated into paired helical filaments in Alzheimer’s disease. J Biochem 99:1807–1810
Ikeda S, Allsop D, Glenner GG (1989) Morphology and distribution of plaque and related deposits in the brains of Alzheimer’s disease and control cases. An immunohistochemical study using amyloid beta-protein antibody. Lab Invest 60:113–122
Imbimbo BP, Ottonello S, Frisardi V et al (2012) Solanezumab for the treatment of mild-to-moderate Alzheimer’s disease. Expert Rev Clin Immunol 8:135–149
Int Veldt BA, Ruttenberg A, Hofman A et al (2001) Nonsteroidal anti-inflammatory drugs and the risk of Alzheimer’s disease. N Engl J Med 345:1515–1521
Iwata N, Higuchi M, Saido TC (2005) Metabolism of amyloid-beta peptide and Alzheimer’s disease. Pharmacol Ther 108:129–148
Iwata N, Tsubuki Y, Takaki K et al (2000) Identification of the major A beta (1-42) degrading catabolic pathway in brain parenchyma: suppression leads to biochemical and pathological deposition. Nature Med 6:143–150
Jack CR, Knopman DS, Jagust WJ et al (2010) Hypothetical model of dynamic biomarkers of the Alzheimer pathological cascade. Lancet Neurol 9:119–128
Jantzen PT, Connor KE, diCarlo G et al (2002) Microglial activation and beta amyloid deposit reduction caused by a nitric oxide-releasing nonsteroidal anti-inflammatory drug in amyloid precursor protein plus presenilin-1 transgenic mice. J Neurosci 22:2246–2254
Janus C, Pearson J, McLaurin J et al (2000) A beta-peptide immunization reduces behavioural impairment and plaques in a model of Alzheimer’s disease. Nature 408:970–982
Jonsson T, Stefansson H, Steinberg S et al (2013) Variant of TREM 2 associated with the risk of Alzheimer’s disease. New Engl J Med 368:107–116
Jorm AF, Korten AE, Henderson AS (1987) The prevalence of dementia: a quantitative integration of the literature. Acta Psychiatr Scand 76:465–470
Kaether C, Haass C, Steiner H (2006) Assembly, trafficking and function of gamma-secretase. Neurodegener Dis 3:275–283
Kakuda N, Akazawa K, Hatsuta H et al (2013) Suspected limited efficacy of γ-secretase inhibitors. Neurobiol Aging 34:1101–1104
Kalback W, Watson MD, Kokjohn TA et al (2002) APP transgenic mice TG2576 accumulate Abeta peptides that are distinct from the chemically modified and insoluble peptides deposited in Alzheimer’s disease senile plaques. Biochemistry 41:922–928
Katzman R (1976) The prevalence and malignancy of Alzheimer’s disease: a major killer. Arch Neurol 33:217–218
Kaufmann WE, Worley PF, Pegg J et al (1996) COX-2, a synaptically induced enzyme, is expressed by excitatory neurons at post synaptic sites in rat cerebral cortex. Proc Natl Acad Sci USA 93:2317–2321
Kim HJ, Chae SC, Lee DK et al (2003) Selective neuronal degeneration induced by soluble oligomeric amyloid beta protein. FASEB J 17:118–120
Klegeris A, Maguire J, McGeer PL (2004) S- but not R-enantiomers of flurbiprofen and ibuprofen reduce human microglial and THP-1 cell toxicity. J Neuroimmunol 152:73–77
Korczyn AD (2008) The amyloid cascade hypothesis. Alzheimer Dementia 4:176–178
Kosik KS, Joachim CL, Selkoe DJ (1986) Microtubule-associated protein tau is a major antigenic component of paired helical filaments in Alzheimer disease. Proc Natl Acad Sci USA 83:4044–4048
Kukar T, Murphy MP, Erikson JL et al (2005) Diverse compounds mimic Alzheimer disease-causing mutations by augmenting Aβ 42 production. Nature Med 11:545–550
Kukar T, Prescott S, Erikson JL et al (2007) Chronic administration of R-flurbiprofen attenuates learning impairments in transgenic amyloid precursor mice. BMC Neurosci 8:54
Kuo YM, Kokjohn TA, Beach TG et al (2001) Comparative analysis of amyloid-beta chemical structure and amyloid plaque morphology of transgenic mouse and Alzheimer’s disease brains. J Biol Chem 276:12991–12998
Laitenen MH, Ngandu T, Rovio S et al (2006) Fat intake at midlife and risk of dementia and Alzheimer’s disease: a population- based study. Dement Geriatr Cogn Disord 22:99–107
Landi F, Cesari M, Onder G et al (2003) Non-steroidal anti-inflammatory drug use and Alzheimer disease in community-dwelling elderly patients. Am J Geriatr Psychiat 11:179–185
Laporte SL, Bollini SS, Lanz TA et al (2012) Structural basis of C-terminal β-amyloid peptide binding by the antibody ponezumab for the treatment of Alzheimer’s disease. J Mol Biol 421:525–536
Lee M, Guo JP, Schwab C et al (2012) Selective inhibition of the membrane attack complex of complement by low molecular weight components of the aurin tricarboxylic acid synthetic complex. Neurobiol Aging 33:2237–2246
Leiketsos GG (2007) Naproxen and celecoxib do not prevent AD in early results from a randomized controlled trial. Neurology 68:1800–1808
Leoutsakos JM, Multhen BO, Breitner JC et al (2011) Effects of non-steroidal anti-inflammatory drug treatments on cognitive decline by phase of pre-clinical Alzheimer disease: findings from the randomized control Alzheimer’s disease anti-inflammatory prevention trial. Int J Geriatr Psychiatry. doi:10:1002/gps.2723
Levi-Lahad E, Wasco W, Poorkaj P et al (1995) Candidate gene for the chromosome 1 familial disease locus. Science 269:973–977
Levy E, Carman MD, Fernandez-Madrid IJ et al (1990) Mutation of the Alzheimer’s disease amyloid gene in hereditary cerebral hemorrhage, Dutch type. Science 248:1124–1126
Li FJ, Shen L, Ji HE (2012) Dietary intakes of vitamin E, vitamin C, and β-carotene and risk of Alzheimer’s disease: a meta-analysis. J Alzheimer’s Dis 31:253–258
Lim GP, Yang F, Chu T et al (2000) Ibuprofen suppresses plaque pathology and inflammation in a mouse model for Alzheimer’s disease. J Neurosci 20:5709–5714
Lim JP, Yang P, Chu T et al (2001) Ibuprofen effects on Alzheimer pathology and open field activity in APPsw transgenic mice. Neurobiol Aging 22:983–991
Lindsay J, Laurin D, Verrault R et al (2002) Risk factors for Alzheimer’s disease: a prospective analysis from the Canadian study of health and aging. Am J Epidemiol 158:445–453
Lorenzo A, Yankner BA (1994) Beta amyloid neurotoxicity requires fibril formation and is inhibited by congo red. Proc Natl Acad Sci USA 125:12243–12247
Lourida I, Soni M, Thompson-Coon J et al (2013) Mediterranean diet, cognitive function and dementia: a systemic review. Epidemiology 24:479–489
Ma K, Thomason LA, McLaurin J (2012) Scyllo-inositol, preclinical, and clinical data for Alzheimer disease. Adv Pharmacol 64:177–212
Maia L, de Mendonca A (2002) Does caffeine intake protect from Alzheimer’s disease? Eur J Neurol, pp 377–382
Mangialasche F, Solomon A, Winblad B et al (2011) Alzheimer’s disease: clinical trials and drug development. Lancet Neurol 9:702–716
Masters CL, Simms G, Weinman NA et al (1985) Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc Natl Acad Sci 82:4245–4249
Mattsson N, Portelius E, Rolstad S et al (2012) Longitudinal cerebrospinal fluid biomarkers over four years in mild cognitive impairment. J Alzheimer’s Dis 30:767–778
May PC, Gitter BD, Waters DC et al (1992) Beta amyloid peptide in vitro toxicity: lot to lot toxicity. Neurobiol Aging 13:605–607
McGeer PL, Akiyama H, Itagaki S, McGeer EG (1989) Activation of the complement pathway in brain tissue of Alzheimer patients. Neurosci Lett 107:341–346
McGeer PL, Akiyama H, Itagaki S, McGeer EG (1989) Immune response in Alzheimer’s disease. Can J Neurol Sci 16:516–527
McGeer PL, McGeer EG (2003) Is there a future for vaccination as a treatment for Alzheimer’s disease. Neurobiol Aging 24:391–395
McGeer PL, McGeer EG, Rogers J, Sibley J (1990) Anti-inflammatory drugs and Alzheimer disease. Lancet 335:107
McGowan E, Eriksen L, Hutton M (2006) A decade of modeling Alzheimer’s disease in transgenic mice. Trends Genet 22:281–289
McKee AC, Cantu RC, Nowinski CJ et al (2009) Chronic traumatic encephalopathy in athletes: progressive tauopathy after repetitive head injury. J Neuropathol Exp Neurol 68:2709–2735
McKee AC, Carreras I, Hossain L et al (2008) Ibuprofen reduces Abeta, hyperphosphorylated tau and memory deficits in Alzheimer mice. Brain Res 1207:225–236
McKee AC, Stein TD, Nowinski CJ et al (2013) The spectrum of disease in chronic traumatic encephalopathy. Brain 136:43–64
Morgan D, Diamond DM, Gottscall PE et al (2000) Abeta peptide vaccination prevents memory loss in an animal model of Alzheimer’s disease. Nature 408:982–985
Morihara T, Chu T, Ubeda O et al (2002) Selective inhibition of Aβ42 production by R-enantiomers. J Neurochem 83:1009–1012
Mullard A (2012) String of Alzheimer’s failures offset by upcoming prevention trials. Nature Rev Drug Discov 11:657–660
Naliivaeva NN, Beckett C, Belyaev ND, Turner AJ (2012) Are amyloid-degrading enzymes viable therapeutic targets in Alzheimer’s disease. J Neurochem 120(Suppl 1):167–185
Nicoll JAR, Wilkinson D, Holmes C et al (2003) Neuropathology of human Alzheimer disease after immunization with amyloid-β peptide: a case report. Nature Med 9:448–452
Okonko OC, Miellke MM, Griffith HR et al (2011) Cerebrospinal fluid profiles and prospective course and outcome in patients with amnestic mild cognitive impairment. Arch Neurol 68:113–119
Ono K, Condron MM, Teplow DB (2009) Structure-neurotoxicity relationships of amyloid beta-protein oligomers. Proc Natl Acad Sci USA 106:14745–14750
Orgogozo JM, Gilman S, Dartigues JF et al (2003) Subacute meningoencephalitis in a subset of patients with AD after Abeta42 immunization. Neurology 61:46–54
Pasaqualletti P, Bonomini C, Dal Forno G et al (2009) A randomized controlled study on the effects of ibuprofen on cognitive progression of Alzheimer’s disease. Aging Clin Exp Res 21:102–110
Postina R (2012) Activation of α-secretase cleavage. J Neurochem 120(Suppl 1):46–54
Prestia A, Caroll A, van der Flier WM et al (2013) Prediction of dementia in MCI patients based on core diagnostic markers for Alzheimer disease. Neurology 80(11):1048–1056
Quinn J, Montine T, Morrow J et al (2003) Inflammation and cerebral amyloidosis are disconnected in an animal model of Alzheimer disease. J Neuroimmunol 137:32–41
Reines SA, Block GA, Morris JC et al (2004) Rofecoxib: no effect on Alzheimer’s disease in a 1-year, randomized, blinded, controlled study. Neurology 62:66–71
Robakis NK, Wisniewski HM, Jenkins EC et al (1987) Chromosome 21q21 sublocalization of gene encoding beta-amyloid-peptide in cerebral vessels and neuritic (senile) plaques of people with Alzheimer disease and Down syndrome. Lancet 1:384–385
Rogers J, Cooper NR, Webster S et al (1992) Complement activation by beta-amyloid in Alzheimer’s disease. Proc Natl Acad Sci USA 89:10016–10020
Rogers J, Kirby LC, Hempelman SC et al (1993) Clinical trial of indomethacin in Alzheimer’s disease. Neurology 43:1609–1611
Roses AD (1993) Apolipoprotein E in neurology. Curr Opin Neurol 9(4):265–270
Salloway S, Sperling R, Karen R et al (2011) A phase 2 randomized trial of ELND005, scyllo-inositol, in mild to moderate Alzheimer disease. Neurology 77:1253–1262
Sambamurti K, Greig NH, Tadanobu U et al (2011) Targets for AD treatment: conflicting messages from γ-secretase inhibitors. J Neurochem 117:359–374
Scharf S, Mander A, Ugoni A et al (1999) A double blind, placebo-controlled trial of diclofenac/misoprostol in Alzheimer’s disease. Neurology 53:197–201
Schenk D, Barbour R, Dunn W et al (1999) Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature 400:173–177
Schneider LS, Lahiri DK (2008) The perils of Alzheimer’s drug development. Curr Alzheimer Res 6:77–78
Schwab C, Hosokawa M, McGeer PL (2004) Transgenic mice overexpressing amyloid beta protein are an incomplete model of Alzheimer disease. Exp Neurol 188:52–64
Selkoe DJ (1991) The molecular pathology of Alzheimer’s disease. Neuron 6:403–409
Shaw LM, Vanderstichele H, Knapik-Czaika M et al (2009) Cerebrospinal fluid biomarker signature in Alzheimer’s disease neuroimaging initiative subjects. Ann Neurol 65(4):403–413
Seppala TT, Nerg O, Koivisto AM et al (2012) CSF biomarkers for Alzheimer disease correlate with brain atrophy findings. Neurology 78(20):11568–11575
Sherrington R, Rogaev EI, Liang Y et al (1995) Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature 375:754–760
Soininen H, West C, Robbins J, Niculescu L (2007) Long term efficacy and safety of celecoxib in Alzheimer’s disease. Dementia Geriatr Cogn Disord 23:8–21
Sperling RA, Aisen PS, Beckett LA et al (2011) Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dementia, pp 280–292
Spillantini MG, Goedert M (2013) Tau pathology and neurodegeneration. Lancet Neurol 12:609–622
Steinerman JR, Irizarry M, Scarmeas N et al (2008) Distinct pools of beta-amyloid in Alzheimer’s disease-affected brain: a clinicopathologic study. Arch Neurol 65:906–912
Stelzman RA, Schnitlen HN, Murtagh FR (1995) An English translation of Alzheimer’s 1907 paper. Clin Anat 8:429–431
Stewart WF, Klawas C, Corrada M, Metter EJ (1997) Risk of Alzheimer disease and duration of NSAID use. Neurology 48:626–632
Sung S, Yang Y, Uryu K et al (2004) Modulation of nuclear factor κB activity by indomethacin influences Aβ levels but not Aβ precursor protein metabolism in a model of Alzheimer’s disease. Am J Pathol 165:2197–2206
Szekely CA, Green RC, Breitner JH (2008) No advantage of Aβ42-lowering NSAIDs for prevention of Alzheimer dementia in six pooled cohort studies. Neurology 70:2291–2298
Tayeb HO, Murray ED, Price BH, Tarazi FI (2013) Bapineuzamab and solanezumab for Alzheimer’s disease: is the amyloid cascade hypothesis still alive? Expert Opin Biol Ther 13:1075–1084
Theis M, Bleiler S (2013) Alzheimer’s disease facts and figures. Alzheimer’s Dem 9:208–245
Toledo JB, Shaw LM, Trojanowski JQ (2013) Plasma amyloid beta measurements: a desired but elusive Alzheimer’s disease marker. Alzheimer’s Res Ther 5:8
Van Broeckhoven C, Haan J, Bakker E et al (1990) Amyloid beta protein precursor gene and hereditary cerebral hemorrhage, Dutch type. Science 248:1120–1122
Van Dam D, Coen K, de Deyn PP (2010) Ibuprofen modifies cognitive disease progression in an Alzheimer’s mouse model. J Psychopharmacol 24:383–388
Van Groen T, Kadish I (2005) Transgenic model mice, effects of potential AD treatments on inflammation and pathology. Brain Res Rev 48:370–378
Van Groen T, Miettinen P, Kadish I (2011) Transgenic AD model mice, effects of potential anti-AD treatments on inflammation. J Alzheimer’s Dis 24:301–313
Vassar R, Bennett BD, Babu-Khan S et al (1999) Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science 286:735–741
Villemagne VL, Burnham S, Bourgeat P et al (2013) Amyloid β deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer’s disease: a prospective cohort study. Lancet Neurol. doi:10.1016/S1474-4442(13)70044-9
Visser PJ, Verhey F, Knol DL et al (2009) Prevalence and prognostic value of CSF markers of Alzheimer’s disease pathology in patients with subjective impairment or mild cognitive impairment in the DESCRIPA study: a prospective cohort study. Lancet Neurol 8:619–627
Vlad SC, Miller DR, Kowall NW, Felson DT (2008) Protective effects of NSAIDs on the development of Alzheimer disease. Neurology 70:1672–1677
Vlassenko AG, Mintun MA, Xiong C et al (2011) Amyloid-beta plaque growth in cognitively normal adults: longitudinal [11C] Pittsburgh compound B data. Ann Neurol 70(5):857–861
Wang PY, Chen JJ, Su HM (2010) Docosohexaenoic acid supplementation of primary hippocampal neurons attenuates the neurotoxicity induced by aggregated amyloid beta protein(42) and up-regulates cytoskeletal protein expression. J Nutr Biochem 31:345–350
Webster SD, Lue F, Brachova L et al (1997) Molecular and cellular characterization of the membrane attack complex C5b-9 in Alzheimer’s disease. Neurobiol Aging 18:415–421
Webster SD, Tenner AJ, Paulos TL et al (1999) The mouse C1q A-chain sequence alters beta-amyloid–induced complement activation. Neurobiol Aging 18:415–421
Weggen S, Erikson JL, Das P et al (2001) A subset of NSAIDs lower amyloidogenic Aβ42 independently of cyclooxygenase activity. Nature 414:212–216
Wisniewski HM, Iqbal K, Banchewr C et al (1989) Cytoskeletal pathology and the formation of beta-amyloid fibers in Alzheimer’s disease. Neurobiol Aging 10:409–414
Wisniewski T, Sadowski M (2008) Preventing β-amyloid fibrillization and deposition: β-sheet breakers and pathological chaperone inhibitors. BMC Neurosci 9(Suppl 2):55
Wolfe MS (2012) γ-Secretase as a target for Alzheimer’s disease. Adv Pharmacol 64:127–153
Wolfson C, Perrault A, Moride Y et al (2002) A case-control analysis of nonsteroidal anti-inflammatory drugs and Alzheimer disease: are they protective? Neuroepidemiology 21:81–86
Wyss-Coray T, Yan F, Lin AH et al (2009) Prominent neurodegeneration and increased plaque formation in complement-inhibited Alzheimer’s mice. Proc Natl Acad Sci USA, pp 10837–10842
Xia W, Yang T, Shankar G et al (2009) A specific enzyme-linked immunosorbent assay for measuring beta-amyloid protein oligomers in human plasma and brain tissue of patients with Alzheimer disease. Arch Neurol 66:190–199
Yan Q, Zhang J, Liu H et al (2003) Anti-inflammatory drug therapy alters beta-amyloid processing and deposition in an animal model of Alzheimer’s disease. J Neurosci 23:7504–7509
Yankner BA, Dawes LR, Fisher S et al (1989) Neurotoxicity of a fragment of the amyloid precursor associated with Alzheimer’s disease. Science 245:417–420
Yasojima K, McGeer EG, McGeer PL (2001) Relationship between beta amyloid peptide generation molecules and neprilysin in Alzheimer disease and normal brain. Brain Res 919:115–121
Yip AG, Green RC, Huyck M et al (2005) Non steroidal anti-inflammatory drug use and Alzheimer’s disease risk: the Mirage study. BMC Geriatr 5:2
Zanjani H, Finch CE, Kemper C et al (2005) Complement activation in very early Alzheimer disease. Alzheimer Dis Assoc Disord 19:55–56
Zandi PP, Anthony JC, Hayden KM et al (2002) Reduced incidence of AD with NSAID but not H2 receptor antagonists. The Cache County study. Neurology 59:880–886
Zetterberg H, Pedersen M, Lind K et al (2007) Intra-individual stability of CSF biomarkers for Alzheimer’s disease over two years. J Alzheimer’s Dis 12:255–260
Zhou J, Fonseca MI, Pisalyaput K, Tenner AJ (2008) Complement C3 and C4 expression in C1q sufficient and deficient mouse models of Alzheimer’s disease. J Neurochem 106:2080–2092
Acknowledgments
This work was supported by contributions from individual British Columbians. The authors declare they are shareholders in Aurin Biotech Inc., a startup biotechnology company with a mandate to develop complement inhibitors to treat human diseases.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
McGeer, P.L., McGeer, E.G. The amyloid cascade-inflammatory hypothesis of Alzheimer disease: implications for therapy. Acta Neuropathol 126, 479–497 (2013). https://doi.org/10.1007/s00401-013-1177-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-013-1177-7