
Abstract
Recent studies continue to find evidence linking Type 2 diabetes (T2D) with Alzheimer's disease (AD), the most common cause of dementia, a general term for memory loss and other cognitive abilities serious enough to interfere with daily life. Insulin resistance or dysfunction of insulin signaling is a universal feature of T2D, the main culprit for altered glucose metabolism and its interdependence on cell death pathways, forming the basis of linking T2D with AD as it may exacerbate Aβ accumulation, tau hyperphosphorylation and devastates glucose transportation, energy metabolism, hippocampal framework and promulgate inflammatory pathways. The current work demonstrates the basic mechanisms of the insulin resistance mediates dysregulation of bioenergetics and progress to AD as a mechanistic link between diabetes mellitus and AD. This work also aimed to provide a potential and feasible zone to succeed in the development of therapies in AD by enhanced hypometabolism and altered insulin signaling.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Alzheimer's is the most common cause of dementia, a general term for memory loss and other cognitive abilities serious enough to interfere with daily life and is recognized as fifth foremost reason of decease for those aged 65 and older [1]. It has no current cure, but treatments for symptoms are available and research continues. Neurotransmitter deficits, degenerated neurons, synaptic dysfunction, extracellular buildup of Amyloid-beta (Aβ) and intracellular neurofibrillary tangles (NFT) are the major crude disfigurements present in AD [2]. To produce Aβ peptides of different lengths such as Aβ38, Aβ40, and Aβ42 due to the active enzymatic component of the gamma-secretase (γ-secretase) complex, presenilins (PS), cleaves amyloid precursor protein (APP) at several sites within the membrane. Insulin resistance is a common phenomenon, closely associated with obesity, and defined as the inability of target tissues to respond normally to insulin. Insulin resistance typically precedes the onset of type 2 diabetes (T2D) by several years. T2D is a risk factor for dementia and for AD, the most common type of dementia. Some epidemiological studies suggest that insulin resistance increases the risk for dementia and AD, even in non-diabetic populations. In vitro and animal studies indicate that insulin resistance can contribute to the pathogenesis of AD through multiple different pathways. Endocrine abnormalities especially diabetes is so common in AD that also regarded as a type of diabetes. Diabetes having an influence on memory processing (recognition and retrieval), morphology of brain (brain atrophy) and synaptic communication is a well demonstrated hazardous aspect that influences pathology of AD [3]. Type 1 diabetes is mainly observed in children and young adults while T2D is more common among adults and is responsible for 90% of the incidences globally [4]. Recent evidence indicates that AD is a brain-specific form of diabetes [5]. T2D is the commonest and imperative co-morbidity of AD, escalating the risk of AD many folds [6]. T2D is exemplified by hyperglycemia, hyperinsulinemia, insulin resistance, metabolic dysfunctions and chronic inflammation and notably all these features are shared by AD [6, 7]. Other key factors as dysfunctional protein O-GlcNAcylation, mitochondrial disparities, oxidative stress, distorted energy metabolism and cholesterol modifications that are link AD [8] and T2D [9, 10]. Hyperglycaemia affects the transduction of insulin signaling which could link to damages tissues and organs, leading to glycation reaction of antioxidant enzymes, and reduction in the activity of SOD and other enzymes [11]. In addition, the hyperinsulinaemia impairment of insulin signaling and insulin resistance are the vital factors that make the sense of keeping insulin at the center stage of both pathologies irrespective of genotype [12]. Insulin also plays an important role in cognition processes and the insulin also has the high intensity in the regions responsible for memory formation and consolidation like hippocampus [13]. Many recent studies have indicated that impaired hippocampus insulin signaling impairs the memory and other executive functions, attributing to the decline of insulin signaling and concurrent development of insulin resistance [14,15,16]. This deliberation advocates a strong link between hyperinsulinemia/insulin resistance and the resultant pathologies like T2D and AD [17]. Peripheral insulin resistance leads to decrease insulin signaling in CNS, followed by alteration in brain metabolism. Increased Aβ toxicity, Tau hyperphosphorylation, oxidative stress and neuroinflammation are attributed to central insulin resistance, which leads to neurodegeneration (Fig. 1). The work provides the basic mechanisms of the insulin resistance mediates dysregulation of bioenergetics and progress to AD as a mechanistic link between diabetes mellitus and AD, providing a potential and feasible zone to succeed in the development of therapies in AD by enhanced hypometabolism and altered insulin signaling. Based on the concept that AD may represent a brain-specific form of diabetes mellitus, the term “type-3 diabetes” indicating AD was made [18,19,20].

Schematic representation of molecular pathways linking insulin resistance and Alzheimer disease
Insulin Signaling in the Central Nervous System
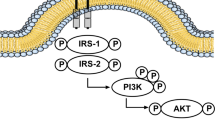
Insulin, hormone that regulates glucose levels in the blood and that is produced by the beta cells of the islets of Langerhans in the pancreas and consists of two polypeptide chains, A (21 amino acids) and B (30 amino acids) connected by disulfide linkages. Insulin initiates its action by binding to implanted glycoprotein receptor formed by two α and two β-subunits [17]. Insulin binding to α-subunit of the receptor fabricate confirmative alterations that lead to its activation and autophosphorylation of several Tyr residues at β-subunit cytosolic region [21, 22]. Autophosphorylated remnants are then acknowledged by insulin receptor substrate (IRS), out of which IRS-1 and IRS-2 are the two major players and the common intermediaries in insulin signal propagation. IRS is ideal and suitable for the configuration of molecular complexes which mediates intracellular signaling pathways. Insulin and Insulin like growth factors (IGF-1) connect to tyrosine kinase receptors, the insulin receptor (IR) and IGF-1. Insulin binding is highest in the olfactory bulb; cerebral cortex and hippocampus besides that insulin receptors are also expressive on endothelial cells of blood brain barrier and are responsible for transport of insulin and IGF-1 through blood–brain barrier (BBB) into CNS [23]. While the exact mechanism of how insulin gets into the brain still remains controversial, insulin circulating in the blood can cross the BBB through a receptor mediated active transport system [23]. This pathway is consistent with studies showing that insulin levels in the cerebrospinal fluid (CSF) increase proportionally with blood insulin after peripheral insulin infusion [21,22,23]. However, the amount of insulin produced in the brain and whether this pool of insulin is physiologically relevant still remains elusive. It is possible that both the centrally and peripherally derived pools of insulin are important for signaling in the brain.
Insulin and IGF-1 are conferred with functions which are important for neuronal survival and maintenance of CNS integrity. Insulin receptors and insulin signaling affect glucose homeostasis, neuronal integrity, cognition, through influencing several receptor mediated mechanisms including Calcium influx, neurotransmitter build up and synaptic connections, apoptosis and neurogenesis [23]. Insulin also regulate expression and levels of gamma aminobutyric acid (GABA), N-methyl-D-aspartate (NMDA) and α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) mediated mechanisms which have strong influence over long-term potentiation (LTP) and long-term depression (LTD). Furthermore, insulin is crucially involved in expansion and preservation of excitatory synapses [24] and dendritic spine formation through activation of phosphatidylinositol-3-kinase (PI3K)/Akt/mammalian target of rapamycin (mTOR) and Ras-related pathways [13, 25] which are integral to insulin signaling [26]. Insulin also influence cell survival by modulating apoptotic pathways and the intermediates involved in apoptotic cascade [27, 28]. Thus insulin through influencing any of these pathways alter the neuronal performance and integrity which may ends up in the defects in learning, memory and other features of AD. Previous studies indicated that brain insulin was equally reduced in AD patients and age-matched controls, indicating that reductions in brain insulin are likely a result of age, not AD [29]. Ultimately, a greater understanding of insulin in the brain relative to the severity of AD and age-matched controls needs to be obtained in order to fully comprehend insulin’s function in healthy and diseased brains. Thus, reduced insulin levels in the CNS can lead to reduced levels of antiamylogenic proteins, and both the overproduction and an impaired clearance of Aβ (Fig. 2).

Some potential insulin pathways and insulin mediated dysfunctional status as the common mediators between T2D and AD
Role of Insulin Resistance in Alzheimer's Disease
Insulin resistance in AD and diabetes can lead to hyperinsulinemia, thereby, saturating insulin-degrading enzyme (IDE) for insulin and Aβ degradation. Recently, many studies indicated that the incidence of AD is higher in T2D patients and obese individuals, implying common mechanisms driving these disorders [12, 30, 31]. Insulin resistance could be a main feature which shared among diabetes, obesity, and AD [32]. The neuronal glucose uptake may not depend on insulin totally, thus the concept of insulin resistance in brain is more related to impaired insulin signaling pathways. The malfunction of insulin signaling pathways and resultant state of hypometabolism observed are considering among factors in altered bioenergetics that connects AD and T2D [6]. The insulin resistant state could lead to compromised neuron functions and cognitive skills accompanied by extreme rise of insulin and relatively declined insulin activity in the periphery as an important predictor of T2D [33, 34]. Consequently, this leads to development of neuritic plaques, hippocampal atrophy, cognitive performance and lower cerebrocortical glucose metabolism which closely may correlate with the memory impairments [7]. A previous study revealed that increased p-Ser312IRS1 manifested in prodromal AD patients that sustained these alterations a decade then, as AD patients [35], suggesting that insulin resistance in AD develops years before clinical manifestations and that neural-derived exosomes carries potential for early AD diagnosis. Due to lack of insulin response, down regulation of insulin receptor, reduced binding of insulin receptors or faulty activation of the insulin signaling cascade that cause the defective brain insulin signaling in AD and T2D. The major consequence of this altered cascade is the decreased neuronal glucose uptake that is manifested as impaired neuroplasticity, neurotransmitter deficits, collapse of bioenergetics mechanism and initiation of fateful inflammatory cascade. Overall the consequences of impaired insulin signaling are attributed to impaired metabolism in brain that may lead to brain malfunction, providing possible explanations for the connection between diabetes, obesity, and AD [14].
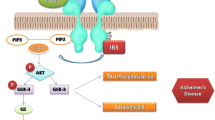
Insulin resistance or dysfunction of insulin signaling is a universal feature of T2D, due to altered glucose metabolism and its interdependence on cell death pathways form the basis of linking T2D with AD. Dysfunctional insulin pathways and resistance of insulin is a status of receptor dysfunction, altered receptor expression, deviations in receptor binding and malfunctioned events in phosphorylation chain or the altered activities related to kinases involved in phosphorylation. At the molecular level, a cell senses insulin through insulin receptors, with the signal propagating through a signaling cascade collectively known as PI3K/Akt/mTOR signaling pathway. Recent studies suggested that the pathway operates as a bistable switch under physiologic conditions for certain types of cells, and insulin response may well be a threshold phenomenon [16, 36, 37]. The pathway's sensitivity to insulin may be blunted by many factors such as free fatty acids, causing insulin resistance (Figs. 3, 4). It also is based on the finding that insulin resistance may be reversed rapidly by exposing cells to mitochondrial uncouples, electron transport chain inhibitors, or mitochondrial superoxide dismutase mimetics [38, 39].

Insulin actions in the central nervous system (CNS), and proposed concequences of insulin resistance in the CNS

Potential molecular mechanisms underlying defective insulin signaling in Alzheimer’s disease (AD). Liraglutide an GLP1-R agonist is able to restore insulin signaling and is a potential therapy for AD, TNFα tumor necrosis factor α, IKKβ IκB kinase, NFκB nuclear factor κB, PKR protein kinase RNA-activated, JNK Janus kinase, IRS-1 insulin receptor substrate, PI3K phosphoinositide 3-kinas, PIP3 phosphatidylinositol (3, 4, 5)-triphosphate, PIP2 phosphatidylinositol-4 5-bisphosphate, AKT protein kinase B, GSK3β glycogen synthase kinase 3β, eIF2α eukaryotic translation initiation factor α, PTEN phosphatase and tensin homolog, GLP1R glucagon-like peptide-1 receptor, p phosphorylation
Interestingly, impaired insulin signaling is present in several transgenic and nontransgenic mouse models of AD. Some previous clinical studies have reported that AD patients could have glucose intolerance, suggesting a bidirectional relationship between the two conditions [40, 41]. There were a reduced levels of IRS-1 associated to the membrane of hippocampal extracts [42] and a decreased activation of IRS-1 and PI3K in the hippocampus and cortex that were observed by 10 months of age [43]. Markers of insulin resistance were also reported in the hypothalamus of APP/PS1 mice [44] since the IRS-1 phosphorylated in serine 616 in the hippocampus at 9 months of age was higher than that of control [45], and increased levels of IRS-1 phosphorylated in serine 636 and 312 in the frontal cortex at 13 months [46] also demonstrated. In combination with peripheral insulin resistance, there were an increased inhibitory phosphorylation of IRS-1 in serine 612 in the hippocampus of 5-month-old tg2576 mice was also reported [43]. Remarkably, the central infusion of Aβ oligomers lead to peripheral insulin resistance, which was further observed in the APP/PS1 and in the 3xTgAD mouse models of AD [47]. Table 1 provides the main mechanisms linking brain insulin/ insulin-like growth factor resistance to AD pathology [41]. To confirm these concepts, further evidence is still required to investigate the mechanisms whereby AD affects the diabetic phenotype.
Hypometabolism in Alzheimer’s Disease
Hypometabolism, characterized by decreased brain glucose consumption, is indispensable for neuronal survival, synaptic connections, maintenance of integrity of BBB. Major preconditioning risk factors such as cardiovascular dysfunction, diabetes, metabolic syndrome, traumatic brain injury, and stroke are shared by the sporadic AD [48,49,50,51]. The quantitative evaluation of reduced glucose metabolism in the AD brain was first performed by arterio-venous difference studies 20–30 years ago [52,53,54]. The continuous and optimum presence of glucose as energy substitute is highly desired in CNS which ultimately depends upon the transportation of glucose across BBB. The reduction in glucose supply may end with the state of reduced energy metabolism which may lead to neuropathological consequences like AD. Several previous studies revealed that glucose hypometabolism is present well before any measurable cognitive dysfunction or AD-specific pathological alterations and therefore represents the early presymptomatic signature of AD development [55,56,57,58]. Also, epidemiological findings strongly verify the fact that affected glucose-energy metabolism and resultant hypometabolic state multiplies the risk of developing AD [59, 60]. This abnormal glucose metabolism seems the main contributor towards the synaptic dysfunction and loss observed in the brains of AD patients [61]. Therefore, altered brain metabolism in T2D is measurable after the onset of dementia symptoms which may be strongly linked with insulin resistance or reduced insulin actions in the brain [14]. The state of insulin resistance, diabetes and metabolic abnormalities could share large features of AD thus nowadays AD is categorically classified as metabolic-cognitive syndrome [62,63,64]. Probably, the insulin resistance may directly lead to accumulation of senile plaques and hyperphosphorylation of tau in AD via inflammatory factors, mitochondrial dysfunction, and oxidative stress, apoptosis, excitotoxicity and overactivation of protein kinases [55]. In addition, compared to mice that ate a normal diet, mice that ate the high-fat, high-sugar diet had significantly higher markers of inflammation, insulin resistance, and cellular stress in area of the hippocampus believed to be involved in AD progression [65]. Nutrition can have a profound effect on brain function, unhealthy diets high in fat and sugar can cause hypothalamic inflammation, which could be linked with the diseases.
In many previous studies focusing on insulin resistance in the AD brain, the important role of insulin in glucose uptake is reviewed [50, 55]. Insulin roles the major determinant for entry of glucoses into brain and the process is facilitated by presence and activated state of glucose transporters, this process of glucose entry and transportation through transporters is hampered by metabolic deformities including insulin resistance [66]. The insulin resistant state and deviations in insulin signaling cascade affect glucose levels through reduced transportthrough decreased glucose transporter 1 (GLUT1) and -3 levels which has been detected in AD brains [67]. The diminished glucose transport directly impacts hyperphosphorylation of tau protein, density of neurofibrillary tangles (NFTs) and hippocampal atrophy [68] thus proving a substantial link between insulin signaling, diminished glucose transport and pathological changes in AD. This altered permeability leads to decreased brain insulin levels and decreased insulin-facilitated neural and glial activity. Conversely, T2D also directly damages BBB, and increase the permeability to a variety of substances [69, 70] and this unchecked entry exit process may lead to infiltration of undesired and toxic substances into brain.
Among factors leading to energy deficiency and oxidative stress, neuro-inflammation, and insulin resistance are characterized by common brain pathologies [71, 72]. Inflammation and provoked inflammatory cascade could be an event in the progression of insulin resistance which are fundamental to pathologies of T2D and AD [19, 73]. The scarcity of glucose and a state of hypometaboilsm created by insulin resistance in CNS is sensed by the glial cells which triggers higher ketone body production, activation of NFκB pathway and reticence or diminished activity of AMP-activated protein kinase [74]. Furthermore, chronic inflammation exacerbates insulin and IGF1 resistance significance contribute to AD [50, 75] through provoking proinflammatory mediators, including tumor necrosis factor-α (TNF), IL-6 and IL-1β [74, 76,77,78]. These inflammatory mediators are also involved in macrophage activation/infiltration into adipose tissue and are also involved in pathophysiology of metabolic disorders [77, 78]. Insulin resistance leads to aberrant activation of c-Jun N-terminal kinase (JNK) which in turn activates inflammatory/stress signaling networks, endoplasmic stress signals, the stress kinases IKK and double-stranded RNA dependent protein kinase. These pathways are in dominance to play a role in hippocampal dysfunction in AD [79]. Insulin and insulin signaling have a strong influence over cellular bioenergetics and impairment of glucose metabolism or insulin signaling directly affect the cell survival. Recent studies of preclinical and clinical on the efficacy of anti-diabetic, insulin-sensitizing drugs on multiple aspects of AD pathology [14, 80] in human patients and animal model that were summarized in Table 2.
Therapeutic Approaches to Insulin Resistance in Alzheimer’s Disease
Diabetes and AD have traditionally been thought to be independent disorders. However, the results of recent epidemiological and basic science investigation have suggested possible associations and some common pathophysiological mechanisms. Insulin resistance is well known as an essential feature of T2D, therefore treatment strategies for T2D, particularly those aimed at improving insulin sensitivity, may also benefit those patients at risk for AD at the early stages. Due to the overlapping yet distinct pathological features among diabetes, insulin resistance and cognitive decline, multitargeted drug therapies along with lifestyle interventions are also explored [81] from the perspective of research in the pharmaceutical industry including nutraceuticals, antioxidant activity, polyphenols [82], omega-3 fatty acids as well as ketogenic diet, lifestyle support and brain-gut connections.
Among nutraceuticals produce curcumin as a brain permeable compound with the ability to target abnormal protein aggregates [83]. Curcumin may also thwart “proapoptotic signaling pathways in primaryhippocampal neuron cultures”. Forthcoming research inimproving bioavailability of curcumin may have the potential to lift the veil on promising naturalsubstances for AD patients. Previous research has also shown the benefit of metformin in mice when coupled withcurcumin and piperine supplementation, particularly regarding enhanced insulin sensitivity, signaling, and better systemic glucose tolerance [83]. However, the anti-inflammatory benefits of fruits and vegetables have been widely publicized for decades, particularly regarding antioxidant action in reducing inflammatory damage [77]. Rodent research has linkedvarious vegetables and fruits as protective “against cognitive and brain neuropathology fromdietary oxidative stress” due to innumerable bioactive constituents like carotenoids, antioxidant vitamins, polyphenols and flavonoids [8]. While current research has identified many different polyphenols from various families of flavonoids, it has been estimated that we have only scratched the surface with the potential therapeutic implications that they provide in vivo [84]. This has significant potential to advance our understanding of proactive approaches toward preventing AD and inhibiting progression. The essential role of omega-3 fatty acids in brain development and maintenance has been well recognized, particularly in the past ten years, yet only recently “have their effects on brain aging been explored” [85]. Diets rich in omega-3 fatty acids and naturally low in omega-6fatty acids may hold the key for nutritional therapy for AD patients [86]. The ketogenic diet may even diminish and clear beta amyloid plaques within the brain, while convalescing damaged mitochondria and reducing universal inflammation [87]. New research has shown that glycated ApoE4 protein and faulty insulin signaling leads not only to impaired energy transport for brain tissues, but also impaired lipid transportation, mainly cholesterol [87, 88]. There is no pharmaceutical intervention that has ever existed that has been more potent in improving overall vasculature throughout the body, than exercise [89]. This also has extensive implications for AD patients and type 2 diabetics thanks to increases in quality of life, neurochemical messaging within the brain, restorative power over insulin resistance, and the ability to clear beta-amyloid plaques in certain individuals [89]. The concept of the gut-brain axis, the bidirectional communication between gut and brain, contributing significantly to the pathogenesis of AD that has been supported by many experimental and clinical studies.
Conclusion
Glucose being an indispensable source of energy and obligate for survival interlinks various pathological mechanisms in T2D and AD as both are aftermaths of glucose metabolism and energy failure that involve disturbance of glucose metabolism by GLUT1 deficiency, O-GlcNAcylation of proteins, disturbed mTOR signaling, mitochondrial dysfunction, and reduced cholinergic transmission, aggregation of toxic Aβ plaques, tau hyperphosphorylation and autophagy. Increasing the knowledge and awareness of the term type 3 diabetes has the potential to pave the way for disease treatment, prevention and possibly even deliver a cure. Currently, there have been no particular treatments with established efficacy in counteracting cognitive decline and/or AD, so the implications of identifying AD as a disorder with an etiology rooted in faulty insulin signaling and irregular energy pathways could be critical in disease management. While the specific mechanisms between AD and all forms of diabetes remain convoluted and unclear, increasing the awareness of AD as a third form of diabetes, T3D has the potential to provide a plethora of proactive and therapeutic strategies to current patients. For now, it seems that the testing of more anti-T2D drugs with beneficial effects against cognitive impairment has a certain promising future.
References
Nguyen TT, Giau VV, Vo TK (2017) Current advances in transdermal delivery of drugs for Alzheimer's disease. Indian J Pharmacol 49(2):145–154
Bedse G et al (2015) Aberrant insulin signaling in Alzheimer's disease: current knowledge. Front Neurosci 9:204–204
Correia SC et al (2012) Insulin signaling, glucose metabolism and mitochondria: major players in Alzheimer's disease and diabetes interrelation. Brain Res 1441:64–78
Duarte JMN (2015) Metabolic alterations associated to brain dysfunction in diabetes. Aging Dis 6(5):304–321
De Felice FG, Lourenco MV, Ferreira ST (2014) How does brain insulin resistance develop in Alzheimer's disease? Alzheimer's Dement 10(1 Supplement):S26–S32
Gabbouj S et al (2019) Altered insulin signaling in Alzheimer's disease brain - special emphasis on PI3K-Akt pathway. Front Neurosci 13:629–629
Talbot K et al (2012) Demonstrated brain insulin resistance in Alzheimer's disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J Clin Invest 122(4):1316–1338
Van Giau V, An SSA, Hulme JP (2018) Mitochondrial therapeutic interventions in Alzheimer's disease. J Neurol Sci 395:62–70
Ghasemi R et al (2013) Brain insulin dysregulation: implication for neurological and neuropsychiatric disorders. Mol Neurobiol 47(3):1045–1065
Nasoohi S, Parveen K, Ishrat T (2018) Metabolic syndrome, brain insulin resistance, and Alzheimer's disease: thioredoxin interacting protein (TXNIP) and inflammasome as core amplifiers. J Alzheimers Dis 66(3):857–885
Martyn JAJ, Kaneki M, Yasuhara S (2008) Obesity-induced insulin resistance and hyperglycemia: etiologic factors and molecular mechanisms. Anesthesiology 109(1):137–148
Baker LD et al (2011) Insulin resistance and Alzheimer-like reductions in regional cerebral glucose metabolism for cognitively normal adults with prediabetes or early type 2 diabetes. Arch Neurol 68(1):51–57
Lee S-H et al (2016) Insulin in the nervous system and the mind: functions in metabolism, memory, and mood. Mol Metab 5(8):589–601
Ferreira LSS et al (2018) Insulin resistance in Alzheimer's disease. Front Neurosci 12:830
Rorbach-Dolata A, Piwowar A (2019) Neurometabolic evidence supporting the hypothesis of increased incidence of type 3 diabetes mellitus in the 21st century. BioMed Res Int 2019:8
Ormazabal V et al (2018) Association between insulin resistance and the development of cardiovascular disease. Cardiovasc Diabetol 17(1):122
Weinstein G et al (2019) Association of metformin, sulfonylurea and insulin use with brain structure and function and risk of dementia and Alzheimer's disease: pooled analysis from 5 cohorts. PLoS ONE 14(2):e0212293
de la Monte SM (2014) Type 3 diabetes is sporadic Alzheimers disease: mini-review. Eur Neuropsychopharmacol 24(12):1954–1960
Caberlotto L et al (2019) Cross-disease analysis of Alzheimer’s disease and type-2 diabetes highlights the role of autophagy in the pathophysiology of two highly comorbid diseases. Sci Rep 9(1):3965
Steen E et al (2005) Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer's disease–is this type 3 diabetes? J Alzheimers Dis 7(1):63–80
Hubbard SR (2013) The insulin receptor: both a prototypical and atypical receptor tyrosine kinase. Cold Spring Harb Perspect Biol 5(3):a008946
Hubbard SR (1997) Crystal structure of the activated insulin receptor tyrosine kinase in complex with peptide substrate and ATP analog. EMBO J 16(18):5572–5581
Bosco D et al (2011) Possible implications of insulin resistance and glucose metabolism in Alzheimer's disease pathogenesis. J Cell Mol Med 15(9):1807–1821
Chiu SL, Chen CM, Cline HT (2008) Insulin receptor signaling regulates synapse number, dendritic plasticity, and circuit function in vivo. Neuron 58(5):708–719
Lee CC, Huang CC, Hsu KS (2011) Insulin promotes dendritic spine and synapse formation by the PI3K/Akt/mTOR and Rac1 signaling pathways. Neuropharmacology 61(4):867–879
Peineau S et al (2007) LTP inhibits LTD in the hippocampus via regulation of GSK3beta. Neuron 53(5):703–717
Kim SJ, Han Y (2005) Insulin inhibits AMPA-induced neuronal damage via stimulation of protein kinase B (Akt). J Neural Transm (Vienna) 112(2):179–191
Tomita T (2016) Apoptosis in pancreatic β-islet cells in Type 2 diabetes. Bosn J Basic Med Sci 16(3):162–179
Frolich L et al (1998) Brain insulin and insulin receptors in aging and sporadic Alzheimer's disease. J Neural Transm (Vienna) 105(4–5):423–438
Kivipelto M et al (2005) Obesity and vascular risk factors at midlife and the risk of dementia and Alzheimer disease. Arch Neurol 62(10):1556–1560
Razay G, Vreugdenhil A, Wilcock G (2006) Obesity, abdominal obesity and Alzheimer disease. Dement Geriatr Cogn Disord 22(2):173–176
Kullmann S et al (2016) Brain insulin resistance at the crossroads of metabolic and cognitive disorders in humans. Physiol Rev 96(4):1169–1209
Lillioja S et al (1993) Insulin resistance and insulin secretory dysfunction as precursors of non-insulin-dependent diabetes mellitus. Prospective studies of Pima Indians. N Engl J Med 329(27):1988–1992
Li J et al (2019) Therapeutic mechanisms of herbal medicines against insulin resistance: a review. Front Pharmacol 10:661
Kapogiannis D et al (2015) Dysfunctionally phosphorylated type 1 insulin receptor substrate in neural-derived blood exosomes of preclinical Alzheimer's disease. FASEB J 29(2):589–596
Fontaine JF et al (2009) MedlineRanker: flexible ranking of biomedical literature. Nucleic Acids Res 37:W141–W146
Wang G (2014) Raison d'être of insulin resistance: the adjustable threshold hypothesis. J R Soc 11(101):20140892–20140892
Nisr RB, Affourtit C (2014) Insulin acutely improves mitochondrial function of rat and human skeletal muscle by increasing coupling efficiency of oxidative phosphorylation. Biochim et Biophys Acta 1837(2):270–276
Sivitz WI, Yorek MA (2010) Mitochondrial dysfunction in diabetes: from molecular mechanisms to functional significance and therapeutic opportunities. Antioxid Redox Signal 12(4):537–577
Bucht G et al (1983) Changes in blood glucose and insulin secretion in patients with senile dementia of Alzheimer type. Acta Med Scand 213(5):387–392
Matioli MNPS, Nitrini R (2015) Mechanisms linking brain insulin resistance to Alzheimer's disease. Dement Neuropsychol 9(2):96–102
Ma QL et al (2009) Beta-amyloid oligomers induce phosphorylation of tau and inactivation of insulin receptor substrate via c-Jun N-terminal kinase signaling: suppression by omega-3 fatty acids and curcumin. J Neurosci 29(28):9078–9089
Velazquez R et al (2017) Central insulin dysregulation and energy dyshomeostasis in two mouse models of Alzheimer's disease. Neurobiol Aging 58:1–13
Ruiz HH et al (2016) Increased susceptibility to metabolic dysregulation in a mouse model of Alzheimer's disease is associated with impaired hypothalamic insulin signaling and elevated BCAA levels. Alzheimers Dement 12(8):851–861
Long-Smith CM et al (2013) The diabetes drug liraglutide ameliorates aberrant insulin receptor localisation and signalling in parallel with decreasing both amyloid-beta plaque and glial pathology in a mouse model of Alzheimer's disease. Neuromol Med 15(1):102–114
Bomfim TR et al (2012) An anti-diabetes agent protects the mouse brain from defective insulin signaling caused by Alzheimer's disease- associated Abeta oligomers. J Clin Invest 122(4):1339–1353
Clarke JR et al (2015) Alzheimer-associated Abeta oligomers impact the central nervous system to induce peripheral metabolic deregulation. EMBO Mol Med 7(2):190–210
Biessels GJ, Reagan LP (2015) Hippocampal insulin resistance and cognitive dysfunction. Nat Rev Neurosci 16(11):660–671
Chakrabarti S et al (2015) Metabolic risk factors of sporadic Alzheimer's disease: implications in the pathology, pathogenesis and treatment. Aging Dis 6(4):282–299
de la Monte SM (2012) Brain insulin resistance and deficiency as therapeutic targets in Alzheimer's disease. Curr Alzheimer Res 9(1):35–66
Patterson C et al (2007) General risk factors for dementia: a systematic evidence review. Alzheimers Dement 3(4):341–347
Hoyer S, Oesterreich K, Wagner O (1988) Glucose metabolism as the site of the primary abnormality in early-onset dementia of Alzheimer type? J Neurol 235(3):143–148
Lying-Tunell U et al (1981) Cerebral blood flow and metabolic rate of oxygen, glucose, lactate, pyruvate, ketone bodies and amino acids. Acta Neurol Scand 63(6):337–350
Ogawa M et al (1996) Altered energy metabolism in Alzheimer's disease. J Neurol Sci 139(1):78–82
Chen Z, Zhong C (2013) Decoding Alzheimer's disease from perturbed cerebral glucose metabolism: implications for diagnostic and therapeutic strategies. Prog Neurobiol 108:21–43
Costantini LC et al (2008) Hypometabolism as a therapeutic target in Alzheimer's disease. BMC Neurosci 9(Suppl 2):S16–S16
Cunnane S et al (2011) Brain fuel metabolism, aging, and Alzheimer's disease. Nutrition 27(1):3–20
Cunnane SC et al (2016) Can ketones help rescue brain fuel supply in later life? Implications for cognitive health during aging and the treatment of Alzheimer's disease. Front Mol Neurosci 9:53–53
Maher PA, Schubert DR (2009) Metabolic links between diabetes and Alzheimer's disease. Expert Rev Neurother 9(5):617–630
Matsuzaki T et al (2010) Insulin resistance is associated with the pathology of Alzheimer disease: the Hisayama study. Neurology 75(9):764–770
Fukuyama H et al (1994) Altered cerebral energy metabolism in Alzheimer's disease: a PET study. J Nucl Med 35(1):1–6
Folch J et al (2018) The implication of the brain insulin receptor in late onset Alzheimer's disease dementia. Pharmaceuticals (Basel) 11(1):11
Kim EJ et al (2005) Glucose metabolism in early onset versus late onset Alzheimer's disease: an SPM analysis of 120 patients. Brain 128(Pt 8):1790–1801
Wang Q et al (2019) Insulin resistance and systemic metabolic changes in oral glucose tolerance test in 5340 individuals: an interventional study. BMC Med 17(1):217
Baranowski BJ, Bott KN, MacPherson REK (2018) Evaluation of neuropathological effects of a high-fat high-sucrose diet in middle-aged male C57BL6/J mice. Physiol Rep 6(11):e13729
Butterfield DA, Halliwell B (2019) Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat Rev Neurosci 20(3):148–160
Dienel GA (2019) Brain glucose metabolism: integration of energetics with function. Physiol Rev 99(1):949–1045
Liu Y et al (2008) Decreased glucose transporters correlate to abnormal hyperphosphorylation of tau in Alzheimer disease. FEBS Lett 582(2):359–364
Yoo DY et al (2016) Chronic type 2 diabetes reduces the integrity of the blood-brain barrier by reducing tight junction proteins in the hippocampus. J Vet Med Sci 78(6):957–962
Prasad S et al (2014) Diabetes mellitus and blood-brain barrier dysfunction: an overview. J Pharmacovigil 2(2):125
Rosales-Corral S et al (2015) Diabetes and Alzheimer disease, two overlapping pathologies with the same background: oxidative stress. Oxid Med Cell Longev 2015:14
Straub RH (2014) Insulin resistance, selfish brain, and selfish immune system: an evolutionarily positively selected program used in chronic inflammatory diseases. Arthr Res Ther 16(Suppl 2):S4–S4
Hemonnot A-L et al (2019) Microglia in Alzheimer disease: well-known targets and new opportunities. Front Aging Neurosci 11:33–41
Erol A (2008) An integrated and unifying hypothesis for the metabolic basis of sporadic Alzheimer's disease. J Alzheimers Dis 13(3):241–253
de la Monte SM (2017) Insulin resistance and neurodegeneration: progress towards the development of new therapeutics for Alzheimer's disease. Drugs 77(1):47–65
Su F, Bai F, Zhang Z (2016) Inflammatory cytokines and Alzheimer's disease: a review from the perspective of genetic polymorphisms. Neurosci Bull 32(5):469–480
Bagyinszky E et al (2017) Role of inflammatory molecules in the Alzheimer's disease progression and diagnosis. J Neurol Sci 376:242–254
Giau VV et al (2018) Gut microbiota and their neuroinflammatory implications in Alzheimer's disease. Nutrients 10(11):1765
Ferreira ST et al (2014) Inflammation, defective insulin signaling, and neuronal dysfunction in Alzheimer's disease. Alzheimers Dement 10(1 Suppl):S76–83
Park CR et al (2000) Intracerebroventricular insulin enhances memory in a passive-avoidance task. Physiol Behav 68(4):509–514
Kandimalla R, Thirumala V, Reddy PH (2017) Is Alzheimer's disease a type 3 diabetes? A critical appraisal. Biochim Biophys Acta Mol Basis Dis 1863(5):1078–1089
Nguyen NH et al (2020) Potential antidiabetic activity of extracts and isolated compound from Adenosma bracteosum (Bonati). Biomolecules 10(2):201
de Matos AM, de Macedo MP, Rauter AP (2018) Bridging type 2 diabetes and Alzheimer's disease: assembling the puzzle pieces in the quest for the molecules with therapeutic and preventive potential. Med Res Rev 38(1):261–324
Ayaz M et al (2019) Flavonoids as prospective neuroprotectants and their therapeutic propensity in aging associated neurological disorders. Front Aging Neurosci 11:155
Canhada S et al (2018) Omega-3 fatty acids' supplementation in Alzheimer's disease: a systematic review. Nutr Neurosci 21(8):529–538
Ajith TA (2018) A recent update on the effects of omega-3 fatty acids in Alzheimer's disease. Curr Clin Pharmacol 13(4):252–260
Broom GM, Shaw IC, Rucklidge JJ (2019) The ketogenic diet as a potential treatment and prevention strategy for Alzheimer's disease. Nutrition 60:118–121
Giau VV et al (2015) Role of apolipoprotein E in neurodegenerative diseases. Neuropsychiatr Dis Treat 11:1723–1737
Frederiksen KS et al (2018) Effects of physical exercise on Alzheimer's disease biomarkers: a systematic review of intervention studies. J Alzheimers Dis 61(1):359–372
De Felice FG et al (2009) Protection of synapses against Alzheimer's-linked toxins: insulin signaling prevents the pathogenic binding of Abeta oligomers. Proc Natl Acad Sci USA 106(6):1971–1976
Lourenco MV et al (2013) TNF-alpha mediates PKR-dependent memory impairment and brain IRS-1 inhibition induced by Alzheimer's beta-amyloid oligomers in mice and monkeys. Cell Metab 18(6):831–843
Craft S et al (2003) Insulin dose-response effects on memory and plasma amyloid precursor protein in Alzheimer's disease: interactions with apolipoprotein E genotype. Psychoneuroendocrinology 28(6):809–822
Reger MA et al (2008) Intranasal insulin administration dose-dependently modulates verbal memory and plasma amyloid-beta in memory-impaired older adults. J Alzheimers Dis 13(3):323–331
Reger MA et al (2006) Effects of intranasal insulin on cognition in memory-impaired older adults: modulation by APOE genotype. Neurobiol Aging 27(3):451–458
Reger MA et al (2008) Intranasal insulin improves cognition and modulates beta-amyloid in early AD. Neurology 70(6):440–448
Benedict C et al (2008) Differential sensitivity of men and women to anorexigenic and memory-improving effects of intranasal insulin. J Clin Endocrinol Metab 93(4):1339–1344
Batista AF et al (2018) The diabetes drug liraglutide reverses cognitive impairment in mice and attenuates insulin receptor and synaptic pathology in a non-human primate model of Alzheimer's disease. J Pathol 245(1):85–100
McClean PL et al (2011) The diabetes drug liraglutide prevents degenerative processes in a mouse model of Alzheimer's disease. J Neurosci 31(17):6587–6594
McClean PL, Holscher C (2014) Liraglutide can reverse memory impairment, synaptic loss and reduce plaque load in aged APP/PS1 mice, a model of Alzheimer's disease. Neuropharmacology 76(Pt A):57–67
Perry T et al (2002) Protection and reversal of excitotoxic neuronal damage by glucagon-like peptide-1 and exendin-4. J Pharmacol Exp Ther 302(3):881–888
Escribano L et al (2010) Rosiglitazone rescues memory impairment in Alzheimer's transgenic mice: mechanisms involving a reduced amyloid and tau pathology. Neuropsychopharmacology 35(7):1593–1604
Funding
This work did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that there is no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Nguyen, T.T., Ta, Q.T.H., Nguyen, T.T.D. et al. Role of Insulin Resistance in the Alzheimer's Disease Progression. Neurochem Res 45, 1481–1491 (2020). https://doi.org/10.1007/s11064-020-03031-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-020-03031-0