
Abstract
NLRP3 (NLRP3: NOD-, LRR-, and pyrin domain-containing protein 3) inflammasome is the best-described inflammasome that plays a crucial role in the innate immune system and a wide range of diseases. The intimate association of NLRP3 with neurological disorders, including neurodegenerative diseases and strokes, further emphasizes its prominence as a clinical target for pharmacological intervention. However, after decades of exploration, the mechanism of NLRP3 activation remains indefinite. This review highlights recent advances and gaps in our insights into the regulation of NLRP3 inflammasome. Furthermore, we present several emerging pharmacological approaches of clinical translational potential targeting the NLRP3 inflammasome in neurological diseases. More importantly, despite small-molecule inhibitors of the NLRP3 inflammasome, we have focused explicitly on Chinese herbal medicine and botanical ingredients, which may be splendid therapeutics by inhibiting NLRP3 inflammasome for central nervous system disorders. We expect that we can contribute new perspectives to the treatment of neurological diseases.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
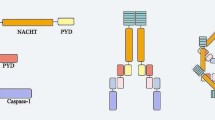
The NLRP3 inflammasome is a protein complex composed of a sensor protein (NLRP3), an adaptor apoptosis-associated speck-like protein containing a CARD (ASC), and a downstream effector (pro-caspase-1) (Fig. 1). Following the activation of the inflammasome sensor molecule by agonists, ASC self-associates into a helical fibrous assembly [1] that forms the so-called ASC speck or pyroptosome [2], which serves as a molecular plateau for caspase-1 activation by proximally induced autocatalysis [3]. Activated caspase-1 promotes the maturation and secretion of several pro-inflammatory cytokines, including interleukin-1β (IL-1β) and interleukin-18 (IL-18). On the one hand, it also triggers cellular pyroptosis capable of clearing pathogens and damaged cells [4]. Depending on the sensor, the main inflammasomes include NLRP3 (NOD-, LRR-, and pyrin domain-containing protein 3), NOD-like receptor family CARD domain-containing protein 4 (NLRC4), interferon-inducible protein AIM2, NLRP1, and others. NLRP3 is a NOD-like receptor family (NLRs) member and is the best-described inflammasome sensor, which plays as a key component of the innate immune system. NLRP3 contains an amino-terminal PYRIN (PYD) domain, a nucleotide-binding NACHT domain, and a carboxy-terminal leucine-rich repeat (LRR) domain (Fig. 1). The NACHT domain carries ATPase activity, and the ATP binding is essential for the function of NLRP3 [5]. Gain-of-function mutations of the Nlrp3 gene result in a dominantly inherited autoinflammatory disease called cryptochrome-associated periodic syndrome (CAPS) [6,7,8]. Recent research reveals that the inactive endogenous full-length NLRP3 is formed in a double-ring cage that is primarily membrane-localized [9]. The location is consistent with previous studies of NLRP3 on various membranous organelles [10]. The double-ring cage is combined by LRR-LRR interactions, while the PYD domain is hidden inside to prevent premature activation [9]. Furthermore, it has been demonstrated that this specific cage configuration is required for the dispersion of the trans-Golgi network (TGN) [9], a common cellular event during NLRP3 activation [11] that we will discuss later. Recently, it has been further suggested that NIMA-related protein kinase 7 (NEK7), a serine–threonine kinase previously involved in mitosis, is required to activate NLRP3 inflammasome [12, 13]. After being triggered by stimuli, NEK7-NLRP3 interacts to form a complex, which is essential for the subsequent recruitment of ASC and the activation of caspase-1. This interaction is thought to occur downstream of reactive oxygen species (ROS) and potassium efflux [12] and is specific to NLRP3 rather than other inflammasome sensors [12, 13].

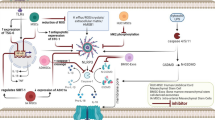
A schematic diagram of mediators and stimulators involved in NLRP3 inflammasome activation and the structure of NLRP3. The canonical activation of NLRP3 inflammasome consists of two steps: priming (signal 1) and activation (signal 2). Signal 1 (left), triggered by the activation of cytokines (such as IL-β, TNF) or pathogen-associated molecular patterns (PAMPs), is to initiate the transcription of NLRP3 and pro-inflammatory genes and upregulate their expressions in cells. The priming step of NLRP3 inflammasome activation provides cells with permission to be activated subsequently. Signal 2 (right) is trigged by various PAMPs and DAMPs. At present, the well-accepted upstream signals which could activate NLRP3 inflammasome comprise K+ efflux, the leakage of cathepsins caused by lysosomal disruption, impaired mitochondrial function, Ca2+ influx and trans-Golgi disassembly. Activation of NLRP3 by agonists subsequently attracts ASC and caspase-1 to assemble into NLRP3 inflammasome, thereby inducing caspase-1 self-cleavage and activation. Activated caspase-1 then promotes the maturation and secretion of pro-inflammatory cytokines including interleukin-1β (IL-1β) and interleukin-18 (IL-18).The non-canonical NLRP3 inflammasome activation is triggered by caspase-11 in mice (human homologs caspase-4 and caspase-5). After sensing LPS, caspase-11 is activated via Toll-like receptor 4 (TLR4), leading to the cleavage of gasdermin D (GSDMD). The cleaved GSMD, N-terminal, forms membrane pores, thereby resulting in potassium efflux and pyroptosis. The dotted line indicates the pathway where the mechanism is not yet established. ASC, apoptosis-associated speck-like protein containing a CARD; CARD, caspase recruitment domain; CLIC, chloride intracellular channel protein; GSDMD, gasdermin D; N- terminal, GSDMD amino-terminal cell death domain; IL-1β, interleukin-1β; IL-18, interleukin-18; IL-1R1, IL-1 receptor type 1; LPS, lipopolysaccharide; LRR, leucine-rich repeat; NEK7, NIMA-related kinase 7; NF-κB, nuclear factor-κB; P2X7, purinergic 2X7 receptor; PtdIns4P, phosphatidylinositol-4-phosphate; PYD, pyrin domain; ROS, reactive oxygen species; TLR, Toll-like receptor; TNF, tumor necrosis factor; TNFR, tumor necrosis factor receptor; TWIK2, two-pore domain weak inwardly rectifying K.+ channel 2
Up to now, NLRP3 inflammasome is the most investigated inflammasome in the central nervous system (CNS). It is mainly studied in microglia but can also be expressed in astrocytes and oligodendrocytes in patients or animal models with neurologic diseases [14, 15]. Regarding CNS, NLRP3 inflammasome is particularly sensitive to endosome injury and responds to various aggregated proteins associated with diseases, such as Aβ [16] or α-synuclein [17]. Many studies suggested the involvement of NLRP3 inflammasomes in neurodegenerative diseases, while the pharmacologic or genetic deletion or inhibition of NLRP3 inflammasomes could improve neurologic function [18]. Previous studies summarized that the use of specific inhibitors of NLRP3 inflammasomes, along with microglial autophagy inducers, could contribute to the elimination of misfolded proteins, the scavenging of impaired mitochondria and ROS in neurodegenerative diseases [19]. Recent studies also highlighted that NLRP3 inflammasome was also engaged in the pathological processes of other neurological disorders, such as ischemic stroke, which have not traditionally been considered as inflammatory in nature [20]. Moreover, a number of inhibitors targeting NLRP3 inflammasomes have demonstrated therapeutic efficacy in animal experiments or in vivo models. This paper reviews the latest developments and gaps regarding NLRP3 inflammasome activation. Meanwhile, we summarize the recent findings on inhibiting the NLRP3 inflammasome signaling pathway in treating neurological diseases. Besides small-molecule inhibitors and microRNA for NLRP3 inflammasomes, we specifically focused on the remarkable studies of herbal medicines, plant-derived ingredients, and drugs used to treat other diseases, which have rarely been systematically reviewed in other similar articles.
Activation of NLRP3 Inflammasome
Canonical NLRP3 Activation
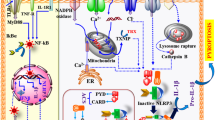
It is well established that two-step procedures stimulate the canonical activation of NLRP3 inflammasome: priming (signal 1) and activation (signal 2) (Fig. 1). Briefly, signal 1 is to initiate the transcription of Nlrp3 and pro-inflammatory genes and upregulate their expressions in cells. This process is involved with pattern recognition receptors (PRRs) such as Toll-like receptors (TLRs), nucleotide-binding oligomerization domain-containing protein 2 (NOD2), and tumor necrosis factor (TNF) receptors induced by various pathogen-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs) [21, 22]. Nuclear factor-κB (NF-κB) is further activated by these receptors, increasing the transcription of the inflammasome components [23]. The activation step (signal 2) occurs following the recognition of an NLRP3 inflammasome stimulus and induces the recruitment of ASC and caspase-1. Unlike other members of the NLR family, the NLRP3 inflammasome responds to various stimuli independent in origin, chemical composition, and structural properties. The stimulants range from intrinsic and extrinsic agents, known as DAMPs and PAMPs. PAMP signals mainly come from invasive pathogens such as bacterial toxins [24]. Lipopolysaccharide (LPS) is commonly thought to be a prototypical PAMP. DAMP signals perceived by NLRP3 inflammasome are usually produced by the damaged body itself, such as triphosphate (ATP), silica crystals [25], amyloid β (Aβ) [16], and α-synuclein [17]. Given the high variability of activators, they are unlikely to interact by directly binding to NLRP3 inflammasome. Recent studies suggested that the NLRP3 inflammasome may perform as a signal integrator, recognizing any molecules or conditions that could induce alterations in homeostasis or pathological state [26]. However, how NLRP3 inflammasome sense and respond to dangerous cellular signals remains indefinite.
Here, we introduced the widely accepted upstream signals and some latest studies. K+ efflux is a common required upstream event for NLRP3 activation [27]. After being simulated by ATP, purinergic 2X7 receptor (P2X7R) promotes K+ efflux with two-pore domain weak inwardly rectifying K+ channel 2 (TWIK2), as well as Ca2+ and Na+ influx [28, 29]. Pore-forming toxins also induce K+ efflux [24]. Meanwhile, Ca2+ flux and K+ efflux are often synergistic during NLRP3 activation [28]. Ca2+ release from the endoplasmic reticulum (ER) promoted by K+ is an upstream signal in NLRP3 inflammasome activation [30]. More particularly, ER stress, resulting in Ca2+ release from the ER lumen, magnifies the activation of NLRP3 inflammasome. Besides potassium and calcium, chloride is also engaged [30]. Cl− efflux is thought to promote ASC aggregation during NLRP3 inflammasome formation, depending on chloride intracellular channel proteins (CLICs) [31]. Lysosomal disruption and the leakage of cathepsins is another common mechanism of NLRP3 activation [25]. Activation caused by phagocytosis of crystalline material or protein aggregates is mediated via this pathway mainly. Meanwhile, lysosomal damage could trigger K+ efflux and Ca2+ influx [27]. Early studies revealed ROS production was necessary for NLRP3 inflammasome activation [32]. In addition, mitochondrial dysfunction and the release of mitochondrial reactive oxygen species (mtROS), cardiolipin, and mitochondrial DNA (mtDNA) into the cytoplasm is an equally critical upstream event independent of K+ efflux and lysosomal disruption involved in NLRP3 activation [33, 34]. Despite mitochondria [35], dysfunction of organelles such as the endoplasmic reticulum [36] and Golgi apparatus [37] are also engaged in NLRP3 inflammasome activation. The Golgi apparatus is definitely the most unexpected and astonishing among all these organelles. Recent studies implicate that the Golgi apparatus is not only an organelle for intracellular sorting but also plays a crucial role in signal transfer during the inner immune response [37]. Chen et al. indicated that the trans-Golgi network (TGN), rather than cis- or medial-Golgi, was disassembly induced by several NLRP3 activators into dispersed trans-Golgi network (dTGN) [11]. dTGN performed as a scaffold that recruited NLRP3 via phosphatidylinositol-4-phosphate (PtdIns4P), promoting NLRP3 inflammasome assembly and the downstream caspase-1 activation. Intriguing, it was further suggested that trans-Golgi disassembly was the common cellular event in two distinct pathways, K+ efflux-dependent and mitochondria-dependent NLRP3 activation, which eventually led to NLRP3 aggregation [11]. Recent studies also suggested that the activation of NLRP3 required the ER-to-Golgi translocation of sterol regulatory element binding protein (SREBP) 2 and SREBP cleavage-activating protein (SCAP) [10]. Another study clarified that NLRP3 stimuli invoked the localization of mitochondria-associated endoplasmic reticulum membranes (MAMs) close to the Golgi membrane [38]. This inter-organelle crosstalk relied on the recruitment of protein kinase domain (PKD) at the Golgi DAG site, which promoted the assembly of NLRP3 oligomers into active inflammasomes [38].
Of note, although researchers have made numerous efforts to understand the upstream events during NLRP3 activation, there is still no definitive conclusion. Many pathways are cross-linked and overlapping, and the results occasionally conflict with others. Further investigation is required.
Non-canonical NLRP3 Activation
Different from the canonical activation, the non-canonical NLRP3 inflammasome activation is triggered by caspase-4 and caspase-5 in humans [39] and caspase-11 in mice [40], respectively (Fig. 1). Generally speaking, upon sensing LPS produced by Gram-negative bacteria, caspase-4, caspase-5, and caspase-11 are activated via Toll-like receptor 4 (TLR4), leading to the cleavage of gasdermin D (GSDMD). The cleaved N-terminal of GSMD then forms membrane pores, thereby resulting in potassium efflux and pyroptosis [41, 42]. Nevertheless, the canonical and non-canonical inflammasome activation shared similar consequences, namely, the release of pro-inflammatory cytokines IL-1β and IL-18. It was reported that the oxidized phospholipid 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphorylcholine (oxPAPC) also induced caspase-11-dependent IL-1β release via this non-canonical pathway [43].
Inhibitors of NLRP3 Inflammasome
Many inhibitors have been developed to investigate the regulatory mechanisms of NLRP3 inflammasomes and their roles in disease pathogenesis. These inhibitors directly or indirectly inhibit NLRP3 inflammasome components or related upstream/downstream signaling events [44]. Among them, the activation step is the favored target for the creation of NLRP3 antagonists, as it should lead to the most direct and specialized suppression. However, the molecular mechanism of inhibition or the precise target has not been fully elucidated for many of these compounds. To date, MCC950 (also known as CP-456773 or CRID3) is the most studied and specific NLRP3 inflammasome inhibitor, as it does not inhibit any other identified inflammasomes such as NLRP1, NLRC4, or AIM2 [45]. Meanwhile, MCC950 could inhibit both the canonical and non-canonical activation of NLRP3 inflammasome by all known stimuli at present. Due to its specificity, it is the most recommended and employed compound to study the relevant events of NLRP3 inflammasome in vitro and in vivo. MCC950 binds to NLRP3 in the NACHT domain and blocks its ability to hydrolyze ATP, thereby preventing it from retaining an active form, thus inhibiting the recruitment to ASC and the cleavage of caspase-1 [46]. It was also suggested that MCC950 could perform its function by inhibiting the chloride efflux following nigericin stimulation [47]. Shockingly, MCC950 shows therapeutic efficacy in a wide range of disease models in vivo, including diabetes [48], neurodegenerative disease [49], traumatic brain injury [50, 51], atherosclerosis [52], steatohepatitis [53], and colitis [32]. Other small-molecule inhibitors of NLRP3 inflammasome include Bay 11–7082 [54], JC-171 [55], and β-hydroxybutyrate (BHB) [56]. Interestingly, many traditional Chinese medicines, botanical ingredients, and drugs used in treating other diseases have also been shown to inhibit the action of NLRP3 inflammasome. We summarize the inhibitors in Table 1 and will discuss these agents later in specific disease models.
Therapeutic Targeting of NLRP3 in Neurological Diseases
Here we overview the recent studies of NLRP3 inflammasome performed in the nervous system and try to assess its potential as a drug target for the treatment of neurological disease, especially in stroke and neurodegenerative diseases such as Alzheimer's disease. Moreover, we have specifically focused on Chinese herbal medicine, botanical ingredients, and drugs used in treating other conditions, which may be splendid therapeutics by inhibiting NLRP3 inflammasome for CNS disorders. Figure 2 presents a schematic diagram of the possible major drivers of NLRP3 inflammasome activation in neurological diseases.

A review of mediators and stimulators of NLRP3 inflammasome activation presently known to be involved mainly in microglia in neurological dysfunction
Ischemic Stroke
Ischemic stroke is a severe life-threatening condition that accounts for almost 87% of all strokes [92]. Although intravenous thrombolysis and thrombectomy have been developed recently, ischemic stroke remains the second leading cause of death globally [93]. The primary complications of strokes, including cognitive impairment and depression [94], consequently carry heavy social and family burdens. However, most neuroprotective agents which proved to be effective in animal studies ended in failure [95, 96]. Therefore, finding new directions for pharmaceutical treatment is urgent and essential.
Previous findings highlighted the importance of NLRP3 inflammasome in mediating the inflammatory response in aseptic tissue injury during post-stroke damage [97, 98]. At the early stage of ischemia, hypoxia and hypoperfusion accompanied by the generation of DAMPs could directly activate NLRP3 inflammasome, exacerbating inner immune response and the injuries of stroke. The generation of ROS induced in ischemia–reperfusion could activate the NLRP3 inflammasome and cause more neural damage [99]. In addition to ROS, other dangerous signals that can trigger NLRP3 activation include energy depletion, acidosis, cathepsin release, mitochondrial DNA oxidation and dysfunction, intracellular Ca2+ accumulation, decreased intracellular K+ concentration, cell swelling, and protein kinase R (PKR) activation [100]. Pro-inflammatory cytokines such as IL-1 β and caspase-1, as well as NLRP3, were highly expressed in cellular models, animal models, and patients with stroke [101,102,103,104]. These massive cytokines and chemokines were highly toxic to neurons. It was well established that inflammatory cascade response mediated by NLRP3 inflammasome contributed to cerebral edema and hemorrhage, blood–brain barrier damage, and more neuronal death [105].
At present, the cell-specific expression and distribution of the NLRP3 inflammasome in cerebral ischemia–reperfusion injury have not been fully elucidated. In the mouse brain, expression of NLRP3, ASC, and caspase-1 has been observed in microglia after LPS stimulation and was not detected in astrocytes, suggesting that microglia may be the main locus engaged in the generation of NLRP3 inflammasome [106]. Microglia is the major innate immune cell population in the brain [107]. Impaired microglia immune functions often lead to overproduction of inflammatory mediators and exacerbate central nervous system damage [108, 109]. Consistent with this, there were signs that NLRP3 was expressed in microglia and vascular endothelial cells but not neurons [57]. In contrast, data also showed that the levels of NLRP3 inflammasome proteins, IL-1β, and IL-18 increased in neurons under ischemic conditions as well as in neurons from stroke patients [88, 103, 110]. Different ischemia models and interventions and the duration of ischemic injury were possible explanations. It was suggested that the activation pattern of NLRP3 inflammasome induced by ischemia–reperfusion injury varied between neurons and glial cells [88].
The NLRP3 inflammasome may act through multiple mechanisms to mediate neuronal and glial cell death in ischemic stroke. The mechanisms include enhancing the production and secretion of pro-inflammatory cytokines such as IL-1β and IL-18 and through the multiplicity of cleaved caspase-1 in mediating neural apoptosis. Consistently, further studies demonstrated that the interference of NLRP3 activation matched the regulation of neuroprotective function. Blockade of NLRP3 reduced the infarction volume and neurovascular complications in the middle cerebral artery occlusion mice model, and inhibited neuronal apoptosis in vivo and in vitro [111]. Compared to the controls, MCC950-treated mice showed significant reductions in infarcts, edema, and hemoglobin content, along with improved neurological deficits [112]. The expression of NLRP3, cleavage products caspase-1, and IL-1β were also decreased in the penumbral region after ischemic. Meanwhile, in parallel, these effects of MCC950 were associated with reduced phosphorylation levels of NFkBp65 and IkBa. Thus the study showed that MCC950 exhibited therapeutic potential in animal models with ischemic stroke. Furthermore, it was indicated that treatment of MCC950 improved cognitive function, neurovascular unit integrity, and neurovascular remodeling in rats after stroke [113]. NLRP3 and IL-1β expressions were also significantly decreased in the hippocampus with MCC950 interference [113]. Recently, it was further suggested that the blood–brain barrier integrity and functional outcome were improved considerably in MCC950-treated mice after ischemic stroke [114]. Moreover, endothelial cell survival was elevated in vivo as well as in vitro experiments with consistent results by inhibiting inflammatory signaling cascades and pyroptosis [114]. Astragaloside IV (AST IV), one of the practical components of the traditional Chinese medicine Astragalus membranaceus, exerted protective effects against cerebral ischemia–reperfusion injury through inhibiting NLRP3 inflammasome-mediated pyroptosis via activating nuclear factor erythroid-2–related factor 2 (Nrf2) signaling [75]. However, the regulatory mechanism of NLRP3 is not well understood. Fann et al. were the first to identify that NF-κB and mitogen-activated protein kinase (MAPK) signaling pathways could regulate the activation of NLRP3 inflammasome in ischemic primary cortical neurons [115]. A recent study indicated that NLRP3 inflammasome might be directly controlled by JAK2/STAT3 signaling pathway after stroke in vitro [116]. NLRP3 inflammasome was also involved in the regulation of autophagy. Yue Liu et al. suggested that miR-135a-5p, highly expressed in M2 phenotype microglia-derived extracellular vesicles, could negatively regulate NLRP3 expression via thioredoxin-interacting protein (TXNIP), thereby reducing neuronal autophagy and ischemic brain damage [83]. Another finding suggested that resveratrol, a Chinese herbal medicine, could alleviate ischemia–reperfusion injury in rats by suppressing NLRP3 inflammasome activation via Sirt1-dependent autophagy activity [72].
Interestingly, some market-approved drugs also present neuroprotection effects via the NLRP3 inflammasome pathway. Lithium (Li+) is used in psychiatry to treat acute mania and acute depression. Recently, it has been reported to exert neuroprotection in stroke patients [117]. It was further suggested that the underlying mechanism of Li+ was related to NLRP3 inflammasomes. Li+ inhibited the activation of NLRP3 through two signaling pathways, AKT/GSK3β/β-catenin and AKT/FoxO3a/β-catenin, which suppressed the production of ROS [88]. Idebenone is a well-used antioxidant as a mitochondrial protectant. Peng et al. found that idebenone could effectively alleviate the loss of mitochondrial membrane potential (Δψm), attenuate mitochondrial dysfunction, inhibit the NLRP3-mtDNA interaction and NLRP3 activation, and subsequently reduce neurological deficits in rats with ischemic stroke [89]. Another recent study reported that verapamil acted as a reliable adjuvant to thrombolytic therapy by reversing the tissue plasminogen activator (tPA)-induced activation of the TXNIP/NLRP3 inflammasome pathway and reducing infarct volume in hyperglycemic stroke [90]. Therefore, targeting upstream and downstream pathways of NLRP3 inflammasome signaling may become a promising therapeutic strategy for ischemic stroke.
Hemorrhagic Stroke
Cerebral hemorrhage is a general term for a range of disruptive brain hemorrhagic disorders with high mortality. Mainly two categories are included, intracerebral hemorrhage (ICH) and subarachnoid hemorrhage (SAH).
Intracerebral Hemorrhage
ICH is the most severe type of stroke. Although the incidence of ICH is less than ischemic stroke [92], it often brings the poorest outcomes and the highest morbidity among stroke subtypes [118]. More grimly, up to half of the survivors will have a recurrent hemorrhagic or ischemic stroke, sudden cognitive decline, or sudden depression within five years after the brain hemorrhage [119]. Controlling blood pressure is the most effective way to slow or possibly prevent this progression [120]. Therefore, searching for effective early treatment is extremely important for patients with ICH. Rapidly formed hematomas could directly lead to brain damage and neurological deficits after the onset of ICH [121]. Thus, it was thought that the removal of the hematoma might be beneficial for ICH patients. However, the surgical trial in intracerebral hemorrhage (STICH) failed to show any overall efficacy of early hematoma clearance compared to initial conservative therapies [122]. Eight years later, the STICH II trial yielded the same results: early surgery could not improve the outcomes of ICH patients in terms of the death rate and disability at six months [123]. Since effective treatments for the primary injury are still lacking, the secondary brain injury caused by metabolites or components of hematoma is of increasing interest to researchers. During this phase, the hematoma triggers a series of pathological changes, including cytotoxicity, apoptosis, oxidative stress, and inflammations, leading to neurological function deterioration [124]. Of note, controlling the inflammatory process may be a potentially beneficial strategy for treating ICH [125].
Converging evidence shows that NLRP3 inflammasome plays a critical role in ICH-induced secondary brain injury and neuroinflammation. Previous studies have demonstrated that NLRP3 inflammasome was involved in the inflammatory response after ICH [62]. The ATP-gated transmembrane cation channel P2X7R was known as a key mediator in the upstream of NLRP3 inflammasome activation and cytokines release [28]. P2X7R and NLRP3 inflammasomes were activated sequentially in rats with collagenase-induced cerebral striatum hemorrhage, which promoted the generation of NLRP3 inflammasome-dependent pro-inflammatory cytokines, such as IL-1β and IL-18, and propelled the onset of brain damage subsequently [62]. Meanwhile, the neuroinflammation after ICH could be alleviated by blue brilliant G (BBG), a selective P2X7R inhibitor, by inhibiting the P2X7R/NLRP3 inflammasome pathway in rats [62]. Researchers further confirmed that activation of NLRP3 inflammasome could enhance inflammatory response, promote neutrophil infiltration, aggravate brain edema, and deteriorate neurological function in mice after ICH [126]. Another research showed that NLRP3 inflammasome was essential for complement-induced neuroinflammation after ICH, which played a key role in ICH-induced neurological dysfunction [127]. As a crucial transcription factor, Nrf2 could suppress the expression of NLRP3 and the subsequent clearance of ROS, thereby mitigating early brain injury [128]. Upregulation of the Nrf2-mediated signaling pathway leading to NLRP3 inactivation by Silymarin could produce a neuroprotective effect in rats with collagenase-induced ICH [76]. Similar results were observed in other recent studies. Intraperitoneal injection of Ghrelin can protect mice from the secondary brain injury after ICH by inhibiting the activation of NLRP3 inflammasome and promoting Nrf2/ARE signaling [129]. Although the relationship between Nrf2 and NLRP3 inflammasomes needs further investigation, these studies provide experimental evidence for the involvement of Nrf2 in the regulation of NLRP3 to some extent. The other underlying mechanisms regulating NLRP3-mediated inflammation in ICH injury included peroxynitrite [62] and mitophagy [130]. A recent study indicated that NLRP3 inflammasome might be regulated via the JAK2-STAT3-A20 pathway after ICH in C57BL/6 mice [131]. Evidence further showed that selective NLRP3 inflammasome inhibitor MCC950 can effectively down-regulate the expression of IL-1β and IL-6 by microglia in mouse models with ICH induced by autologous blood and bacterial collagenase [58]. Moreover, the intervention of MCC950 presented significant neuroprotective effects. Intraperitoneally injected MCC950 (10 mg/kg) in mice after ICH reduced cell death in brain tissue, maintained the integrity of the blood–brain barrier, and improved long-term prognosis [58]. Interestingly, the beneficial effects of MCC950 were eliminated after microglia depletion [58].
In addition to small-molecule inhibitors, post-transcriptional modification by specific RNA interference (RNAi) or microRNA (miRNA) on the expression of NLRP3 showed equivalent neuroprotection in animal models with ICH. NLRP3 RNAi could inhibit the inflammatory response induced by erythrocytelysis, alleviate microglia-mediated neuronal injury, and improve neurological dysfunction after ICH [132]. Previous research has suggested that miR-223 can down-regulate NLRP3 and suppress inflammation, thus reducing brain edema and improving neurological function [84]. Moreover, a recent study showed that miR-152 could inhibit the TXNIP-mediated activation of NLRP3 inflammasome, thereby attenuating neuroinflammation, brain injury, and neurological deficits in the collagenase-induced ICH rats [85].
Resveratrol, a plant polyphenol complex, has been shown to induce autophagy and inhibit mitochondrial damage in macrophages, thereby inhibiting NLRP3 inflammasome activation, NLRP3-mediated IL-1β release, and pyroptosis [133]. Several studies have shown that resveratrol can reduce neuroinflammation and alleviate secondary brain injury after ICH [134]. As discussed in other parts of our article, resveratrol already showed beneficial effects in Alzheimer’s disease [73], cerebral ischemia [72], and SAH [74], which were all closely associated with the regulation of NLRP3. Nevertheless, whether resveratrol also exerts anti-inflammatory effects through the inhibition of the NLRP3 inflammasome remains unclear in these studies of ICH and requires further research. In conclusion, early inhibition targeting the NLRP3 may be a potential strategy for the treatment of ICH.
Subarachnoid Hemorrhage
Subarachnoid hemorrhage (SAH), mainly caused by aneurysms rupture, has a mortality and disability rate of up to 25–50% [135] while only accounts for 3% of all strokes [92]. Early brain injury and delayed cerebral ischemia are two major factors affecting functional outcomes after SAH. Previous and recent research on SAH emphasizes the importance of inflammation in aneurysm formation and rupture [136, 137]. Chronic inflammation is also thought to be a promising drug therapy target to stabilize unruptured intracranial aneurysms or prevent the formation of unruptured intracranial aneurysms [138]. A previous study observed that the P2X7R/cryopyrin inflammatory axis might promote the production of IL-1 β/IL-18 after SAH by activating caspase-1, thus promoting the formation of neuroinflammation [139]. Recent research showed that in the rat model with SAH, melatonin treatment could attenuate early brain injury, which was closely associated with the inhibition of NLRP3 inflammasome activation [91]. The pro-inflammatory cytokine levels and ROS production were also significantly reduced. Evidence also proved that MCC950, a selective NLRP3 inhibitor, could alleviate early brain injury caused by SAH in animal models [140, 141]. Intraperitoneal injection of MCC950 with a dose of 10 mg/kg reduced neuroinflammation and improved SAH-induced neurological dysfunction in Sprague–Dawley rats [141]. Resveratrol, as mentioned above, also protected rats and the primary cultured cortical neurons from early brain injury after SAH, at least in part through suppressing the NLRP3 inflammasome signaling pathway [74]. It was also suggested that Pterostilbene, a polyphenol mainly derived from blueberries and grapes, could reduce early brain injury possibly through the inhibition of NLRP3 inflammasome and Nox2-related oxidative stress [77]. Moreover, Schisandrin B significantly improved the neurological function in rats with SAH, which was related to the inhibition of NLRP3 and neuroinflammation [142]. Inhibition of NLRP3 alleviated cerebral edema, microthrombosis, tight junction disruption, and microglial activation but also prevented middle cerebral artery vasospasm during the delayed period and attenuated sensorimotor disturbances caused by SAH [140]. Furthermore, suppression of the NLRP3 inflammasome pathway would promote neurogenesis action following SAH and improve the impaired delayed neurological dysfunction [143, 144]. However, the mechanism needs further studies. It has been demonstrated that NLRP3-dependent neuroinflammation could be mediated by the TGR5/cAMP/PKA signaling pathway [144]. In experimental models of SAH induced by endovascular perforation, the NLRP3 inflammasome could be activated by triggering receptor expressed on myeloid cells 1 (TREM-1) and mediate microglial pyroptosis [145].
Alzheimer Disease
Alzheimer’s disease (AD) is the primary dementia disease in the world, which is characterized by progressive cognitive and behavioral disorders. According to the latest report of Alzheimer’s Disease International (ADI), up to 6.5 million Americans (> age 65) are suffering from Alzheimer’s dementia today. An estimated $321 billion of total payments were cost [146]. However, in contrast to the severity of the disease, despite more than a century of exploration, there have been few breakthroughs in drug development for Alzheimer’s disease, and many promising drug developments failed [147]. A new direction for drug exploration is required.
The converging pathological features of AD are extracellular senile plaques formed by amyloid ß (Aβ) protein, intracellular neurofibrillary tangles composed of the hyperphosphorylation of tau protein, and the loss of neurons [148]. These pathological changes are closely related to innate immune abnormalities in the brain [149]. Furthermore, it has been identified that NLRP3 activation contributes to AD without equivocation. The expression of NLRP3 and downstream inflammatory cytokines (IL-1β and IL-18) was elevated in peripheral monocytes in severe AD patients [150]. Similar elevated expression patterns were observed in the temporal cerebral cortex of AD patients by Mahmoudiasl et al. [151]. Almost two decades ago, Aβ, as a kind of DAMPs, had been identified as the direct activator of the NLRP3 inflammasome, leading to the maturation and release of IL-1β for the first time [16]. The mechanism involved included lysosomal damage and cathepsin B release [16]. Subsequently, considerable research was carried out. It was further suggested that the activation of NLRP3 inflammasome in microglia exacerbated Aβ pathology [59] by prion-like ASC-speck cross-seeding [152]. Despite fibrillar Aβ, it was reported recently that lower molecular weight Aβ oligomers and protofibrils could also trigger the activation of NLRP3, suggesting that innate immune response induced by Aβ species may occur before the initiation of Aβ deposition [153]. NLRP3 inflammasome deficiency would reduce Aβ deposition in the amyloid precursor protein/presenilin 1 (APP/PS1) model of AD [49, 59]. Furthermore, neuroinflammation induced by activation of NLRP3 inflammasome impaired Aβ clearance in microglia [154].
Presently, Aβ-activated NLRP3 inflammasomes participating in the pathogenesis of AD are convinced, while the relationship between tau and NLRP3 inflammasome is more limited studied. Recently, it was confirmed that the activation of NLPR3 in microglia could promote the pathological changes of tau protein, which was conducive to the onset of AD [155]. Meanwhile, NLRP3 could be activated by tau oligomers and monomers in microglia directly and mediate Aβ-induced tau pathology. Interestingly, tau protein also participated in the priming step for the activation of NLRP3 inflammasome [156, 157]. NLRP3-deficient mice showed decreased tau pathology along with improved cognitive function. Further research demonstrated that aggregated tau could activate the NLRP3 inflammasome in primary microglia [158]. Tau-induced pathological responses can be alleviated in tau transgenic mice deficient for ASC or by the treatment of NLRP3 inhibitors [158]. The latest study further emphasized the importance of NLRP3 inflammasome and suggested that the Drp1-HK1-NLRP3 signal axis may play a vital role in the white matter degeneration in AD [159]. Dynamin-related protein 1 (Drp1) is a mitochondrial fission guanosine triphosphatase. Knock-down of Drp1 in mature oligodendrocytes-specific heterozygous could inhibit the activation of NLRP3 inflammasome, correct myelin degeneration and axonal loss, and improve cognitive function. White matter loss often presents early in the course of AD and is hypothesized to be the initial affair in AD pathology. The findings may provide new ideas for the treatment of AD.
Given that the NLRP3 inflammasome signaling axis may play an essential role in the pathogenesis of AD, targeting NLRP3 as a potential treatment for AD attracted more attention and has been discussed extensively. Here, we will primarily discuss the latest research, especially the pharmaceutical research of Chinese traditional herb-derived compounds.
Numerous pieces of research demonstrated that MCC950 could reduce Aβ-induced pathological events and improve cognitive function. MCC950 can inhibit the activation of NLRP3 inflammasome in microglia, ameliorate the impairment of synaptic plasticity, and reduce the accumulation of Aβ [160]. Meanwhile, the inactivation of NLRP3 inflammasome mediated by MCC950 could attenuate the reactivity of microglia induced by Aβ1-42 oligomers and improve memory impairment [161]. MCC950 could also completely suppress the immune response mediated by the NLRP3 inflammasome pathway which was induced by fibrils and low molecular weight Aβ aggregates [153]. In addition, the activation of IL-1β induced by tau aggregates was also prevented by MCC950, as well as the tau-mediated pathological changes [158]. However, the phase II clinical trial of MCC950 was suspended due to the possible hepatotoxicity in rheumatoid arthritis [162]. Drug improvement based on MCC950 to reduce the side effect may be the next potential strategy. Bay 11–7082 is another inhibitor of the NF-κB/NLRP3 pathway. The pre-administration of Bay 11–7082 can block the activation of the NLRP3 inflammasome and attenuate neuronal damage and cognitive dysfunction in aged rats [63]. In APP23 mice treated with kainic acid (KA), BAY 11–7082 protected them from KA-induced neuronal degeneration and Aβ deposition, thereby improving their cognitive function [64]. VX-765 is a reversible caspase-1 inhibitor that has been found to cross the blood–brain barrier and exert neuroprotective effects. In the mouse model of AD, treatment with VX-765 could reduce neuroinflammation and deposition of Aβ plaques, ultimately improving the neurocognitive function [67]. Unfortunately, the clinical trial of VX-765 was halted due to its hepatotoxicity and immunosuppression [162]. The more inspiring discovery came from fenamate non-steroidal anti-inflammatory drugs (NSAIDs), compounds already approved by the FDA for other treatments, which could also act as selective inhibitors of NLRP3 inflammasome [87]. The results showed that fenamate NSAIDs could inhibit NLRP3 activation by blocking the volume-regulated anion channel (VRAC), independently of cyclooxygenase (COX) enzymes. Importantly, treatment with mefenamic acid protected the mice from Aβ-induced memory deficits and NLRP3-mediated neuroinflammation in two animal models of AD [87]. These encouraging findings in animal studies may provide a more rapid breakthrough in clinical trials. Other NLRP3 inflammasome-specific inhibitors, such as inzomelid (Inflazome/Roche) and NT-0167 (NodThera/Roche), are currently undergoing clinical trials. A recent finding showed that miR-22 expression levels were lower in peripheral blood of AD patients than in healthy subjects. Moreover, miR-22 attenuated cognitive dysfunction in AD mice by targeting GSDMD to modulate glial cell pyroptosis, inhibit the activation of NLRP3 inflammasome, and decrease inflammatory cytokine release [86].
In addition, many plant extracts and traditional Chinese medicines have also been shown to have neuroprotective effects in AD by modulating the NLRP3 inflammasome pathway. Dl-3-n-butylphthalide (Dl-NBP) is the active ingredient extracted from Chinese herbal celery seeds. It is not only a commonly used drug for the treatment of ischemic stroke but also a novel therapeutic approach for neurodegenerative diseases. TXNIP has been demonstrated a key role in the activation of the NLRP3 inflammasome [163]. In APP/PS1 mice, Dl-NBP treatment could suppress TXNIP-NLRP3 interaction, inhibit NLRP3 inflammasome-mediated inflammatory damage by up-regulating Nrf2, ameliorate neuronal apoptosis, and thereby reduce oxidative stress damage [78]. Achyranthes bidentate is a traditional Chinese medicine with anti-inflammatory and antioxidant features and has been used for the treatment of dementia for a long time. A recent study showed that Achyranthes bidentate polypeptide fraction κ (ABPPκ) decreased Aβ oligomer-induced IκBα phosphorylation and NLRP3 expression in BV2 microglia. In vivo, pre-treatment of ABPPk reduced the expression of NLRP3, cleaved caspase-1, and ASC in the brain, promoted the polarization of M2 phenotype in microglia, and thus attenuated the memory impairment and the loss of hippocampal neurons in mice [80]. Resveratrol is a polyphenol compound derived from natural plants mainly in red grape skins and wine. It was reported that resveratrol could reverse the mitochondrial dysfunction by decreasing the expression of NF-κB/NLRP3/IL-1β, ameliorate learning and memory impairment, and reduce the neural injury in the hippocampus in the Aβ1–42-induced mouse model of AD [73]. Another recent study highlighted the crucial role of the low-density lipoprotein receptor-related protein 1 (LRP1) in the regulation of the endocytosis of tau and its spread [164]. Polyphenol (LSP) extracted from lychee seed could inhibit NLRP3 inflammasome-mediated neuroinflammation in vitro and improve cognitive function in APP/PS1 mice. It was further clarified that the molecular mechanism of LSP was mediated by up-regulating the expression of the LRP1/AMPK signaling pathway and enhancing the microglial autophagy [81]. In conclusion, considering their good safety and fewer side effects, patients with AD may benefit from NLRP3-targeting traditional Chinese herbal medicines, but it requires more research.
Parkinson’s Disease
It is suggested that the NLRP3 inflammasome may play a critical role in the pathogenesis of Parkinson's disease (PD) [68]. α-Synuclein (αSyn) is the pathologic feature protein of PD whose dysfunction is closely related to PD pathogenesis. Neuronal uptake of αSyn leads to impaired mitochondrial function [165] and the following release of excess mitochondrial reactive oxygen species (mtROS), which could directly activate the NLRP3 inflammasome in macrophages and microglia [166]. It was further clarified that αSyn could potently trigger the LPS-independent priming and activation of NLRP3 inflammasome assembly in microglia both in vivo and in vitro models of PD [17]. Expression of the NLRP3 inflammasome component was generally increased in PD animal models [18, 71], as well as in brain tissues [18] and peripheral blood samples [167, 168] from PD patients. Nlrp3-knockout mice suffered less inflammatory cascade response and hazardous neurotoxicity than Nlrp3-expressing wild-type ones [71]. Moreover, NLRP3 deletion markedly reduced motor dysfunction and dopaminergic neurodegeneration in the MPTP-induced mouse model of PD [71]. Similar results were observed in the MCC950-treated mouse. The administration of MCC950 in various animal PD models inhibited the activation of NLRP3 inflammasome and efficiently attenuated motor impairment, nigrostriatal dopaminergic degeneration, and the aggregation of α-synaptic nucleoproteins [18]. Inhibition of the downstream of the NLRP3/caspase-1/IL-1β pathway by caspase-1 knockout or caspase-1 inhibitor also alleviated dopaminergic neuronal loss and dyskinesia in murine models under PD conditions [68, 69]. In addition, the blockade of IL-1 receptors by IL-1 receptor antagonist (IL-1Ra) significantly reduced the degeneration of dopaminergic neurons and motor deficits in vivo [71]. Dl-NBP has been used in the treatment of acute ischemic stroke in China. Interestingly, both in vivo and in vitro, Dl-NBP inhibited NLRP3 inflammasome activation and α-Syn aggregation, thereby suppressing neuroinflammation, ameliorating mitochondrial damage, and rescuing dopaminergic neurons [79]. Furthermore, Perillyl Alcohol, a monoterpene compound found in the essential oils of plants, could support neuronal survival and improve behavioral activities in the mouse via inhibiting the NLRP3 inflammasome activation, thereby elevating levels of various antioxidant enzymes and restoring levels of dopamine and other neurotransmitters in the striatum [82]. These latest studies highlight the role of NLRP3 inflammasome as a potential target for PD treatment.
Multiple Sclerosis
Previous studies demonstrated that in patients with relapse-remitting multiple sclerosis (RRMS), the expression levels of NLRP3 and IL-1β were significantly different in responders and non-responders with the administration of interferon beta (IFN-β) treatment [169]. However, this result is still controversial. Some studies came to similar conclusions [170], and others came to the opposite ones. In contrast, Sunny Malhotra et al.’s findings were against the involvement of polymorphisms located in the Nlrp3 gene and the response to IFN-β in multiple sclerosis (MS) patients [171]. Recently, it was shown that the presence of IL-1β signaling in peripheral blood cells of patients with primary progressive MS (PPMS) rather than relapsed clinical types [172]. This IL-1β signature was related to the upregulation of an overactive NLRP3 inflammasome. More importantly, NLRP3 inflammasome was suggested to act as a pathogenic factor in MS. PPMS patients with higher IL-1β levels in blood cells had accelerated disease progression and required walking-help ten years earlier than those with lower IL-1β levels [172].
Experimental autoimmune encephalomyelitis (EAE) is an ideal disease model of human MS. Earlier research indicated that MCC950 treatment could reduce IL-1β generation in vivo and attenuate the severity of EAE [45]. The results were reaffirmed by Malhotra et al. Mice treated with MCC950 were associated with less disease severity in EAE and reduced LPS-induced axonal damage [172]. Oral treatment with MCC950 (50 mg/kg/day) halted disease relapses and reversed MS-associated central neuropathic pain in the relapsing–remitting EAE mouse model [173]. More interestingly, NLRP3 expression was observed in astrocytes, microglia, and neurons, although only microglia and astrocytes displayed IL-1β immunoreactivity [172]. Another small-molecule NLRP3 inflammasome inhibitor, JC-171, has been identified to reduce NLRP3-mediated IL-1β secretion in a dose-dependent manner by disrupting NLRP3-ASC interactions in both in vitro and in vivo EAE models [55]. Manoalide is a newly identified selective NLRP3 inhibitor that acts by blocking the NEK7–NLRP3 interaction [70]. Manoalide treatment could relieve the inflammation and attenuate the pathogenesis of EAE in mice [70].
Other Neurological Disorders
Traumatic brain injury (TBI)
TBI provokes an inflammatory cascade including potassium and chloride efflux, ATP transferred to the extracellular space, alteration of calcium signaling, mitochondrial injury, and ROS release, all of which eventually trigger the NLRP3 inflammasome activation [174]. Treatment of MCC950 in animals at the acute stages of TBI could reduce microglial activation, maintain the integrity of the blood–brain barrier, attenuate NLRP3-mediated neuroinflammation, and improve neurological outcomes [50, 51]. Of note, the efficacy of MCC950 was inhibited in microglia-depleted TBI animals [50]. BAY 11–7082, which selectively inhibits the NLRP3 inflammasome through interaction with NLRP3 ATPase, significantly attenuated inflammatory infiltration and damage in the cortex and hippocampus of murine within 24 h after TBI [65]. The synergistic effect of Bay 11–7082 and dexmedetomidine was further found in inhibiting NLRP3/caspase-1 axis activity and improving neurological dysfunction following TBI [175]. The study also indicated that the knockout of NLRP3 presented overlapping outcomes. NLRP3-deficient mice showed preserved cognitive function and less severe brain damage compared to wild-type mice [65].
Epilepsy
NLRP3 inflammasome activation has been reported to contribute to the pathogenesis of seizures. The elevated expression of NLRP3 inflammasome was observed in the hippocampus of different epilepsy murine models [176, 177] and in the hippocampus of patients with mesial temporal lobe epilepsy [178]. NLRP3 knockdown in amygdala kindling-induced status epilepticus rat models was associated with reduced expression of IL-1β and caspase-1, along with improved seizure severity [177]. It was further clarified that NLRP3-knockout also protected mouse and cellular epilepsy models from neuronal apoptosis [60]. Similar results were observed in MCC950-treated cellular models of epilepsy [60]. Unfortunately, as previously described, phase II clinical trials of two caspase-1-inhibiting peptidomimetic prodrugs, VX-740 and VX-765, for the treatment of arthritis, epilepsy, and psoriasis were terminated due to liver toxicity [162].
Others
In murine cardiac arrest (CA) and cardiopulmonary resuscitation (CPR) models, MCC950-treated mice showed improvements in functional recovery and survival rates [179]. The conclusion was further reconfirmed in another study by intraperitoneal injection of MCC950 (10 mg/kg) after the return of spontaneous circulation (ROSC) in models of CA. Compared with the control group, cerebral microcirculation, cerebral edema, 24-h survival rate, and neurological deficits were remarkably improved in animals treated with MCC950 [180]. In addition, NLRP3-mediated neuronal pyroptosis contributed to the pathologic process of sepsis-associated encephalopathy (SAE). Administration of MCC950 could reverse these effects and rescue cognitive deficits in mouse models of SAE [61]. MCC950 could also attenuate inflammation, neuronal apoptosis, and mitochondrial disorders via the Nrf2 pathway in SAE mice [181]. Nevertheless, the underlying mechanisms of NLRP3 in those diseases require more investigation.
Conclusion and Prospect for the Future
Activation of NLRP3 inflammasome is a pathological hallmark common to numerous immune-mediated and degenerative neurological diseases. NLRP3 has been shown to impact various neurological disease models in mice, highlighting the potential application of NLRP3-targeted therapy for these diseases. Several compounds interfering with the NLRP3 inflammasome pathway have exhibited therapeutic effects in preclinical trials. Due to the genetic and biological intricacy of neurological disorders such as AD and PD, a single approach may not be efficient for their treatments. Thus inflammasome inhibitors may be able to work in conjunction with other drugs and support their therapeutic outcomes. Considering the safety and fewer side effects, traditional Chinese medicines, as well as plant-derived ingredients also offer innovative perspectives and alternative options for intractable conditions. However, despite decades of research, our understanding of NLRP3 inflammasome in central nervous system diseases is still lacking. A better appreciation of the physiological mechanisms regulating NLRP3 inflammasome will lead to a better assessment of the therapeutic potential of inflammasome inhibitors in human neurological disorders. We also expect that targeting NLRP3 inflammasome of the neurological diseases will be applied to the clinical treatment soon, so as to benefit more patients.
Data Availability
Data sharing not applicable to this article as no datasets were generated or analyzed during the current study.
Abbreviations
- αSyn:
-
α-Synuclein
- ABPPκ:
-
Achyranthes bidentate polypeptide fraction κ
- AD:
-
Alzheimer’s disease
- APP/PS1:
-
Amyloid precursor protein/presenilin 1
- ASC:
-
Apoptosis-associated speck-like protein containing a CARD
- ATP:
-
Triphosphate
- Aβ:
-
Amyloid β
- CA:
-
Cardiac arrest
- CAPS:
-
Cryptochrome-associated periodic syndrome
- CLICs:
-
Chloride intracellular channel proteins
- CNS:
-
Central nervous system
- COX:
-
Cyclooxygenase
- CPR:
-
Cardiopulmonary resuscitation
- DAMPs:
-
Damage-associated molecular patterns
- Dl-NBP:
-
Dl-3-n-butylphthalide
- Drp1:
-
Dynamin-related protein 1
- dTGN:
-
Dispersed trans-Golgi network
- EAE:
-
Experimental autoimmune encephalomyelitis
- ER:
-
Endoplasmic reticulum
- FDA:
-
Food and Drug Administration
- GSDMD:
-
Gasdermin D
- ICH:
-
Intracerebral hemorrhage
- IFN-β:
-
Interferon beta
- IL-1Ra:
-
IL-1 receptor antagonist
- IL-1β:
-
Pro-interleukin-1β
- IL-18:
-
Interleukin-18
- KA:
-
Kainic acid
- LPS:
-
Lipopolysaccharide
- LRP1:
-
Low-density lipoprotein receptor-related protein 1
- LRR:
-
Leucine-rich repeat
- LSP:
-
Polyphenol
- MAMs:
-
Mitochondria-associated endoplasmic reticulum membranes
- MAPK:
-
Mitogen-activated protein kinase
- miRNA:
-
MicroRNA
- MPTP:
-
1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- MS:
-
Multiple sclerosis
- mtDNA:
-
Mitochondrial DNA
- mtROS:
-
Mitochondrial reactive oxygen species
- NEK7:
-
NIMA-related protein kinase 7
- NF-κB:
-
Nuclear factor-κB
- NLRC4:
-
NLR family CARD domain-containing protein 4
- NLRP3:
-
NOD-, LRR-, and PYRIN domain-containing protein 3
- NOD2:
-
Nucleotide-binding oligomerization domain-containing protein 2
- Nrf2:
-
Erythroid-2–related factor 2
- NSAIDs:
-
Non-steroidal anti-inflammatory drugs
- oxPAPC:
-
Oxidized phospholipid 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphorylcholine
- P2X7:
-
Purinergic 2X7 receptor
- PAMPs:
-
Pathogen-associated molecular patterns
- PD:
-
Parkinson’s disease
- PKD:
-
Protein kinase domain
- PPMS:
-
Primary progressive multiple sclerosis
- PRRs:
-
Pattern recognition receptors
- PtdIns4P:
-
Phosphatidylinositol-4-phosphate
- RNAi:
-
RNA interference
- ROS:
-
Reactive oxygen species
- ROSC:
-
Return of spontaneous circulation
- RRMS:
-
Relapse-remitting multiple sclerosis
- SAE:
-
Sepsis-associated encephalopathy
- SAH:
-
Subarachnoid hemorrhage
- SCAP:
-
SREBP cleavage-activating protein
- SREBP:
-
Sterol regulatory element binding protein
- TBI:
-
Traumatic brain injury
- TGN:
-
trans-Golgi network
- TLR4:
-
Toll-like receptor 4
- TLRs:
-
Toll-like receptors
- TNF:
-
Tumor necrosis factor
- tPA:
-
Tissue plasminogen activator
- TREM-1:
-
Receptor expressed on myeloid cells 1
- TWIK2:
-
Two-pore domain weak inwardly rectifying K + channel 2
- TXNIP:
-
Thioredoxin-interacting protein
- VRAC:
-
Volume-regulated anion channel
References
Lu A, Magupalli VG, Ruan J, Yin Q, Atianand MK, Vos MR, Schroder GF, Fitzgerald KA et al (2014) Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell 156(6):1193–1206. https://doi.org/10.1016/j.cell.2014.02.008
Fernandes-alnemri T, Wu J, Yu J, Datta P, Miller B, Jankowski W, Rosenberg S, Zhang J et al (2007) The pyroptosome: a supramolecular assembly of ASC dimers mediating inflammatory cell death via caspase-1 activation. Cell Death Differ 14(9):1590–1604
Hoss F, Rodriguez-alcazar JF, Latz E (2017) Assembly and regulation of ASC specks. Cell Mol Life Sci 74(7):1211–1229. https://doi.org/10.1007/s00018-016-2396-6
Broz P, Dixit VM (2016) Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol 16(7):20–407. https://doi.org/10.1038/nri.2016.58
Duncan JA, Bergstralh DT, Wang Y, Willingham SB, Ye Z, Zimmermann AG, Ting JP (2007) Cryopyrin/NALP3 binds ATP/dATP, is an ATPase, and requires ATP binding to mediate inflammatory signaling. Proc Natl Acad Sci U S A 104(19):6–8041
Hoffman HM, Mueller JL, Broide DH, Wanderer AA, Kolodner RD (2001) Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nat Genet 29(3):301–305
Aksentijevich I, Nowak M, Mallah M, Chae JJ, Watford WT, Hofmann SR, Stein L, Russo R et al (2002) De novo CIAS1 mutations, cytokine activation, and evidence for genetic heterogeneity in patients with neonatal-onset multisystem inflammatory disease (NOMID): a new member of the expanding family of pyrin-associated autoinflammatory diseases. Arthritis Rheum 46(12):3340–3348
Aganna E, Martinon F, Hawkins PN, Ross JB, Swan DC, Booth DR, Lachmann HJ, Bybee A et al (2002) Association of mutations in the NALP3/CIAS1/PYPAF1 gene with a broad phenotype including recurrent fever, cold sensitivity, sensorineural deafness, and AA amyloidosis. Arthritis Rheum 46(9):2445–2452
Andreeva L, David L, Rawson S, Shen C, Pasricha T, Pelegrin P, Wu H (2021) NLRP3 cages revealed by full-length mouse NLRP3 structure control pathway activation. Cell 184(26):6299-6312.e22. https://doi.org/10.1016/j.cell.2021.11.011
Guo C, Chi Z, Jiang D, Xu T, Yu W, Wang Z, Chen S, Zhang L et al (2018) Cholesterol homeostatic regulator SCAP-SREBP2 integrates NLRP3 inflammasome activation and cholesterol biosynthetic signaling in macrophages. Immunity 49(5):842-856.e7. https://doi.org/10.1016/j.immuni.2018.08.021
Chen J, Chen ZJ (2018) PtdIns4P on dispersed trans-Golgi network mediates NLRP3 inflammasome activation. Nature 564(7734):71–76. https://doi.org/10.1038/s41586-018-0761-3
He Y, Zeng MY, Yang D, Motro B, Nunez G (2016) NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature 530(7590):354–357. https://doi.org/10.1038/nature16959
Shi H, Wang Y, Li X, Zhan X, Tang M, Fina M, Su L, Pratt D et al (2016) NLRP3 activation and mitosis are mutually exclusive events coordinated by NEK7, a new inflammasome component. Nat Immunol 17(3):250–258. https://doi.org/10.1038/ni.3333
Maturana CJ, Aguirre A, Saez JC (2017) High glucocorticoid levels during gestation activate the inflammasome in hippocampal oligodendrocytes of the offspring. Dev Neurobiol 77(5):625–642. https://doi.org/10.1002/dneu.22409
Johann S, Heitzer M, Kanagaratnam M, Goswami A, Rizo T, Weis J, Troost D, Beyer C (2015) NLRP3 inflammasome is expressed by astrocytes in the SOD1 mouse model of ALS and in human sporadic ALS patients. Glia 63(12):2260–2273. https://doi.org/10.1002/glia.22891
Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, Fitzgerald KA, Latz E et al (2008) The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol 9(8):65–857. https://doi.org/10.1038/ni.1636
Panicker N, Sarkar S, Harischandra DS, Neal M, Kam T, Jin H, Saminathan H, Langley M et al (2019) Fyn kinase regulates misfolded alpha-synuclein uptake and NLRP3 inflammasome activation in microglia. J Exp Med 216(6):1411–1430. https://doi.org/10.1084/jem.20182191
Gordon R, Albornoz EA, Christie DC, Langley MR, Kumar V, Mantovani S, Robertson AAB, Butler MS et al (2018) Inflammasome inhibition prevents alpha-synuclein pathology and dopaminergic neurodegeneration in mice. Sci Transl Med 10(465). https://doi.org/10.1126/scitranslmed.aah4066
Wu A, Zhou X, Qiao G, Yu L, Tang Y, Yan L, Qiu W, Pan R et al (2021) Targeting microglial autophagic degradation in NLRP3 inflammasome-mediated neurodegenerative diseases. Ageing Res Rev 65:101202. https://doi.org/10.1016/j.arr.2020.101202
Hong P, Gu R, Li F, Xiong X, Liang W, You Z, Zhang H (2019) NLRP3 inflammasome as a potential treatment in ischemic stroke concomitant with diabetes. J Neuroinflammation 16(1):121. https://doi.org/10.1186/s12974-019-1498-0
Franchi L, Eigenbrod T, Nunez G (2009) Cutting edge: TNF-alpha mediates sensitization to ATP and silica via the NLRP3 inflammasome in the absence of microbial stimulation. J Immunol 183(2):6–792. https://doi.org/10.4049/jimmunol.0900173
Xing Y, Yao X, Li H, Xue G, Guo Q, Yang G, An L, Zhang Y et al (2017) Cutting edge: TRAF6 mediates TLR/IL-1R signaling-induced nontranscriptional priming of the NLRP3 inflammasome. J Immunol 199(5):1561–1566. https://doi.org/10.4049/jimmunol.1700175
Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, Macdonald K, Speert D, Fernandes-alnemri T, Wu J et al (2009) Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol 183(2):787–91. https://doi.org/10.4049/jimmunol.0901363
Greaney AJ, Leppla SH, Moayeri M (2015) Bacterial exotoxins and the inflammasome. Front Immunol 6:570. https://doi.org/10.3389/fimmu.2015.00570
Hornung V, Bauernfeind F, Halle A, Samstad EO, Kono H, Rock KL, Fitzgerald KA, Latz E (2008) Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol 9(8):56–847. https://doi.org/10.1038/ni.1631
Liston A, Masters SL (2017) Homeostasis-altering molecular processes as mechanisms of inflammasome activation. Nat Rev Immunol 17(3):208–214. https://doi.org/10.1038/nri.2016.151
Munoz-planillo R, Kuffa P, Martinez-colon G, Smith BL, Rajendiran TM, Nunez G (2013) K(+) efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 38(6):1142–1153. https://doi.org/10.1016/j.immuni.2013.05.016
Di A, Xiong S, Ye Z, Malireddi RKS, Kometani S, Zhong M, Mittal M, Hong Z et al (2018) The TWIK2 potassium efflux channel in macrophages mediates NLRP3 inflammasome-induced inflammation. Immunity 49(1):56-65.e4. https://doi.org/10.1016/j.immuni.2018.04.032
Katsnelson MA, Lozada-soto KM, Russo HM, Miller BA, Dubyak GR (2016) NLRP3 inflammasome signaling is activated by low-level lysosome disruption but inhibited by extensive lysosome disruption: roles for K+ efflux and Ca2+ influx. Am J Physiol Cell Physiol 311(1):83-C100. https://doi.org/10.1152/ajpcell.00298.2015
Murakami T, Ockinger J, Yu J, Byles V, Mccoll A, Hofer AM, Horng T (2012) Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc Natl Acad Sci U S A 109(28):11282–11287. https://doi.org/10.1073/pnas.1117765109
Green JP, Yu S, Martin-sanchez F, Pelegrin P, Lopez-castejon G, Lawrence CB, Brough D (2018) Chloride regulates dynamic NLRP3-dependent ASC oligomerization and inflammasome priming. Proc Natl Acad Sci U S A 115(40):9371-E9380. https://doi.org/10.1073/pnas.1812744115
Dostert C, Petrilli V, Bruggen RV, Steele C, Mossman BT, Tschopp J (2008) Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science 320(5876):674–7. https://doi.org/10.1126/science.1156995
Zhou R, Yazdi AS, Menu P, Tschopp J (2011) A role for mitochondria in NLRP3 inflammasome activation. Nature 469(7329):221–225. https://doi.org/10.1038/nature09663
Iyer SS, He Q, Janczy JR, Elliott EI, Zhong Z, Olivier AK, Sadler JJ, Knepper-adrian V et al (2013) Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity 39(2):311–323. https://doi.org/10.1016/j.immuni.2013.08.001
Liu Q, Zhang D, Hu D, Zhou X, Zhou Y (2018) The role of mitochondria in NLRP3 inflammasome activation. Mol Immunol 103:115–124. https://doi.org/10.1016/j.molimm.2018.09.010
Zhou Y, Tong Z, Jiang S, Zheng W, Zhao J, Zhou X (2020) The roles of endoplasmic reticulum in NLRP3 inflammasome activation. Cells-Basel 9(5). https://doi.org/10.3390/cells9051219
Tao Y, Yang Y, Zhou R, Gong T (2020) Golgi apparatus: an emerging platform for innate immunity. Trends Cell Biol 30(6):467–477. https://doi.org/10.1016/j.tcb.2020.02.008
Zhang Z, Meszaros G, He W, Xu Y, Magliarelli HDF, Mailly L, Mihlan M, Liu Y et al (2017) Protein kinase D at the Golgi controls NLRP3 inflammasome activation. J Exp Med 214(9):2671–2693. https://doi.org/10.1084/jem.20162040
Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, Hu L, Shao F (2014) Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 514(7521):187–192. https://doi.org/10.1038/nature13683
Kayagaki N, Warming S, Lamkanfi M, Walle LV, Louie S, Dong J, Newton K, Qu Y et al (2011) Non-canonical inflammasome activation targets caspase-11. Nature 479(7371):117–121. https://doi.org/10.1038/nature10558
Kayagaki N, Stowe IB, Lee BL, O’rourke K, Anderson K, Warming S, Cuellar T, Haley B et al (2015) Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 526(7575):666–671. https://doi.org/10.1038/nature15541
Baker PJ, Boucher D, Bierschenk D, Tebartz C, Whitney PG, D’silva DB, Tanzer MC, Monteleone M et al (2015) NLRP3 inflammasome activation downstream of cytoplasmic LPS recognition by both caspase-4 and caspase-5. Eur J Immunol 45(10):26–2918. https://doi.org/10.1002/eji.201545655
Zanoni I, Tan Y, Gioia MD, Broggi A, Ruan J, Shi J, Donado CA, Shao F et al (2016) An endogenous caspase-11 ligand elicits interleukin-1 release from living dendritic cells. Science 352(6290):1232–6. https://doi.org/10.1126/science.aaf3036
Baldwin AG, Brough D, Freeman S (2016) Inhibiting the inflammasome: a chemical perspective. J Med Chem 59(5):1691–1710. https://doi.org/10.1021/acs.jmedchem.5b01091
Coll RC, Robertson AAB, Chae JJ, Higgins SC, Munoz-planillo R, Inserra MC, Vetter I, Dungan LS et al (2015) A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med 21(3):248–255. https://doi.org/10.1038/nm.3806
Coll RC, Hill JR, Day CJ, Zamoshnikova A, Boucher D, Massey NL, Chitty JL, Fraser JA et al (2019) MCC950 directly targets the NLRP3 ATP-hydrolysis motif for inflammasome inhibition. Nat Chem Biol 15(6):556–559. https://doi.org/10.1038/s41589-019-0277-7
Jiang H, He H, Chen Y, Huang W, Cheng J, Ye J, Wang A, Tao J et al (2017) Identification of a selective and direct NLRP3 inhibitor to treat inflammatory disorders. J Exp Med 214(11):3219–3238. https://doi.org/10.1084/jem.20171419
Zhai Y, Meng X, Ye T, Xie W, Sun G, Sun X (2018) Inhibiting the NLRP3 inflammasome activation with MCC950 ameliorates diabetic encephalopathy in db/db mice. Molecules 23(3). https://doi.org/10.3390/molecules23030522
Dempsey C, Araiz AR, Bryson KJ, Finucane O, Larkin C, Mills EL, Robertson AAB, Cooper MA et al (2017) Inhibiting the NLRP3 inflammasome with MCC950 promotes non-phlogistic clearance of amyloid-beta and cognitive function in APP/PS1 mice. Brain Behav Immun 61:306–316. https://doi.org/10.1016/j.bbi.2016.12.014
Xu X, Yin D, Ren H, Gao W, Li F, Sun D, Wu Y, Zhou S et al (2018) Selective NLRP3 inflammasome inhibitor reduces neuroinflammation and improves long-term neurological outcomes in a murine model of traumatic brain injury. Neurobiol Dis 117:15–27. https://doi.org/10.1016/j.nbd.2018.05.016
Ismael S, Nasoohi S, Ishrat T (2018) MCC950, the selective inhibitor of nucleotide oligomerization domain-like receptor protein-3 inflammasome, protects mice against traumatic brain injury. J Neurotrauma 35(11):1294–1303. https://doi.org/10.1089/neu.2017.5344
Heijden TVD, Kritikou E, Venema W, Duijn JV, Santbrink PJV, Slutter B, Foks AC, Bot I et al (2017) NLRP3 inflammasome inhibition by MCC950 reduces atherosclerotic lesion development in apolipoprotein E-deficient mice-brief report. Arterioscler Thromb Vasc Biol 37(8):1457–1461. https://doi.org/10.1161/ATVBAHA.117.309575
Mridha AR, Wree A, Robertson AAB, Yeh MM, Johnson CD, Rooyen DMV, Haczeyni F, Teoh NC et al (2017) NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. J Hepatol 66(5):1037–1046. https://doi.org/10.1016/j.jhep.2017.01.022
Juliana C, Fernandes-alnemri T, Wu J, Datta P, Solorzano L, Yu J, Meng R, Quong AA et al (2010) Anti-inflammatory compounds parthenolide and Bay 11–7082 are direct inhibitors of the inflammasome. J Biol Chem 285(13):9792–9802. https://doi.org/10.1074/jbc.M109.082305
Guo C, Fulp JW, Jiang Y, Li X, Chojnacki JE, Wu J, Wang X, Zhang S (2017) Development and characterization of a hydroxyl-sulfonamide analogue, 5-chloro-N-[2-(4-hydroxysulfamoyl-phenyl)-ethyl]-2-methoxy-benzamide, as a novel NLRP3 inflammasome inhibitor for potential treatment of multiple sclerosis. ACS Chem Neurosci 8(10):2194–2201. https://doi.org/10.1021/acschemneuro.7b00124
Youm Y, Nguyen KY, Grant RW, Goldberg EL, Bodogai M, Kim D, D’agostino D, Planavsky N et al (2015) The ketone metabolite beta-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat Med 21(3):263–269. https://doi.org/10.1038/nm.3804
Yang F, Wang Z, Wei X, Han H, Meng X, Zhang Y, Shi W, Li F et al (2014) NLRP3 deficiency ameliorates neurovascular damage in experimental ischemic stroke. J Cerebral Blood Flow Metab 34(4):660–667. https://doi.org/10.1038/jcbfm.2013.242
Ren H, Kong Y, Liu Z, Zang D, Yang X, Wood K, Li M, Liu Q (2018) Selective NLRP3 (pyrin domain-containing protein 3) inflammasome inhibitor reduces brain injury after intracerebral hemorrhage. Stroke 49(1):184–192. https://doi.org/10.1161/STROKEAHA.117.018904
Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-saecker A, Griep A, Axt D et al (2013) NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 493(7434):674–678. https://doi.org/10.1038/nature11729
Shen K, Mao Q, Yin X, Zhang C, Jin Y, Deng A, Gu Z, Chen B (2018) NLRP3 inflammasome activation leads to epileptic neuronal apoptosis. Curr Neurovasc Res 15(4):276–281. https://doi.org/10.2174/1567202616666181122165540
Fu Q, Wu J, Zhou X, Ji M, Mao Q, Li Q, Zong M, Zhou Z et al (2019) NLRP3/caspase-1 pathway-induced pyroptosis mediated cognitive deficits in a mouse model of sepsis-associated encephalopathy. Inflammation 42(1):306–318. https://doi.org/10.1007/s10753-018-0894-4
Feng L, Chen Y, Ding R, Fu Z, Yang S, Deng X, Zeng J (2015) P2X7R blockade prevents NLRP3 inflammasome activation and brain injury in a rat model of intracerebral hemorrhage: involvement of peroxynitrite. J Neuroinflammation 12:190. https://doi.org/10.1186/s12974-015-0409-2
Liu P, Gao T, Li T, Yang Y, Xu Y, Xu Z, Mi W (2021) Repeated propofol exposure-induced neuronal damage and cognitive impairment in aged rats by activation of NF-kappaB pathway and NLRP3 inflammasome. Neurosci Lett 740:135461. https://doi.org/10.1016/j.neulet.2020.135461
Ruan Y, Qiu X, Lv Y, Dong D, Wu X, Zhu J, Zheng X (2019) Kainic acid induces production and aggregation of amyloid beta-protein and memory deficits by activating inflammasomes in NLRP3- and NF-kappaB-stimulated pathways. Aging 11(11):3795–3810. https://doi.org/10.18632/aging.102017
Irrera N, Pizzino G, Calo M, Pallio G, Mannino F, Fama F, Arcoraci V, Fodale V et al (2017) Lack of the Nlrp3 inflammasome improves mice recovery following traumatic brain injury. Front Pharmacol 8:459. https://doi.org/10.3389/fphar.2017.00459
Inoue M, Williams KL, Oliver T, Vandenabeele P, Rajan JV, Miao EA, Shinohara ML (2012) Interferon-beta therapy against EAE is effective only when development of the disease depends on the NLRP3 inflammasome. Sci Signal 5(225):ra38. https://doi.org/10.1126/scisignal.2002767
Flores J, Noel A, Foveau B, Lynham J, Lecrux C, Leblanc AC (2018) Caspase-1 inhibition alleviates cognitive impairment and neuropathology in an Alzheimer’s disease mouse model. Nat Commun 9(1):3916. https://doi.org/10.1038/s41467-018-06449-x
Mao Z, Liu C, Ji S, Yang Q, Ye H, Han H, Xue Z (2017) The NLRP3 inflammasome is involved in the pathogenesis of Parkinson’s disease in rats. Neurochem Res 42(4):1104–1115. https://doi.org/10.1007/s11064-017-2185-0
Qiao C, Zhang L, Sun X, Ding J, Lu M, Hu G (2017) Caspase-1 deficiency alleviates dopaminergic neuronal death via inhibiting caspase-7/AIF pathway in MPTP/p mouse model of Parkinson’s disease. Mol Neurobiol 54(6):4292–4302. https://doi.org/10.1007/s12035-016-9980-5
Li C, Lin H, He H, Ma M, Jiang W, Zhou R (2022) Inhibition of the NLRP3 inflammasome activation by manoalide ameliorates experimental autoimmune encephalomyelitis pathogenesis. Front Cell Dev Biol 10:822236. https://doi.org/10.3389/fcell.2022.822236
Lee E, Hwang I, Park S, Hong S, Hwang B, Cho Y, Son J, Yu J (2019) MPTP-driven NLRP3 inflammasome activation in microglia plays a central role in dopaminergic neurodegeneration. Cell Death Differ 26(2):213–228. https://doi.org/10.1038/s41418-018-0124-5
He Q, Li Z, Wang Y, Hou Y, Li L, Zhao J (2017) Resveratrol alleviates cerebral ischemia/reperfusion injury in rats by inhibiting NLRP3 inflammasome activation through Sirt1-dependent autophagy induction. Int Immunopharmacol 50:208–215. https://doi.org/10.1016/j.intimp.2017.06.029
Qi Y, Shang L, Liao Z, Su H, Jing H, Wu B, Bi K, Jia Y (2019) Intracerebroventricular injection of resveratrol ameliorated Abeta-induced learning and cognitive decline in mice. Metab Brain Dis 34(1):257–266. https://doi.org/10.1007/s11011-018-0348-6
Zhang X, Wu Q, Zhang Q, Lu Y, Liu J, Li W, Lv S, Zhou M et al (2017) Resveratrol attenuates early brain injury after experimental subarachnoid hemorrhage via inhibition of NLRP3 inflammasome activation. Front Neurosci 11:611. https://doi.org/10.3389/fnins.2017.00611
Xiao L, Dai Z, Tang W, Liu C, Tang B (2021) Astragaloside IV alleviates cerebral ischemia-reperfusion injury through NLRP3 inflammasome-mediated pyroptosis inhibition via activating Nrf2. Oxid Med Cell Longev 2021:9925561. https://doi.org/10.1155/2021/9925561
Yuan R, Fan H, Cheng S, Gao W, Xu X, Lv S, Ye M, Wu M et al (2017) Silymarin prevents NLRP3 inflammasome activation and protects against intracerebral hemorrhage. Biomed Pharmacother 93:308–315. https://doi.org/10.1016/j.biopha.2017.06.018
Liu H, Zhao L, Yue L, Wang B, Li X, Guo H, Ma Y, Yao C et al (2017) Pterostilbene attenuates early brain injury following subarachnoid hemorrhage via inhibition of the NLRP3 inflammasome and Nox2-related oxidative stress. Mol Neurobiol 54(8):5928–5940. https://doi.org/10.1007/s12035-016-0108-8
Wang C, Xu Y, Wang X, Guo C, Wang T, Wang Z (2019) Dl-3-n-Butylphthalide inhibits NLRP3 inflammasome and mitigates Alzheimer’s-like pathology via Nrf2-TXNIP-TrX axis. Antioxid Redox Signal 30(11):1411–1431. https://doi.org/10.1089/ars.2017.7440
Que R, Zheng J, Chang Z, Zhang W, Li H, Xie Z, Huang Z, Wang H et al (2021) Dl-3-n-Butylphthalide rescues dopaminergic neurons in Parkinson’s disease models by inhibiting the NLRP3 inflammasome and ameliorating mitochondrial impairment. Front Immunol 12:794770. https://doi.org/10.3389/fimmu.2021.794770
Ge X, Wang Y, Yu S, Cao X, Chen Y, Cheng Q, Ding F (2021) Anti-inflammatory activity of a polypeptide fraction from Achyranthes bidentate in amyloid beta oligomers induced model of Alzheimer’s disease. Front Pharmacol 12:716177. https://doi.org/10.3389/fphar.2021.716177
Qiu W, Pan R, Tang Y, Zhou X, Wu J, Yu L, Law BY, Ai W et al (2020) Lychee seed polyphenol inhibits Abeta-induced activation of NLRP3 inflammasome via the LRP1/AMPK mediated autophagy induction. Biomed Pharmacother 130:110575. https://doi.org/10.1016/j.biopha.2020.110575
Ahmed S, Panda SR, Kwatra M, Sahu BD, Naidu V (2022) Perillyl alcohol attenuates NLRP3 inflammasome activation and rescues dopaminergic neurons in experimental in vitro and in vivo models of Parkinson’s disease. ACS Chem Neurosci 13(1):53–68. https://doi.org/10.1021/acschemneuro.1c00550
Liu Y, Li Y, Xiao L, Chen L, Zheng S, Zeng E, Xu C (2021) Extracellular vesicles derived from M2 microglia reduce ischemic brain injury through microRNA-135a-5p/TXNIP/NLRP3 axis. Lab Investig 101(7):837–850. https://doi.org/10.1038/s41374-021-00545-1
Yang Z, Zhong L, Xian R, Yuan B (2015) MicroRNA-223 regulates inflammation and brain injury via feedback to NLRP3 inflammasome after intracerebral hemorrhage. Mol Immunol 65(2):267–276. https://doi.org/10.1016/j.molimm.2014.12.018
Hu L, Zhang H, Wang B, Ao Q, He Z (2020) MicroRNA-152 attenuates neuroinflammation in intracerebral hemorrhage by inhibiting thioredoxin interacting protein (TXNIP)-mediated NLRP3 inflammasome activation. Int Immunopharmacol 80:106141. https://doi.org/10.1016/j.intimp.2019.106141
Han C, Guo L, Yang Y, Guan Q, Shen H, Sheng Y, Jiao Q (2020) Mechanism of microRNA-22 in regulating neuroinflammation in Alzheimer’s disease. Brain Behav 10(6):01627. https://doi.org/10.1002/brb3.1627
Daniels MJD, Rivers-auty J, Schilling T, Spencer NG, Watremez W, Fasolino V, Booth SJ, White CS et al (2016) Fenamate NSAIDs inhibit the NLRP3 inflammasome and protect against Alzheimer’s disease in rodent models. Nat Commun 7:12504. https://doi.org/10.1038/ncomms12504
Chen B, Zhang M, Ji M, Zhang D, Chen B, Gong W, Li X, Zhou Y et al (2022) The neuroprotective mechanism of lithium after ischaemic stroke. Commun Biol 5(1):105. https://doi.org/10.1038/s42003-022-03051-2
Peng J, Wang H, Gong Z, Li X, He L, Shen Q, Pan J, Peng Y (2020) Idebenone attenuates cerebral inflammatory injury in ischemia and reperfusion via dampening NLRP3 inflammasome activity. Mol Immunol 123:74–87. https://doi.org/10.1016/j.molimm.2020.04.013
Ismael S, Nasoohi S, Yoo A, Mirzahosseini G, Ahmed HA, Ishrat T (2021) Verapamil as an adjunct therapy to reduce tPA toxicity in hyperglycemic stroke: implication of TXNIP/NLRP3 inflammasome. Mol Neurobiol 58(8):3792–3804. https://doi.org/10.1007/s12035-021-02384-z
Cao S, Shrestha S, Li J, Yu X, Chen J, Yan F, Ying G, Gu C et al (2017) Melatonin-mediated mitophagy protects against early brain injury after subarachnoid hemorrhage through inhibition of NLRP3 inflammasome activation. Sci Rep 7(1):2417. https://doi.org/10.1038/s41598-017-02679-z
Tsao CW, Aday AW, Almarzooq ZI, Alonso A, Beaton AZ, Bittencourt MS, Boehme AK, Buxton AE et al (2022) Heart Disease and Stroke Statistics-2022 update: a report from the American Heart Association. Circulation 145(8):153-e639. https://doi.org/10.1161/CIR.0000000000001052
Campbell BCV, Silva DAD, Macleod MR, Coutts SB, Schwamm LH, Davis SM, Donnan GA (2019) Ischaemic stroke. Nat Rev Dis Primers 5(1):70. https://doi.org/10.1038/s41572-019-0118-8
Pendlebury ST, Rothwell PM (2009) Prevalence, incidence, and factors associated with pre-stroke and post-stroke dementia: a systematic review and meta-analysis. Lancet Neurol 8(11):1006–1018. https://doi.org/10.1016/S1474-4422(09)70236-4
O’collins VE, Macleod MR, Donnan GA, Horky LL, Worp BHVD, Howells DW (2006) 1,026 experimental treatments in acute stroke. Ann Neurol 59(3):467–477. https://doi.org/10.1002/ana.20741
Guekht A, Skoog I, Edmundson S, Zakharov V, Korczyn AD (2017) ARTEMIDA Trial (a randomized trial of efficacy, 12 months international double-blind actovegin): a randomized controlled trial to assess the efficacy of actovegin in poststroke cognitive impairment. Stroke 48(5):1262–1270. https://doi.org/10.1161/STROKEAHA.116.014321
Fann DY, Lee S, Manzanero S, Chunduri P, Sobey CG, Arumugam TV (2013) Pathogenesis of acute stroke and the role of inflammasomes. Ageing Res Rev 12(4):66–941. https://doi.org/10.1016/j.arr.2013.09.004
Savage CD, Lopez-castejon G, Denes A, Brough D (2012) NLRP3-inflammasome activating DAMPs stimulate an inflammatory response in glia in the absence of priming which contributes to brain inflammation after injury. Front Immunol 3:288
Minutoli L, Puzzolo D, Rinaldi M, Irrera N, Marini H, Arcoraci V, Bitto A, Crea G et al (2016) ROS-mediated NLRP3 inflammasome activation in brain, heart, kidney, and testis ischemia/reperfusion injury. Oxid Med Cell Longev 2016:2183026. https://doi.org/10.1155/2016/2183026
Gong Z, Pan J, Shen Q, Li M, Peng Y (2018) Mitochondrial dysfunction induces NLRP3 inflammasome activation during cerebral ischemia/reperfusion injury. J Neuroinflammation 15(1):242. https://doi.org/10.1186/s12974-018-1282-6
Barrington J, Lemarchand E, Allan SM (2017) A brain in flame; do inflammasomes and pyroptosis influence stroke pathology? Brain Pathol 27(2):205–212. https://doi.org/10.1111/bpa.12476
Denes A, Pinteaux E, Rothwell NJ, Allan SM (2011) Interleukin-1 and stroke: biomarker, harbinger of damage, and therapeutic target. Cerebrovasc Dis 32(6):27–517. https://doi.org/10.1159/000332205
Fann DY, Lee SY, Manzanero S, Tang SC, Gelderblom M, Chunduri P, Bernreuther C, Glatzel M et al (2013) Intravenous immunoglobulin suppresses NLRP1 and NLRP3 inflammasome-mediated neuronal death in ischemic stroke. Cell Death Dis 4:790. https://doi.org/10.1038/cddis.2013.326
Fann DY, Santro T, Manzanero S, Widiapradja A, Cheng Y, Lee S, Chunduri P, Jo D et al (2014) Intermittent fasting attenuates inflammasome activity in ischemic stroke. Exp Neurol 257:114–119. https://doi.org/10.1016/j.expneurol.2014.04.017
Wang Q, Tang XN, Yenari MA (2007) The inflammatory response in stroke. J Neuroimmunol 184(1–2):53–68
Gustin A, Kirchmeyer M, Koncina E, Felten P, Losciuto S, Heurtaux T, Tardivel A, Heuschling P et al (2015) NLRP3 inflammasome is expressed and functional in mouse brain microglia but not in astrocytes. PLoS ONE 10(6):0130624. https://doi.org/10.1371/journal.pone.0130624
Prinz M, Erny D, Hagemeyer N (2017) Ontogeny and homeostasis of CNS myeloid cells. Nat Immunol 18(4):385–392. https://doi.org/10.1038/ni.3703
Salter MW, Stevens B (2017) Microglia emerge as central players in brain disease. Nat Med 23(9):1018–1027. https://doi.org/10.1038/nm.4397
Vilalta A, Brown GC (2018) Neurophagy, the phagocytosis of live neurons and synapses by glia, contributes to brain development and disease. FEBS J 285(19):3566–3575. https://doi.org/10.1111/febs.14323
Wang C, Zhang L, Li G, Shi Y, Li J, Zhang X, Wang Z, Ding F et al (2014) Mulberroside A protects against ischemic impairment in primary culture of rat cortical neurons after oxygen-glucose deprivation followed by reperfusion. J Neurosci Res 92(7):54–944. https://doi.org/10.1002/jnr.23374
Ye X, Shen T, Hu J, Zhang L, Zhang Y, Bao L, Cui C, Jin G et al (2017) Purinergic 2X7 receptor/NLRP3 pathway triggers neuronal apoptosis after ischemic stroke in the mouse. Exp Neurol 292:46–55. https://doi.org/10.1016/j.expneurol.2017.03.002
Ismael S, Zhao L, Nasoohi S, Ishrat T (2018) Inhibition of the NLRP3-inflammasome as a potential approach for neuroprotection after stroke. Sci Rep 8(1):5971. https://doi.org/10.1038/s41598-018-24350-x
Ward R, Li W, Abdul Y, Jackson L, Dong G, Jamil S, Filosa J, Fagan SC et al (2019) NLRP3 inflammasome inhibition with MCC950 improves diabetes-mediated cognitive impairment and vasoneuronal remodeling after ischemia. Pharmacol Res 142:237–250. https://doi.org/10.1016/j.phrs.2019.01.035
Bellut M, Papp L, Bieber M, Kraft P, Stoll G, Schuhmann MK (2021) NLPR3 inflammasome inhibition alleviates hypoxic endothelial cell death in vitro and protects blood-brain barrier integrity in murine stroke. Cell Death Dis 13(1):20. https://doi.org/10.1038/s41419-021-04379-z
Fann DY, Lim Y, Cheng Y, Lok K, Chunduri P, Baik S, Drummond GR, Dheen ST et al (2018) Evidence that NF-kappaB and MAPK signaling promotes NLRP inflammasome activation in neurons following ischemic stroke. Mol Neurobiol 55(2):1082–1096. https://doi.org/10.1007/s12035-017-0394-9
Zhu H, Jian Z, Zhong Y, Ye Y, Zhang Y, Hu X, Pu B, Gu L et al (2021) Janus kinase inhibition ameliorates ischemic stroke injury and neuroinflammation through reducing NLRP3 inflammasome activation via JAK2/STAT3 pathway inhibition. Front Immunol 12:714943. https://doi.org/10.3389/fimmu.2021.714943
Mohammadianinejad SE, Majdinasab N, Sajedi SA, Abdollahi F, Moqaddam MM, Sadr F (2014) The effect of lithium in post-stroke motor recovery: a double-blind, placebo-controlled, randomized clinical trial. Clin Neuropharmacol 37(3):73–78. https://doi.org/10.1097/WNF.0000000000000028
Giang KW, Mandalenakis Z, Dellborg M, Lappas G, Eriksson P, Hansson P, Rosengren A (2018) Long-term risk of hemorrhagic stroke in young patients with congenital heart disease. Stroke 49(5):1155–1162. https://doi.org/10.1161/STROKEAHA.117.020032
Rosand J (2021) Preserving brain health after intracerebral haemorrhage. Lancet Neurol 20(11):879–880. https://doi.org/10.1016/S1474-4422(21)00339-2
Salman RA, Law ZK, Bath PM, Steiner T, Sprigg N (2018) Haemostatic therapies for acute spontaneous intracerebral haemorrhage. Cochrane Database Syst Rev 4:005951. https://doi.org/10.1002/14651858.CD005951.pub4
Keep RF, Hua Y, Xi G (2012) Intracerebral haemorrhage: mechanisms of injury and therapeutic targets. Lancet Neurol 11(8):31–720. https://doi.org/10.1016/S1474-4422(12)70104-7
Mendelow AD, Gregson BA, Fernandes HM, Murray GD, Teasdale GM, Hope DT, Karimi A, Shaw MDM et al (2005) Early surgery versus initial conservative treatment in patients with spontaneous supratentorial intracerebral haematomas in the International Surgical Trial in Intracerebral Haemorrhage (STICH): a randomised trial. Lancet 365(9457):387–397. https://doi.org/10.1016/S0140-6736(05)17826-X
Mendelow AD, Gregson BA, Rowan EN, Murray GD, Gholkar A, Mitchell PM (2013) Early surgery versus initial conservative treatment in patients with spontaneous supratentorial lobar intracerebral haematomas (STICH II): a randomised trial. Lancet 382(9890):397–408. https://doi.org/10.1016/S0140-6736(13)60986-1
Xi G, Keep RF, Hoff JT (2006) Mechanisms of brain injury after intracerebral haemorrhage. Lancet Neurol 5(1):53–63
Shao A, Zhu Z, Li L, Zhang S, Zhang J (2019) Emerging therapeutic targets associated with the immune system in patients with intracerebral haemorrhage (ICH): from mechanisms to translation. EBioMedicine 45:615–623. https://doi.org/10.1016/j.ebiom.2019.06.012
Ma Q, Chen S, Hu Q, Feng H, Zhang JH, Tang J (2014) NLRP3 inflammasome contributes to inflammation after intracerebral hemorrhage. Ann Neurol 75(2):19–209. https://doi.org/10.1002/ana.24070
Yao S, Cao F, Chen J, Chen W, Fan R, Li G, Zeng Y, Jiao S et al (2017) NLRP3 is required for complement-mediated caspase-1 and IL-1beta activation in ICH. J Mol Neurosci 61(3):385–395. https://doi.org/10.1007/s12031-016-0874-9
Luo Y, Reis C, Chen S (2019) NLRP3 inflammasome in the pathophysiology of hemorrhagic stroke: a review. Curr Neuropharmacol 17(7):582–589. https://doi.org/10.2174/1570159X17666181227170053
Cheng Y, Chen B, Xie W, Chen Z, Yang G, Cai Y, Shang H, Zhao W (2020) Ghrelin attenuates secondary brain injury following intracerebral hemorrhage by inhibiting NLRP3 inflammasome activation and promoting Nrf2/ARE signaling pathway in mice. Int Immunopharmacol 79:106180. https://doi.org/10.1016/j.intimp.2019.106180
Zheng S, Jian D, Gan H, Wang L, Zhao J, Zhai X (2021) FUNDC1 inhibits NLRP3-mediated inflammation after intracerebral hemorrhage by promoting mitophagy in mice. Neurosci Lett 756:135967. https://doi.org/10.1016/j.neulet.2021.135967
Ji N, Wu L, Shi H, Li Q, Yu A, Yang Z (2022) VSIG4 Attenuates NLRP3 and ameliorates neuroinflammation via JAK2-STAT3-A20 pathway after intracerebral hemorrhage in mice. Neurotox Res 40(1):78–88. https://doi.org/10.1007/s12640-021-00456-5
Yuan B, Shen H, Lin L, Su T, Zhong S, Yang Z (2015) Recombinant adenovirus encoding NLRP3 RNAi attenuate inflammation and brain injury after intracerebral hemorrhage. J Neuroimmunol 287:5–71. https://doi.org/10.1016/j.jneuroim.2015.08.002
Chang Y, Ka S, Hsu W, Chen A, Chao LK, Lin C, Hsieh C, Chen M et al (2015) Resveratrol inhibits NLRP3 inflammasome activation by preserving mitochondrial integrity and augmenting autophagy. J Cell Physiol 230(7):1567–1579. https://doi.org/10.1002/jcp.24903
Cai J, Liu W, Lu F, Kong W, Zhou X, Miao P, Lei C, Wang Y (2018) Resveratrol attenuates neurological deficit and neuroinflammation following intracerebral hemorrhage. Exp Ther Med 15(5):4131–4138. https://doi.org/10.3892/etm.2018.5938
Kamp MA, Steiger H, Lieshout JHV (2020) Experimental aneurysmal subarachnoid hemorrhage: tiding over. Transl Stroke Res 11(1):1–3. https://doi.org/10.1007/s12975-019-00726-7
Murakami K, Koide M, Dumont TM, Russell SR, Tranmer BI, Wellman GC (2011) Subarachnoid hemorrhage induces gliosis and increased expression of the pro-inflammatory cytokine high mobility group box 1 protein. Transl Stroke Res 2(1):72–79. https://doi.org/10.1007/s12975-010-0052-2
Suzuki H (2019) Inflammation: a good research target to improve outcomes of poor-grade subarachnoid hemorrhage. Transl Stroke Res 10(6):597–600. https://doi.org/10.1007/s12975-019-00713-y
Frosen J, Cebral J, Robertson AM, Aoki T (2019) Flow-induced, inflammation-mediated arterial wall remodeling in the formation and progression of intracranial aneurysms. Neurosurg Focus 47(1):21. https://doi.org/10.3171/2019.5.FOCUS19234
Chen S, Ma Q, Krafft PR, Hu Q, Rolland W, Sherchan P, Zhang J, Tang J et al (2013) P2X7R/cryopyrin inflammasome axis inhibition reduces neuroinflammation after SAH. Neurobiol Dis 58:296–307. https://doi.org/10.1016/j.nbd.2013.06.011
Dodd WS, Noda I, Martinez M, Hosaka K, Hoh BL (2021) NLRP3 inhibition attenuates early brain injury and delayed cerebral vasospasm after subarachnoid hemorrhage. J Neuroinflammation 18(1):163. https://doi.org/10.1186/s12974-021-02207-x
Luo Y, Lu J, Ruan W, Guo X, Chen S (2019) MCC950 attenuated early brain injury by suppressing NLRP3 inflammasome after experimental SAH in rats. Brain Res Bull 146:320–326. https://doi.org/10.1016/j.brainresbull.2019.01.027
Chen S, Ding Y, Shi S, Tu X (2022) Schisandrin B inhibits NLRP3 inflammasome pathway and attenuates early brain injury in rats of subarachnoid hemorrhage. Chin J Integr Med. https://doi.org/10.1007/s11655-021-3348-z
Zuo Y, Wang J, Liao F, Yan X, Li J, Huang L, Liu F (2018) Inhibition of heat shock protein 90 by 17-AAG reduces inflammation via P2X7 receptor/NLRP3 inflammasome pathway and increases neurogenesis after subarachnoid hemorrhage in mice. Front Mol Neurosci 11:401. https://doi.org/10.3389/fnmol.2018.00401
Hu X, Yan J, Huang L, Araujo C, Peng J, Gao L, Liu S, Tang J et al (2021) INT-777 attenuates NLRP3-ASC inflammasome-mediated neuroinflammation via TGR5/cAMP/PKA signaling pathway after subarachnoid hemorrhage in rats. Brain Behav Immun 91:587–600. https://doi.org/10.1016/j.bbi.2020.09.016
Xu P, Hong Y, Xie Y, Yuan K, Li J, Sun R, Zhang X, Shi X et al (2021) TREM-1 exacerbates neuroinflammatory injury via NLRP3 inflammasome-mediated pyroptosis in experimental subarachnoid hemorrhage. Transl Stroke Res 12(4):643–659. https://doi.org/10.1007/s12975-020-00840-x
2022 Alzheimer's disease facts and figures. Alzheimer's & dementia: the journal of the Alzheimer's Association, 2022. https://doi.org/10.1002/alz.12638
Long JM, Holtzman DM (2019) Alzheimer disease: an update on pathobiology and treatment strategies. Cell 179(2):312–339. https://doi.org/10.1016/j.cell.2019.09.001
Hampel H, Nistico R, Seyfried NT, Levey AI, Modeste E, Lemercier P, Baldacci F, Toschi N et al (2021) Omics sciences for systems biology in Alzheimer’s disease: state-of-the-art of the evidence. Ageing Res Rev 69:101346. https://doi.org/10.1016/j.arr.2021.101346
Hur J, Frost GR, Wu X, Crump C, Pan SJ, Wong E, Barros M, Li T et al (2020) The innate immunity protein IFITM3 modulates gamma-secretase in Alzheimer’s disease. Nature 586(7831):735–740. https://doi.org/10.1038/s41586-020-2681-2
Saresella M, Rosa FL, Piancone F, Zoppis M, Marventano I, Calabrese E, Rainone V, Nemni R et al (2016) The NLRP3 and NLRP1 inflammasomes are activated in Alzheimer’s disease. Mol Neurodegener 11:23. https://doi.org/10.1186/s13024-016-0088-1
Ahmed ME, Iyer S, Thangavel R, Kempuraj D, Selvakumar GP, Raikwar SP, Zaheer S, Zaheer A (2017) Co-localization of glia maturation factor with NLRP3 inflammasome and autophagosome markers in human Alzheimer’s disease brain. J Alzheimer’s Dis 60(3):1143–1160. https://doi.org/10.3233/JAD-170634
Venegas C, Kumar S, Franklin BS, Dierkes T, Brinkschulte R, Tejera D, Vieira-saecker A, Schwartz S et al (2017) Microglia-derived ASC specks cross-seed amyloid-beta in Alzheimer’s disease. Nature 552(7685):355–361. https://doi.org/10.1038/nature25158
Luciunaite A, Mcmanus RM, Jankunec M, Racz I, Dansokho C, Dalgediene I, Schwartz S, Brosseron F et al (2020) Soluble Abeta oligomers and protofibrils induce NLRP3 inflammasome activation in microglia. J Neurochem 155(6):650–661. https://doi.org/10.1111/jnc.14945
Tejera D, Mercan D, Sanchez-caro JM, Hanan M, Greenberg D, Soreq H, Latz E, Golenbock D et al (2019) Systemic inflammation impairs microglial Abeta clearance through NLRP3 inflammasome. EMBO J 38(17):101064. https://doi.org/10.15252/embj.2018101064
Ising C, Venegas C, Zhang S, Scheiblich H, Schmidt SV, Vieira-saecker A, Schwartz S, Albasset S et al (2019) NLRP3 inflammasome activation drives tau pathology. Nature 575(7784):669–673. https://doi.org/10.1038/s41586-019-1769-z
Panda C, Voelz C, Habib P, Mevissen C, Pufe T, Beyer C, Gupta S, Slowik A (2021) Aggregated Tau-PHF6 (VQIVYK) potentiates NLRP3 inflammasome expression and autophagy in human microglial cells. Cells-Basel 10(7). https://doi.org/10.3390/cells10071652
Zhao Y, Tan S, Huang Z, Shan F, Li P, Ning Y, Ye S, Zhao Z et al (2021) NLRP3 inflammasome-dependent increases in high mobility group box 1 involved in the cognitive dysfunction caused by Tau-overexpression. Front Aging Neurosci 13:721474. https://doi.org/10.3389/fnagi.2021.721474
Stancu I, Cremers N, Vanrusselt H, Couturier J, Vanoosthuyse A, Kessels S, Lodder C, Brone B et al (2019) Aggregated Tau activates NLRP3-ASC inflammasome exacerbating exogenously seeded and non-exogenously seeded Tau pathology in vivo. Acta Neuropathol 137(4):599–617. https://doi.org/10.1007/s00401-018-01957-y
Zhang X, Wang R, Hu D, Sun X, Fujioka H, Lundberg K, Chan ER, Wang Q et al (2020) Oligodendroglial glycolytic stress triggers inflammasome activation and neuropathology in Alzheimer's disease. Sci Adv 6(49). https://doi.org/10.1126/sciadv.abb8680
Qi Y, Klyubin I, Cuello AC, Rowan MJ (2018) NLRP3-dependent synaptic plasticity deficit in an Alzheimer’s disease amyloidosis model in vivo. Neurobiol Dis 114:24–30. https://doi.org/10.1016/j.nbd.2018.02.016
Fekete C, Vastagh C, Denes A, Hrabovszky E, Nyiri G, Kallo I, Liposits Z, Sarvari M (2019) Chronic amyloid beta oligomer infusion evokes sustained inflammation and microglial changes in the rat hippocampus via NLRP3. Neuroscience 405:35–46. https://doi.org/10.1016/j.neuroscience.2018.02.046
Mackenzie SH, Schipper JL, Clark AC (2010) The potential for caspases in drug discovery. Curr Opin Drug Discov Dev 13(5):568–576
Yoshihara E (2020) TXNIP/TBP-2: a master regulator for glucose homeostasis. Antioxidants 9(8). https://doi.org/10.3390/antiox9080765
Rauch JN, Luna G, Guzman E, Audouard M, Challis C, Sibih YE, Leshuk C, Hernandez I et al (2020) LRP1 is a master regulator of tau uptake and spread. Nature 580(7803):381–385. https://doi.org/10.1038/s41586-020-2156-5
Reeve AK, Ludtmann MHR, Angelova PR, Simcox EM, Horrocks MH, Klenerman D, Gandhi S, Turnbull DM et al (2015) Aggregated alpha-synuclein and complex I deficiency: exploration of their relationship in differentiated neurons. Cell Death Dis 6:1820. https://doi.org/10.1038/cddis.2015.166
Sarkar S, Malovic E, Harishchandra DS, Ghaisas S, Panicker N, Charli A, Palanisamy BN, Rokad D et al (2017) Mitochondrial impairment in microglia amplifies NLRP3 inflammasome proinflammatory signaling in cell culture and animal models of Parkinson’s disease. NPJ Parkinson’s Dis 3:30. https://doi.org/10.1038/s41531-017-0032-2
Fan Z, Pan Y, Zhang Z, Yang H, Yu S, Zheng Y, Ma J, Wang X (2020) Systemic activation of NLRP3 inflammasome and plasma alpha-synuclein levels are correlated with motor severity and progression in Parkinson’s disease. J Neuroinflammation 17(1):11. https://doi.org/10.1186/s12974-019-1670-6
Wang X, Chi J, Huang D, Ding L, Zhao X, Jiang L, Yu Y, Gao F (2020) alpha-synuclein promotes progression of Parkinson’s disease by upregulating autophagy signaling pathway to activate NLRP3 inflammasome. Exp Ther Med 19(2):931–938. https://doi.org/10.3892/etm.2019.8297
Malhotra S, Rio J, Urcelay E, Nurtdinov R, Bustamante MF, Fernandez O, Oliver B, Zettl U et al (2015) NLRP3 inflammasome is associated with the response to IFN-beta in patients with multiple sclerosis. Brain 138(Pt 3):52–644. https://doi.org/10.1093/brain/awu388
Imani D, Azimi A, Salehi Z, Rezaei N, Emamnejad R, Sadr M, Izad M (2018) Association of nod-like receptor protein-3 single nucleotide gene polymorphisms and expression with the susceptibility to relapsing-remitting multiple sclerosis. Int J Immunogenet 45(6):329–336. https://doi.org/10.1111/iji.12401
Malhotra S, Sorosina M, Rio J, Peroni S, Midaglia L, Villar LM, Alvarez-cermeno JC, Schroeder I et al (2018) NLRP3 polymorphisms and response to interferon-beta in multiple sclerosis patients. Mult Scler 24(11):1507–1510. https://doi.org/10.1177/1352458517739137
Malhotra S, Costa C, Eixarch H, Keller CW, Amman L, Martinez-banaclocha H, Midaglia L, Sarro E et al (2020) NLRP3 inflammasome as prognostic factor and therapeutic target in primary progressive multiple sclerosis patients. Brain 143(5):1414–1430. https://doi.org/10.1093/brain/awaa084
Khan N, Kuo A, Brockman DA, Cooper MA, Smith MT (2018) Pharmacological inhibition of the NLRP3 inflammasome as a potential target for multiple sclerosis induced central neuropathic pain. Inflammopharmacology 26(1):77–86. https://doi.org/10.1007/s10787-017-0401-9
O’brien WT, Pham L, Symons GF, Monif M, Shultz SR, Mcdonald SJ (2020) The NLRP3 inflammasome in traumatic brain injury: potential as a biomarker and therapeutic target. J Neuroinflammation 17(1):104. https://doi.org/10.1186/s12974-020-01778-5
Zheng B, Zhang S, Ying Y, Guo X, Li H, Xu L, Ruan X (2018) Administration of Dexmedetomidine inhibited NLRP3 inflammasome and microglial cell activities in hippocampus of traumatic brain injury rats. Biosci Rep 38(5). 10.1042/BSR20180892
He Q, Jiang L, Man S, Wu L, Hu Y, Chen W (2018) Curcumin reduces neuronal loss and inhibits the NLRP3 inflammasome activation in an epileptic rat model. Curr Neurovasc Res 15(3):186–192. https://doi.org/10.2174/1567202615666180731100224
Meng X, Tan L, Tan M, Jiang T, Tan C, Li M, Wang H, Yu J (2014) Inhibition of the NLRP3 inflammasome provides neuroprotection in rats following amygdala kindling-induced status epilepticus. J Neuroinflammation 11:212. https://doi.org/10.1186/s12974-014-0212-5
Toscano ECDB, Vieira ELM, Dias BBR, Caliari MV, Goncalves AP, Giannetti AV, Siqueira JM, Suemoto CK et al (2021) NLRP3 and NLRP1 inflammasomes are up-regulated in patients with mesial temporal lobe epilepsy and may contribute to overexpression of caspase-1 and IL-beta in sclerotic hippocampi. Brain Res 1752:147230. https://doi.org/10.1016/j.brainres.2020.147230
Jiang M, Li R, Lyu J, Li X, Wang W, Wang Z, Sheng H, Zhang W et al (2020) MCC950, a selective NLPR3 inflammasome inhibitor, improves neurologic function and survival after cardiac arrest and resuscitation. J Neuroinflammation 17(1):256. https://doi.org/10.1186/s12974-020-01933-y
Zheng G, Xu J, He F, Hu J, Ge W, Ji X, Wang C, Bradley JL et al (2021) Effects of NLRP3 inflammasome blockade on postresuscitation cerebral function in a rat model of cardiopulmonary resuscitation. Biomed Pharmacother 143:112093. https://doi.org/10.1016/j.biopha.2021.112093
Xie K, Zhang Y, Wang Y, Meng X, Wang Y, Yu Y, Chen H (2020) Hydrogen attenuates sepsis-associated encephalopathy by NRF2 mediated NLRP3 pathway inactivation. Inflamm Res 69(7):697–710. https://doi.org/10.1007/s00011-020-01347-9
Funding
The study was supported by the Scientific Research Project of Hunan Provincial Health Commission (202117010086) and Natural Science Foundation of Hunan Province, China (Grant No. 2021JJ40832).
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Wenfang He had the idea for the article and prepared the draft of the manuscript. Literature search and data analysis were performed by Yanjun Zhong and Chenfang Wu. Jinxiu Li and Zhiping Hu reviewed and critically revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics Approval
This is not applicable.
Consent to Participate
This is not applicable.
Consent for Publication
This is not applicable.
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
He, W., Hu, Z., Zhong, Y. et al. The Potential of NLRP3 Inflammasome as a Therapeutic Target in Neurological Diseases. Mol Neurobiol 60, 2520–2538 (2023). https://doi.org/10.1007/s12035-023-03229-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-023-03229-7