
Abstract
Objective
Sepsis-associated encephalopathy (SAE) is a major cause of mortality worldwide. Oxidative stress, inflammatory response and apoptosis participate in the pathogenesis of SAE. Nuclear factor erythroid 2-related factor 2 (Nrf2) and nucleotide-binding oligomerization domain-like receptor containing pyrin domain 3 (NLRP3) pathway is involved in oxidative stress and inflammatory response. We reported that hydrogen gas protected against sepsis in wild-type (WT) but not Nrf2 knockout (KO) mice. Therefore, it is vital to identify the underlying cause of hydrogen gas treatment of sepsis-associated encephalopathy.
Methods
SAE was induced in WT and Nrf2 KO mice by cecal ligation and puncture (CLP). As a NLRP3 inflammasome inhibitor, MCC950 (50 mg/kg) was administered by intraperitoneal (i.p.) injection before operation. Hydrogen gas (H2)-rich saline solution (5 mL/kg) was administered by i.p. injection at 1 h and 6 h after sham and CLP operations. Brain tissue was collected to assess the NLRP3 and Nrf2 pathways by western blotting, reverse transcription-polymerase chain reaction (RT-PCR) and immunofluorescence.
Results
SAE increased NLRP3 and Nrf2 expression in microglia. MCC950 inhibited SAE-induced NLRP3 expression, interleukin (IL)-1β and IL-18 cytokine release, neuronal apoptosis and mitochondrial dysfunction. SAE increased NLRP3 and caspase-1 expression in WT mice compared to Nrf2 KO mice. Hydrogen increased Nrf2 expression and inhibited the SAE-induced expression of NLRP3, caspase-1, cytokines IL-1β and IL-18, neuronal apoptosis, and mitochondrial dysfunction in WT mice but not Nrf2 KO mice.
Conclusion
SAE increased NLRP3 and Nrf2 expression in microglia. Hydrogen alleviated inflammation, neuronal apoptosis and mitochondrial dysfunction via inhibiting Nrf2-mediated NLRP3 pathway.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Sepsis-associated encephalopathy (SAE), one of the main causes of mortality worldwide, is characterized by not only cognitive dysfunction and psychiatric disorders [1] but also negative effects on the central nervous system of sepsis survivors. Despite many advances in technical support for drug therapy and organ function, there has been no significant decrease in sepsis mortality over the past 3 decades [2, 3]. Therefore, it is vital to understand the physiopathology, and molecular basis of SAE for a potential therapeutic strategy.
Nucleotide-binding oligomerization domain-like receptor containing pyrin domain 3 (NLRP3) inflammasome is an intracellular multiprotein complex composed of NLRP3, ASC and caspase-1 [4]. This complex is responsible for the activation of caspase-1 and maturation of proinflammatory cytokines interleukin (IL)-1β and IL-18 [5]. In addition, mitochondrial dysfunction and mitochondrial reactive oxygen species (mtROS) generation have been shown to be crucial for NLRP3 inflammasome activation [6, 7]. Moreover, the inhibition of mtROS production in endothelial cells decreases NLRP3 inflammasome-mediated pyroptosis and concomitant IL-1β release and prevents the occurrence and development of SAE. Recent studies have consistently associated NLRP3 inflammasome with cognitive deficits in SAE [8,9,10]. Therefore, in sepsis or other diseases, mitochondria function is inhibited by the NLRP3 inflammasome.
In recent years, hydrogen and hydrogen-rich saline have been shown to play a protective role in many diseases by mediating antioxidative stress, the anti-inflammatory response and antiapoptotic processes involved in sepsis and sepsis-induced organ injury, ischaemia/reperfusion (I/R) injury, neurodegenerative diseases and traumatic brain injury [11]. In addition, our team reported that hydrogen inhalation significantly decreased mortality in septic mice and improved vital organ injury. In addition, hydrogen protected against sepsis through regulating antioxidative stress and the anti-inflammatory response in various tissues [12]. Accumulating evidence indicates that neuroinflammation participates in the physiopathology of SAE [13, 14]. The mechanism by which sepsis induces excessive neuroinflammatory activation remains to be revealed.
Nuclear factor erythroid 2-related factor 2 (Nrf2) is a well-known transcription factor that plays a critical role in the antioxidant stress system and drives the expression of antioxidant response element (ARE) gene. Our previous studies have confirmed that hydrogen has a protective effect against brain injury via the activation of Nrf2 pathway and that the expression of its downstream protein heme oxygenase-1 (HO-1) is increased in sepsis [15]. Intriguingly, published results showed that Nrf2-ARE signalling pathway activation is a critical regulatory factor in NLRP3 inflammation activation [16]. Therefore, we focused on the role of Nrf2 and NLRP3 in hydrogen-treated sepsis and further attempted to investigate the relationship between Nrf2 and NLRP3 when hydrogen was used to alleviate SAE.
Materials and methods
Animals and surgical procedures
All experimental animal procedures were approved by the Animal Management Committee of Tianjin Medical University (permit number: 2019-X1-04) and carried out in accordance with the “Policies on the Use of Animal and Humans in Neuroscience Research”. Male WT and Nrf2 knockout (KO) mice (20–25 g) were procured from the Better Biotechnology Company (Nanjing, China). The mice were acclimatized for one week and fed standard rodent chow and water in a standard environment of 22 °C and a 12 h light/dark cycle.
SAE was induced by cecal ligation and puncture (CLP) as previously described [17]. In brief, under anaesthesia conditions, the caecum was carefully exposed after a 1 cm abdominal median incision, and the ileocecal valve was found, ligated on the distal three-quarters of caecum and punctured with a 22-gauge needle. Faecal contents were extruded into peritoneal cavity. Then, the caecum was returned to abdomen, and the incision was sutured step-by-step. Control animals underwent the same procedure without ligation and puncture. The operated mice were revived by subcutaneous (s.c.) injection of 1 ml of 0.9% normal saline.
Hydrogen-rich saline
Hydrogen gas was dissolved in 0.9% NaCl for 4 h under high pressure (0.4 MPa) to reach a supersaturated state. The saturated hydrogen-rich saline was kept in an aluminium bag with no dead volume and stored at 4 °C under atmospheric pressure. The hydrogen-rich saline was sterilized by γ-radiation. The H2 concentration of in saline was detected with a needle-type H2 sensor (Unisense A/S, Aarhus, Denmark). To maintain a stable 0.6 mmol/L H2 concentration, hydrogen-rich saline was freshly prepared once a week.
Experimental design
Experiment 1: The SAE model was produced by proceeding CLP operation. To determine the dynamic changes of NLRP3 inflammasome in brain tissues, mice were sacrificed at 0 h, 2 h, 12 h and 24 h after CLP operation. A portion of cerebral cortex tissues was collected to detect NLRP3 inflammasome expression by western blot analysis. Another part of cerebral cortex tissue was collected for NLRP3 and IBA1 expression analysis by immunofluorescence.
Experiment 2: Male C57BL/6 mice were divided into three groups: Con group, SAE group, and SAE + MCC950 group. MCC950 (China Peptides Co., Ltd., China) was used to inhibit NLRP3. MCC950 (50 mg/kg BW) was administered by intraperitoneal injection before operation. At 12 h after operation, cerebral cortex tissue was collected and used to measure NLRP3 mRNA expression by reverse transcription-polymerase chain reaction (RT-PCR); NLRP3, ASC, IBA1, Bcl-2 and Bax expression by western blotting; NLRP3 and IBA1 expression by immunofluorescence; IL-1β and IL-18 cytokine expression by enzyme-linked immunosorbent assay (ELISA); neuronal apoptosis by terminal deoxynucleotidyl transferase dUTP nick end labelling (TUNEL); and mitochondrial function (the mitochondrial membrane potential (MMP), respiratory control ratio (RCR), and ATP and reactive oxygen species (ROS) levels).
Experiment 3: To detect Nrf2 in brain tissue, mice were sacrificed at 12 h after CLP operation. A portion of cerebral cortex tissue was collected and used to detect Nrf2 expression by western blot analysis. Another part of cerebral cortex tissue was collected for Nrf2 and IBA1 expression analysis by immunofluorescence.
Experiment 4: Wild-type (WT) and Nrf2 KO mice were randomly divided into two groups: Con group and SAE group. At 12 h after operation, cerebral cortex tissues were collected to detect measure NLRP3 and cleaved caspase-1 (referred to as caspase-1 for short) protein and mRNA expression by western blotting and RT-PCR.
Experiment 5: WT and Nrf2 KO mice were randomly divided into three groups: Con group, SAE group and SAE + hydrogen-rich saline group. H2-rich saline (5 mL/kg) was injected i.p. at 1 h and 6 h after sham or CLP operation. At 1 day, 3 days, 5 days and 7 days after operation, Y maze testing was performed to test memory ability. At 12 h after operation, cerebral cortex tissue was collected to detect Nrf2 mRNA expression to make sure the inhibition ratio (results were shown in the supplemental figure), NLRP3 inflammasome and caspase-1 by western blotting assay, IL-1β and IL-18 by ELISA assay, and measure brain histological changes by haematoxylin and eosin (H&E) staining; neuronal apoptosis by TUNEL assay; Nrf2, NLRP3 and cleaved caspase-1 protein expression by western blotting; IL-1β and IL-18 cytokine levels by ELISA; and mitochondrial function (MMP, RCR, and ATP and ROS levels).
Immunofluorescence analysis
Mice were perfused with 4% paraformaldehyde when experiment was performed, and brain tissue was collected to detect IBA1 and NLRP3 or IBA1 and Nrf2. Brain tissue was immersed, embedded, sectioned, deparaffinized, and rehydrated, following which antigen retrieval was performed, and the tissue was blocked. Slides were incubated with primary antibody against IBA1 (1:500, Abcam, Cambridge, UK), NLRP3 (1:200, Abcam, Cambridge, UK), or Nrf2 (1:200, Abcam, Cambridge, UK) overnight at 4℃. Then, the slides were incubated in secondary antibody (1 : 1000, DyLight 488-labelled IgG or DyLight 594 AffiniPure IgG, EarthOx, USA) for 2 h at room temperature. DAPI was used to stain each section, and coverslips were mounted onto the slides. The tissues were then examined with a fluorescence microscope (Olympus, Japan). At least ten fields of view were analysed, and the ratio of DAPI-positive cells was calculated and recorded.
Western blot analysis
Proteins were extracted from brain tissue at different time points after operation. The extracted protein was quantified using a BCA protein assay kit (Thermo Fisher Scientific, Inc., USA). Proteins were loaded into each lane of a 12–15% SDS polyacrylamide gel, separated, and transferred to polyvinylidene difluoride (PVDF) membranes (Millipore, USA), following which the PVDF membranes were blocked with blocking buffer (5% nonfat dry milk) for 1 h and incubated with specific primary antibodies against Nrf2 (1:1000, no. 62352, Abcam, Cambridge, UK), NLRP3 (1:500, no. 4207, Abcam, Cambridge, UK), ASC (1:1000, no. 155970, Abcam, Cambridge, UK), cleaved caspase-1 (1:1000, no. sc-22166, Santa Cruz, US), IBA1 (1:1000, no. 15690), Bax (1:1000, no. 32503, Abcam, Cambridge, UK), Bcl-2 (1:1000, no. 185002, Abcam, Cambridge, UK) and β-actin (1:2000, no. 8227, Abcam, Cambridge, UK) overnight at 4 °C. After washing, the membranes were incubated with secondary antibody (1:6000, no.10183, Abcam, Cambridge, UK) for 2 h at room temperature. The membranes were exposed with a UVP bioimaging system. Blot densities were analysed with Quantity One System software. The results are expressed as the ratio of target protein to β-actin.
RT-PCR analysis
Total RNA was extracted from brain tissues using TRIzol reagent (Thermo Fisher, Waltham, USA) according to the manufacturer’s protocol. cDNA was synthesized using a QuantiTect Reverse Transcription Kit (Qiagen). Relative NLRP3 and caspase-1 mRNA expression was determined with SYBR Premix Ex Taq™ (TaKaRa, Tokyo, Japan) on a real-time detection instrument (Bio-Rad, USA). The PCR procedure was as follows: 5 min at 95 °C, 40 cycles of 15 s at 95 °C, and 60 °C for 1 min. Quantified data were normalized to β-actin, which was used as a control. The following primers were used: Nrf2 forward: 5′- TCTATGTGCCTCCAAAGG -3′, reverse, 5′-CTCAGCATGATGGACTTGGA-3′; NLRP3 forward: 5′- GTGAAACAAAACGTGCCTTAGAA -3′, reverse, 5′- GGAGGGCTTGATAGCAGTGAA -3′; caspase-1 forward: 5′- GAAGAACAGAACAAAGAAGATGGCACA -3′, reverse, 5′- AGCTCCAACCCTCGGAGAAAGAT -3′; and β-actin forward: 5′- CTGTGCCCATCTACGAGGGCTAT -3′, reverse, 5′- TTTGATGTCACGCACGATTTCC -3’.
Cytokine analysis
Brain tissue was collected and homogenized to detect the cytokines IL-1β and IL-18. IL-1β and IL-18 levels were quantified using commercial ELISA kits according to the manufacturers’ instructions. (R&D Systems Europe, Oxford, UK).
Brain histopathological detection
Brain tissue was collected after perfusion with 4% paraformaldehyde in PBS when the experiments were performed and then fixed in the same fixative for 24 h at 4 °C. The tissue was then embedded in paraffin and cut into 5-µm-thick coronal sections. The sections were stained with H&E and examined using a microscope (BioRevo BZ-9000, Keyence Japan). Histopathology was scored via the presence of neuronal degeneration, vascular oedema and perivascular oedema. Each feature was graded with a score of 0–3 according to the degree of injury; thus, the total score was calculated on a nine-point scale.
Y maze testing
Y maze test was performed in a Y-shaped maze (including three arms that were 40 cm long, 10 cm wide and 12 cm high) to assess memory [15]. The arms were called start arm, other arm and novel arm. Y maze test was divided into two trials. In the first trial, the novel arm was blocked, and the mice could move freely across the start arm and the other arm for 10 min. In the second trial, the novel arm was also open, and the mice was allowed to move freely for 5 min. A ceiling-mounted video camera and Any-Maze behaviour tracking software were used to record and analyse the data, respectively, to calculate the percentage of alternation and the number of times the mice entered the novel arm.
Preparation of mitochondria
After the experiment was performed, brain tissue was collected to detect mitochondrial function. According to previous methods [18], tissues were cut into tiny pieces, and washed with mitochondrial isolation buffer to remove traces of blood and homogenized. After centrifugation to remove cell debris, the supernatant was obtained and further centrifuged. The mitochondrial pellet was then resuspended and washed with respiration buffer on ice. The protein concentration was measured with a BCA protein assay kit. The obtained mitochondria were used to detect mitochondrial function within 4 h. The RCR, MMP, and ROS and ATP contents were detected to reflect mitochondrial function.
Determination of mitochondrial respiratory function
RCR
Mitochondrial respiratory function was measured with a Clark-type oxygen electrode. According to previous methods [19], mitochondrial state 3 and state 4 respiration was recorded in mice from a different litter, and the ratio of state 3 to state 4 mitochondrial respiration was regarded as the mitochondrial RCR, which reflects mitochondrial respiratory function.
MMP analysis
According to the manufacturer’s instructions, MMP was measured using fluorescent dye JC-1 (Beyotime, China). Mitochondria were incubated with JC-1 dye for 30 min at 37 °C in the dark. The intensity of red fluorescence and green fluorescence can be detected by multifunctional enzyme labelling instrument (EnSpire, PerkinElmer, Massachusetts, USA). And the ratio of red-to-green fluorescence was calculated and used to reflect the MMP.
ROS assay
According to the manufacturer’s instructions, ROS production in brain tissue was determined using the oxygen radical-sensitive DCFH-DA probe (Beyotime, China). Mitochondria were incubated with DCFH-DA solution for 30 min at 37 ℃. Then, ROS were detected by flow cytometry (BD Biosciences).
ATP assay
Brain tissue was collected, lysed on ice and centrifuged at 10,000×g for 10 min at 4 °C. The supernatant ATP level was detected in accordance with the manufacturer’s instructions using a commercial ATP assay kit (Beyotime, China).
Apoptosis assay
Neuronal apoptosis was observed by TUNEL assay. To observe fragmented DNA in nucleus indicating neuronal apoptosis, the Dead End TM fluorometric TUNEL system (Roche, US) was applied. The nuclei of apoptotic cells were stained pink. DNAs in all cells was stained blue with DAPI.
Statistical analysis
All data were analysed with GraphPad Prism 5.0 Software (San Diego, CA, USA). All values are expressed as means ± SD. Statistical analyses were performed by ANOVA with Tukey–Kramer post hoc analysis. A P value < 0.05 indicated statistical significance.
Results
Expression of NLRP3 inflammasome at different time points in SAE mice
NLRP3 inflammasome plays a critical role in mediating the innate immune defence against pathogenic infection. We detected the expression of NLRP3 and downstream ASC at different time points after CLP operation in brain tissue by western blotting and immunofluorescence. Western blot results showed that NLRP3 expression was increased until 24 h after operation, and peak NLRP3 expression was observed at 12 h after operation (Fig. 1a, b, P < 0.05). In contrast, there was no significant difference in ASC expression (Fig. 1a and C, P > 0.05). Immunofluorescence results demonstrated that CLP induced an increase in IBA1 (a marker of activated microglia)- and NLRP3-positive cells in brain tissue (Fig. 1d and e, P < 0.05). These data suggested that SAE induced NLRP3 inflammasome activation in the microglial cells of brain tissue.

Expression of NLRP3 in the brain tissue of SAE mice. The SAE model was produced by CLP operation. Brain tissues was collected to detect NLRP3 inflammasome (a and b, n = 3) and ASC adaptor protein (a and c, n = 3) expression by western blotting, and staining for NLRP3 and IBA1 (d and e, n = 3) by immunofluorescence at 0 h, 2 h, 12 h, and 24 h after operation. Data are expressed as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001
MCC950 inhibited NLRP3 and microglial activation in the cerebral cortex of SAE mice
To analyse the effect of MCC950 on NLRP3 inflammasome in brain tissue of septic mice and assess the presence of NLRP3 in microglial cells, we determined NLRP3 inflammasome and ASC expression and the expression of microglial cell activation marker IBA1 by western blotting and immunofluorescence. Sepsis induced an increase in NLRP3 inflammasome protein and mRNA as well as IBA1 (a microglial cell marker) protein expression in brain tissue (Fig. 2a–c e, P < 0.05), but ASC expression was not different between Con group and SAE group (Fig. 2a and d, P > 0.05). As expected, MCC950 inhibited the expression of NLRP3 protein and mRNA as well as IBA1 protein in SAE + MCC950 group than in SAE group (Fig. 2a–c e, P < 0.05). Compared with Con group, the number of NLRP3- and IBA1-positive cells was higher in SAE group, while MCC950 clearly decreased the number of NLRP3- and IBA1-positive cells in SAE + MCC950 group compared with that in SAE group (Fig. 2f and G, P < 0.05). These results indicated that MCC950 inhibited NLRP3 inflammasome and microglial cell activation induced by SAE.

Effect of MCC950 on NLRP3 pathway in the brain tissues of SAE mice. The SAE model was produced by CLP operation. MCC950 (50 mg/kg BW) was administered by intraperitoneal injection before operation. Brain tissue was collected to detect NLRP3 inflammasome (a and b, n = 3), ASC adaptor protein (a and c, n = 3) and IBA1 (a marker of microglia activation) (a and d, n = 3) expression by western blotting and staining for NLRP3 and IBA1 (e and f, n = 3) by immunofluorescence at 12 h after operation. Data are expressed as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001
MCC950 prevented excessive release of cytokines IL-1β and IL-18 and apoptosis induced by SAE
Inflammatory cytokines are the final common pathway in the pathophysiology of brain dysfunction in SAE. We observed the release of cytokines IL-1β and IL-18 in SAE mice after MMC950 administration. SAE induced the release of substantially more IL-1β and IL-18 cytokines compared with Con group (Fig. 3a and b, P < 0.05). Compared with SAE group, MCC950 inhibited the release of above cytokines in the SAE + MCC950 group.

Effect of MCC950 on cytokines and neuronal apoptosis in the brain tissue of SAE mice. The SAE model was produced by CLP operation. MCC950 (50 mg/kg BW) was administered by intraperitoneal injection before operation. Brain tissue was collected to detect cytokines IL-1β (a, n = 6) and IL-18 (b, n = 6) by ELISA, Bcl-2 and Bax (c and d, n = 3) expression by western blotting and apoptosis by TUNEL assay (e and f, n = 3) at 12 h after operation. Data are expressed as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001
Inflammation is responsible for the induction of neural cell apoptosis. In addition to cytokines, we measured neuronal apoptosis in brain tissue. SAE induced cell apoptosis, as indicated by an increase in the number of TUNEL-positive cells (Fig. 3e and f, P < 0.05) and the Bax/Bcl-2 ratio (Fig. 3c and d, P < 0.05). MCC950 alleviated this increase in the number of TUNEL-positive cells and Bax/Bcl-2 ratio (Fig. 3c–f, P < 0.05). These results suggested that MCC950 not only relieved NLRP3 inflammasome activation but also inhibited cytokine release and neuronal apoptosis induced by SAE.
MCC950 improved mitochondrial dysfunction in SAE mice
To investigate the effect of MCC950 on mitochondrial dysfunction and injury in SAE mice, we tested the following indicators of mitochondrial function: MMP, mitochondrial RCR, and ATP and ROS content. SAE stimulated a decrease in MMP and RCR, reduced ATP, and increased ROS release (Fig. 4a–d, P < 0.05). MCC950 administration visibly increased the MMP, RCR and ATP content while reducing the ROS content in SAE mice (Fig. 4a–d, P < 0.05).

MCC950 attenuated mitochondrial dysfunction induced by SAE in mice. The SAE model was produced by CLP operation. MCC950 (50 mg/kg BW) was administered by intraperitoneal injection before operation. Brain tissue was collected to detect MMP (a, n = 6), RCR (a, n = 6), ATP content (a, n = 6) and ROS release (a, n = 6) 12 h after operation. Data are expressed as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001
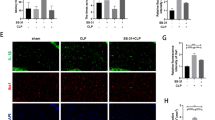
Nrf2 activation in microglial cells from the cerebral cortex of SAE mice
Nrf2 plays a critical role in the antioxidant stress system and drives the gene expression of ARE. As shown in Fig. 5, we assessed the expression of Nrf2 protein in the cerebral cortex of SAE mice by western blotting and immunofluorescence. Western blotting showed that Nrf2 expression was increased after SAE stimulation (Fig. 5a and b, P < 0.05). Immunofluorescence experiments showed the same trend as western blotting and indicated that the number of Nrf2- and IBA-positive cells was increased compared with Con group (Fig. 5c and d, P < 0.05). These results suggested that SAE induced Nrf2 activation in microglial cells of cerebral cortex in SAE mice.

Expression of Nrf2 in the brain tissue of SAE mice. The SAE model was produced by CLP operation. Brain tissue was collected to detect Nrf2 expression (a and b, n = 3) by western blotting and staining for Nrf2 and IBA1 (c and d, n = 3) by immunofluorescence at 12 h after the operation. Data are expressed as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001
Nrf2 depletion aggravated NLRP3 inflammasome pathway activation in SAE mice
To test the effect of Nrf2 on NLRP3 after CLP, we measured the protein and mRNA expression of NLRP3 and downstream caspase-1 in SAE mice. We found that SAE increased NLRP3 and cleaved-caspase protein levels and NLRP3 and caspase-1 mRNA expression in WT mice (Fig. 6a–d, P < 0.05). Consistently, the effect of SAE on NLRP3 and caspase-1 mRNA and protein levels were more pronounced in Nrf2 KO mice but not in WT mice (Fig. 6a–d, P < 0.05).

Expression of NLRP3 in WT and Nrf2 knockout mice. The SAE model was produced by CLP operation in wild-type and Nrf2 knockout mice. Brain tissue was collected to detect NLRP3 inflammasome (a and b, n = 3) and cleaved caspase-1 (a and c, n = 3) protein levels by western blotting as well as NLRP3 (d, n = 3) and caspase-1 (e, n = 3) mRNA levels by RT-PCR at 12 h after operation. Data are expressed as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001
Hydrogen-rich saline alleviated NLRP3 inflammasome activation via Nrf2 pathway
To assess the effect of hydrogen on NLRP3 inflammasome pathway in SAE mice, we detected NLRP3 and downstream caspase-1, IL-1β and IL-18. SAE increased NLRP3 activation and the maturation of caspase-1, IL-1β and IL-18 in WT mice (Fig. 7a–d, P < 0.05). Hydrogen administration attenuated the expression of NLRP3, caspase-1, IL-1β and IL-18 induced by SAE in WT mice.

Effect of hydrogen on Nrf2 and NLRP3 in WT and Nrf2 knockout mice. The SAE model was produced by CLP operation in wild-type and Nrf2 knockout mice. H2-rich saline (5 mL/kg) was i.p. injected at 1 and 6 h after operation. Brain tissue was collected to detect the expression of NLRP3 (a and b, n = 3) and cleaved caspase-1 (a and c, n = 3) by western blotting and the cytokines IL-1β (d, n = 6) and IL-18 (e, n = 6) by ELISA at 12 h after operation. Data are expressed as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001
We used WT and Nrf2 KO mice to further detect the effect of Nrf2 on NLRP3 inflammasome in SAE after hydrogen treatment. SAE induced an increase in NLRP3, caspase-1, IL-1β and IL-18 levels in Nrf2 KO mice (Fig. 7a–d, P < 0.05). Nrf2 knockout abrogated the inhibitory effect of hydrogen on NLRP3 inflammasome activation and cytokine maturation stimulated by SAE in Nrf KO mice (Fig. 7a–d, P < 0.05). These data supported the notion that hydrogen alleviated NLRP3 inflammasome activation in SAE mice via Nrf2 pathway.
Hydrogen-rich saline alleviated brain histological injury, neuronal apoptosis and memory function via Nrf2 pathway
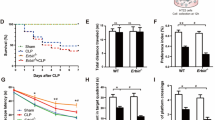
As shown in Fig. 8, in WT mice, hydrogen alleviated the brain histological injury, the number of apoptotic cells and increased the percentage of alternation and duration in novel arm which reflected the memory function in SAE mice (Fig. 8a–e, P < 0.05). However, there is no difference between in SAE group and in SAE + H2 group in Nrf KO mice (Fig. 8a–e, P > 0.05). When compared with SAE + H2 group in WT mice, the brain histological injury, the number of apoptotic cells and the percentage of alternation and duration in novel arm were deteriorated in SAE + H2 group in Nrf KO mice. These results supported that H2 alleviated brain histological injury, neuronal apoptosis and memory function via Nrf2 pathway.

Effect of hydrogen on brain histological injury, neuronal apoptosis and memory function in WT and Nrf2 knockout mice. The SAE model was produced by CLP operation in wild-type and Nrf2 knockout mice. H2-rich saline (5 mL/kg) was i.p. injected at 1 and 6 h after operation. At 12 h after operation, cerebral cortex tissue was collected to detect histological changes by H&E staining (a, b); neuronal apoptosis by TUNEL assay (c, d). At 1 day, 3 days, 5 days and 7 days after operation, Y maze testing was performed to test memory ability (e, f). Data are expressed as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001
Hydrogen-rich saline alleviated mitochondrial dysfunction via the Nrf2 pathway
SAE led to mitochondrial dysfunction, as shown by a decline in MMP and RCR, reduced ATP release and increased ROS release compared to WT and Nrf KO mice in the SAE group (Fig. 9a–d, P < 0.05). Hydrogen-rich saline increased the MMP, RCR, and ATP release and alleviated the change in ROS release induced by SAE in WT mice but not Nrf2 KO mice (Fig. 9a–d). Nrf2 knockout abolished the hydrogen-mediated alleviation of SAE-induced mitochondrial dysfunction. These results indicated that hydrogen-rich saline alleviated mitochondrial dysfunction via Nrf2 pathway in SAE mice.

Effect of hydrogen on mitochondrial function in WT and Nrf2 knockout mice. The SAE model was produced by CLP operation in wild-type and Nrf2 knockout mice. H2-rich saline solution (5 mL/kg) was i.p. injectedat 1 and 6 h after operation. Brain tissues were collected to detect MMP (a, n = 6), RCR (b, n = 6), ATP content (c, n = 6) and ROS release (d, n = 6) 12 h after the operation. Data are expressed as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001
Discussion
Sepsis accompanied by brain dysfunction is called SAE, which is characterized by long-term cognitive disorder and functional disability in sepsis survivors [20]. The pathogenesis of SAE is a multifactorial network of processes and elements, including oxidative damage, inflammatory cytokines, apoptosis and mitochondrial dysfunction [21]. Oxidative stress can aggravate the expression of proinflammatory cytokines and apoptosis in the pathogenesis of SAE. In this study, we found that SAE mice exhibited not only mitochondrial dysfunction, as shown by a decline in MMP and RCR, decreased ATP release and excessive ROS release, but also decreased antiapoptotic protein expression, increased proapoptotic expression and an increased proportion of apoptotic cells. In addition, SAE induced excessive release of cytokines IL-1β and IL-18. These data indicated that SAE is closely associated with inflammation, apoptosis and oxidative stress.
During the pathogenesis of SAE, the innate immune response plays a key role, especially in microglial activation, which participates in regulating oxidative damage and inflammation [14]. Activation of NLRP3 inflammasome, a multiprotein cytosolic complex, causes the maturation of pro-caspase-1, which in turn leads to the production and release of the inflammatory cytokines IL-1β and IL-18 [22]. Several reports showed that NLRP3 inflammasome, which is associated with inflammatory responses in SAE [8], activated inflammation and promoted the excessive release of cytokines, therefore, causing brain injury in brain ischaemia/reperfusion [23]. Our present data indicated that activation of NLRP3 inflammasome and its expression along with that of the downstream adaptor protein ASC peaked at approximately 12 h after operation rather than 2 h or 24 h in SAE mice. An immunofluorescence assay indicated that the number of NLRP3- and IBA1-positive cells was increased, suggesting that NLRP3 activation was increased in the microglia of brain tissue from SAE mice. The inhibition of NLRP3 expression by MCC950 significantly decreased the NLRP3 inflammasome pathway, microglial cell activation, and neuronal apoptosis and improved mitochondrial dysfunction induced by SAE.
Nrf2 is a key transcription factor that maintains intracellular redox balance by modulating the transcription of antioxidant genes [24]. Previous studies verified that Nrf2 upregulation attenuated IL-1β release by Nrf2-mediated NQO-1, glutamate cysteine ligase expression, and HO-1 expression [25, 26]. Nrf2 activation promotes ROS-detoxifying enzyme expression and represents a survival pathway [27]. Furthermore, the lack of Nrf2 results in elevated levels of ROS, which are detrimental to normal cellular function and promote cell death. Nrf2 and NLRP3 inflammasome were reported to play a critical role in inflammatory diseases [28, 29] and have also become useful targets of anti-inflammatory interventions for the treatment of lung injury [29, 30], lupus nephritis [31], and SAE [8]. As shown by our results, SAE induced Nrf2 and microglial activation; moreover, Nrf2 expression was increased in the microglia of brain tissue, as shown by immunofluorescence. An increasing number of publications have reported that Nrf2 activation can block inflammasome activation [32]. To detect the mechanism of Nrf2, Nrf2 KO mice were used to study the relationship between NLRP3 and Nrf2 in SAE. SAE induced NLRP3 protein and mRNA expression in WT mice, and NLRP3 protein and mRNA expression were higher in Nrf2 KO mice than in WT mice. In addition, the expression of caspase-1, IL-1β and IL-18, which are downstream of NLRP3, exhibited the same trend. These data demonstrated that Nrf2 is required for the inhibition of inflammasome and its components, including caspase-1, IL-1β and IL-18, in SAE mice.
Hydrogen was previously thought to be a conservative gas that could not be absorbed easily due to its low solubility. Recent studies have reported that H2 exerts antiapoptotic, anti-inflammatory and antioxidative stress effects in diseases such as sepsis, multiple organ dysfunction syndrome (MODS), lipopolysaccharide (LPS)-induced acute lung injury, and stroke [33,34,35,36,37]. Our study and previous studies have showed that hydrogen can regulate Nrf2 pathway activation and protect against traumatic brain injury [38], sepsis [39], intestinal injury [40], etc. In addition, hydrogen was shown to have a protective effect against sepsis via Nrf2 pathway and improved the sepsis survival rate in WT mice but not Nrf2 KO mice [40, 41]. As shown by the present results, SAE induced Nrf2 expression in brain tissue, and hydrogen-rich saline further increased Nrf2 expression in WT mice but not Nrf2 KO mice. Moreover, NLRP3 and its downstream proteins caspase-1, IL-1β and IL-18 were elevated in both WT and Nrf2 KO mice in SAE group. Hydrogen-rich saline reversed the excessive activation of the NLRP3 pathway and NLRP3 expression induced by SAE in WT mice but not Nrf2 KO mice. Furthermore, we also investigated the effect of Nrf2 on mitochondrial function, brain injury, apoptosis and cognitive function in hydrogen-rich saline-treated SAE mice. We found that SAE induced mitochondrial dysfunction, brain injury, apoptosis and cognitive dysfunction, which could be improved in WT mice but not Nrf2 KO mice by hydrogen treatment. These data indicated that hydrogen regulated NLRP3 inflammasome, mitochondrial and cognitive dysfunction, and brain injury via Nrf2 pathway.
In conclusion, SAE not only brought about mitochondrial dysfunction, microglial activation, inflammatory response, apoptosis, brain injury and cognitive dysfunction but also stimulated Nrf2 and NLRP3 pathway activation. In addition, hydrogen plays a protective role against SAE through Nrf2-mediated NLRP3 pathway inhibition. Our results indicate a strategy to reduce the activity of inflammatory corpuscles and suggest that Nrf2 is a potential target for the use of hydrogen to treat NLRP3-related diseases, such as SAE.
Abbreviations
- ANOVA:
-
Analysis of variance
- ARE:
-
Antioxidant response element
- CLP:
-
Cecal ligation and puncture
- DAPI:
-
4′, 6-Diamidino-2-phenylindole
- ELISA:
-
Enzyme-linked immune-sorbent assay
- HO-1:
-
Heme oxygenase-1
- IL:
-
Interleukin
- I/R:
-
Ischemia–reperfusion
- MMP:
-
Mitochondrial membrane potential
- mtROS:
-
Mitochondrial reactive oxygen species
- Nrf2:
-
Nuclear factor erythroid 2-related factor 2
- NLRP3:
-
Nucleotide-binding oligomerization domain-like receptor containing pyrin domain 3
- RT-PCR:
-
Reverse transcription-polymerase chain reaction
- PVDF:
-
Polyvinylidene difluoride
- RCR:
-
Respiratory control ratio
- ROS:
-
Reactive oxygen species
- SD:
-
Standard deviation
- SAE:
-
Sepsis-associated encephalopathy
- SDS:
-
Sodium dodecyl sulfate
- TUNEL:
-
Terminal deoxynucleotidyl transferase dUTP nick end labelling
- WT:
-
Wild type
References
Pytel P, Alexander JJ. Pathogenesis of septic encephalopathy. Curr Opin Neurol. 2009;22:283–7.
Angus DC, Linde-Zwirble WT, Lidicker J, Clermont G, Carcillo J, Pinsky MR. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit Care Med. 2001;29:1303–10.
Dellinger RP, Levy MM, Carlet JM, Bion J, Parker MM, Jaeschke R, et al. Surviving Sepsis Campaign: international guidelines for management of severe sepsis and septic shock: 2008. Crit Care Med. 2008;36:296–32727.
Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140:821–32.
Mariathasan S, Weiss DS, Newton K, McBride J, O'Rourke K, Roose-Girma M, et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. 2006;440:228–32.
Sarkar S, Malovic E, Harishchandra DS, Ghaisas S, Panicker N, Charli A, et al. Mitochondrial impairment in microglia amplifies NLRP3 inflammasome proinflammatory signaling in cell culture and animal models of Parkinson's disease. NPJ Parkinsons Dis. 2017;3:30.
Hoyt LR, Randall MJ, Ather JL, DePuccio DP, Landry CC, Qian X, et al. Mitochondrial ROS induced by chronic ethanol exposure promote hyper-activation of the NLRP3 inflammasome. Redox Biol. 2017;12:883–96.
Fu Q, Wu J, Zhou XY, Ji MH, Mao QH, Li Q, et al. NLRP3/caspase-1 pathway-induced pyroptosis mediated cognitive deficits in a mouse model of sepsis-associated encephalopathy. Inflammation. 2019;42:306–18.
Xu XE, Liu L, Wang YC, Wang CT, Zheng Q, Liu QX, et al. Caspase-1 inhibitor exerts brain-protective effects against sepsis-associated encephalopathy and cognitive impairments in a mouse model of sepsis. Brain, behavior, and immunity 2019.
Zong MM, Zhou ZQ, Ji MH, Jia M, Tang H, Yang JJ. Activation of β2-Adrenoceptor Attenuates Sepsis-Induced Hippocampus-Dependent Cognitive Impairments by Reversing Neuroinflammation and Synaptic Abnormalities. Frontiers in cellular neuroscience 2019.
George JF, Agarwal A. Hydrogen: another gas with therapeutic potential. Kidney Int. 2010;77:85–7.
Xie K, Liu L, Yu Y, Wang G. Hydrogen gas presents a promising therapeutic strategy for sepsis. Biomed Res Int. 2014;2014:807635.
Ji MH, Qiu LL, Tang H, Ju LS, Sun XR, Zhang H, et al. Sepsis-induced selective parvalbumin interneuron phenotype loss and cognitive impairments may be mediated by NADPH oxidase 2 activation in mice. J Neuroinflammation. 2015;12:182.
Michels M, Vieira AS, Vuolo F, Zapelini HG, Mendonca B, Mina F, et al. The role of microglia activation in the development of sepsis-induced long-term cognitive impairment. Brain Behav Immun. 2015;43:54–9.
Liu L, Xie K, Chen H, Dong X, Li Y, Yu Y, et al. Inhalation of hydrogen gas attenuates brain injury in mice with cecal ligation and puncture via inhibiting neuroinflammation, oxidative stress and neuronal apoptosis. Brain Res. 2014;1589:78–92.
Xu LL, Wu YF, Yan F, Li CC, Dai Z, You QD, et al. 5-(3,4-Difluorophenyl)-3-(6-methylpyridin-3-yl)-1,2,4-oxadiazole (DDO-7263), a novel Nrf2 activator targeting brain tissue, protects against MPTP-induced subacute Parkinson's disease in mice by inhibiting the NLRP3 inflammasome and protects PC12 cells against oxidative stress. Free Radic Biol Med. 2019;134:288–303.
Sui DM, Xie Q, Yi WJ, Gupta S, Yu XY, Li JB, et al. Resveratrol protects against sepsis-associated encephalopathy and inhibits the NLRP3/IL-1β axis in microglia. Mediat Inflamm. 2016;216:104557.
Wu J, Zhang M, Hao S, Jia M, Ji M, Qiu L, et al. Mitochondria-targeted peptide reverses mitochondrial dysfunction and cognitive deficits in sepsis-associated encephalopathy. Mol Neurobiol. 2015;52:783–91.
Zhang X, Zhang Z, Yang Y, Suo Y, Liu R, Qiu J, et al. Alogliptin prevents diastolic dysfunction and preserves left ventricular mitochondrial function in diabetic rabbits. Cardiovasc Diabetol. 2018;17:160.
Gofton TE, Young GB. Sepsis-associated encephalopathy. Nat Rev Neurol. 2012;8:557–66.
Wang P, Hu Y, Yao D, Li Y. Omi/HtrA2 regulates a mitochondria-dependent apoptotic pathway in a murine model of septic encephalopathy. Cell Physiol Biochem. 2018;49:2163–73.
Davis BK, Wen H, Ting JP. The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu Rev Immunol. 2011;29:707–35.
De Nardo D, Latz E. NLRP3 inflammasomes link inflammation and metabolic disease. Trends Immunol. 2011;32:373–9.
Jaramillo MC, Zhang DD. The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes Dev. 2013;27:2179–91.
Liu X, Zhang X, Ding Y, Zhou W, Tao L, Lu P, et al. Nuclear Factor E2-related factor-2 negatively regulates NLRP3 inflammasome activity by inhibiting reactive oxygen species-induced NLRP3 priming. Antioxid Redox Signal. 2017;26:28–43.
Lv H, Liu Q, Wen Z, Feng H, Deng X, Ci X. Xanthohumol ameliorates lipopolysaccharide (LPS)-induced acute lung injury via induction of AMPK/GSK3beta-Nrf2 signal axis. Redox Biol. 2017;12:311–24.
Taguchi K, Motohashi H, Yamamoto M. Molecular mechanisms of the Keap1-Nrf2 pathway in stress response and cancer evolution. Genes Cells. 2011;16:123–40.
Ahmed SM, Luo L, Namani A, Wang XJ, Tang X. Nrf2 signaling pathway: pivotal roles in inflammation. Biochim Biophys Acta Mol Basis Dis. 2017;1863:585–97.
Wang Z, Zhang A, Meng W, Wang T, Li D, Liu Z, et al. Ozone protects the rat lung from ischemia-reperfusion injury by attenuating NLRP3-mediated inflammation, enhancing Nrf2 antioxidant activity and inhibiting apoptosis. Eur J Pharmacol. 2018;835:82–93.
Liu Q, Ci X, Wen Z, Peng L. Diosmetin alleviates lipopolysaccharide-induced acute lung injury through activating the Nrf2 pathway and inhibiting the NLRP3 inflammasome. Biomol Ther (Seoul). 2018;26:157–66.
Ka SM, Lin JC, Lin TJ, Liu FC, Chao LK, Ho CL, et al. Citral alleviates an accelerated and severe lupus nephritis model by inhibiting the activation signal of NLRP3 inflammasome and enhancing Nrf2 activation. Arthritis Res Ther. 2015;17:331.
Maier NK, Leppla SH, Moayeri M. The cyclopentenone prostaglandin 15d-PGJ2 inhibits the NLRP1 and NLRP3 inflammasomes. J Immunol. 2015;194:2776–855.
Chen HG, Xie KL, Han HZ, Wang WN, Liu DQ, Wang GL, et al. Heme oxygenase-1 mediates the anti-inflammatory effect of molecular hydrogen in LPS-stimulated RAW 264.7 macrophages. Int J Surg. 2013;11:1060–6.
Xie K, Fu W, Xing W, Li A, Chen H, Han H, et al. Combination therapy with molecular hydrogen and hyperoxia in a murine model of polymicrobial sepsis. Shock. 2012;38:656–63.
Xie K, Yu Y, Pei Y, Hou L, Chen S, Xiong L, et al. Protective effects of hydrogen gas on murine polymicrobial sepsis via reducing oxidative stress and HMGB1 release. Shock. 2010;34:90–7.
Hayashida K, Sano M, Ohsawa I, Shinmura K, Tamaki K, Kimura K, et al. Inhalation of hydrogen gas reduces infarct size in the rat model of myocardial ischemia-reperfusion injury. Biochem Biophys Res Commun. 2008;373:30–5.
Jiang H, Yu P, Qian DH, Qin ZX, Sun XJ, Yu J, et al. Hydrogen-rich medium suppresses the generation of reactive oxygen species, elevates the Bcl-2/Bax ratio and inhibits advanced glycation end product-induced apoptosis. Int J Mol Med. 2013;31:1381–7.
Yuan J, Wang D, Liu Y, Chen X, Zhang H, Shen F, et al. Hydrogen-rich water attenuates oxidative stress in rats with traumatic brain injury via Nrf2 pathway. J Surg Res. 2018;228:238–46.
Chen H, Xie K, Han H, Li Y, Liu L, Yang T, et al. Molecular hydrogen protects mice against polymicrobial sepsis by ameliorating endothelial dysfunction via an Nrf2/HO-1 signaling pathway. Int Immunopharmacol. 2015;28:643–54.
Yu Y, Yang Y, Bian Y, Li Y, Liu L, Zhang H, et al. Hydrogen gas protects against intestinal injury in wild type but not NRF2 knockout mice with severe sepsis by regulating HO-1 and HMGB1 release. Shock. 2017;48:364–70.
Yu Y, Yang Y, Yang M, Wang C, Xie K, Yu Y. Hydrogen gas reduces HMGB1 release in lung tissues of septic mice in an Nrf2/HO-1-dependent pathway. Int Immunopharmacol. 2019;69:11–8.
Acknowledgements
This study was supported by a grant from the Natural Science Foundation of Tianjin (18JCYBJC93700 to Hongguang Chen; 17JCYBJC24800 to Keliang Xie); Science and Technology Support Key Program Affiliated to the Key Research and Development Plan of Tianjin Science and Technology Project (18YFZCSY00560 to Keliang Xie); National Natural Science Foundation of China (81601667 to Hongguang Chen; 81671888 to Yonghao Yu; 81772043, 81971879 to Keliang Xie), Beijing, China.
Author information
Authors and Affiliations
Contributions
KX designed the research, drafted and revised the manuscript; YZ YY and HC performed the animal model and sample collection and drafted the manuscript; XM, YW and YW performed the detection of sample. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no relevant conflicts of interest.
Additional information
Responsible Editor: John Di Battista.
Publisher's Note
Publisher's Note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Xie, K., Zhang, Y., Wang, Y. et al. Hydrogen attenuates sepsis-associated encephalopathy by NRF2 mediated NLRP3 pathway inactivation. Inflamm. Res. 69, 697–710 (2020). https://doi.org/10.1007/s00011-020-01347-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00011-020-01347-9