
Abstract
Alzheimer’s disease (AD) is one of the most prevalent neurodegenerative disorders. The etiology and pathology of AD are complicated, variable, and yet to be completely discovered. However, the involvement of inflammasomes, particularly the NLRP3 inflammasome, has been emphasized recently. NLRP3 is a critical pattern recognition receptor involved in the expression of immune responses and has been found to play a significant role in the development of various immunological and neurological disorders such as multiple sclerosis, ulcerative colitis, gout, diabetes, and AD. It is a multimeric protein which releases various cytokines and causes caspase-1 activation through the process known as pyroptosis. Increased levels of cytokines (IL-1β and IL-18), caspase-1 activation, and neuropathogenic stimulus lead to the formation of proinflammatory microglial M1. Progressive researches have also shown that besides loss of neurons, the pathophysiology of AD primarily includes amyloid beta (Aβ) accumulation, generation of oxidative stress, and microglial damage leading to activation of NLRP3 inflammasome that eventually leads to neuroinflammation and dementia. It has been suggested in the literature that suppressing the activity of the NLRP3 inflammasome has substantial potential to prevent, manage, and treat Alzheimer’s disease. The present review discusses the functional composition, various models, signaling molecules, pathways, and evidence of NLRP3 activation in AD. The manuscript also discusses the synthetic drugs, their clinical status, and projected natural products as a potential therapeutic approach to manage and treat NLRP3 mediated AD.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
Alzheimer’s disease (AD) is one of the most prevalent neurodegenerative disorders. It has been rising significantly over the years among the elderly population over the years [1]. The most common clinical symptoms of AD include memory impairment, personality and behavioral abnormalities, difficulty in execution, and apraxia as well as other neuropsychiatric conditions [2, 3]. Progressive researches have shown that besides loss of neurons, the pathophysiology of AD primarily includes amyloid beta (Aβ) accumulation, formation, and deposition of neurofibrillary tangles (NFTs) that have hyperphosphorylated tau proteins, generation of oxidative stress, and microglial damage leading to activation of major protein NLR family pyrin domain containing 3 (NLRP3) inflammasome that eventually leads to neuroinflammation and dementia. When compared to a healthy brain, the brains of Alzheimer’s patients exhibit a substantial deposition of tau protein and interaction with abnormal Aβ proteins [4]. These alterations are closely linked with abnormalities in innate immune system within the brain, wherein inflammatory response by glial cells and damage to blood vessels in a non-specific manner plays a key role [5, 6]. The etiology and pathophysiology of AD are complicated, variable, and yet to be completely discovered. However, the involvement of inflammasomes, particularly the NLRP3 inflammasome, has been emphasized recently [7, 8]. NLRP3 is a critical pattern recognition receptor involved in the expression of immune responses and has been found to play a significant role in the development of various immunological and neurological disorders such as multiple sclerosis, ulcerative colitis, gout, diabetes, and AD [9, 10]. NLRP3 is a multimeric protein which releases various cytokines and causes caspase-1 activation through the process known as pyroptosis. Increased levels of cytokines (IL-1β and IL-18), caspase-1 activation, and neuropathogenic stimulus lead to the formation of proinflammatory microglial M1. Further, the inhibition of M1 microglial activation is caused by the formation of M2 phenotype which is anti-inflammatory in nature. This process is the indication of inactivation of NLRP3 inflammasome and can lead to reduction in the pathogenesis of AD and could have potential as a target for the management and treatment of this disease. Recently, various published evidence have strengthened the pathological involvement of NLRP3 inflammasome in the etiology of AD, and hence, several attempts were made to develop or repurpose drug candidates that can inhibit the NLRP3 mediated AD. The importance of NLRP3 in neurological disorders can be validated from the fact that currently a number of synthetic NLRP3 inhibitors are being investigated in clinical trials to bring them from bench to bedside. However, the synthetic agents have exhibited a few limitations such as the lack of well-validated safety profile, lower solubility, impermeability in crossing BBB, and unwanted adverse effect. Researchers have, thus, also explored the NLRP3 inhibitory activity of clinically established drugs such as statins and various natural products. The current manuscript describes the functional composition, various models, signaling molecules, pathways, and evidence of NLRP3 activation in AD. The manuscript also discusses the synthetic drugs their clinical status and projected natural products as a potential therapeutic approach to manage and treat NLRP3 mediated AD.
STRUCTURE AND FUNCTION OF NLRP3 INFLAMMASOME
Inflammasomes are also referred as multi-protein complexes that identify the foreign pathogens as well as the host’s own distress signals. The structure of inflammasomes consists of a molecular receptor protein (including classical NLR as well as non-classical NLR), an adaptor protein (generally ASC), and caspase-1 precursor protein. The ASC protein, or apoptosis-associated speck-like protein, comprises a pyrin domain (PYD) at the N-terminus and a cysteine aspartic proteinase recruitment domain (CARD) at the C-terminus. CARD functions as a recruitment effector protein that may be identified and connected to the CARD domain of pro-caspase-1 [11]. Distinct molecular receptor proteins form distinct inflammasomes [12, 13]. Inflammasomes, upon identification of distress signals, trigger the recruitment and activation of proinflammatory pro-caspase-1 enzyme. NLRP3 inflammasome is composed of NLRP3 protein, ASC protein, and caspase-1 precursor protein and plays the crucial role in the pathogenesis of various pathogenic conditions including AD [14]. NLRP3 is a pattern recognition receptor protein with a leucine-rich repeat domain and a carboxyl terminal; however, it lacks the CARD domain and hence cannot bind directly to caspase-1 precursor. Rather, it primarily binds to ASC protein via PYD-PYD linkage, which then binds to pro-caspase-1 via CARD-CARD linkage. Pro-caspase-1 initially forms a tetramer and subsequently forms heterodimer having enzymatic properties [15]. Caspase-1 protein has the ability to convert inactive precursors of IL-1β and IL-18 into their corresponding active matured forms to perform a range of non-specific inflammatory actions [16]. NLRP3 inflammasome activation can lead to the emergence of a variety of pathogenic conditions such as gout, multiple sclerosis, diabetes, and AD [17]. Thus, understanding the function, activation, and mechanism of action of NLRP3 inflammasome has ample clinical significance in the prevention and treatment of neuroinflammatory diseases.
MECHANISM OF ACTIVATION OF NLRP3 INFLAMMASOME
Various internal and external signals such as pathogens associated molecular patterns (PAMPs), calcium pyrophosphate, silica, palmitate, sodium urate, cholesterol, and lipopolysaccharides have been associated with the activation of NLRP3 inflammasome. Some danger-associated molecular patterns (DAMPs) like hyperglycemia, hypercalcemia, hypokalemia, ATP, reactive oxygen species (ROS), and cathepsin B also activate this inflammasome [18]. Recently, sodium channels, specifically dysregulated epithelial sodium channels, have also been found to play important role in activating NLRP3 inflammasome [19].
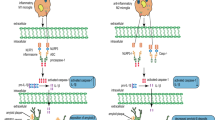
Currently, three theories have been stated to explain the mechanism by which NLRP3 inflammasome is activated by foreign signals [20]. The first one is the theory of lysosomal damage, which states that, when PAMPs are engulfed by the macrophage cells, they destabilize the phagocytes and cause lysosomal acidification. As a result, the wall of the lysosome ruptures and cathepsin B is released in the cytoplasm. Cathepsin B potentially degrades the inhibitory protein of NLRP3 and thus activates the NLRP3 inflammasome [21]. To upregulate the expression of NLRP3, macrophage cells must be subjected to initial stimuli that triggers the activation of transcription factor NF-kB via Toll-like receptor (TLRs) ligands and cytokine receptors. Molecules that influence NF-kB activity can indirectly alter NLRP3 priming. The activated NF-kB promotes the transcription, translation, and subsequent post-translational modifications of the NLRP3 mRNA in order to complete the priming of NLRP3 [22]. Another theory states that ROS play a key role in the activation of NLRP3 inflammasome. Increased ROS level due to oxidative cell stress promotes the auto-oxidation of thioredoxin protein (TRX) that is bound to thioredoxin interaction protein (TXNIP) under normal conditions. The separated TXNIP then binds to NLRP3 protein, relying on the ASC domain to recruit pro-caspase-1 and completes the assembly, thereby leading to NLRP3 inflammasome activation. Activation signals, such as elevated glucose levels, ATP, and so on, typically activate the inflammasome in this manner [23, 24]. According to the mitochondrial DNA hypotheses, NLRP3 inflammasome is activated by mitochondrial deoxyribonucleic acid (mtDNA), which is released from the damaged mitochondria when attacked by PAMPs and DAMPs. This type of activation method is facilitated by increased calcium influx, potassium outflux, cytotoxin-mediated cell membrane breakdown, binding of ATP to ligand-gated purinergic ion channel 7 receptor (P2X7R), and microbial toxin intervention in the mitochondrial micropore as shown in Fig. 1 [25, 26].

Mechanism of NLRP3 activation and neuroinflammation.
K+ Efflux and NLRP3 Activation
According to ion channel hypothesis, NLRP3 activation occurs via efflux of K+ ion which it can be induced by various activators of NLRP3 such as ATP, uric acid, and nigericin [27]. Reduced intracellular K+ concentration is a trigger activation of inflammasome of NLRP3, and its high concentration outside the cell produces opposite effect by blocking it. K+ efflux also mediates the release of IL-1β from monocytes and macrophages and potentiates the conversion of pro-IL-1β to its active form [28, 29]. Mechanistically, endogenous membrane pore formation via pannexin-1 hemichannel and P2X7 ATP-gated ion channel upon activation in response to extracellular ATP initiates the swift outflow of K+ ion [30, 31]. Study has showed that asbestos and aluminum hydroxide silica that promote the efflux of K+ are unable to stimulate NLRP3 activation, unless it is pre-stimulated by LPS microbial ligands or any other stimulus [32, 33]. Additionally, compounds such as GB111-NH2, CL097, and imiquimod can induce NLRP3 without K+ efflux [34, 35]. Consequently, K+ efflux may not be a strong and independent factor for the NLRP3 activation.
Ca2+ Mobilization and NLRP3 Activation
Calcium mobilization is prevalent in many signaling pathways. Numerous studies have shown the pathogenic role of Ca2+ in NLRP3 activation. Enzyme phospholipase C hydrolyzes PIP2 (phosphatidylinositol 4,5-bisphosphate) to IP3 (inositol 1,4,5-triphosphate) [36]. IP3 afterwards triggers Ca2+ efflux from the endoplasmic reticulum (ER) through IP3 receptor (IP3R) and increases the intracellular [37]. Intracellular Ca2+ level is also increased in response to opening of voltage-gated Ca2+ channel. Extensive researches have been carried out, and many conclusions have been deduced to establish the positive correlation between intracellular Ca2+ level and NLRP3 activation [38]. Mechanistically, increased intracellular Ca2+ level causes mitochondrial dysfunction, increases the level of ROS, promotes lysosomal degradation, and potentiates the nuclear translocation of NF-kB that cumulatively leads to NLRP3 activation as shown in Fig. 1. The pathogenic role of Ca2+ in the NLRP3 activation was also confirmed in the study where chelator of Ca2+ “BAPTA-AM” pyroptosis [39, 40]. Additionally, 2APB (2-aminoethoxy diphenylborinate) which is a IP3 receptor antagonist and Ca2+ blocker inhibit calcium influx and thus mitigate NLRP3 activation.
Na+ and Cl− Efflux and NLRP3 Activation
Na+ and Cl− efflux and NLRP3 activation have been reported in various published reports. Preventing Na+ influx through the reduction of extracellular Na+ ion concentration decreases NLRP3 inflammasome activation [28]. Increment in the Na+ concentration inside the cell causes swelling of the cell and inflow of water which further decreases the K+ ion concentration inside the cell and potentiates the NLRP3 activation [28]. An increase in the intracellular Cl− ion concentration also upregulates the secretion and maturation of pro-IL-1β, and increase in the extracellular concentration of Cl− downregulates IL-1β activation and secretion [41]. Studies have revealed that Cl− channel inhibitors such as flufenamic acid, 5-nitro-2-(3-phenylpropylamino) acid, indanyloxyacetic acid, benzoic acid, mefenamic acid, and 4,40-isothiocyanato-2,20-stilbenedisulfonic acid prevent NLRP3 inflammasome activation and hence strengthen the role of this ion in the NLRP3 activation [42]. However, more detailed investigation is needed to understand how Na+ and Cl− efflux trigger activation of the NLRP3 inflammasome.
ROS and NLRP3 Activation
ROS production via NADPH (nicotinamide adenine dinucleotide phosphate) oxidase and mitochondria is one of the key regulators in the NLRP3 activation [33, 43, 44]. The production of ROS by NADPH oxidase can also be stimulated by DAMPs, PAMPs, phagocytosis, and ATP [33]. There are various agonists of NLRP3 that induce ROS generation via mitochondria, and this connects the role of ROS in NLRP3 activation [45]. The activation of NLRP3 is regulated by the ROS production that was proven by blocking of NLRP3 activation by various agonists through suppressing p22 subunit of NADPH oxidase, ROS scavengers, and antioxidants [33]. However, ROS-inducing substances such as cytokines cannot directly activate the NLRP3 inflammasome and, hence, suggest that activation of NLRP3 can be processed by ROS [46]. Moreover, inhibitors of ROS and their high concentration prevent the primary step of activation of NLRP3, but direct NLRP3 activation is not affected by them [47]. An association was also found between K + concentration and ROS in the NLRP3 activation [48]. Interaction is undefined between them, but either lower intracellular K+ concentration is sufficient or lower intracellular K+ concentration triggers the generation of ROS that in turn activates NLRP3. NLRP3 agonists also trigger the TXNIP association with NLRP3 which is ROS-dependent NLRP3 ligand in macrophage [49]. As the level of ROS is increased intracellularly, TXNIP dissociates in ROS-dependent manner from oxidized thioredoxin (TRX) which interacts with NLRP3 (specifically LRR) leading to its activation [49]. Cyclic GMP-AMP synthase (cGAS) and stimulator of interferon genes (STING) are also reported to be activated upon NAD + depletion and consequently cause DNA damage [50].
It is further important to highlight that mitochondrion, through their respiratory activities, produces reactive oxygen species and is thus entwined with activation of NLRP3 inflammasome [51]. Studies reported that mtDNA is induced through TLR signaling and causes activation of NLRP3 inflammasome [52]. These findings suggest that there is involvement of mtROS, mtDNA and mitochondrial dysfunction in activating NLRP3 inflammasome. Cardiolipin, Mitofusin 2, and mitochondrial antiviral signaling protein (MAVS) are the molecules of the mitochondria other than mtROS and mtDNA that respond to the NLRP3 stimuli. MAVS is required for the activation of NLRP3 inflammasome by nigericin, poly (I:C), and ATP [53, 54]. Mitofusin 2 associates with NLRP3 directly in a viral infection. Cardiolipin on the other hand binds with the LRR part of the NLRP3 inflammasome, and disruption in its expression decreases the inflammasome activation [55].
The Lysosomal Rupture and NLRP3 Activation
Cholesterol crystals, asbestos alum, silica, calcium crystals, amyloid-β, and MSU are categorized as particulate matter (PM). They damage the lysosomes, cause spilling of their contents into the cytosol, and activate the NLRP3 inflammasome [56, 57]. Studies have also shown that silica, Aβ, and asbestos undergo endocytosis by macrophages and lead to instability of the lysosome resulting in their rupture or damage and cause release of cathepsin B proteinase into the cytoplasm that is directly involved in NLRP3 activation [45, 58, 59]. Until now, the exact mechanism of lysosomal damage associated NLRP3 inflammasome activation is not known. However, based on the published reports, it was inferred that monosodium urate releases a large amount of Na+ ions and eventually decreases intracellular K+ outflow concentration [28]. Damage of lysosomes also triggers K+ outflow via pore formation and can activate NLRP3 inflammasome [31, 60]. It was also reported that the release of lysosomal enzymes for phagocytosis of particulate matter causes the activation of the NLRP3 [40]. Additionally, increased intracellular Ca2+ ion-induced, macrophage-induced, and dysfunctional mitochondria-induced OS causes lysosomal rupture and activates NLRP3 inflammasome [33]. The involvement of lysosomal damage and released cathepsin B, in the activation of NLRP3, was confirmed when use of cathepsin B inhibitor, “CA-074Me,” inhibited the NLRP3 activation and subsequent release of IL-1β [33].
TLRs AND mTOR SIGNALING PATHWAY IN NLRP3 ACTIVATION
A highly expressed PRR, i.e., TLRs, play a crucial role in innate immunity [61]. MyD88 and TIRAP-like adaptors constitute TLR pathway and mediate various sub-pathways that causes the production of multiple cytokines such as pro-IL-1β, TNF-ɑ, and IL-6 via downstream kinases such as TRAF6 and IRAK1/4 [62, 63]. This further facilitates inflammatory responses [64, 65]. TLR pathways play a crucial role in the regulation of NLRP3 inflammasome activation via canonical MyD88-IRAK1/4-TRAF6-TAK1-IKK-NF-κB pathway [62, 66, 67]. At post-translational levels, triggered TLRs cause MyD88/TRIFTARF6-TAK1-JNKs pathway activation, which targets NLRP3 by phosphorylation at Ser198 residue. Following this, the deubiquitination of NLRP3 results in NLRP3-ASC assembly [68, 69]. Hence, NLRP3 phosphorylation via TLR is a trigger for the activation of NLRP3 inflammasome [68, 69]. NLRP3 inflammasome is completely activated only when transcriptional upregulation and post-translational modification work together [70]. Moreover, TLR signaling also triggers alternative pathway of NLRP3 inflammasome activation via LPS stimulation [71].
mTOR is a serine/threonine protein kinase and an essential catalytic subunit, member of PI3K-related family of kinases. It is known to facilitate the activation of NLRP3 inflammasome [72, 73]. mTOR consists of two protein complexes, mTORC1 and mTORC2, and can recognize PPRs and play a vital role in innate immune system [72, 73]. It has been investigated that transcription of gene can be regulated by mTOR through NF-kB pathway and can be inhibited by inhibiting mTORC1 (Rapamysin) [74, 75]. An increase mitochondrial ROS was also observed via mTOR, resulting in the activation of NLRP3 inflammasome [76]. As per published reports, it was found that mTORC2 causes NLRP3 inflammation activation and also catalyzes SGK1 phosphorylation causing a K+ efflux [70, 77]. Hence, it can be concluded that the regulation of NLRP3 and pro-IL-1β expression as well as that of other components is upregulated by mTOR signaling via either AKT1-IKK-NF-κB pathway or by recognizing ROS and cation efflux [70].
MAPK is a another serine/threonine protein kinase that can recognize various PPRs such as oxidative stress and osmotic imbalance and regulates the inflammatory responses via NLRP3 activation [70, 78, 79]. Wang et al. investigated that TXNIP activation was associated due to ROS production via MAPK pathway [80, 81]. Thus, NLRP3 expression as well as pro-IL-1β expression and their activation has been found to be enhanced by MAPK signaling pathway [82, 83].
NEGATIVE REGULATION OF NLRP3 INFLAMMASOME ACTIVATION
The NLRP3 inflammasome may benefit the human body by aiding resistance to external microbial infection and intrinsic tissue damage, when activated appropriately and adequately. However, overactivation, on the other hand, might typically have deleterious effects on the cellular as well as bodily health. Presently, numerous mechanisms have been reported that negatively regulate the NLRP3 inflammasome activation via binding and blocking the receptor and autophagy or by disrupting the expression of genes [84]. For instance, miR-223 can downregulate the NLRP3 inflammasome activation via the inhibition of NLRP3 gene expression. T cells and interferons also negatively regulate the expression of P2X7R and influence the STAT signaling pathway. In a study, Lamkanfi and Dixit [5] reported that treatment of in vitro cultured cells with hydrogen peroxide led to the activation of NLRP3 inflammasome and decreased signal transduction along with phosphorylation of STAT serine 727 and inhibited by pre-treatment with BAPTA-AM (a selective calcium chelator). Furthermore, dopamine, β-hydroxybutyric acid, nitric oxide, and unsaturated fatty acids have also been found to suppress the activity of NLRP3 inflammasome. Elevated microtubule-associated protein 1A/1B-light chain 3 (LC3B), an autophagy protein, can cause reduction of ROS levels and prevent NLRP3 inflammasome activation [85].
The presence of NLRP3 in the mitochondria is imperative to activate and moderate its activity as mitochondrial adaptor proteins are crucial for triggering the activation. Acetylation potentially mediates the actin-dependent transfer of mitochondrial ASC, leading to the interaction between the mitochondrial ASC and NLRP3 in the endoplasmic reticulum. Autophagy-related proteins possess the ability to protect the mitochondria as well as prevent the activation of NLRP3 inflammasome. RIPK2 protein suppresses the action of the NLRP3 inflammasome in influenza virus infection by increasing mitochondrial autophagy [86].
Thus, negative regulation of the NLRP3 inflammasome may be mediated via a variety of unique and interesting pathways. The production of IL-1 is inhibited by the type 1 IFN signaling pathway, which negatively affects the inflammasome activation. In others, effector T cells suppress the innate immune response by lowering the neutrophil infiltration in an antigen-dependent manner, thereby reducing NLRP3 inflammasome activation.
Signaling Pathways Inhibiting NLRP3 Inflammasome Activation
Autophagy and NLRP3 Inhibition
During stress condition of cells such as damaged organelles, ATG complexes carry out the recycle and removal of unwanted substances by the formation of autolysosomes. The targets are enveloped in the autophagosomes which are later fused with lysosomes and degraded by autolysosomes, and the process is known as autophagy [87, 88]. Over the years, autophagy has been found to be associated with various inflammatory disorders [89], and a potential increase in the severity was seen in these diseases in cases of abnormal inflammasome activation [90]. Homeostasis is a crucial aspect, and autophagy maintains this in the innate immunity, signified by various studies in which comparatively more IL-1β was produced in ATG16L1-lacking macrophages followed by inflammasome activation [91, 92]. Inflammasome activation requires second signals like ROS which can be prevented by the action of autophagy via NOD2-RIPK2-ULK1 pathway and PERK-eIF2α-ATF4-ATGs pathway [93, 94]. The evidence suggests that increased autophagy is directly related to the inhibition of inflammasome activation [95].
cAMP-PKA Signaling Pathway and NLRP3 Inhibition
Over several years, studies have shown that serine/threonine kinase can inhibit IL-1β production and hence NLRP3 inflammasome activation [96]. It has been reported this pathway is involved in both canonical and non-canonical pathways and is also involved in the modifications both at transcription and post-translational levels [96, 97]. The mechanism involves the phosphorylation of proteins and ubiquitination resulting in NLRP3 degradation induced by dopamine and prostaglandin-like products [97, 98].
It is well-known that phosphorylation at Ser295 residue is caused by the binding of cAMP with PKA. This cAMP is produced by PGE2 through a series of steps where it activates adenyl cyclase after binding to the receptor converting ATP into cAMP. cAMP is also produced by the binding of bile acid with TGR5 and that of L-adrenaline with ADRA2B. The cAMP produced by L-adrenaline results in direct phosphorylation of caspase-11 and results in collapse of inflammasome assembly [99,100,101]. During transcription, the inhibition of NF-κB signaling by this pathway causes a decrease in NLRP3 expression as well as pro-IL-β [102, 103].
AMPK Signaling Pathway and NLRP34 Inhibition
When the AMP:ATP ratio is increased inside the cell, then this serine/threonine kinase is activated and is responsible for the suppression of inflammatory responses [65, 104]. AMPK interferes with the IKK/IκB/NF-κB signaling pathway and is also known to reduce the release of ROS in cells [65, 104]. Antioxidant effect of this signaling pathway involved the modulation of oxidation of NAD(P)H for the production of ROS in mitochondria, thereby inhibiting the inflammasome activation [65, 104]. Mechanistically, this pathway acts by suppressing the IκB phosphorylation and by inhibiting the NAD(P)H oxidase [65, 104]. Apart from this, AMPK-GSK3β-Nrf2 pathway inhibits inflammasome activation by interfering with the inflammasome assembly [65, 104].
Post-Translational Modifications of NLRP3 to Inhibit NLRP3 Activation
Ubiquitination
In ubiquitination (brief introduction, what is ubiquitination and all), Lopez-Castejon et al. report that NLRP3’s ubiquitination activates it, while the prevention of its deubiquitiantion through b-AP15 (DUB inhibitor) suppresses it [8]. Schorn et al. also reported that G5 (isopeptidase/DUB inhibitor) blocks NLRP3 inflammasome activation via the inhibition of ubiquitination [41]. They also discovered that protein BRCC3 is the enzyme in mice, and BRCC36 is the enzyme in humans that is involved in deubiquitination [41]. It was later found that FBXL2 (F-box L2) is a ubiquitin ligase and regulates the ubiquitination and degradation of the NLRP3 inflammasome by proteasomes [105]. Later studies discovered that MARCH7 regulates the polyubiquitination (K48-linked) and inhibition of the NLRP3 inflammasome and that TRIM31 acts in a similar fashion [106, 107].
Phosphorylation and Other Post-Translational Modifications
Protein phosphorylation was observed in various signaling pathways and also in activating the NLRP3 inflammasome [108]. Yan et al. observed that phosphorylation of NLRP3 is done at Ser295 by PKA (Protein Kinase A) and has a negative effect on the NLRP3 activation as it inhibits the ATPase activity of NLRP3 [97]. Guo et al. also observed that the TGR5 pathway is activated by bile acids which eventually results in PKA activation. This pathway happens at the same site, i.e., Ser295 [109]. Phosphorylation of NLRP3 also plays an inhibitory role in its activation that implies that protein phosphates have a modulatory role in its activation [110]. Pathways other than phosphorylation and ubiquitination were also observed to have a regulatory role in activating the NLRP3 inflammasome [100, 101].
Regulation by NLRP3 Interacting Partners
Various proteins such as TXNIP, Nek7, microtubule affinity-regulating kinase 4 (MARK4), heat shock protein 90 (Hsp90), and its co-chaperone SGT1tat interact with NLRP3 and have been reported to modulate the activation of NLRP3 inflammasome [111]. As discussed previously, the ROS which are produced by the stimuli of NLRP3 oxidize thioredoxin and lead to its dissociation from thioredoxin-interacting protein. This results in TXNIP interacting with the NLRP3 inflammasome and facilitating its activation [13]. It was also observed that TXNIP is non-essential in activating the NLRP3 inflammasome when it is triggered by Aβ, monosodium urate, and ATP [112]. It was further reported by Lu et al. that activation of the NLRP3 inflammasome is regulated by PKR [113]. The lack of PKR decreases the maturation of pro-IL-1β and pro-IL-18 and the activation of caspase-1 [113]. Nek7 is vital in the regulation of the NLRP3 activation as the catalytic residue of Nek7 interacts with the LRR and NOD part of the NLRP3 inflammasome (and it also upregulates ASC speck formation as well as activation of caspase-1 and oligomerization of NLRP3) [114, 115]. Many studies have revealed the role of Nek7 in the regulation of the activation of the NLRP3 inflammasome, but the exact pathway of regulation is yet to be known. It has also been reported that Hsp90 prevents the destruction of NLRP3 by shielding it from autophagy and proteasomes, and SGT1 gene forms complex with NLRP3 and keeps it in an inactive [116]. GBP5 (guanylate binding protein 5) is important in the activation of the NLRP3 inflammasome upon exposure to pathogenic bacteria, ATP, and particulate matter [117]. GBP5 facilitates ASC oligomerization which is regulated by NLRP3 [117]. Other researches showed that GBP5 is non-essential for the activation in macrophages [118]. So far, various studies corroborate that GBP5 facilitates AIM2 inflammasome activation and not NLRP3, and more research is needed to substantiate GBP5 in NLRP3 inflammasome activation [118].
EVIDENCE FOR THE ROLE OF NLRP3 ACTIVATION IN ALZHEIMER’S DISEASE
As per the published report, we came across the fact that NLRP3 inflammasome activation promotes the development of AD in 2 ways. Firstly, by regulating the level of neurotoxins that cause neuronal degeneration and by promoting Aβ deposition, resulting in a self-perpetuating feedback mechanism that contributes to the pathogenesis of AD. Based on the published report, it was reported that the activation of NLRP3 inflammasome may be mediated by the aggregation of fibrillar Aβ or oligomers and protofibrils of Aβ having low molecular weight [119]. It was observed that increased expression of genes of caspase-1 and NLRP3 facilitates the production of lesions in APP/PS-1 transgenic mice brains [120] and reduction in the concentration of IL1β as well as caspase-1 facilitates the Aβ clearance[121]. Further studies have shown that the NLRP3 inflammasome’s expression also limits the microglial phagocytosis of Aβ and, thus, promotes Aβ accumulation and synergizes the onset and progression of AD lesions [122].
In one of the clinical studies by Ahmed et al., the correlation between NLRP3 inflammasome and autophagy lysosome-labeled A0205 protein, glial maturation factor (GMF) and p-tau protein were investigated using immunohistochemistry by comparing autopsy brains of human AD patients with normal individuals of the same age. It was found that the neuroinflammation caused by NLRP3 inflammasomes was increased by GMF, which further dampened the removal of aggregated protein via autophagy signaling pathway [123]. Kim et al. reported that NLRP3 and NLRP1 inflammasomes were triggered by Aβ or LPS in peripheral monocytes of AD patients [124]. It has also been observed that a key element in the development of AD neuroinflammation is translocation of peripheral monocytes over the blood–brain barrier, and this even was potentiated by the NLRP3 activation.
Furthermore, the induction of NLRP3 inflammasome can also enhance pathological hyperphosphorylated tau protein production that is permissive towards the initiation and progression of AD [125] This suggested that inactivation of NLRP3 inflammasome can help in inhibiting the hyperphosphorylation and accumulation of tau in tau-related disorders as well as corroborates the Aβ cascade theory of AD pathogenesis [126]. Numerous research findings have shown that neuroinflammatory plaques built up in the brains of AD patients secrete neurotoxins, employ microglial cells to phagocytose the A \(\beta\) aggregate, and release proinflammatory substances, consequently initiating the damage of neighboring tissues and amplifying the toxic effects of Aβ [127]. In AD mouse models, it was observed that activated NLRP3 inflammasome within the cell promotes M1 phenotype microglial activation, leading to Aβ accumulation and higher cognitive impairment [17]. On the other hand, microglial cells with inactivated NLRP3 inflammasome are skewed towards M2 phenotype, leading to decreased extracellular Aβ accumulation and prevention of synaptic nerve cell dysfunction [128].
The overexpression of TXNIP is associated with AD pathogenesis, and this was affirmed by significant upregulation of TXNIP mRNA when their components were investigated using Western blot technique, immunohistochemistry, and PCR in the human cerebral cortex autopsy. Moreover, close association was asserted between the overexpression of TXNIP protein and pathogenesis of AD, and hence, TXNIP which is correlated with NLRP3 activation can serve as a molecular connection between AD and chronic immunological inflammation and may be a potential target for therapeutics [129, 130].
Besides, Lewy body pathologies and increased levels of interleukin-1β in the brains of AD patients are also suspected to be the final product of activated inflammasome action [131, 132]. It was further reported that IL-1β expression was markedly reduced in the microglial cells when treated with cathepsin B inhibitor or lysosomal inhibitor cytochalasin D. This indicates that NLRP3 inflammasome may be activated by Aβ fibrils [122].
In an attempt to comprehend the role of NLRP3 in the development of AD, Heneka et al. hybridized NLRP3 knockout mice with transgenic APP/PS1 mice. It was observed that caspase cleavage was suppressed in the hybrid brain, while the total IL-1β concentration in the hybrid breed was comparable to wild mice, indicating that NLRP3 and IL-1β have a role in the pathogenesis of Alzheimer’s disease [133]. In another study, in NLRP3 gene knockout AD mice, wild-type mice, and APP/PS1/NLRP3 hybrid, it was observed that APP/PS1/NLRP3 hybrid knockout mice exhibited significant reduction in spatial and memory impairment as compared to the APP/PS1 mice, which displayed serious spatial memory deficiencies. LTP (long-term potentiation) is believed to be positively connected with memory and negatively with the intensity of AD. When the plasticity of synapses was investigated within the hippocampus of mice, it was found that mice with activated NLRP3 or caspase-1 prevented LTP suppression and, hence, signified the pathogenic role of NLRP3 pathways in learning and memory [26]. Additionally, it has been reported that in AD, extracellular Aβ is endocytosed by microglia leading to the rupture of lysosome and release of cathepsin B; processing NLRP3 mediated caspase-1 activation [10]. In addition to it, the formation of ROS accelerated by Aβ is also involved in this process [30, 134]. The interaction of NLRP3 occurs directly with fibrils and Aβ oligomers that help in the promotion of ASC and NLRP3 interaction inside the cell’s free environment [134]. The first signal of NLRP3 activation is induced by Aβ aggregation where Aβ binds with CD36 which is a class B scavenger receptor associated with TLR3 and TLR4 [135, 136]. This results in activating NLRP3 and translocation of NF-κB to the nucleus from the cytoplasm leading to NLRP3 and pro-IL-1β transcription and their subsequent activation [30]. Furthermore, E64d and CA-074Me are cathepsin B inhibitors found to lower memory loss and depreciate Aβ plaques [137]. All these studies indicated that NLRP3 inflammasome activation plays key role in the progression of Alzheimer’s disease as shown in Fig. 2.

Mechanism of Alzheimer’s disease pathogenesis via NLRP3 activation.
INHIBITION OF NLRP3 ACTIVATION AS AN EMERGING THERAPEUTIC APPROACH IN THE MANAGEMENT AND TREATMENT OF ALZHEIMER’S DISEASE
Based on various published report, it can be suggested that suppressing the activity of the NLRP3 inflammasome has substantial potential to prevent, manage, and treat Alzheimer’s disease. B-hydroxybutyrate (BHB), a ketone that efficiently crosses the BBB and inhibits NLRP3 inflammasome activation in human monocytes, was found to be present in lower amounts in AD patients [138]. However, its increased levels exhibited better cognitive functioning among AD patients [138]. In another study, treatment of APP/PS1 transgenic AD mice with BHB led to a significant decrease in the number as well as volume of plaques present in its cerebral cortex, thereby suggesting BHB or BH derivatives as potential candidates to prevent or treat AD [139]. As discussed previously, neuroinflammation plays a key role in the onset of AD, and the increased expression of IL-1β, which is the main inducer of neuroinflammation, has been observed in the brain of AD patients. Thus, inhibiting the NLRP3 inflammasome activation can improve the abnormal behavior in AD [140, 141]. The application of Dapansutrile (OLT1177), a NLRP3 specific inhibitor, is reported to enhance the memory as well as learning ability, normalize AD markers in plasma, restore the synaptic plasticity, and decrease the pathological plaques in the cerebral cortex, as well as reduce the activity of brain glial cells. Thus, based on this study, it was concluded that OLT1177 can be a potential drug candidate for the treatment of AD in the future.
Park et al. [119] reported that diacetyl-p-phenylenediamine, a synthetic compound containing benzene ring, triggers microglia phagocytosis and enhanced the cognitive performance in transgenic AD mice by altering the NF-kB signaling pathway and by reducing the expression of NLRP3 inflammasome [119]. Studies also indicated the potential application of various herb extracts like radix scrophulariae (RS) and pterostilbene; a phytoconstituent derived from grapes and strawberries reduced Aβ-induced neuroinflammation within the microglial cell in the treatment of AD by the inhibition of NLRP3 inflammasome activation [123, 142]. Kim et al. showed that RS treatment in 5XFAD mice inhibited IL-1β production, decreased the expression of NLRP3 protein, and showed a significant reduction in the caspase-1 activity [124]. RS also influences neuroinflammation and inhibited the expression of the BACE1 protein and formation of Aβ plaque [124]. Mirzaei et al. showed that fresh coconut oil antagonizes the Aβ-induced toxicity and showed neuroprotective activity by reducing NLRP3 inflammasome activation [126].
Hong et al. showed that progesterone (PG) exhibited potential neuroprotective activity by reducing the Aβ-induced NLRP3 inflammasome activation and by increasing autophagy in astrocytes [127]. Another study showed that stavudine (D4T), an inhibitor of nucleoside reverse transcriptase, downregulated the production of caspase-1 and IL-18, reduced the NLRP3 assembly, increased Aβ autophagy, and showed potent anti-Alzheimer’s effect [138]. Another potential therapeutic molecule for drug development in AD is TSG or 2,3,5,4-tetrahydroxy stilbene-2-O-b-D-glucoside that reduces LPS-induced neuroinflammation via inhibition of NLRP3 inflammasome activity as well as modulates mitochondrial autophagy in the microglia.
SYNTHETIC DRUGS AS NLRP3 INHIBITORS
Glyburide, also known as BIIBO93, is a synthetic sulfonylurea medicament. It is extensively employed in the USA to treat diabetes (type 2) [143]. Glyburide has also been studied in clinical trials for the possible therapeutic effect against stroke [144]. Glyburide does not inhibit the release of IL-1β via NLRP1 or NLRC4 pathway but does NLRP3 inflammasome-specific inhibition [44]. Glyburide also inhibits caspase-1 activation and secretion of 1L-1β when studied against the stimuli which acts independently from P2X7 receptor [44]. Additionally, glyburide has exhibited inhibitory potential in vivo [127] and in vitro studies, but this inhibitory potential requires a large dose and hence causes hypoglycemia [145]. 16,673–34-0 was synthesized during the synthesis of glyburide. Fischer and Schulze-Osthoff showed that 16,673–34-0 inhibits the formation of the NLRP3 inflammasome in primary adult rat cardiomyocytes and murine macrophages but did not affect the inflammasomes of NLRC4 or AIM2 [49, 58]. It was found that 16,673–34-0 exhibited inhibitory potential in the events at a later stage of the process (ASC binding or conformational changes) [49, 58]. Its exact mechanism of action is not known, and further research is required to establish its potential. JC124 derived from glyburide was modified by Kuwar et al. to regulate the hypoglycemic activity of glyburide. JC124 showed enhanced anti-inflammatory potential when investigated against traumatic brain injury. Treatment with JC124 also reduced caspase-1, TNFα, ASC, immature IL-1β, and inducible nitric oxide synthase expression and, thus, inhibited the activation of the NLRP3 inflammasome [146]. JCC124 also exhibited protective action against transgenic AD and acute MI mouse models [147, 148]. FCC11A-2 or (1-ethyl-5-methyl-2-phenyl-1H-benzo[d]imidazole) is a small compound and NLRP3 inhibitor. In a study, Liu et al. showed the NLRP3 inflammasome inhibition property of this synthetic compound FCC11A-2 in THP-1 cells and mouse model of DSS-induced colitis suppressed the release of IL-1β and IL-18. It also incumbered proximity-induced pro-caspase-1 autocleavage leading to reduced levels of active caspase-1 via NF-B activation [149]. Parthenolide is a sesquiterpene lactone present in plants, and it is widely used as a medicinal herb in a variety of inflammatory disorders due to its potent anti-inflammatory effects [150]. It suppresses the activation of caspase-1 by alkylating its cysteine residues. It also interferes directly with the activity of ATPase of NLRP3 via modification of cystein [60]. Nevertheless, due to its limited solubility as well as bioavailability, its water-soluble analogs, VX-740 and VX-765, are presently being studied [151, 152]. VX-765 and pralnacasan/VX-740 are peptidomimetic compounds and are analogs of parthenolide and act by inhibiting caspase-1, acting as. These compounds are pro-drug and hence, converted to their aldo-acids by plasma esterases and block caspase-1 by modifying the catalytic cysteine fragments left in the caspase-1 active site [135, 152, 153]. VX-740 exhibited promising results in the treatment of osteoarthritis and rheumatoid arthritis models and showed anti-inflammatory potential during clinical phase I and phase II trials [154]. However, this compound showed severe hepatic toxicity after long-term use [155]. Apart from VX-740, VX-765 also showed potential to decrease IL-1β/18 in the dermatitis mouse model of rheumatoid arthritis. Recent researches also showed that VX-765 mitigated the severity of AD and improved the cognitive function in mice [156].
Bay 11–7082, which is a phenyl vinyl sulfone, has been reported to inhibit the NF-κB pathway via inhibition of IKKβ kinase activity. In one of the study, Bay 11–7082 inhibited ASC pyroptosome organization and NLRP3 inflammasome activity in NG5 cells via binding to the ATPase domain. Bay 11–7082 specifically inhibits the NLRP3 inflammasome [60]. Preclinical study in mice and dogs demonstrated that they are non-mutagenic, have acceptable pharmacokinetics properties, and are well-tolerated [157]. Vinyl sulfonate/sulfone molecules like Bay 11–7082 and others give hope and structures for improvements in the future and can be used for the management and treatment of AD. BHB is a ketone metabolite and has extensively been studied in the inhibitory potential of NLRP3 inflammasome. Youm et al. analyzed the NLRP3 inflammasome blocking property of BHB, and the outcome of the study showed significant reduction in the level of IL-1b/18 in response to NLRP3 inflammasome activation. BHB acts by blocking the NLRP3 inflammasome activation without interfering with the inhibition of glycolysis, ROS, autophagy, and AMP-activated protein kinase [158]. These studies concrete the fact that increased BHB concentration can decrease the seriousness of inflammatory disease which are regulated by NLRP3. MCC950 is a diaryl sulfonylurea compound, being one of the most effective and specific NLRP3 inflammasome inhibitors. It is currently under great consideration for the treatment of NLRP3-mediated various disease condition including AD. MCC950 interacts with the NACHT domain and blocks ATP hydrolysis (necessary for NLRP3 oligomerization) and, thus, specifically reduces NLRP3-induced ASC oligomerization and activation, keeping NLRP3 in an inactive state [148, 150]. Earlier, it was found that MCC950 inhibit the maturation of IL-1β by caspase-1, but Coll et al. reported that MCC950 has the ability to block canonical as well as non-canonical activation of NLRP3 inflammasome and decreases IL-1β generation by inhibiting ASC oligomerization in human macrophages and mice [48]. Another recent study found that MCC950 binds directly to the NLRP3 NACHT domain’s Walker B region and inhibits ATP hydrolysis and ameliorated the assembly of NLRP3 inflammasomes [159]. MCC950 has been shown to reduce pulmonary and cutaneous inflammation in mice, and further in vivo studies in mouse model of human multiple sclerosis indicated that MCC950 reduced the severity of experimental autoimmune encephalomyelitis (EAE) [136]. In another experiment on mouse model of Parkinson’s, oral administration of MCC950 was found to reverse dopaminergic degeneration [154]. Also, MCC950 promotes cognitive function and clearance of Aβ via suppression of NLRP3 inflammasome [160]. MCC950 intracerebral injection inhibited exogenous tau pathology. This study showed that MCC950 is a potent drug for AD’s treatment. CY-09 was first identified by Jiang et al. and has a direct and notable inhibitory activity on the inflammasome of NLRP3 in mice models [45]. CY-09 inhibited MSU-mediated inflammation and activation of caspase-1 in bone marrow-derived macrophages. CY-09 prevents the binding of ATP with NLRP3 by removing the Walker A motif of NLRP3 but doesn’t affect RIG-1, NLRP1, NOD2, and NLRC4. This inhibitory activity is independent of ubiquitination of NLRP3. CY-09 exhibited a notable pharmacokinetic profile along with increased stability, oral bioavailability, and safety along with remarkable therapeutic and preventive potential in the mice models of CAPS, gout, and T2D. Further research is needed to clearly understand its whole potential [45].
It is well established that for the function of NLRP3 inflammation, ASC is a necessary adaptor protein, and by inhibiting ASC, asbestos, silica, monosodium urate crystals (MSU), or pathogen-induced activation of caspase-1 and secretion of IL-1β can be inhibited [33, 161, 162]. Also, the deposition of Aβ is promoted by ASC. Hence, the deficiency of ASC can reduce the deposition of Aβ. Through direct interaction with ASC, PYD-only proteins (POPs), POP1 and POP2, suppress the activation of inflammasome and disrupt PYD connections between ASC and NLRP3. POPs can also the inhibit activation of NF-kB and could be a way to “fine-tune” the activation of NLRP3 inflammasome in AD. 3,4-Methylenedioxy-β-nitrostyrene (MNS) is a synthetic small molecule and NLRP3 inhibitor. He et al. showed that MNSbinds to the NACHT and LRR domains of NLRP3 and downregulates ATPase activity. The downregulating of the ATPase activity implies that MNStargets the NLRP3 cysteine(s). It has no effect on the NLRC4 and AIM2 inflammasomes and hence acts as a specific NLRP3 inhibitor. Tranilist (TR), a tryptophan metabolite analog, has also been identified as an NLRP3 inflammasome inhibitor. TR is comparatively safer and showed high tolerance level in clinical studies [136, 143]. In one of the studies, it was found that TR inhibited the intrinsic NLRP3-ASC interaction but had no effect on the NLRP3-NEK7 interactions, suggesting that it directly targets NLRP3 by binding to the NLRP3 NACHT motif and hinders the NLRP3-NLRP3 interactions [45]. However, TR does not interfere with the NLRP3 inflammasome’s upstream signaling events such as the expression of NLRP3 or pro-IL-1β, K + , and Cl− efflux, ROSgeneration, or mitochondrial dysfunction. Additionally, TR also showed potent effect against gout disease and type 2 diabetes mellitus mice models [137]. Thus, owing to its high safety and efficacy in clinical use, TR may be considered as a potential therapeutic agent in the treatment of NLRP3-mediated disorders. OLT117 is a β-sulfonyl nitrile, which is currently under evaluation in phase II clinical trials against degenerative arthritis [137]. Marchetti et al. studied the MSU and zymosan-induced arthritic and suppressed IL-6 and IL-1β secretion and has the potency to reduce infiltration of neutrophils and swelling of joints. OLT117 blocks the non-canonical and canonical as well as inhibits ATPase activity of NLRP3. OLT1177 prevents the secretion of IL-1β by reducing the activity of caspase-1 in the monocytes derived from cryopyrin-associated periodic syndrome patients. OLT1177 is a promising compound in the treatment of diseases related to the NLRP3 inflammasome.
Apart from the specific NLRP3 inhibitor, non-specific inhibitors such as NSAIDs, autophagic modulator, and ILs have also been studied for possible NLRP3 inhibitory potency. In a hybrid model of APP/PS1 mice, increased deposition of Aβ plaque was found that indicated AD pathophysiological features. Hence, the use of NSAIDs showed reduced pathogenesis of AD through its anti-inflammatory potential via NLRP3 inhibition [163]. Ibuprofen showed reduced area and number of deposited Aβ, inhibited microglial activation, and activated IL-1β [143]. Moreover, NF-κB mediated transcription of NLRP3 and pro-IL-1β production can be inhibited by using NSAIDs. Autophagy and autophagy-related proteins are the emerging therapeutic interventions for the management and treatment of AD. As discussed previously, autophagy is a natural cellular self-protective mechanism in which pathogens, aggregated proteins, or damaged organelles are gathered in intracellular autophagosomes and eventually digested by hydrolytic enzymes in the lysosomes. Numerous published evidence have showed that the pathogenesis of AD and Aβ clearance is impaired by inappropriate autophagy [164]. Autophagy has been shown to modulate the activation of inflammasome by removing stimuli, activating inflammasome and degrading inflammasome contents [165]. Furthermore, autophagy stimulated/promoted the degeneration of extracellular Aβ fibrils and also helped in the inhibition of the activation of NLRP3 inflammasome induced by Aβ [166]. As autophagy is a key part in releasing IL-1β, it can be inhibited by using Rapamycin, which is an autophagy agonist [127]. Based numerous published studies, it was found that inhibiting NLRP3 and its progressive constituents can be set as the potential therapeutic cure for AD. These can be inhibited by blocking IL-1β signals via IL‑1β antibodies and IL‑1R antagonists [163]. Currently, the soluble decoy receptor rilonacept, the IL-1R antagonist anakinra, and neutralizing monoclonal anti-IL-1β antibody canakinumab have been investigated against IL-1β-mediated neuroinflammation and AD [165, 166]. Multiple non-inflammatory and inflammatory disorders have been successfully treated with anakinra to specifically suppress the activity of IL-1β, including familial Mediterranean fever, rheumatoid arthritis, gout, cryopyrin-associated periodic syndrome, heart failure, type 2 diabetes mellitus, and AD [127, 143, 163]. Furthermore, several researchers have suggested that IL-1β could be a possible therapy method for Alzheimer’s disease. IL-1Ra deletion mice, for example, show increased neuronal damage, and microglial activation when it gets exposure to exogenous Aβ and treated with its inhibitor showed significant anti-Alzheimer’s effect [167]. The long-term treatment of 3Tg-AD mice with an IL-1R blocking antibody improved cognition, reduced cerebral inflammation, and alleviated tau pathology and lowered fAβ levels [168]. As a matter of case, IL-1β inhibitors can be useful to some extent, but they might not be tolerated by everyone. Additional detail of various other NLRP3 is shown in Table 1.
NATURAL PRODUCTS AS A POTENT NLRP3 INHIBITORS
Curcumin
Curcumin is a yellow lipid-soluble polyphenolic compound derived from the rhizomes of turmeric (Curcuma longa) and from the plants such as ginger and used extensively in the food and cosmetic industries as a coloring and flavoring agent [124]. However, it has lately attracted interest due to its numerous pharmacological qualities such as anti-inflammatory, antibacterial, and antioxidant properties, as well as its therapeutic potential in neurological, cardiovascular, pulmonary, metabolic, cancer, and autoimmune disorders [124]. Recent studies revealed that hypoxia or glucose deficiency significantly increased IL-1β and glutamate production in the hippocampus of mice [169]. It was found that curcumin administration to the mouse lowered the production of IL-1β in the hippocampus [169]. Curcumin also reduced glutamate-induced phosphorylation of PERK and IRE1α, which are transmembrane detectors of ER stress that trigger inflammatory responses in AD [169]. Furthermore, curcumin prevented glutamate-induced ROS production and inhibited glutamate-induced TXNIP expression in neuroblastoma cell lines and thus found to be involved in the inhibition of NLRP3-mediated neuroinflammation [169]. Moreover, curcumin blocked the activation of caspase-3 and hindered mitochondrial functions in glutamate-stimulated hippocampus and thus inhibited glutamate-induced cell death [169]. Thus, the above findings concluded that curcumin stimulates AMPK, which limits glutamate-induced ER stress and ROS generation, thereby suppressing TXNIP-induced activation of NLRP3 inflammasome, eventually reducing the production of IL-1β in the hippocampus and minimizing brain damage [169]. In another study, curcumin substantially suppressed the production of IL-1β induced by LPS with nigericin, MSU, aluminum, or ATP [170]. Its pre-treatment decreased nigericin-induced intracellular potassium depletion and reduced lysosome disruption and cathepsin B leakage, as well as inhibited the release of HMGB-1 (high mobility group box 1) and hence inhibited NLRP3 inflammasome activation [170]. These findings show that decrease in secreted mature IL-1β is predominantly due to the suppression of NLRP3 inflammasome activation, and curcumin was found to be potential inhibitor of NLRP3 inflammasome.
Aloe Vera
Aloe vera have various medicinal uses; provide hydration to the skin; promote wound healing; and exhibited antioxidant, anti-diabetic, anti-inflammatory, antineoplastic, and anti-microbial properties [171]. There are more than 75 active moieties including immunomodulatory components present in the Aloe vera gel plant. Treatment with Aloe vera remarkably decreased IL-1β synthesis (induced by LPS) in monocyte-derived macrophages and THP-1 cells and downregulated IL-1β, TNF-α, and IL-6 level in colorectal mucosa human model and in sepsis mouse model [172,173,174]. It also blocked caspase-1, pro-IL-1β, and NLRP3 expressions along with P2X7 receptor induced by LPS [172]. Aloe vera also prevented the activation of p38, ERk, NF-κB, and JNK signaling mechanisms [172].
Oridonin
Oridonin, an herbal compound derived from Rabdosia rubescens, has been used as a medicinal extract in traditional Chinese medicine since time immemorial [155, 175]. It has been linked to a multitude of anticancer actions, including cell cycle arrest, inhibition of angiogenesis, and induction of apoptosis [164, 165]. It has been reported that oridonin inhibits NF-kB as well as MAPK activation and suppresses the production of inflammasome-independent proinflammatory cytokines [127, 166]. Moreover, it has potential beneficial effects in case of sepsis, colitis, and neuroinflammation [176]. Upon treatment with oridonin in a RAW 264.7 cell, NLRP3 inflammasome assembly and activation were blocked [177]. Upon treatment in mice with acute lung injury induced by LPS, NLRP3/ASC/ IL-1β protein levels were reduced [178]. In the mice with induced traumatic brain injury, oridonin treatment led to the reduced levels of NLRP3/Casp1 genes, along with IL-1B and IL-18 level in the cerebral cortex [178]. He et al. revealed that oridonin forms a covalent bond with the cysteine 279 moiety of the NLRP3 NACHT domain and oppose the NLRP3-NEK7 interaction, thereby preventing the subsequent NLRP3 inflammasome activation. Thus, future research may validate it as a potential NLRP3 inflammasome inhibitor.
Isoliquiritigenin
Isoliquiritigenin is a chalcone-structured flavonoid derived from the roots as well as rhizomes of Glycyrrhiza uralensis. It is a natural sweetening agent and medicinal food that exhibit antioxidant and anti-inflammatory properties [179, 180]. It’s action in the suppression of NLRP3 inflammasome was first reported in 2014 [181]. This compound is specific towards NLRP3 inflammasome because it showed no impact on other inflammasomes [181]. It worked by inducing the inhibition in oligomerization of ASC and, hence, suppressed inflammasome activation [181]. In the mice eWAT, treatment with this drug helped in lowering the levels of gene expression for NLRP3/casp1/ and IL-1β [181]. In another study, 0.02% w/w of this drug had no impact on body weight increase, but it was observed to reduce the food intake [182]. In ICH rat model, isoliquiritigenin administration was found to reduce neurological deficits along with the reduction in the levels of NLRP3/casp1 gene expression and IL-1β and IL-18 level in the brain [181]. This compound was also found to mitigate the NF-κB signaling pathway to facilitate Nrf2 gene transcription in the injured brain [70].
Silibilin
Silibilin (SB) is a natural flavonoid and a polyphenolic compound extracted from Silybum marianum and has anti-inflammatory and hepatoprotective properties [183]. Upon activation of NLRP3 inflammasome through LPS and ATP, reduced autocleavage of casp1 was observed on treatment with SB [184]. SB has been reported to reduce the formation of inflammasome complexes and production of ROS but shown to have no effect on the expression level of proteins (ASC, caspase-1, and NLRP3). Upon incubation for 3 h with RAW 264.7 cells, SB blocked NF-κB signaling; activation of NLRP3 inflammasome and release of IL-1β were observed [184, 185]. Additionally, in other study, upon administering SB orally to mice with liver steatosis, blocked casp1 cleavage was observed along with reduced NLRP3 levels [186].
Cardamonin
Cardamonin is a flavonoid and seed extract of Alpinia katsumadai, which is a traditional Chinese herb of ginger family that has been used to treat various disorders of inflammation [187]. It is shown to inhibit the release of IL-1β and autocleavage of caspase-1 during activation of NLRP3 inflammasome in a preclinical study [13]. It is found to be highly specific towards NLRP3 inflammasome activation and blocks the inflammasome assembly [130]. Additionally, in another study, upon intraperitoneal administration of cardamonin 1 h before injecting LPS, the levels of IL-1β were reduced, and efficacy was comparable with MCC950 [130, 153].
Caffeic Acid Phenethyl Ester
Caffeic acid phenethyl ester (CAPE) is a natural polyphenolic compound obtained from the honeybee propolis and possess potent anti-inflammatory and antioxidant properties [5]. CAPE inhibits the oligomerization of ASC, release of IL-1β, and autocleavage of caspase-1 during activation of NLRP3 inflammasome [85]. CAPE is not bound to the CARD of ASC or PYD of NLRP3 but to the PYD of ASC and found to bind with Lys24 and Glu13 of ASC via hydrogen bonds and via lipophilic interaction with Leu45 and Lys21 [85]. Further, in the in vivo study, treatment with CAPE decreased the NLRP3 protein level in a dose-dependent manner. It was also found that CAPE inhibits the activity of NLRP3 inflammasome in human aortic valve interstitial cells (AVICs) and also suppress the deubiquitination of NLRP3 [188].
Ginsenosides
Ginsenosides is extracted from ginseng (Panax roots) and used traditionally against various diseases like gastric ulcer, diabetes, AD, and cancer [189]. Currently, 30 ginsenosides have been identified, and Rb1 and Rg3 are found to be potent anti-inflammatory agents and inhibitor of NLRP3 inflammasome [190]. Rb1 (Korean ginseng) is extracted from Panax ginseng and is hydrolyzed by intestinal bacteria to produce compound K (CK) [191]. On administering CK orally in db/db mice, improvement of tolerance towards glucose, sensitivity towards insulin, and improved memory as well as cognitive function were observed [192]. Reduction in the levels of NLRP3/casp1/ IL-1β/ASC was also found in the db/db mice hippocampus on treatment with CK [192]. Recently, it has been observed that Rg3 is highly specific towards NLRP3 inflammasome activation [193]. In LPS-induced NLRP3 activation, autocleavage of casp1 was blocked, and IL-1β and IL18 levels were reduced upon treatment with Rg3 [193]. Pre-treatment in the mice with Rg3 before induction of endotoxic shock via LPS also resulted in suppressed IL-1β [193]. Studies have shown that Rg3 also worked by blocking NLRP3-NEK7 interaction [194]. Studies have shown that ginsenoside PPT (ginseol K-g1) also can inhibit the activation of NLRP3 inflammasome and reduced the levels of IL-1β in serum along with blockage of autocleavage of caspase-1 and oligomerization of ASC [127]. In another study, on acute peritonitis model of mice induced by MSU, neutrophil incursion and IL-1β level in fluids of peritoneal lavage were attenuated by PPT [127]. Additionally, PPT was found to reduce the NLRP3 IL-1β and caspase-1 levels in fibrotic livers and alleviate hepatic fibrosis induced by thioacetamide [128].
EGCG
Epigallocatechin-3-gallate or EGCG is a well-known polyphenol derived from green tea and exhibits immense antioxidant as well as neuroprotective properties [195]. Studies showed that it inhibits the activation of NFκB, thereby reducing the expression of various inflammatory substances including iNOS, IL-6, TNFα, and MMPs [195]. In a study conducted on unilateral ureteral obstruction mice model, EGCG was found to inhibit renal inflammation associated with NLRP3 inflammasome action along with reduced caspase-1, IL-1β, and NFκB pathway as well as NLRP3 expression [196]. The advantageous properties of EGCG have also been demonstrated in contrast-induced nephropathy rat models recently [196]. It increased the expression of HO-1 and Nrf2 and reduced oxidative stress and NLRP3 expression as well as IL-1β secretion [196].
Genipin
Genipin is water-soluble aglycone and works as a cross-linking bifunctional reagent. It is isolated from Gardenia jasminoides and has protective effects on the gallbladder and the liver [197]. It exhibits anti-bleeding, anti-swelling, anti-inflammatory, anti-hypertensive, and neuroprotective potency [197]. Genipin prevents the leakage of protons regulated by uncoupling protein 2 (UCP2) which is activated by superoxide and mitochondrial UCP2. Increased expression of UCP2 results in the increased expression of NLRP3 in human THP-1 [197]. Treatment with genipin decreases the levels of IL-1β in ATP-treated LPS-primed cells [197]. Genipin prevents the activation of caspase-1 and IL-1β and the oligomerization of ASC which is dependent on NLRP3 but does not suppress immature caspase-1 and IL-1β and ASC [197].
Ginseng
Ginseng is a ginsenoside containing compound that inhibits the NFκB signaling pathway and decreases the production of inflammatory cytokines [198]. It is amphiphilic in nature and alters the functions as well as the fluidity of plasma membrane by intercalating with it [199]. The extract of red ginseng (RGE), obtained by steaming and drying of fresh ginseng root, was found to inhibit nigericin-, ATP-, and aluminum-induced secretion of IL-1β in LPS-primed bone marrow-derived macrophages (BMDMs) and THP-1 cells [200]. It also improved the fatality rate in LPS-induced septic shock mice models via reduction in the expression of iNOS and subsequent production of NO [199, 200]. Similar to ginsenosides, it also decreases the secretion of IL-1β in double-stranded DNA-transfected macrophage cells [200]. Moreover, it acts as a potent NLRP3 inflammasome inhibitor and decreases the secretion of the active form of caspase-1 [200].
Mangiferin
Mangiferin, a polyphenol, water-soluble compound, has a C-glycosyl xanthone skeleton. It is derived from mango trees and component of a few medicinal herbs [201]. It exhibits antiviral, analgesic, anti-microbial, anti-diabetic, anti-inflammatory, neuroprotective, and anti-sclerotic tendencies [201]. Mangiferin lessens the release of IL-1β in human endothelial cells when exposed to high glucose [201]. It inhibits the production of ROS and reduced ER stress, phosphorylation of IRE1 and the expression of TXNIP [202]. It also reduced the production of IL-1β and expression of NLRP3 [202]. Mangiferin blocked the expression of iNOS, increased the expression of HO-1 and Nrf2, and hence, exhibited antioxidant effect.
Propolis
Propolis is a resinous compound produced by bees (Apis mellifera L.) by combining enzymes and secretions of their hypopharyngeal glands with plant buds and wax [203]. Raw propolis contain various compounds like amino acids, terpenoids, sugars, polyphenols, and steroids, and this composition varies greatly with the geography and collecting season as well as with the botanical origin [203]. Propolis is known for its anti-inflammatory and neuroprotective properties [203]. Artepillin C, a key physiologically active phenolic component found in green propolis from Southeast Brazil, was evaluated in PMA-stimulated RAW264.7 macrophages [203, 204]. The outcome of the study showed that it considerably decreased the release of ROS which is an activator of NLRP3 [203, 204]. Moreover, it considerably contributed to the regulation of chemokine-mediated inflammation via reduction in the release of IL-12p40 amd IL-1β as well as TNFα [203, 204]. Moreover, propolis was also found to inhibit Legionella pneumophila-induced IL-1β secretion in LPS-primed BMDMs [203, 204]. These results indicate that propolis may be useful in regulating the inhibition of NLRP3 inflammasome, and future studies are to understand the molecular mechanism behind it.
Quercetin
Quercetin is a flavonoid which exhibits antioxidant, anti-inflammatory, and neuroprotective potential [205]. It is found in fruits, vegetables, and herb beverages and prevents diabetic kidney and inhibits inflammation [205]. It has been reported that quercetin improves the renal dysfunction and hyperuricemia induced by fructose and mitigates the release of IL-1β [205, 206]. It was reported that the activity of caspase-1, NLRP3, and ASC expression is stimulated by fructose and quercetin inhibits these activities [206]. As discussed previously, the inhibition of NLRP3 activation is regulated by ROS and TXNIP, and quercetin has been reported to act as a potent antioxidant and hence inhibits the NLRP3 activation [207, 208]. In a study of a spinal cord injury model, it was observed that administration of quercetin promoted recovery via inhibition of NLRP3 activation [209]. Further research is needed to understand the mechanism of action of quercetin as how it acts. However, molecular mechanism showed that quercetin decreases the level of TNFα, IL-1β, and IL-18 and reduces the synthesis of ROS along with a decrease in the expression of ASC, caspase-1, and NLRP3 [209].
Resveratrol
Resveratrol is a polyphenolic compound derived from grape skin and mulberry and exists in two isomeric forms, viz., cis and trans [210]. The cis-resveratrol is produced spontaneously during the fermentation of grapes by isomerization of the trans isomer by yeast isomerases and can be obtained by exposing the trans isomer to sunlight [210]. Studies revealed that the trans isomer exhibits a broad spectrum of pharmacological effects, such as antioxidant, anti-inflammatory, anticarcinogenic, antiplatelet aggregation, anti-aging immune-modulatory, and neuroprotective effect [210]. One of the well-known functions of resveratrol is to promote the activation of Sirt1 (sirtuin 1), a deacetylase that alters and deregulates inflammatory genes [210]. It negatively regulates the NLRP3 inflammasome by activating AMPK [211]. Study have also showed that pre-treatment with cis-resveratrol significantly reduced the IL-1β production [212]. According to Huang, the inhibitory action of cis-resveratrol on the secretion of IL-1β may be related to decreased expression of the purinergic receptor P2X7R and the ER stress marker GRP78 that contributes to reduced ROS generation and decreased caspase-1 and caspase-4 activation [212]. It was also found that the cis isomer suppressed the expression of COX-2 that might be due to the decreased PGE2 generation [212]. In another study, resveratrol was found to increase the level of cellular cAMP that inhibit the inflammatory reactions as well as NLRP3 inflammasome activation [213]. Yang et al. reported that treatment with resveratrol for 4 weeks decreased the expression NLRP3 inflammasome, IL-1, IL-6, and TNF [214]. It was also reported that resveratrol’s regulatory effects on the NLRP3 inflammasome were possibly caused by the cumulative effects of Sirt1 and Sirt6 [214]. Recently, a study revealed that resveratrol therapy efficiently reduced the maturation of caspase-1 and IL-1β in response to TLR1/2 agonist Pam3CSK4 as well as NLRP3-inflammasome inducers such as nigericin, silica, ATP, and MSU [214]. Resveratrol administration also inhibited the acetylated α-tubulin accumulation, hence reducing the proximal localization of NLRP3 and ASC in macrophages [215]. In a recent study, it was found that resveratrol hindered the ATP-induced activation of NLRP3 and IL-1β generation in BV2 cell line as well as in septic mouse model [215]. Another research study showed that resveratrol induces cellular autophagy via AMPK activation and protects cells from ROS-induced NLRP3-mediated inflammatory damage [216]. Thus, it was concluded that resveratrol can potentially suppress the NLRP3 inflammasome activation of both in vivo and in vitro, with decreased mitochondrial ROS generation being the possible mechanism behind it.
Sulforaphane
Sulforaphane (SFN) is an isothiocyanate found in mustard, broccoli, brussel sprouts, radish, cabbage, and other cruciferous vegetables [217]. It is produced from the cell walls of plants by the action of enzyme myrosinase [217]. It is reported to be effective in various neurological, cardiovascular, and other diseases [217]. SFN primarily activates the Nrf2 antioxidant and suppresses NFκB signaling pathway [218, 219]. It also downregulates the processing of IL-1β by inflammasome complexes of NLRP3, AIM2, NLRC4, and NLRP1 in bone marrow-derived macrophages [220]. Treatment with SFN decreased IL-1β synthesis which was stimulated by MSU crystals in a peritonitis model of gout and hence showed the inhibitory effect on NLRP3 inflammasome [221, 222]. Studies have also shown that SFN is a promising compound in the treatment of AD as it crosses the blood–brain barrier and reduced the level of Aβ [221, 222]. It was reported that sulforaphane notably downregulates IL-1β synthesis and the expression of NLRP3 in macrophages of Aβ1-42 peptide-stimulated THP-1 cells and saturated fatty acids [222, 223]. In conclusion, SFN reduces the expression of caspase-1, ASC, and IL-1β and NLRP3 and acted as a potent anti-Alzheimer’s agent.
Curculiginis Rhizoma
Curculiginis rhizoma (EX) is the desiccated rhizome extract of Curculigo orchioides Gaertn, and Epimedii folium is the desiccated leaf extract of Epimedium brevicornu Maxim [117]. A variety of pharmacological effects such as antioxidant, neuroprotective, and anti-inflammatory are exhibited by EX [117] The level of IL-1β, TNF-α, IL-6, and other proinflammatory cytokines was found to be decreased in the cortex and hippocampus when treated with these compounds [117]. Moreover, EX ameliorated the phosphorylation of MyD88, MAPKs, cathepsin B, NF-κB, and NLRP3 inflammasome expression and also increased glutathione, glutathione peroxidase, superoxide dismutase, and catalase [117].
Virgin Coconut Oil
Virgin coconut oil (VCO) has shown significant effect against AD as it reverses the oxidative stress and the expression of NLRP3 gene towards normal [118, 224]. It also exhibited antioxidant and anti-inflammatory potential [118, 224]. In a study, extra-virgin olive oil was found to protect the mice against the onset of β-amyloid-induced neurotoxic manifestations and restored normal BBB activity [225]. The presence of polyphenolic oleocanthal in VCO reduced the deposition of Aβ, decreased inflammation, restored normal BBB activity, and lowered the level of proinflammatory cytokines in the brain [225]. Additionally, VCO inhibited NACHT, LRR, and NLRP3 inflammasome and induces autophagy, thereby slowing potent anti-Alzheimer’s effect [225].
Lychee Seeds
Lychee seeds (LS) comprises of proanthocyanidins and polyphenols such as catechin, quercetin, and rutin that inhibited NF-κB pathway and prevent neuronal cell death triggered by Aβ and the microglial neuroinflammatory response [142, 226]. These polyphenols appreciably prevented the activation of the NLRP3 inflammasome in bEnd.3 cells (Aβ25–35-induced) and prevented IL-1β, NLRP3, caspase-1, and p62 expression in PS1/APP mice. LS also triggered the autophagy via AMPK/mTOR/ULK1 pathway and enhanced memory and spatial learning [142, 226]. Further, research showed that LSPs (lychee seeds polyphenols) suppressed the release of IL-1β, caspase-1 cleavage and NLRP3 and ASC expression in BV-2 cells (Aβ1–42-induced) [142, 226].
Picrorhiza kurroa
A number of research studies have demonstrated the beneficial therapeutic effects of Picrorhiza kurroa Bentham (PK), which is a common Ayurvedic medicinal herb [124]. PK possess potent anti-allergic, antioxidant, and anti-inflammatory as well as anti-asthmatic properties [124]. It has been reported that PK mitigated the microglial-induced neuro-inflammation and suppressed the leucine-rich repeat (LRR), nucleotide-binding oligomerization domain, and activation of NLRP3 inflammasome in AD mouse model. Additionally, PK reduced the β-secretase 1 expression which is involved in the production of Aβ [124].
Baicalin
Baicalin is a flavonoid compound derived from the roots of Scutellaria baicalensis Georgi and exhibits potent anti-inflammatory and neuroprotective potential [169]. Oral administration of baicalin to APP/PS1 transgenic mice over a period of 33 days considerably lowered the spatial memory impairment in Morris water maze test and in passive avoidance test [169]. It also reduced the activation of microglial cells and decreased the expression of IL-1β, IL-18, and iNOS against Aβ-induced neurotoxicity [169]. These findings suggested that the protective action of baicalin against Alzheimer’s disease is related to the dysregulation of microglia-induced neuroinflammation via inhibition of NLRP3 inflammasome and the TLR4/NF-kB signaling pathway[169].
Flavocoxid
Flavocoxid is a combination of pure catechin and baicalin [142]. It possesses potent anti-inflammatory and neuroprotective property potential and suppresses the peroxidase activity of COX-2 enzymes [142]. Its neuroprotective potential is related with regulating the IL-1β production and improvement of cognitive functions [142]. Along with a notable flavocoxid, it also reduced the Aβ plaques, reduced neuron apoptosis, and mitigated the loss of neurons in the hippocampus [142]. Apart from the aforementioned discussed phytoconstituents, dihydromyricetin, astaxanthin, and tenuigenin have also showed significant potential in the inhibition of NLRP3 inflammasome as shown in Table 2 (Fig. 3) [169, 170, 226].

Mechanism of action of natural products against NLRP3 activation and Alzheimer’s disease.
CLINICAL TRIALS AND PATENTS OF NLRP3 INFLAMMASOME INHIBITORS IN THE PHARMACEUTICAL INDUSTRY PIPELINE
Based on the pathological involvement of NLRP3 and potential pharmacological attributes of various NLRP3 inhibitors, various NLRP3 inflammasomes are under clinical trials and also under developmental stage [62]. Novartis in partnership with IFM Therapeutics designed IFM2427 [63] that possess NLRP3 inhibitory activity [63, 66]. IFM2427 is in the phase I clinical trials and is designed while targeting gout, steatohepatitis (non-alcoholic), and atherosclerosis-like inflammatory diseases [62]. NodThera and Olatec Therapeutics released the structure of their inhibitor OLT1177, i.e., dapansutrile [68, 227, 228]. Dapansutrile is a potential COVID-19 treatment drug and is in the phase II trials [67, 96, 98].
Recently, patent has been granted to NodThera for the NLRP3 inhibitors which are derivatives of carbamoyl [229]. A potential action was shown in the preclinical trials by NT-0167 by significantly inhibiting the release of IL-1β and was reported by NodThera [67]. There has been a report that urea ester produgs having membrane permeability act as NLRP3 antagonists in PBMC cells and inhibit the release of IL-1β [99, 100]. Inflazome biotech has been granted patents from the USA and Europe for Somalix and Inzomelid, which are potent and specific NLRP3 inflammasome antagonists. Somalix is peripheral targeting and is in phase II trials, whereas Inzomelid is a CNS-targeting and recently passed phase I of the clinical trials [66, 102, 103]. O’Neill and his co-workers have received patent for MCC950, MCC7840, and other sulfonylureas NLRP3 antagonists. The universities of Queensland and Cadila have designed a novel array of potent NLRP3 antagonist derivatives of N-cyano sulfoximine urea, and these derivatives have significant oral bioavailability. More details of various clinical trials on NLRP3 inhibitors are discussed in Table 3.
CHALLENGES AND FUTURE PERSPECTIVE
AD, one of the major neurodegenerative diseases, is the most common form of dementia. Typical symptoms of AD are characterized by loss of memory, impairment of cognition, depression, and behavioral disturbance. The pathological hallmarks of AD in the brain include deposition of extracellular amyloid plaques, cleavage products of the amyloid precursor protein, (APP), and neurofibrillary tangles containing intracellular fibrillar aggregates of the microtubule-associated protein tau. For many years, numerous studies have shown Aβ as a crucial DAMP-promoting neuroinflammation through multiple molecular pathways. However, Aβ-inducedNLRP3 inflammasome activation was first described in 2008. Using the primary microglia as an in vitro model, their study has reported that NLRP3 inflammasome recruitment was involved in phagocytosis of Aβ and subsequent lysosomal damage and release of cathepsin B.
Since their discovery about two decades ago, the inflammasome, a molecular platform triggering the activation of inflammatory molecules (e.g., caspases and processing of pro-IL), has garnered particular attention in modulating innate responses AD. Importantly, an over-activated inflammasome complex can initiate a destructive environment and the subsequent onset of AD. Although the role of NLRPs (especially NLRP3) has been implicated in neuroinflammation, as discussed extensively in numerous studies, a significant knowledge gap that remains is the characterization of drugs, including their efficacy and how they can be used to target neurodegenerative diseases. Evidence suggests that HIV-1 infection leads to chronic neural inflammation, as in other neurodegenerative diseases, since the virus and its components can prime and assemble the components of the NLRP3 inflammasome, which ultimately leads to pyroptosis and inflammation. The role of these NLRPs plays a pivotal role in the development of neuroHIV, AD, PD, and MS, but further studies are needed for expanding the realm of applying key molecular targets as efficacious therapeutics.
To date, numerous promising NLRP3 drugs for neurological disorders have been developed and tested; however, the biggest challenge for their clinical translation is their permeability at the BBB junction. Thus, newer strategies, including drugs, are needed to overcome the limitations of drug transport across anatomical barriers (BBB) to have the desired therapeutic efficacy without inducing systemic or CNS toxicity. In recent times, nanotechnology has shown great potential in delivering drugs across the BBB for many neurological manifestations and has significantly increased the quality of patients’ lives. However, there are reports available that have shown how the nanomaterial’s physiological properties (material types, nanoparticle size, surface functionalization, etc.) can themselves lead to NLRP3 inflammasome activation 186 in the CNS. Therefore, it is imperative to develop or repurpose safe existing drugs/small molecules that will have better target specificity, high lipophilicity, good partition coefficiency, and BBB penetrability.
Cannabinoid receptor 2 (CB2R) has been documented as a therapeutic target in inflammation‐associated afflictions. Shao et al. [230] has explained the reason for the stimulation of the anti-inflammatory CB2R leading to the repression of NLRP3 inflammasome kindling and stimulation (in a manner analogous to autophagy induction) in murine BV2 microglia treated with LPS and ATP as well as in a murine model of EAE. CB2R stimulation attenuates the intensity of murine EAE. Hence, CB2R agonists like HU‐308 may be promising effective agents for intervening in NLRP3 inflammasome‐related diseases via autophagy induction.
Currently, various small synthetic molecules, such as MCC950, AAV2-hIL-10, zonisamide, and JNJ7777120, and natural products, such as resveratrol, tanshinone-I, and apocynin, have been explored for inhibiting microglial activation. However, these small molecules and natural products are limited by fast hepatic metabolism and poor BBB permeation. Nanotechnology offers the advantage over the conventional therapeutic approaches by stabilizing drugs to cross the BBB and exhibit a superior pharmacotherapeutic effect. However, this is not always the case. For example, nanocarriers sometimes exhibit unintended interactions with the proteins and tissues due to their size, surface morphology, and neurotoxicity. Cationic and metallic NPs, such as gold, silver, titanium dioxide, and silica NPs, interact with the proteins, disrupt cellular structures, alter cell membrane permeability, and exhibit neurotoxicity. To overcome this problem, incorporating antioxidants into the NPs is proposed as an alternative approach. Unlike metallic NPs, polymeric NPs are more stable and offer controlled release and selective targeting profiles. However, these nanocarriers aggregate and exhibit neurotoxicity. Thus, attempts have been made to control their size, charge, and morphology to control these unwanted aggregatory properties. Additionally, using PEG for surface coating and designing specific ligand approach will also offer selective targeting to NLRP3 and bypass the interaction with cellular components within the CNS.
Apart from the small molecules, natural products, and nanocarrier-based therapeutic approach, miRNA has also been emerged as an alternate tool to target NLRP3 in AD. In general, RNA polymerase II is responsible for the transcription of miRNA or pri-miRNA, upon which ribonuclease Drosha further acts, and pri-miRNA is converted into pre-miRNA. This process occurs in the nucleus, and once pre-miRNA is formed, it moves out of the nucleus and into the cytoplasm, in which it is cleaved and mature miRNA is produced. The mature mRNA gets incorporated or loaded into RNA-induced silencing complex (RISC) and argonaute. Finally, these miRNAs are involved in the silencing of mRNA. MicroRNAs may hand out one more opportunity for suppressing inflammasomes. These endogenous non‐coding RNAs are 20–23 nt long and associated with the 3 untranslated parts (3 UTR) of protein‐coding mRNAs to control their translation. MicroRNA‐223 attaches to a conserved location in the 3 UTR of the NLRP3 transcript, inhibiting protein synthesis and consequently blocking NLRP3 inflammasome activation and IL‐1β generation. A lack of microRNA‐223 is associated with neutrophilia, spontaneous lung inflammation, and heightened proclivity to an endotoxin challenge in mice. A few other microRNAs have been documented to be players in the stimulation of the NLRP3 inflammasome, such as microRNA‐155, microRNA‐377, and microRNA‐133a1. Decreasing the concentrations of such agents may prove beneficial in the intervention of inflammasome associated disorders.
However, the use of many of the necked miRNA was associated with fast degradation by RNAse, problems in crossing biological membranes, rapid clearance, and thermal instability when administered. Hence, techniques, such as chemical modification, encapsulating them in suitable nanocarriers, and using cationic polymers, have been used to overcome these limitations. Currently, a few nanocarrier-based (lipid-based) miRNAs, such as MRX34, miR-34a, and let-7, are under clinical investigation for possible use in various disease conditions.
Thus, based on the in-depth literature survey, available clinical evidence, and completed clinical trials, we suggest that a safe and effective nanocarrier system should be developed for the targeted delivery of various synthetic and natural drugs in addition to miRNA. Genomic expression of mRNA in addition to pathway enrichment analysis should be done to identify selective targets for miRNA. Furthermore, to avoid the toxicity and off-targeted side effects and to achieve cell-/target-specific delivery of drugs and miRNA, low-dose combinations of miRNA and natural drugs can be used. This novel drug delivery system may pave the way for clinical treatment in the coming years.
CONCLUSION
NLRP3 inflammasome plays an important role in the neuroinflammatory response associated with AD. Although the NLRP3 inflammasome has been the most intensively investigated inflammasome in the past decade, a unified mechanism for NLRP3 inflammasome activation has not been determined. Recently, Nek7 is identified as a critical NLRP3 regulator. Regardless of the complexity of the pathway, many molecules and drugs targeting the NLRP3 inflammasome have been discovered, providing a new direction for the treatment of AD. The activation/inhibition mechanism of NLRP3 inflammasome and its regulation of the functional state of brain microglia and the pathophysiological process of Alzheimer’s disease remain to be further studied. Further clinical trials are necessary to confirm the role of NLRP3 inhibitors in AD. Research on the neuroinflammation mechanism in AD is beneficial to the prevention, treatment, and management of AD patients.
Availabilty of Data and Materials
Not applicable.
References
Honig, L.S., B. Vellas, M. Woodward, M. Boada, R. Bullock, M. Borrie, et al. 2018. Trial of solanezumab for mild dementia due to Alzheimer’s disease. New England Journal of Medicine 378: 321–330. https://doi.org/10.1056/NEJMOA1705971.
Yamazaki, Y., N. Zhao, T.R. Caulfield, C.C. Liu, and G. Bu. 2019. Apolipoprotein E and Alzheimer disease: Pathobiology and targeting strategies. Nature Reviews. Neurology 15: 501–518. https://doi.org/10.1038/S41582-019-0228-7.
Querfurth, H.W., and F.M. LaFerla. 2010. Alzheimer’s disease. New England Journal of Medicine 362: 329–344. https://doi.org/10.1056/NEJMRA0909142.
Stephenson, J., E. Nutma, P. van der Valk, and S. Amor. 2018. Inflammation in CNS neurodegenerative diseases. Immunology 154: 204–219. https://doi.org/10.1111/IMM.12922.
Lamkanfi, M., and V.M. Dixit. 2009. Inflammasomes: Guardians of cytosolic sanctity. Immunological Reviews 227: 95–105. https://doi.org/10.1111/J.1600-065X.2008.00730.X.
Heneka, M.T., M.P. Kummer, A. Stutz, A. Delekate, S. Schwartz, A. Vieira-Saecker, et al. 2013. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 493: 674–678. https://doi.org/10.1038/NATURE11729.
Lamkanfi, M., and V.M. Dixit. 2014. Mechanisms and functions of inflammasomes. Cell 157: 1013–1022. https://doi.org/10.1016/J.CELL.2014.04.007.
Halle, A., V. Hornung, G.C. Petzold, C.R. Stewart, B.G. Monks, T. Reinheckel, et al. 2008. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nature Immunology 9: 857–865. https://doi.org/10.1038/NI.1636.
Meyer-Luehmann, M., T.L. Spires-Jones, C. Prada, M. Garcia-Alloza, A. De Calignon, A. Rozkalne, et al. 2008. Rapid appearance and local toxicity of amyloid-beta plaques in a mouse model of Alzheimer’s disease. Nature 451: 720–724. https://doi.org/10.1038/NATURE06616.
Pétrilli, V., S. Papin, C. Dostert, A. Mayor, F. Martinon, and J. Tschopp. 2007. Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death and Differentiation 14: 1583–1589. https://doi.org/10.1038/SJ.CDD.4402195.
Savage, C. D., G. Lopez-Castejon, A. Denes, and D. Brough 2012. NLRP3-inflammasome activating DAMPs stimulate an inflammatory response in glia in the absence of priming which contributes to brain inflammation after injury. Frontiers Immunology 3. https://doi.org/10.3389/FIMMU.2012.00288/FULL.
Suzuki, T., K. Kohyama, K. Moriyama, M. Ozaki, S. Hasegawa, T. Ueno, et al. 2020. Extracellular ADP augments microglial inflammasome and NF-κB activation via the P2Y12 receptor. European Journal of Immunology 50: 205–219. https://doi.org/10.1002/EJI.201848013.
Zhou, R., A. Tardivel, B. Thorens, I. Choi, and J. Tschopp. 2010. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nature Immunology 11: 136–140. https://doi.org/10.1038/NI.1831.
Murphy, N., T.R. Cowley, J.C. Richardson, D. Virley, N. Upton, D. Walter, et al. 2012. The neuroprotective effect of a specific P2X7 receptor antagonist derives from its ability to inhibit assembly of the NLRP3 inflammasome in glial cells. Brain Pathology 22: 295–306. https://doi.org/10.1111/J.1750-3639.2011.00531.X.
Wu, A., X. Zhou, G. Qiao, L. Yu, Y. Tang, and L. Yan. 2021. Targeting microglial autophagic degradation in NLRP3 inflammasome-mediated neurodegenerative diseases. Elsevier 2147:101–2.
Bai, H., Q.F. Zhang, J.J. Duan, D.J. Yu, and L.J. Liu. 2018. Downregulation of signal transduction and STAT3 expression exacerbates oxidative stress mediated by NLRP3 inflammasome. Neural Regeneration Research 13: 2147. https://doi.org/10.4103/1673-5374.241470.
Sharma, D., and T.D. Kanneganti. 2016. The cell biology of inflammasomes: Mechanisms of inflammasome activation and regulation. Journal of Cell Biology 213: 617–629. https://doi.org/10.1083/JCB.201602089.
Gold, M., and J. El Khoury. 2015. β-amyloid, microglia, and the inflammasome in Alzheimer’s disease. Semin Immunopathol 37: 607–611. https://doi.org/10.1007/S00281-015-0518-0.
Scambler, T., H.H. Jarosz-Griffiths, S. Lara-Reyna, S. Pathak, J. Wong, J. Holbrook, et al. 2019. ENaC-mediated sodium influx exacerbates NLRP3-dependent inflammation in cystic fibrosis. Elife 8. https://doi.org/10.7554/ELIFE.49248.
Shimada, K., T. Crother, J. Karlin, and J. Dagvadorj. 2012. Immunity NC-, 2012 undefined. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Elsevier n.d.
Wu, P.J., Y.F. Hung, H.Y. Liu, and Y.P. Hsueh. 2017. Deletion of the inflammasome sensor Aim2 mitigates Aβ deposition and microglial activation but increases inflammatory cytokine expression in an Alzheimer disease mouse model. NeuroImmunoModulation 24: 29–39. https://doi.org/10.1159/000477092.
Lučiūnaitė, A., R.M. Mcmanus, M. Jankunec, I. Rácz, C. Dansokho, I. Dalgėdienė, et al. 2019. Soluble Aβ oligomers and protofibrils induce NLRP3 inflammasome activation in microglia. Wiley Online Libr 155: 650–661. https://doi.org/10.1111/jnc.14945.
Furman, D., J. Chang, L. Lartigue, C. Bolen. 2017. Medicine FH-N, 2017 U. Expression of specific inflammasome gene modules stratifies older individuals into two extreme clinical and immunological states. NatureCom.
Friker, L., H. Scheiblich, and I. Hochheiser. 2020. Reports RB-C, 2020 U. β-amyloid clustering around ASC fibrils boosts its toxicity in microglia. Elsevier.
Ahmed, M.E., S. Iyer, R. Thangavel, D. Kempuraj, G.P. Selvakumar, S.P. Raikwar, et al. 2017. Co-localization of glia maturation factor with NLRP3 inflammasome and autophagosome markers in human Alzheimer’s disease brain. J Alzheimer’s Dis 60: 1143–1160. https://doi.org/10.3233/JAD-170634.
Saresella, M., F. La Rosa, F. Piancone, M. Zoppis, I. Marventano, E. Calabrese, et al. 2016. The NLRP3 and NLRP1 inflammasomes are activated in Alzheimer’s disease. Molecular Neurodegeneration 11: 23–37. https://doi.org/10.1186/S13024-016-0088-1.
Marchetti, C., S. Toldo, J. Chojnacki, E. Mezzaroma, K. Liu, F.N. Salloum, et al. 2015. Pharmacologic inhibition of the NLRP3 inflammasome preserves cardiac function after ischemic and nonischemic injury in the mouse. Journal of Cardiovascular Pharmacology 66: 1–8. https://doi.org/10.1097/FJC.0000000000000247.
Muñoz-Planillo, R., P. Kuffa, G. Martínez-Colón, B.L. Smith, T.M. Rajendiran, and G. Núñez. 2013. K+ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 38: 1142–1153. https://doi.org/10.1016/J.IMMUNI.2013.05.016.
Walev, I., J. Klein, M. Husmann, A. Valeva, S. Strauch, H. Wirtz, et al. 2000. Potassium regulates IL-1 beta processing via calcium-independent phospholipase A2. The Journal of Immunology 164: 5120–5124. https://doi.org/10.4049/JIMMUNOL.164.10.5120.
Marchetti, C., J. Chojnacki, S. Toldo, E. Mezzaroma, N. Tranchida, S.W. Rose, et al. 2014. A novel pharmacologic inhibitor of the NLRP3 inflammasome limits myocardial injury after ischemia-reperfusion in the mouse. Journal of Cardiovascular Pharmacology 63: 316–322. https://doi.org/10.1097/FJC.0000000000000053.
Nakanishi, A., N. Kaneko, H. Takeda, T. Sawasaki, S. Morikawa, W. Zhou, et al. 2018. Amyloid β directly interacts with NLRP3 to initiate inflammasome activation: identification of an intrinsic NLRP3 ligand in a cell-free system. Inflamm Regen 38. https://doi.org/10.1186/S41232-018-0085-6.
Yin, J., F. Zhao, J.E. Chojnacki, J. Fulp, W.L. Klein, S. Zhang, et al. 2018. NLRP3 inflammasome inhibitor ameliorates amyloid pathology in a mouse model of Alzheimer’s disease. Molecular Neurobiology 55: 1977–1987. https://doi.org/10.1007/S12035-017-0467-9.
Fulp, J., L. He, S. Toldo, Y. Jiang, A. Boice, C. Guo, et al. 2018. Structural insights of benzenesulfonamide analogues as NLRP3 inflammasome inhibitors: Design, synthesis, and biological characterization. Journal of Medicinal Chemistry 61: 5412–5423. https://doi.org/10.1021/ACS.JMEDCHEM.8B00733.
Groß, C.J., R. Mishra, K.S. Schneider, G. Médard, J. Wettmarshausen, D.C. Dittlein, et al. 2016. K+ efflux-independent NLRP3 inflammasome activation by small molecules targeting mitochondria. Immunity 45: 761–773. https://doi.org/10.1016/J.IMMUNI.2016.08.010.
Sanman, L.E., Y. Qian, N.A. Eisele, T.M. Ng, W.A. van der Linden, and D.M. Monack, et al. 2016. Disruption of glycolytic flux is a signal for inflammasome signaling and pyroptotic cell death. Elife 5. https://doi.org/10.7554/ELIFE.13663.
Clapham, D.E., 2007. Calcium signaling. Cell 1047–58.
Murakami, T., J. Ockinger, J. Yu, V. Byles, A. McColl, A.M. Hofer, et al. 2012. Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc Natl Acad Sci U S A 109: 11282–11287. https://doi.org/10.1073/PNAS.1117765109/-/DCSUPPLEMENTAL.
Lee, G.S., N. Subramanian, A.I. Kim, I. Aksentijevich, R. Goldbach-Mansky, D.B. Sacks, et al. 2012. The calcium-sensing receptor regulates the NLRP3 inflammasome through Ca2+ and cAMP. Nat 492:123–7. https://doi.org/10.1038/nature11588.
Feldmeyer, L., M. Keller, G. Niklaus, D. Hohl, S. Werner, and H.D. Beer. 2007. The inflammasome mediates UVB-induced activation and secretion of interleukin-1β by keratinocytes. Current Biology 17: 1140–1145. https://doi.org/10.1016/J.CUB.2007.05.074.
Chu, J., L.M. Thomas, S.C. Watkins, L. Franchi, G. Núñez, and R.D. Salter. 2009. Cholesterol-dependent cytolysins induce rapid release of mature IL-1β from murine macrophages in a NLRP3 inflammasome and cathepsin B-dependent manner. Journal of Leukocyte Biology 86: 1227–1238. https://doi.org/10.1189/JLB.0309164.
Schorn, C., B. Frey, K. Lauber, C. Janko, M. Strysio, H. Keppeler, et al. 2011. Sodium overload and water influx activate the NALP3 inflammasome. Journal of Biological Chemistry 286: 35–41. https://doi.org/10.1074/JBC.M110.139048.
Verhoef, P.A., S.B. Kertesy, K. Lundberg, J.M. Kahlenberg, and G.R. Dubyak. 2005. Inhibitory effects of chloride on the activation of caspase-1, IL-1β secretion, and cytolysis by the P2X7 receptor. The Journal of Immunology 175: 7623–7634. https://doi.org/10.4049/JIMMUNOL.175.11.7623.
Boxer, M.B., M. Shen, D.S. Auld, J.A. Wells, and C.J. Thomas. 2014. A small molecule inhibitor of caspase 1. Probe Reports from NIH Mol Libr Prog.
Juliana, C., T. Fernandes-Alnemri, J. Wu, P. Datta, L. Solorzano, J.W. Yu, et al. 2010. Anti-inflammatory compounds parthenolide and Bay 11–7082 are direct inhibitors of the inflammasome. Journal of Biological Chemistry 285: 9792–9802. https://doi.org/10.1074/JBC.M109.082305.
Coll, R.C., J.R. Hill, C.J. Day, A. Zamoshnikova, D. Boucher, N.L. Massey, et al. 2019. MCC950 directly targets the NLRP3 ATP-hydrolysis motif for inflammasome inhibition. Nature Chemical Biology 15: 556–559. https://doi.org/10.1038/S41589-019-0277-7.
Lamkanfi, M., T.-D. Kanneganti, L. Franchi, and G. Núñez. 2007. Caspase-1 inflammasomes in infection and inflammation. Journal of Leukocyte Biology 82: 220–225. https://doi.org/10.1189/JLB.1206756.
MacKenzie, S.H., J.L. Schipper, and A.C. Clark. 2010. The potential for caspases in drug discovery. Current Opinion in Drug Discovery & Development 13: 568.
Rudolphi, K., N. Gerwin, N. Verzijl, P. van der Kraan, and W. van den Berg. 2003. Pralnacasan, an inhibitor of interleukin-1beta converting enzyme, reduces joint damage in two murine models of osteoarthritis. Osteoarthritis and Cartilage 11: 738–746. https://doi.org/10.1016/S1063-4584(03)00153-5.
Fischer, U., and K. Schulze-Osthoff. 2005. Apoptosis-based therapies and drug targets. Cell Death and Differentiation 12 (Suppl 1): 942–961. https://doi.org/10.1038/SJ.CDD.4401556.
Hou, Y., Y. Wei, S. Lautrup, B. Yang, Y. Wang, S. Cordonnier, et al. 2021. NAD+ supplementation reduces neuroinflammation and cell senescence in a transgenic mouse model of Alzheimer’s disease via cGAS-STING. Proc Natl Acad Sci USA 118. https://doi.org/10.1073/PNAS.2011226118/-/DCSUPPLEMENTAL.
Van Bruggen, R., M.Y. Köker, M. Jansen, M. Van Houdt, D. Roos, T.W. Kuijpers, et al. 2010. Human NLRP3 inflammasome activation is Nox1-4 independent. Blood 115: 5398–5400. https://doi.org/10.1182/BLOOD-2009-10-250803.
Zhou, R., A.S. Yazdi, P. Menu, and J. Tschopp. 2010. A role for mitochondria in NLRP3 inflammasome activation. Nat 4697329;469:221–5. https://doi.org/10.1038/nature09663.
Shimada, K., T.R. Crother, J. Karlin, J. Dagvadorj, N. Chiba, S. Chen, et al. 2012. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 36: 401–414. https://doi.org/10.1016/J.IMMUNI.2012.01.009.
Park, S., C. Juliana, S. Hong, P. Datta, I. Hwang, T. Fernandes-Alnemri, et al. 2013. The mitochondrial antiviral protein MAVS associates with NLRP3 and regulates its inflammasome activity. The Journal of Immunology 191: 4358–4366. https://doi.org/10.4049/JIMMUNOL.1301170.
Subramanian, N., K. Natarajan, M.R. Clatworthy, Z. Wang, and R.N. Germain. 2013. The adaptor MAVS promotes NLRP3 mitochondrial localization and inflammasome activation. Cell 153: 348–361. https://doi.org/10.1016/J.CELL.2013.02.054.
Iyer, S.S., Q. He, J.R. Janczy, E.I. Elliott, Z. Zhong, A.K. Olivier, et al. 2013. Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity 39: 311–323. https://doi.org/10.1016/J.IMMUNI.2013.08.001.
Martinon, F., V. Pétrilli, A. Mayor, A. Tardivel, and J. Tschopp. 2006. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 440: 237–241. https://doi.org/10.1038/NATURE04516.
Flores, J., A. Noël, B. Foveau, J. Lynham, C. Lecrux, and A.C. LeBlanc. 2018. Caspase-1 inhibition alleviates cognitive impairment and neuropathology in an Alzheimer’s disease mouse model. Nat Commun 9. https://doi.org/10.1038/S41467-018-06449-X.
Franchi, L., R. Muñoz-Planillo, and G. Núñez. 2012. Sensing and reacting to microbes through the inflammasomes. Nature Immunology 13: 325–332. https://doi.org/10.1038/NI.2231.
Perregaux, D.G., P. Mcniff, R. Laliberte, N. Hawryluk, H. Peurano, E. Stam E, et al. 2001. Identification and characterization of a novel class of interleukin-1 post-translational processing inhibitors. ASPET.
Medzhitov, R. 2007. Recognition of microorganisms and activation of the immune response. Nature 449: 819–826. https://doi.org/10.1038/NATURE06246.
Kawai, T., and S. Akira. 2010. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nature Immunology 11: 373–384. https://doi.org/10.1038/NI.1863.
Kawai, T., and S. Akira. 2011. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 34: 637–650. https://doi.org/10.1016/J.IMMUNI.2011.05.006.
Ouslimani, N., J. Peynet, D. Bonnefont-Rousselot, P. Thérond, A. Legrand, and J.L. Beaudeux. 2005. Metformin decreases intracellular production of reactive oxygen species in aortic endothelial cells. Metabolism 54: 829–834. https://doi.org/10.1016/J.METABOL.2005.01.029.
Wang, S., M. Zhang, B. Liang, J. Xu, Z. Xie, C. Liu, et al. 2010. AMPKalpha2 deletion causes aberrant expression and activation of NAD(P)H oxidase and consequent endothelial dysfunction in vivo: Role of 26S proteasomes. Circulation Research 106: 1117–1128. https://doi.org/10.1161/CIRCRESAHA.109.212530.
Kang, L.L., D.M. Zhang, C.H. Ma, J.H. Zhang, K.K. Jia, J.H. Liu, et al. 2016. Cinnamaldehyde and allopurinol reduce fructose-induced cardiac inflammation and fibrosis by attenuating CD36-mediated TLR4/6-IRAK4/1 signaling to suppress NLRP3 inflammasome activation. Sciientific Report 6. https://doi.org/10.1038/SREP27460.
Hu, X., Q. Chi, Q. Liu, D. Wang, Y. Zhang, S. Li. 2019. Atmospheric H 2 S triggers immune damage by activating the TLR-7/MyD88/NF-κB pathway and NLRP3 inflammasome in broiler thymus. Chemosphere 237. https://doi.org/10.1016/J.CHEMOSPHERE.2019.124427.
Fernandes-Alnemri, T., S. Kang, C. Anderson, J. Sagara, K.A. Fitzgerald, and E.S. Alnemri. 2013. Cutting edge: TLR signaling licenses IRAK1 for rapid activation of the NLRP3 inflammasome. The Journal of Immunology 191: 3995–3999. https://doi.org/10.4049/JIMMUNOL.1301681.
Song, N., Z.S. Liu, W. Xue, Z.F. Bai, Q.Y. Wang, J. Dai, et al. 2017. NLRP3 phosphorylation is an essential priming event for inflammasome activation. Molecular Cell 68: 185-197.e6. https://doi.org/10.1016/J.MOLCEL.2017.08.017.
Swanson, K.V., M. Deng, and J.P.Y. Ting. 2019. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nature Reviews Immunology 19: 477–489. https://doi.org/10.1038/S41577-019-0165-0.
Gaidt, M.M., T.S. Ebert, D. Chauhan, T. Schmidt, J.L. Schmid-Burgk, F. Rapino, et al. 2016. Human monocytes engage an alternative inflammasome pathway. Immunity 44: 833–846. https://doi.org/10.1016/J.IMMUNI.2016.01.012.
Saxton, R.A., and D.M. Sabatini. 2017. mTOR signaling in growth, metabolism, and disease. Cell 168: 960–976. https://doi.org/10.1016/J.CELL.2017.02.004.
Kim, J., and K.L. Guan. 2019. mTOR as a central hub of nutrient signalling and cell growth. Nature Cell Biology 21: 63–71. https://doi.org/10.1038/S41556-018-0205-1.
Dhingra, R., H. Gang, Y. Wang, A.K. Biala, Y. Aviv, V. Margulets, et al. 2013. Bidirectional regulation of nuclear factor-κB and mammalian target of rapamycin signaling functionally links Bnip3 gene repression and cell survival of ventricular myocytes. Circulation. Heart Failure 6: 335–343. https://doi.org/10.1161/CIRCHEARTFAILURE.112.000061.
Temiz-Resitoglu, M., S.P. Kucukkavruk, D.S. Guden, P. Cecen, A.N. Sari, B. Tunctan, et al. 2017. Activation of mTOR/IκB-α/NF-κB pathway contributes to LPS-induced hypotension and inflammation in rats. European Journal of Pharmacology 802: 7–19. https://doi.org/10.1016/J.EJPHAR.2017.02.034.
Li, X., X. Zhang, Y. Pan, G. Shi, J. Ren, H. Fan, et al. 2018. mTOR regulates NLRP3 inflammasome activation via reactive oxygen species in murine lupus. Acta Biochimica et Biophysica Sinica (Shanghai) 50: 888–896. https://doi.org/10.1093/ABBS/GMY088.
Choi, Y.J., K.M. Moon, K.W. Chung, J.W. Jeong, D. Park, D.H. Kim, et al. 2016. The underlying mechanism of proinflammatory NF-κB activation by the mTORC2/Akt/IKKα pathway during skin aging. Oncotarget 7:52685–94. https://doi.org/10.18632/ONCOTARGET.10943.
Rezatabar, S., A. Karimian, V. Rameshknia, H. Parsian, M. Majidinia, T.A. Kopi, et al. 2019. RAS/MAPK signaling functions in oxidative stress, DNA damage response and cancer progression. Journal of Cellular Physiology 234: 14951–14965. https://doi.org/10.1002/JCP.28334.
McCubrey, J.A., M.M. LaHair, and R.A. Franklin. 2006. Reactive oxygen species-induced activation of the MAP kinase signaling pathways. Antioxidants & Redox Signaling 8: 1775–1789. https://doi.org/10.1089/ARS.2006.8.1775.
Wang, W., X.Q. Ding, T.T. Gu, L. Song, J.M. Li, Q.C. Xue, et al. 2015. Pterostilbene and allopurinol reduce fructose-induced podocyte oxidative stress and inflammation via microRNA-377. Free Radical Biology & Medicine 83: 214–226. https://doi.org/10.1016/J.FREERADBIOMED.2015.02.029.
Zhou, R., A.S. Yazdi, P. Menu, and J. Tschopp. 2011. A role for mitochondria in NLRP3 inflammasome activation. Nature 469: 221–226. https://doi.org/10.1038/NATURE09663.
Gan, P., Z. Gao, X. Zhao, and G. Qi. 2016. Surfactin inducing mitochondria-dependent ROS to activate MAPKs, NF-κB and inflammasomes in macrophages for adjuvant activity. Scientific Reports 6. https://doi.org/10.1038/SREP39303.
Zhao, W., L. Ma, C. Cai, and X. Gong. 2019. Caffeine inhibits NLRP3 inflammasome activation by suppressing MAPK/NF-κB and A2aR signaling in LPS-induced THP-1 macrophages. International Journal of Biological Sciences 15: 1571–1581. https://doi.org/10.7150/IJBS.34211.
Panicker, N., S. Sarkar, D.S. Harischandra, M. Neal, T.I. Kam, H. Jin, et al. 2019. Fyn kinase regulates misfolded α-synuclein uptake and NLRP3 inflammasome activation in microglia. Journal of Experimental Medicine 216: 1411–1430. https://doi.org/10.1084/JEM.20182191.
Heneka, M.T., M.P. Kummer, A. Stutz, A. Delekate, S. Schwartz, A. Vieira-Saecker, et al. 2012. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 4937434;493:674–8. https://doi.org/10.1038/nature11729.
Ota, M., J. Matsuo, I. Ishida, H. Takano, Y. Yokoi, H. Hori, et al. 2019. Effects of a medium-chain triglyceride-based ketogenic formula on cognitive function in patients with mild-to-moderate Alzheimer’s disease. Neuroscience Letters 690: 232–236. https://doi.org/10.1016/J.NEULET.2018.10.048.
Shibutani, S.T., T. Saitoh, H. Nowag, C. Münz, and T. Yoshimori. 2015. Autophagy and autophagy-related proteins in the immune system. Nature Immunology 16: 1014–1024. https://doi.org/10.1038/NI.3273.
Levine, B., and G. Kroemer. 2019. Biological functions of autophagy genes: A disease perspective. Cell 176: 11–42. https://doi.org/10.1016/J.CELL.2018.09.048.
Nguyen, H.T.T., P. Lapaquette, M.A. Bringer, and A. Darfeuille-Michaud. 2013. Autophagy and Crohn’s disease. Journal of Innate Immunity 5: 434–443. https://doi.org/10.1159/000345129.
Spalinger, M.R., S. Lang, C. Gottier, X. Dai, D.J. Rawlings, A.C. Chan, et al. 2017. PTPN22 regulates NLRP3-mediated IL1B secretion in an autophagy-dependent manner. Autophagy 13: 1590–1601. https://doi.org/10.1080/15548627.2017.1341453.
Saitoh, T., N. Fujita, M.H. Jang, S. Uematsu, B.G. Yang, T. Satoh, et al. 2008. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature 456: 264–268. https://doi.org/10.1038/NATURE07383.
Lee, J., H.R. Kim, C. Quinley, J. Kim, J. Gonzalez-Navajas, R. Xavier, et al. 2012. Autophagy suppresses interleukin-1β (IL-1β) signaling by activation of p62 degradation via lysosomal and proteasomal pathways. Journal of Biological Chemistry 287: 4033–4040. https://doi.org/10.1074/JBC.M111.280065.
Lupfer, C., P.G. Thomas, P.K. Anand, P. Vogel, S. Milasta, J. Martinez, et al. 2013. Receptor interacting protein kinase 2-mediated mitophagy regulates inflammasome activation during virus infection. Nature Immunology 14: 480–488. https://doi.org/10.1038/NI.2563.
Shi, C.S., K. Shenderov, N.N. Huang, J. Kabat, M. Abu-Asab, K.A. Fitzgerald, et al. 2012. Activation of autophagy by inflammatory signals limits IL-1β production by targeting ubiquitinated inflammasomes for destruction. Nature Immunology 13: 255–263. https://doi.org/10.1038/NI.2215.
Seveau, S., J. Turner, M.A. Gavrilin, J.B. Torrelles, L. Hall-Stoodley, J.S. Yount, et al. 2018. Checks and balances between autophagy and inflammasomes during infection. Journal of Molecular Biology 430: 174–192. https://doi.org/10.1016/J.JMB.2017.11.006.
Brandwein, S.R. 1986. Regulation of interleukin 1 production by mouse peritoneal macrophages. Effects of arachidonic acid metabolites, cyclic nucleotides, and interferons. The Journal of Biological Chemistry 261:8624–32. https://doi.org/10.1016/S0021-9258(19)84425-3.
Yan, Y., W. Jiang, L. Liu, X. Wang, C. Ding, Z. Tian, et al. 2015. Dopamine controls systemic inflammation through inhibition of NLRP3 inflammasome. Cell 160: 62–73. https://doi.org/10.1016/J.CELL.2014.11.047.
Taskén, K., and E.M. Aandahl. 2004. Localized effects of cAMP mediated by distinct routes of protein kinase A. Physiological Reviews 84: 137–167. https://doi.org/10.1152/PHYSREV.00021.2003.
Chen, R., L. Zeng, S. Zhu, J. Liu, H.J. Zeh, G. Kroemer, et al. 2019. cAMP metabolism controls caspase-11 inflammasome activation and pyroptosis in sepsis. Science Advances 5. https://doi.org/10.1126/SCIADV.AAV5562.
Guo, C., S. Xie, Z. Chi, J. Zhang, Y. Liu, L. Zhang, et al. 2016. Bile acids control inflammation and metabolic disorder through inhibition of NLRP3 inflammasome. Immunity 45: 802–816. https://doi.org/10.1016/J.IMMUNI.2016.09.008.
Mortimer, L., F. Moreau, J.A. MacDonald, and K. Chadee. 2016. NLRP3 inflammasome inhibition is disrupted in a group of auto-inflammatory disease CAPS mutations. Nature Immunology 17: 1176–1186. https://doi.org/10.1038/NI.3538.
Ollivier, V., G.C.N. Parry, R.R. Cobb, D. De Prost, and N. Mackman. 1996. Elevated cyclic AMP inhibits NF-kappaB-mediated transcription in human monocytic cells and endothelial cells. Journal of Biological Chemistry 271: 20828–20835. https://doi.org/10.1074/JBC.271.34.20828.
Chen, B.-C., C.-C. Liao, M.-J. Hsu, Y.-T. Liao, C.-C. Lin, J.-R. Sheu, et al. 2006. Peptidoglycan-induced IL-6 production in RAW 264.7 macrophages is mediated by cyclooxygenase-2, PGE2/PGE4 receptors, protein kinase A, I kappa B kinase, and NF-kappa B. Journal of Immunology 177:681–93. https://doi.org/10.4049/JIMMUNOL.177.1.681.
Steinberg, G.R., and B.E. Kemp. 2009. AMPK in health and disease. Physiological Reviews 89: 1025–1078. https://doi.org/10.1152/PHYSREV.00011.2008.
Hornung, V., F. Bauernfeind, A. Halle, E.O. Samstad, H. Kono, K.L. Rock, et al. 2008. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nature Immunology 9: 847–856. https://doi.org/10.1038/NI.1631.
Lopez-Castejon, G., N.M. Luheshi, V. Compan, S. High, R.C. Whitehead, S. Flitsch, et al. 2013. Deubiquitinases regulate the activity of caspase-1 and interleukin-1β secretion via assembly of the inflammasome. Journal of Biological Chemistry 288: 2721–2733. https://doi.org/10.1074/JBC.M112.422238.
Py, B.F., M.S. Kim, H. Vakifahmetoglu-Norberg, and J. Yuan. 2013. Deubiquitination of NLRP3 by BRCC3 critically regulates inflammasome activity. Molecular Cell 49: 331–338. https://doi.org/10.1016/J.MOLCEL.2012.11.009.
Han, S.H., T.B. Lear, J.A. Jerome, S. Rajbhandari, C.A. Snavely, D.L. Gulick, et al. 2015. Lipopolysaccharide primes the NALP3 inflammasome by inhibiting its ubiquitination and degradation mediated by the SCFFBXL2 E3 ligase. Journal of Biological Chemistry 290: 18124–18133. https://doi.org/10.1074/JBC.M115.645549.
Song, H., B. Liu, W. Huai, Z. Yu, W. Wang, J. Zhao, et al. 2016. The E3 ubiquitin ligase TRIM31 attenuates NLRP3 inflammasome activation by promoting proteasomal degradation of NLRP3. Nature Communication 7. https://doi.org/10.1038/NCOMMS13727.
Sandall, C.F., and J.A. MacDonald. 2019. Effects of phosphorylation on the NLRP3 inflammasome. Archives of Biochemistry and Biophysics 670: 43–57. https://doi.org/10.1016/J.ABB.2019.02.020.
Spalinger, M.R., S. Kasper, C. Gottier, S. Lang, K. Atrott, S.R. Vavricka, et al. 2016. NLRP3 tyrosine phosphorylation is controlled by protein tyrosine phosphatase PTPN22. The Journal of Clinical Investigation 126: 1783–1800. https://doi.org/10.1172/JCI83669.
Masters, S.L., A. Dunne, S.L. Subramanian, R.L. Hull, G.M. Tannahill, F.A. Sharp, et al. 2010. Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1β in type 2 diabetes. Nature Immunology 11: 897–904. https://doi.org/10.1038/NI.1935.
Lu, B., T. Nakamura, K. Inouye, J. Li, Y. Tang, P. Lundbäck, et al. 2012. Novel role of PKR in inflammasome activation and HMGB1 release. Nature 488: 670–674. https://doi.org/10.1038/NATURE11290.
He, Y., M.Y. Zeng, D. Yang, B. Motro, and G. Núñez. 2016. NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature 530: 354–357. https://doi.org/10.1038/NATURE16959.
Shi, H., Y. Wang, X. Li, X. Zhan, M. Tang, M. Fina, et al. 2016. NLRP3 activation and mitosis are mutually exclusive events coordinated by NEK7, a new inflammasome component. Nature Immunology 17: 250–258. https://doi.org/10.1038/NI.3333.
Mayor, A., F. Martinon, T. De Smedt, V. Pétrilli, and J. Tschopp. 2007. A crucial function of SGT1 and HSP90 in inflammasome activity links mammalian and plant innate immune responses. Nature Immunology 8: 497–503. https://doi.org/10.1038/NI1459.
Meunier, E., M.S. Dick, R.F. Dreier, N. Schürmann, D.K. Broz, S. Warming, et al. 2014. Caspase-11 activation requires lysis of pathogen-containing vacuoles by IFN-induced GTPases. Nature 509: 366–370. https://doi.org/10.1038/NATURE13157.
Meunier, E., P. Wallet, R.F. Dreier, S. Costanzo, L. Anton, S. Rühl, et al. 2015. Guanylate-binding proteins promote activation of the AIM2 inflammasome during infection with Francisella novicida. Nature Immunology 16: 476–484. https://doi.org/10.1038/NI.3119.
Park, M.H., M. Lee, G. Nam, M. Kim, J. Kang, B.J. Choi, et al. 2019. N, N′-Diacetyl-p-phenylenediamine restores microglial phagocytosis and improves cognitive defects in Alzheimer’s disease transgenic mice. Proc Natl Acad Sci U S A 116: 23426–23436. https://doi.org/10.1073/PNAS.1916318116/VIDEO-8.
Qiu, W.Q., R. Pan, Y. Tang, X.G. Zhou, J.M. Wu, L. Yu, et al. 2020. Lychee seed polyphenol inhibits Aβ-induced activation of NLRP3 inflammasome via the LRP1/AMPK mediated autophagy induction. Biomedicine & Pharmacotherapy 130: 110575. https://doi.org/10.1016/J.BIOPHA.2020.110575.
Aricioglu, F., C.S. Ozkartal, T. Bastaskin, E. Tüzün, C. Kandemir, S. Sirvanci, et al. 2019. Antidepressant-like effects induced by chronic blockade of the purinergic 2X7 receptor through inhibition of non-like receptor protein 1 inflammasome in chronic unpredictable mild stress model of depression in rats. Clin Psychopharmacol Neurosci 17: 261–272. https://doi.org/10.9758/CPN.2019.17.2.261.
Chen, D., H. Gao, C. Peng, S. Pei, A. Dai, X. Yu, et al. 2020. Quinones as preventive agents in Alzheimer’s diseases: Focus on NLRP3 inflammasomes. AcademicOupCom 72: 1481–1490. https://doi.org/10.1111/jphp.13332.
Wang, C., Y. Xu, X. Wang, C. Guo. 2019. TW-A& redox, 2019 undefined. Dl-3-n-Butylphthalide inhibits NLRP3 inflammasome and mitigates Alzheimer’s-like pathology via Nrf2-TXNIP-TrX axis. LiebertpubCom 30:1411–31. https://doi.org/10.1089/ars.2017.7440.
Kim, N., J. Do, I.G. Ju, S.H. Jeon, J.K. Lee, and M.S. Oh. 2020. Picrorhiza kurroa prevents memory deficits by inhibiting NLRP3 inflammasome activation and BACE1 expression in 5xFAD mice. Neurotherapeutics 17: 189–199. https://doi.org/10.1007/S13311-019-00792-7.
Gu, Y., J. Wu, C. Faucheu, J.L. Lalanne, A. Diu, D.J. Livingston, et al. 1995. Interleukin-1β converting enzyme requires oligomerization for activity of processed forms in vivo. EMBO Journal 14: 1923–1931. https://doi.org/10.1002/J.1460-2075.1995.TB07184.X.
Mirzaei, F., M. Khazaei, A. Komaki, I. Amiri, and C. Jalili. 2018. Virgin coconut oil (VCO) by normalizing NLRP3 inflammasome showed potential neuroprotective effects in amyloid-β induced toxicity and high-fat diet fed rat. Food and Chemical Toxicology 118: 68–83. https://doi.org/10.1016/J.FCT.2018.04.064.
Hong, Y., Y. Liu, D. Yu, M. Wang, and Y. Hou. 2019. The neuroprotection of progesterone against Aβ-induced NLRP3-caspase-1 inflammasome activation via enhancing autophagy in astrocytes. International Immunopharmacology 74: 105669. https://doi.org/10.1016/J.INTIMP.2019.05.054.
Wang, J., R. Qiu, L. Yuan, F. Meng, and Q. Tang. 2015. Analysis on the Alpinia katsumadai components of Zingiberaceae plants and their functions on myeloma resistance. Pakistan Journal of Pharmaceutical Sciences 28: 1065–1068.
Murtaza, G., S. Karim, M.R. Akram, S.A. Khan, S. Azhar, A. Mumtaz, et al. 2014. Caffeic acid phenethyl ester and therapeutic potentials. BioMed Research International. https://doi.org/10.1155/2014/145342.
Wang, Z., G. Xu, Y. Gao, X. Zhan, N. Qin, S. Fu, et al. 2019. Cardamonin from a medicinal herb protects against LPS-induced septic shock by suppressing NLRP3 inflammasome. Acta Pharm Sin B 9: 734. https://doi.org/10.1016/J.APSB.2019.02.003.
Lee, H.E., G. Yang, N.D. Kim, S. Jeong, Y. Jung, J.Y. Choi, et al. 2016. Targeting ASC in NLRP3 inflammasome by caffeic acid phenethyl ester: a novel strategy to treat acute gout. Scientific Reports 6:1–11. https://doi.org/10.1038/srep38622.
Guo, H., J.B. Callaway, and J.P.Y. Ting, 2015. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nature Medicine 217:21:677–87. https://doi.org/10.1038/nm.3893.
Jiang, J., X. Sun, M. Akther, M.L. Lian, L.H. Quan, S. Koppula, et al. 2020. Ginsenoside metabolite 20(S)-protopanaxatriol from Panax ginseng attenuates inflammation-mediated NLRP3 inflammasome activation. The Journal of Ethnopharmacology 2020;251. https://doi.org/10.1016/J.JEP.2020.112564.
Eren, E., and N. Özören, 2019. The NLRP3 inflammasome: a new player in neurological diseases. Turkish Journal Biology = Turk Biyol Derg 43:349–59. https://doi.org/10.3906/BIY-1909-31.
Jiang, H., H. He, Y. Chen, W. Huang, J. Cheng, J. Ye, et al. 2017. Identification of a selective and direct NLRP3 inhibitor to treat inflammatory disorders. Journal of Experimental Medicine 214: 3219–3238. https://doi.org/10.1084/JEM.20171419.
Platten, M., P.P. Ho, S. Youssef, P. Fontoura, H. Garren, E.M. Hur, et al. 2005. Treatment of autoimmune neuroinflammation with a synthetic tryptophan metabolite. Science 310: 850–855. https://doi.org/10.1126/SCIENCE.1117634.
Toldo, S., and A. Abbate. 2018. The NLRP3 inflammasome in acute myocardial infarction. Nature Reviews. Cardiology 15: 203–214. https://doi.org/10.1038/NRCARDIO.2017.161.
Song, J., Z.Y. Cui, L.H. Lian, X. Han, L.S. Hou, G. Wang, et al. 2020. 20 S-Protopanaxatriol ameliorates hepatic fibrosis, potentially involving FXR-mediated inflammatory signaling cascades. Journal of Agriculture and Food Chemistry 68: 8195–8204. https://doi.org/10.1021/ACS.JAFC.0C01978.
Shippy, D.C., C. Wilhelm, P.A. Viharkumar, T.J. Raife, and T.K. Ulland. 2020. β-Hydroxybutyrate inhibits inflammasome activation to attenuate Alzheimer’s disease pathology. Journal of Neuroinflammation 17: 1–12. https://doi.org/10.1186/S12974-020-01948-5/FIGURES/4.
La Rosa, F., M. Saresella, I. Marventano, F. Piancone, E. Ripamonti, N. Al-Daghri, et al. 2019. Stavudine reduces NLRP3 inflammasome activation and modulates amyloid-β autophagy. Journal of Alzheimer’s Disease 72: 401–412. https://doi.org/10.3233/JAD-181259.
Lonnemann, N., S. Hosseini, C. Marchetti, D.B. Skouras, D. Stefanoni, A. D’Alessandro, et al. 2020. The NLRP3 inflammasome inhibitor OLT1177 rescues cognitive impairment in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A 117: 32145–32154. https://doi.org/10.1073/PNAS.2009680117/-/DCSUPPLEMENTAL.
Qiu, W.Q,, R. Pan, Y. Tang, X.G. Zhou, J.M. Wu, L. Yu, et al. 2020. Lychee seed polyphenol inhibits Aβ-induced activation of NLRP3 inflammasome via the LRP1/AMPK mediated autophagy induction. Biomed Pharmacother 130. https://doi.org/10.1016/J.BIOPHA.2020.110575.
Lim, G.P., F. Yang, T. Chu, P. Chen, W. Beech, B. Teter, et al. 2000. Ibuprofen suppresses plaque pathology and inflammation in a mouse model for Alzheimer’s disease. Journal of Neuroscience 20: 5709–5714. https://doi.org/10.1523/JNEUROSCI.20-15-05709.2000.
Deplano, S., H.T. Cook, R. Russell, L. Franchi, S. Schneiter, G. Bhangal, et al. 2013. P2X7 receptor-mediated Nlrp3-inflammasome activation is a genetic determinant of macrophage-dependent crescentic glomerulonephritis. Journal of Leukocyte Biology 93: 127–134. https://doi.org/10.1189/JLB.0612284.
Schroder, K., and J. Tschopp. 2010. The inflammasomes. Cell 140: 821–832. https://doi.org/10.1016/J.CELL.2010.01.040.
Kerr, I.D., J.H. Lee, C.J. Farady, R. Marion, M. Rickert, M. Sajid, et al. 2009. Vinyl sulfones as antiparasitic agents and a structural basis for drug design. Journal of Biological Chemistry 284: 25697–25703. https://doi.org/10.1074/JBC.M109.014340.
Huang, Y., H. Jiang, Y. Chen, X. Wang, Y. Yang, J. Tao, et al. 2018. Tranilast directly targets NLRP3 to treat inflammasome-driven diseases. EMBO Mol Med 10. https://doi.org/10.15252/EMMM.201708689.
Esser, N., N. Paquot, and A.J. Scheen. 2015. Anti-inflammatory agents to treat or prevent type 2 diabetes, metabolic syndrome and cardiovascular disease. Expert Opinion on Investigational Drugs 24: 283–307. https://doi.org/10.1517/13543784.2015.974804.
Youm, Y.H., K.Y. Nguyen, R.W. Grant, E.L. Goldberg, M. Bodogai, D. Kim, et al. 2015. The ketone metabolite β-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nature Medicine 21: 263–269. https://doi.org/10.1038/NM.3804.
Ozaki, E., M. Campbell, and S.L. Doyle. 2015. Targeting the NLRP3 inflammasome in chronic inflammatory diseases: Current perspectives. Journal of Inflammation Research 8: 15–27. https://doi.org/10.2147/JIR.S51250.
O’Neill, L.A.J. 1996. Interleukin I receptors and signal transduction. Biochemical Society Transactions 24: 207–211. https://doi.org/10.1042/BST0240207.
Akash, M.S.H., Q. Shen, K. Rehman, and S. Chen. 2012. Interleukin-1 receptor antagonist: A new therapy for type 2 diabetes mellitus. Journal of Pharmaceutical Sciences 101: 1647–1658. https://doi.org/10.1002/JPS.23057.
Coll, R.C., A.A.B. Robertson, J.J. Chae, S.C. Higgins, R. Muñoz-Planillo, M.C. Inserra, et al. 2015. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nature Medicine 21: 248–257. https://doi.org/10.1038/NM.3806.
Gordon, R., E.A. Albornoz, D.C. Christie, M.R. Langley, V. Kumar, S. Mantovani, et al. 2018. Inflammasome inhibition prevents α-synuclein pathology and dopaminergic neurodegeneration in mice. Science Translational Medicine 10. https://doi.org/10.1126/SCITRANSLMED.AAH4066.
Kuo, L.M., C.Y. Kuo, C.Y. Lin, M.F. Hung, J.J. Shen, and T.L. Hwang. 2014. Intracellular glutathione depletion by oridonin leads to apoptosis in hepatic stellate cells. Molecules 19: 3327–3344. https://doi.org/10.3390/MOLECULES19033327.
Fujita, E., Y. Nagao, K. Kaneko, S. Nakazawa, and H. Kuroda. 1976. The antitumor and antibacterial activity of the Isodon diterpenoids. Chem Pharm Bull (Tokyo) 24: 2118–2127. https://doi.org/10.1248/CPB.24.2118.
Zhao, G., T. Zhang, X. Ma, K. Jiang, H. Wu, C. Qiu, et al. 2017. Oridonin attenuates the release of pro-inflammatory cytokines in lipopolysaccharide-induced RAW264.7 cells and acute lung injury. Oncotarget 8:68153–64. https://doi.org/10.18632/ONCOTARGET.19249.
Guzman, M.L., R.M. Rossi, S. Neelakantan, X. Li, C.A. Corbett, D.C. Hassane, et al. 2007. An orally bioavailable parthenolide analog selectively eradicates acute myelogenous leukemia stem and progenitor cells. Blood 110: 4427–4435. https://doi.org/10.1182/BLOOD-2007-05-090621.
Konneh, M. 1998. Tranilast Kissei Pharmaceutical. Drugs 1:141–6.
Abbate, A., M.C. Kontos, J.D. Grizzard, G.G.L. Biondi-Zoccai, B.W. Van Tassell, R. Robati, et al. 2010. Interleukin-1 blockade with anakinra to prevent adverse cardiac remodeling after acute myocardial infarction (Virginia Commonwealth University Anakinra Remodeling Trial [VCU-ART] Pilot study). American Journal of Cardiology 105: 1371–1377. https://doi.org/10.1016/J.AMJCARD.2009.12.059.
Craft, J.M., D.M. Watterson, E. Hirsch, and L.J. Van Eldik. 2005. Interleukin 1 receptor antagonist knockout mice show enhanced microglial activation and neuronal damage induced by intracerebroventricular infusion of human beta-amyloid. Journal of Neuroinflammation 2. https://doi.org/10.1186/1742-2094-2-15.
Kitazawa, M., D. Cheng, M.R. Tsukamoto, M.A. Koike, P.D. Wes, V. Vasilevko, et al. 2011. Blocking IL-1 signaling rescues cognition, attenuates tau pathology, and restores neuronal β-catenin pathway function in an Alzheimer’s disease model. The Journal of Immunology 187: 6539–6549. https://doi.org/10.4049/JIMMUNOL.1100620.
Xiang, Z., L. Ho, S. Yemul, Z. Zhao, P. Pompl, K. Kelley, et al. 2002. Cyclooxygenase-2 promotes amyloid plaque deposition in a mouse model of Alzheimer’s disease neuropathology. Gene Expression 10: 271–278. https://doi.org/10.3727/000000002783992352.
Zare-Shahabadi, A., E. Masliah, G.V.W. Johnson, and N. Rezaei. 2015. Autophagy in Alzheimer’s disease. Reviews in the Neurosciences 26: 385–395. https://doi.org/10.1515/REVNEURO-2014-0076.
Harris, J., T. Lang, J.P.W. Thomas, M.B. Sukkar, N.R. Nabar, and J.H. Kehrl. 2017. Autophagy and inflammasomes. Molecular Immunology 86: 10–15. https://doi.org/10.1016/J.MOLIMM.2017.02.013.
Cho, M.H., K. Cho, H.J. Kang, E.Y. Jeon, H.S. Kim, H.J. Kwon, et al. 2014. Autophagy in microglia degrades extracellular β-amyloid fibrils and regulates the NLRP3 inflammasome. Autophagy 10: 1761–1775. https://doi.org/10.4161/AUTO.29647.
Dinarello, C.A., A. Simon, and J.W.M. Van Der Meer. 2012. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nature Reviews. Drug Discovery 11: 633–652. https://doi.org/10.1038/NRD3800.
Van Opdenbosch, N., and M. Lamkanfi. 2019. Caspases in cell death, inflammation, and disease. Immunity 50: 1352–1364. https://doi.org/10.1016/J.IMMUNI.2019.05.020.
Jin, X., M.Y. Liu, D.F. Zhang, X. Zhong, K. Du, P. Qian, et al. 2019. Baicalin mitigates cognitive impairment and protects neurons from microglia-mediated neuroinflammation via suppressing NLRP3 inflammasomes and TLR4/NF-κB signaling pathway. CNS Neuroscience & Therapeutics 25: 575–590. https://doi.org/10.1111/CNS.13086.
Bitto, A., D. Giuliani, G. Pallio, N. Irrera, E. Vandini, F. Canalini, et al. 2017. Effects of COX1-2/5-LOX blockade in Alzheimer transgenic 3xTg-AD mice. Inflammation Research 66: 389–398. https://doi.org/10.1007/S00011-017-1022-X.
Hamman, J.H. 2008. Composition and applications of Aloe vera leaf gel. Molecules 13: 1599–1616. https://doi.org/10.3390/MOLECULES13081599.
Budai, M.M., A. Varga, S. Milesz, J. Tozsér, and S. Benko. 2013. Aloe vera downregulates LPS-induced inflammatory cytokine production and expression of NLRP3 inflammasome in human macrophages. Molecular Immunology 56: 471–479. https://doi.org/10.1016/J.MOLIMM.2013.05.005.
Langmead, L., R.J. Makins, and D.S. Rampton. 2004. Anti-inflammatory effects of aloe vera gel in human colorectal mucosa in vitro. Alimentary Pharmacology & Therapeutics 19: 521–527. https://doi.org/10.1111/J.1365-2036.2004.01874.X.
Yun, N., C.H. Lee, and S.M. Lee. 2009. Protective effect of Aloe vera on polymicrobial sepsis in mice. Food and Chemical Toxicology 47: 1341–1348. https://doi.org/10.1016/J.FCT.2009.03.013.
Kadota, S., P. Basnet, E. Ishii, T. Tamura, and T. Namba. 1997. Antibacterial activity of trichorabdal A from Rabdosia trichocarpa against Helicobacter pylori. Zentralbl Bakteriol 286: 63–67. https://doi.org/10.1016/S0934-8840(97)80076-X.
Zhang, Z.Y., R. Daniels, and H.J. Schluesener. 2013. Oridonin ameliorates neuropathological changes and behavioural deficits in a mouse model of cerebral amyloidosis. Journal of Cellular and Molecular Medicine 17: 1566–1576. https://doi.org/10.1111/JCMM.12124.
Yang, H., H. Lv, H. Li, X. Ci, and L. Peng, 2019. Oridonin protects LPS-induced acute lung injury by modulating Nrf2-mediated oxidative stress and Nrf2-independent NLRP3 and NF-κB pathways. Cell Community Signal 17. https://doi.org/10.1186/S12964-019-0366-Y.
Yan, C., H. Yan, J. Mao, Y. Liu, L. Xu, H. Zhao, et al. 2020. Neuroprotective effect of oridonin on traumatic brain injury via inhibiting NLRP3 inflammasome in experimental mice. Frontiers Neuroscience 14. https://doi.org/10.3389/FNINS.2020.557170.
Shibata, S. 2000. A drug over the millennia: Pharmacognosy, chemistry, and pharmacology of licorice. Yakugaku Zasshi 120: 849–862. https://doi.org/10.1248/YAKUSHI1947.120.10_849.
Peng, F., Q. Du, C. Peng, N. Wang, H. Tang, X. Xie, et al. 2015. A review: The pharmacology of isoliquiritigenin. Phytotherapy Research 29: 969–977. https://doi.org/10.1002/PTR.5348.
Honda, H., Y. Nagai, T. Matsunaga, N. Okamoto, Y. Watanabe, K. Tsuneyama, et al. 2014. Isoliquiritigenin is a potent inhibitor of NLRP3 inflammasome activation and diet-induced adipose tissue inflammation. Journal of Leukocyte Biology 96: 1087–1100. https://doi.org/10.1189/JLB.3A0114-005RR.
Lee, Y., E.Y. Kwon, M.S. Choi. 2018. Dietary isoliquiritigenin at a low dose ameliorates insulin resistance and NAFLD in diet-induced obesity in C57BL/6J mice. The International Journal of Molecular Sciences 19. https://doi.org/10.3390/IJMS19103281.
Abenavoli, L., A.A. Izzo, N. Milić, C. Cicala, A. Santini, and R. Capasso. 2018. Milk thistle (Silybum marianum): A concise overview on its chemistry, pharmacological, and nutraceutical uses in liver diseases. Phytotherapy Research 32: 2202–2213. https://doi.org/10.1002/PTR.6171.
Zhang, B., B. Wang, S. Cao, Y. Wang, and D. Wu. 2017. Silybin attenuates LPS-induced lung injury in mice by inhibiting NF-κB signaling and NLRP3 activation. International Journal of Molecular Medicine 39: 1111–1118. https://doi.org/10.3892/IJMM.2017.2935.
Matias, M.L., V.J. Gomes, M. Romao-Veiga, V.R. Ribeiro, P.R. Nunes, G.G. Romagnoli, et al. 2019. Silibinin downregulates the NF-κB pathway and NLRP1/NLRP3 inflammasomes in monocytes from pregnant women with preeclampsia. Molecules 24: 1548. https://doi.org/10.3390/MOLECULES24081548.
Zhang, B., D. Xu, L. She, Z. Wang, N. Yang, R. Sun, et al. 2018. Silybin inhibits NLRP3 inflammasome assembly through the NAD +/SIRT2 pathway in mice with nonalcoholic fatty liver disease. The FASEB Journal 32: 757–767. https://doi.org/10.1096/FJ.201700602R.
Zhou, R., A. Yazdi, P. Menu, and J.T. Nature. 2011. undefined. A role for mitochondria in NLRP3 inflammasome activation. Nature.
Cholerton, B., L.D. Baker, G.S. Watson, M.A. Reger, S.T. Henderson, C. Hale, et al. 2004. Effects of β-hydroxybutyrate on cognition in memory-impaired adults. Elsevier 25: 311–314. https://doi.org/10.1016/S0197-4580(03)00087-3.
Hofseth, L.J., and M.J. Wargovich. 2007. Inflammation, cancer, and targets of ginseng. Journal of Nutrition 137: 183S-185S. https://doi.org/10.1093/JN/137.1.183S.
Yi, Y.S. 2019. Roles of ginsenosides in inflammasome activation. Journal of Ginseng Research 43: 172–178. https://doi.org/10.1016/J.JGR.2017.11.005.
Akao, T., H. Kida, M. Kanaoka, M. Hattori, and K. Kobashi. 1998. Intestinal bacterial hydrolysis is required for the appearance of compound K in rat plasma after oral administration of ginsenoside Rb1 from Panax ginseng. Journal of Pharmacy and Pharmacology 50: 1155–1160. https://doi.org/10.1111/J.2042-7158.1998.TB03327.X.
Li, C.W., M.Z. Deng, Z.J. Gao, Y.Y. Dang, G.D. Zheng, X.J. Yang, et al. 2020. Effects of compound K, a metabolite of ginsenosides, on memory and cognitive dysfunction in db/db mice involve the inhibition of ER stress and the NLRP3 inflammasome pathway. Food & Function 11: 4416–4427. https://doi.org/10.1039/C9FO02602A.
Shi, Y., H. Wang, M. Zheng, W. Xu, Y. Yang, F. Shi, et al. 2020. Ginsenoside Rg3 suppresses the NLRP3 inflammasome activation through inhibition of its assembly. Wiley Online Library 34:208–21. https://doi.org/10.1096/fj.201901537R.
Boulangé, C.L., A.L. Neves, J. Chilloux, J.K. Nicholson, and M.E. Dumas. 2016. Impact of the gut microbiota on inflammation, obesity, and metabolic disease. Genome Medicine 8: 42. https://doi.org/10.1186/S13073-016-0303-2.
Shin, H.Y., S.H. Kim, H.J. Jeong, S.Y. Kim, T.Y. Shin, J.Y. Um, et al. 2007. Epigallocatechin-3-gallate inhibits secretion of TNF-alpha, IL-6 and IL-8 through the attenuation of ERK and NF-kappaB in HMC-1 cells. International Archives of Allergy and Immunology 142: 335–344. https://doi.org/10.1159/000097503.
Gao, Z., Y. Han, Y. Hu, C. Wu, Y. Wang, and X. Zhang, et al. 2016. Targeting HO-1 by epigallocatechin-3-gallate reduces contrast-induced renal injury via anti-oxidative stress and anti-inflammation pathways. PLoS One 11. https://doi.org/10.1371/JOURNAL.PONE.0149032.
Rajanbabu, V., L. Galam, J. Fukumoto, J. Enciso, P. Tadikonda, T.N. Lane, et al. 2015. Genipin suppresses NLRP3 inflammasome activation through uncoupling protein-2. Cellular Immunology 297: 40–45. https://doi.org/10.1016/J.CELLIMM.2015.06.002.
Lu, J.-M., Q. Yao, and C. Chen. 2009. Ginseng compounds: An update on their molecular mechanisms and medical applications. Current Vascular Pharmacology 7: 293–302. https://doi.org/10.2174/157016109788340767.
Kim, J., H. Ahn, B.C. Han, S.H. Lee, Y.W. Cho, C.H. Kim, et al. 2014. Korean red ginseng extracts inhibit NLRP3 and AIM2 inflammasome activation. Immunology Letters 158: 143–150. https://doi.org/10.1016/J.IMLET.2013.12.017.
Yoon, S.J., J.Y. Park, S. Choi, J.B. Lee, H. Jung, T.D. Kim, et al. 2015. Ginsenoside Rg3 regulates S-nitrosylation of the NLRP3 inflammasome via suppression of iNOS. Biochemical and Biophysical Research Communications 463: 1184–1189. https://doi.org/10.1016/J.BBRC.2015.06.080.
Song, J., J. Li, F. Hou, X. Wang, and B. Liu. 2015. Mangiferin inhibits endoplasmic reticulum stress-associated thioredoxin-interacting protein/NLRP3 inflammasome activation with regulation of AMPK in endothelial cells. Metabolism 64: 428–437. https://doi.org/10.1016/J.METABOL.2014.11.008.
Pan, C.W., Z.Z. Pan, J.J. Hu, W.L. Chen, G.Y. Zhou, W. Lin, et al. 2016. Mangiferin alleviates lipopolysaccharide and D-galactosamine-induced acute liver injury by activating the Nrf2 pathway and inhibiting NLRP3 inflammasome activation. European Journal of Pharmacology 770: 85–91. https://doi.org/10.1016/J.EJPHAR.2015.12.006.
Matsuno, T., S.K. Jung, Y. Matsumoto, M. Saito, and J. Morikawa. 1997. Preferential cytotoxicity to tumor cells of 3,5-diprenyl-4-hydroxycinnamic acid (artepillin C) isolated from propolis. Anticancer Research 17: 3565–3568.
Hori, J.I., D.S. Zamboni, D.B. Carrão, G.H. Goldman, and A.A. Berretta. 2013. The inhibition of inflammasome by brazilian propolis (EPP-AF). Evid Based Complement Alternate Medicine. https://doi.org/10.1155/2013/418508.
Wang, C., Y. Pan, Q.Y. Zhang, F.M. Wang, and L.D. Kong. 2012. Quercetin and allopurinol ameliorate kidney injury in STZ-treated rats with regulation of renal NLRP3 inflammasome activation and lipid accumulation. PLoS ONE 7: e38285. https://doi.org/10.1371/JOURNAL.PONE.0038285.
Hu, Q.H., X. Zhang, Y. Pan, Y.C. Li, and L.D. Kong. 2012. Allopurinol, quercetin and rutin ameliorate renal NLRP3 inflammasome activation and lipid accumulation in fructose-fed rats. Biochemical Pharmacology 84: 113–125. https://doi.org/10.1016/J.BCP.2012.03.005.
Wang, W., C. Wang, X.Q. Ding, Y. Pan, T.T. Gu, M.X. Wang, et al. 2013. Quercetin and allopurinol reduce liver thioredoxin-interacting protein to alleviate inflammation and lipid accumulation in diabetic rats. British Journal of Pharmacology 169: 1352–1371. https://doi.org/10.1111/BPH.12226.
Zhang, X., J.H. Zhang, X.Y. Chen, Q.H. Hu, M.X. Wang, R. Jin, et al. 2015. Reactive oxygen species-induced TXNIP drives fructose-mediated hepatic inflammation and lipid accumulation through NLRP3 inflammasome activation. Antioxidants & Redox Signaling 22: 848–870. https://doi.org/10.1089/ARS.2014.5868.
Jiang, W., Y. Huang, N. Han, F. He, M. Li, Z. Bian, et al. 2016. Quercetin suppresses NLRP3 inflammasome activation and attenuates histopathology in a rat model of spinal cord injury. Spinal Cord 54: 592–596. https://doi.org/10.1038/SC.2015.227.
Inoue, H., and R. Nakata. 2015. Resveratrol targets in inflammation. Endocrine, Metabolic & Immune Disorders: Drug Targets 15: 186–195. https://doi.org/10.2174/1871530315666150316120316.
Kulkarni, S.S., and C. Cantó. 2015. The molecular targets of resveratrol. Biochimica et Biophysica Acta 1852: 1114–1123. https://doi.org/10.1016/J.BBADIS.2014.10.005.
Huang, T.T., H.C. Lai, Y.B. Chen, L.G. Chen, Y.H. Wu, and Y.F. Ko, et al. 2014. cis-Resveratrol produces anti-inflammatory effects by inhibiting canonical and non-canonical inflammasomes in macrophages. Innate Immunity 20:735–50. https://doi.org/10.1177/1753425913507096.
Yang, S.J., and Y. Lim. 2014. Resveratrol ameliorates hepatic metaflammation and inhibits NLRP3 inflammasome activation. Metabolism 63: 693–701. https://doi.org/10.1016/J.METABOL.2014.02.003.
Misawa, T., M. Takahama, T. Kozaki, H. Lee, J. Zou, T. Saitoh, et al. 2013. Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nature Immunology 14: 454–460. https://doi.org/10.1038/NI.2550.
Sui, D.M., Q. Xie, W.J. Yi, S. Gupta, X.Y. Yu, and J.B. Li, et al. 2016. Resveratrol protects against sepsis-associated encephalopathy and inhibits the NLRP3/IL-1β axis in microglia. Mediators of Inflammation. https://doi.org/10.1155/2016/1045657.
Wu, J., X. Li, G. Zhu, Y. Zhang, M. He, and J. Zhang. 2016. The role of resveratrol-induced mitophagy/autophagy in peritoneal mesothelial cells inflammatory injury via NLRP3 inflammasome activation triggered by mitochondrial ROS. Experimental Cell Research 341: 42–53. https://doi.org/10.1016/J.YEXCR.2016.01.014.
Houghton, C.A., R.G. Fassett, and J.S. Coombes. 2013. Sulforaphane: Translational research from laboratory bench to clinic. Nutrition Reviews 71: 709–726. https://doi.org/10.1111/NURE.12060.
Dinkova-Kostova, A.T., W.D. Holtzclaw, R.N. Cole, K. Itoh, N. Wakabayashi, Y. Katoh, et al. 2002. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc Natl Acad Sci U S A 99: 11908–11913. https://doi.org/10.1073/PNAS.172398899.
Heiss, E., C. Herhaus, K. Klimo, H. Bartsch, and C. Gerhäuser. 2001. Nuclear factor kappa B is a molecular target for sulforaphane-mediated anti-inflammatory mechanisms. Journal of Biological Chemistry 276: 32008–32015. https://doi.org/10.1074/JBC.M104794200.
Alfieri, A., S. Srivastava, R.C.M. Siow, D. Cash, M. Modo, M.R. Duchen, et al. 2013. Sulforaphane preconditioning of the Nrf2/HO-1 defense pathway protects the cerebral vasculature against blood-brain barrier disruption and neurological deficits in stroke. Free Radical Biology & Medicine 65: 1012–1022. https://doi.org/10.1016/J.FREERADBIOMED.2013.08.190.
Manolagas, S.C. 2010. From estrogen-centric to aging and oxidative stress: A revised perspective of the pathogenesis of osteoporosis. Endocrine Reviews 31: 266–300. https://doi.org/10.1210/er.2009-0024.
An, Y.W., K.A. Jhang, S.Y. Woo, J.L. Kang, and Y.H. Chong. 2016. Sulforaphane exerts its anti-inflammatory effect against amyloid-β peptide via STAT-1 dephosphorylation and activation of Nrf2/HO-1 cascade in human THP-1 macrophages. Neurobiology of Aging 38: 1–10. https://doi.org/10.1016/J.NEUROBIOLAGING.2015.10.016.
Yang, G., H.E. Lee, and J.Y Lee. 2016. A pharmacological inhibitor of NLRP3 inflammasome prevents non-alcoholic fatty liver disease in a mouse model induced by high fat diet. Scientific Reports 6. https://doi.org/10.1038/SREP24399.
Man, S.M., R. Karki, R.K.S. Malireddi, G. Neale, P. Vogel, M. Yamamoto, et al. 2015. The transcription factor IRF1 and guanylate-binding proteins target activation of the AIM2 inflammasome by Francisella infection. Nature Immunology 16: 467–475. https://doi.org/10.1038/NI.3118.
Lan, Z., G. Xie, M. Wei, P. Wang, P. and L. Chen. 2017. The protective effect of Epimedii Folium and Curculiginis Rhizoma on Alzheimer’s disease by the inhibitions of NF-κB/MAPK pathway and NLRP3 inflammasome. Oncotarget 8:43709–20. https://doi.org/10.18632/ONCOTARGET.12574.
Xiong, R., X.G. Zhou, Y. Tang, J.M. Wu, Y.S. Sun, J.F. Teng, JF, et al. 2005. Lychee seed polyphenol protects the blood-brain barrier through inhibiting Aβ(25–35)-induced NLRP3 inflammasome activation via the AMPK/mTOR/ULK1-mediated autophagy in bEnd.3 cells and APP/PS1 mice. Phytotherapy Research 35:954–73. https://doi.org/10.1002/PTR.6849.
Su, Q., L. Li, Y. Sun, H. Yang, Z. Ye, and J. Zhao. 2018. Effects of the TLR4/Myd88/NF-κB signaling pathway on NLRP3 inflammasome in coronary microembolization-induced myocardial injury. Cellular Physiology and Biochemistry 47: 1497–1508. https://doi.org/10.1159/000490866.
Okada, M., A. Matsuzawa, A. Yoshimura, and H. Ichijo. 2014. The lysosome rupture-activated TAK1-JNK pathway regulates NLRP3 inflammasome activation. Journal of Biological Chemistry 289: 32926–32936. https://doi.org/10.1074/JBC.M114.579961.
Sokolowska, M., L.-Y. Chen, Y. Liu, A. Martinez-Anton, H.-Y. Qi, C. Logun, et al. 2015. Prostaglandin E2 inhibits NLRP3 inflammasome activation through EP4 receptor and intracellular cyclic AMP in human macrophages. The Journal of Immunology 194: 5472–5487. https://doi.org/10.4049/JIMMUNOL.1401343.
Shao, B.Z., Wei, W., Ke, P., Xu, Z.Q., Zhou, J.X., Liu, C. 2014. Activating cannabinoid receptor 2 alleviates pathogenesis of experimental autoimmune encephalomyelitis via activation of autophagy and inhibiting NLRP3 inflammasome. CNS neuroscience & therapeutics 20(12): 1021–1028. https://doi.org/10.1111/cns.12349. PMID: 25417929; PMCID: PMC6492996.
Author information
Authors and Affiliations
Contributions
The work was conceptualized and designed by M. Mumtaz Alam, M. Shaquiquzzaman, Syed Ehtaishamul Haque, Mohammad Ahmed Khan, and Suhel Parvez. The first draft was prepared by Barkha Sharma, Garvit Satija1, Anish Madan, and Mansi Garg. The manuscript was reviewed by Suruchi Khanna, Prachi Tiwari, Ashif Iqubal, Mohammad Ahmed Khan, and M. Mumtaz Alam. The revised draft was prepared by Barkha Sharma, Garvit Satija1, Anish Madan, Mansi Garg, Ashif Iqubal, and Mohammad Ahmed Khan. The final version was reviewed by M. Mumtaz Alam, M. Shaquiquzzaman, Syed Ehtaishamul Haque, Suruchi Khanna, and Prachi Tiwari.
Corresponding authors
Ethics declarations
Ethics Approval
This is a review paper and does not require any ethical approval.
Consent for Publication
This is a review manuscript and used already published data.
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Sharma, B., Satija, G., Madan, A. et al. Role of NLRP3 Inflammasome and Its Inhibitors as Emerging Therapeutic Drug Candidate for Alzheimer’s Disease: a Review of Mechanism of Activation, Regulation, and Inhibition. Inflammation 46, 56–87 (2023). https://doi.org/10.1007/s10753-022-01730-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10753-022-01730-0