
Abstract
The inflammasome adapter ASC links activated inflammasome sensors to the effector molecule pro-caspase-1. Recruitment of pro-caspase-1 to ASC promotes the autocatalytic activation of caspase-1, which leads to the release of pro-inflammatory cytokines, such as IL-1β. Upon triggering of inflammasome sensors, ASC assembles into large helical fibrils that interact with each other serving as a supramolecular signaling platform termed the ASC speck. Alternative splicing, post-translational modifications of ASC, as well as interaction with other proteins can perturb ASC function. In several inflammatory diseases, ASC specks can be found in the extracellular space and its presence correlates with poor prognosis. Here, we review the role of ASC in inflammation, and focus on the structural mechanisms that lead to ASC speck formation, the regulation of ASC function during inflammasome assembly, and the importance of ASC specks in disease.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The innate immune system has developed strategies to defend the body from pathogens and other health threats based on the recognition of pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) by germline-encoded pattern-recognition receptors (PRRs) [1, 2]. Detection of PAMPs or DAMPs by PRRs results in many cases in the activation of the nuclear factor kappa-light-chain enhancer of activated B-cells (NF-κB), a master transcription factor that leads to the upregulation of pro-inflammatory genes. NF-κB activity is tightly regulated by ubiquitination and phosphorylation. These mechanisms are controlled by the IκB kinase (IKK) complex, which consists of the canonical IKKs (IKKα and IKKβ) and the NF-κB essential regulator (NEMO). The IKK-related kinases TANK-binding kinase 1 (TBK1) and IKK-inducible (IKKi) can also regulate the NF-κB pathway [1, 3]. Excessive or maintained inflammation can cause chronic or systemic diseases. Inflammasomes are multimolecular signaling platforms that play a key role in the promotion of inflammation. They are responsible for the generation of the bioactive cleaved form of the pro-inflammatory cytokines interleukin (IL)-1β and IL-18. The transcription of pro-IL-1β, as well as other inflammasome components, is induced by the activation of NF-κB, whereas pro-IL-18 is constitutively expressed [4]. Inflammasome formation requires, in most cases, a sensor protein, an adapter protein, and an effector protein [4, 5]. ASC (apoptosis-associated speck-like protein containing a CARD) functions as the central adapter for the formation of most inflammasomes. In this review, we provide an overview on the role of ASC as an adapter during inflammasome assembly. We focus on the structural details of ASC speck formation, the regulation of ASC activity, and the role of ASC specks in disease.
ASC expression pattern
The gene PYCARD (PYD and CARD domain containing) was first cloned by Masumoto et al. [6]. It contains three exons that encode a 22 kDa protein composed of 195 amino acids (aa) termed ASC. ASC is highly expressed in the spleen and lungs, but also in the digestive tract, skin, urinary bladder, and bone marrow. Peripheral blood leukocytes show high expression of ASC. Among them, CD14+ monocytes have the highest expression levels of ASC, whereas polymorphonuclear cells and CD3+ T lymphocytes express lower levels of ASC [7, 8]. Furthermore, ASC is constitutively expressed in most cell lines of the myeloid lineage (e.g., THP-1, HL-60, or U937), but it is not expressed in the murine macrophage cell line Raw 264.7. Other cell lines, such as Jurkat T cells, Burkitt’s lymphoma, or HeLa, also lack ASC expression [6, 9].
The subcellular localization of the inactive ASC is controversial. Whereas most of the publications reported that in resting cells ASC is distributed throughout the cytosol [6, 8] (or throughout the cytosol and the nucleus [10]), others reported that ASC is mainly present in the nucleus [11, 12]. The reason for this discrepancy could be a result of ASC overexpression or of different antibodies used.
ASC function
ASC was originally described to function as a pro-apoptotic protein [6, 8], and further studies have reported that ASC plays a key role in cell death, which is of major importance in cancer. On the other hand, ASC also enhances inflammation, since its co-expression with pro-caspase-1 induces IL-1β secretion [13, 14]. Under certain conditions, inflammation can promote tumorigenesis [15]. However, the relationship between the role of ASC in cancer and its effect in eliciting inflammation remains unknown.
In 2000, Conway et al. identified TMS1 (target of methylation-induced silencing-1) as a tumor suppressor gene that undergoes downregulation upon methylation of its 5′ CpG island [8, 16]. The TMS1 CpG island remains unmethylated in normal breast tissue, but it was found methylated (and thereby silenced) in 11 out of 27 studied primary breast carcinomas [8]. Later on, PYCARD and TMS1 were reported to be the same gene. ASC expression has been shown to be upregulated during inflammation. For example, pro-inflammatory stimuli, such as IL-1, lipopolysaccharide (LPS), interferon-γ, or tumor necrosis factor (TNF)-α, as well as Fas receptor activation, induce or increase ASC expression [17, 18]. Whereas methylation of the PYCARD CpG island is linked to loss of ASC expression in cancer cells, it has not yet been assessed whether the PYCARD CpG island controls ASC expression after pro-inflammatory stimuli.
ASC function seems to be cell type dependent. In a tumor-induced model, ASC performs pro-inflammatory functions in myeloid cells, whereas it acts as a tumor suppressor in epidermal keratinocytes [19]. However, other studies propose that keratinocytes can assemble an inflammasome and release pro-inflammatory cytokines upon different stimuli [20–22]. The function of ASC seems to be tightly regulated, and it can also vary depending on the developmental state of a cancer. In primary melanoma, ASC expression decreases cell viability, whereas in metastatic melanoma, ASC functions in an adverse way [23]. In this review, we will focus on the pro-inflammatory role of ASC as a part of the inflammasomes.
ASC as an inflammasome adapter
ASC is composed of two conserved domains: a pyrin domain (PYD) and a caspase recruitment domain (CARD), which are connected by an unstructured linker region. Both domains function to promote homotypic interactions. Thus, ASC functions as the central adapter for the formation of inflammasomes [13].
Inflammasomes are multimolecular signaling platforms that promote the cleavage of pro-caspase-1 and the maturation of the pro-inflammatory cytokines IL-1β and IL-18 [4]. Their formation is organized in a hierarchy and requires, in most cases, a sensor protein, an adapter protein, and an effector protein [4, 5]. Whereas ASC always acts as the adapter, and caspase-1 as the effector, different proteins can function as the sensor. The inflammasome sensor proteins belong to either the NLR (nucleotide-binding oligomerization domain (NOD)-like receptor) family, or to the HIN200 (hematopoietic interferon-inducible nuclear antigens with 200 amino-acid repeats) family. NLRP1 (NOD-, leucine-rich repeat (LRR)-, PYD-containing 1), NLRP3, NLRP6, and NLRC4 (NOD-, LRR-, CARD-containing 4) constitute members of the NLR family. AIM2 (absent in melanoma 2) is a member of the HIN200 family. The protein pyrin can also function as sensor and assemble inflammasomes [4, 5, 24]. Inflammasome sensor proteins recognize different PAMPs and DAMPs and act as a seed for the recruitment of ASC, which is mediated by the interaction between the PYDs of both proteins [4, 5]. NLRC4 exemplifies an exception from this process. Since it contains a CARD domain, NLRC4 can either directly recruit pro-caspase-1 or use ASC as a linker protein via CARD–CARD interactions [4, 25]. Sensor seed formation induces ASC to form long filaments through oligomerization with successive ASC proteins via homotypic interactions of the ASC PYD domain. Pro-caspase-1 contains a CARD that functions as a pro-domain. It allows for the recruitment of pro-caspase-1 to the ASC filament, as well as for the oligomerization with other pro-caspase-1 molecules. By this mechanism, several units of pro-caspase-1 can be recruited to the ASC speck structure and auto-catalyze their cleavage at two positions, generating thereby one p20 and one p10 subunit per molecule of pro-caspase-1. Consecutively, two subunits of p20 and two of p10 form an active heterotetramer that will process pro-IL-1β [26].
Active caspase-1 cleaves pro-IL-1β and pro-IL-18 and induces the release of their mature forms, which exert potent pro-inflammatory effects [4, 5, 27]. Inflammasome assembly furthermore results in an inflammatory type of cell death termed pyroptosis, which requires gasdermin-D cleavage [28, 29]. The structure formed by ASC aggregates that leads to pyroptosis has also been termed pyroptosome [10].
Inflammasome-related diseases
Inflammasome activation can cause a pronounced inflammatory reaction, which may induce the development of inflammatory diseases if inflammasome function is not properly regulated. For instance, more than a hundred sequence variants have been associated with heterozygous autosomal gain-of-function mutations in the inflammasome sensor NLRP3 [30]. Patients with these mutations suffer from cryopyrin-associated periodic syndromes (CAPS) and related disorders. They develop episodes of both systemic and local inflammation that are clinically different depending on the type of mutation [5, 31]. Furthermore, uncontrolled NLRP3 inflammasome activation is thought to play a role in multifactorial disorders, such as atherosclerosis, diabetes, gout, Parkinson’s disease, and Alzheimer’s disease [5]. Familial Mediterranean fever (FMF) is another autoinflammatory syndrome related to inflammasome hyperactivity. FMF patients present homozygous mutations in the Mediterranean fever (MEFV) gene, which encodes for pyrin. They suffer from idiopathic and self-limited bouts of local and systemic inflammation that last for 2–4 days, and may develop secondary amyloidosis as the main complication of the disease [32]. Other inflammasome sensors, including AIM2 or NLRC4, have also been associated with inflammatory diseases [5].
ASC structure and polymerization
ASC is comprised of two well-defined structural domains (the PYD and the CARD), which are connected by a 23 aa linker region (Fig. 1a). The linker features some local structure, but restrains flexibility and inter-domain dynamics between PYD and CARD [33]. The linker positions them in a back-to-back orientation, which keeps them structurally independent and allows binding to multiple partners [33, 34]. Both the PYDs at the N terminus and the CARD at the C terminus of ASC belong to the death domain (DD) superfamily, which is one of the largest protein domain families. Most DD superfamily members participate in cell death and inflammation, and characteristically form homotypic interactions. These interactions are typically not only restricted to the formation of dimers, but rather promote the assembly of oligomeric signaling platforms [35]. The death fold is characterized by six antiparallel helices (α1–α6) that are connected by loops (Fig. 1b). The overall three-dimensional structure of a death fold is roughly globular [36, 37]. Although the sequence similarities between the PYD and the other DD superfamily members [DD, CARD, and death effector domain (DED)] are limited, the ASC–PYD adopts a classical six helix bundle fold [38]. The same overall structure of their N-terminal PYDs was shown by crystallization and NMR for the NLRP3-PYD, AIM2-PYD, and others [39–44].

NMR structure of ASC (PDB ID: 2KN6) [33]. a Cartoon diagram of ASC, showing the PYD, the flexible linker and the CARD. b Cartoon diagram of the typical DD fold of the ASC–PYD. Indicated are the six α-helices (α1 to α6)
ASC assembles into filaments
ASC is soluble as a monomer at low pH and in solution with chaotropic agents, but it can form long and well-defined fibrils at higher pH [45]. Similarly, at physiological potassium concentration ASC remains soluble, whereas reducing the potassium to half-physiological concentration leads to ASC oligomerization at 37 °C but not at 4 °C. This phenomenon could be verified in a cell-free system with recombinant ASC [10].
The supramolecular structure of the ASC filament was analyzed by a combined approach based on cryo-EM and NMR spectroscopy by two different groups [45, 46] (Fig. 2). The ASC fibrils appear to be highly conserved between human and mouse [45], and both filaments represent hollow fibrils with an inner diameter of 20 Å and an outer diameter of 90 Å [45, 46]. Both human and mouse fibrils show a C3 point group symmetry with a roughly 53° right-handed rotation and a rise of around 14 Å per rotation (53° rotation, 14.2 Å rise in mouse; 52.9° rotation, 13.9 Å rise in human) [45, 46] (Fig. 2e).

ASC speck structure at different levels of complexity. a Confocal laser scanning microscopy image of a pyroptotic macrophage that expresses ASC-mCherry and has assembled an ASC speck (red). The cell was counterstained for nucleus (blue) and membranes (green). b High-resolution stimulated emission depletion (STED) image of an ASC speck. It shows distinct ASC fibrils emerging from the condensed, tightly cross-linked core of the ASC speck. c, d Schematic view of an ASC speck, containing the inflammasome sensor NLRP3 (green), which clusters and forms a seed for the prion-like polymerization of ASC into fibrils through homotypic interactions of its PYD (blue). The CARDs of ASC (red) form clusters on the surface of the PYD fibrils. These CARD clusters can either interact with other ASC–CARDs and thereby crosslink the ASC fibrils into a condensed speck, or they can form a seed for the assembly of polymeric CARD fibrils of pro-caspase-1. After polymerization of pro-caspase-1, the close proximity of several caspases leads to self-cleavage into the mature caspase-1, which is released from the speck. e Schematic view of the three-start ASC–PYD helix, indicating the direction of further polymerization and the three strands. f Detailed scheme of the three interfaces that mediate ASC–PYD interactions within the ASC fibril. The intra-strand interface I contributes the most to ASC PYD–PYD interactions, followed by the smaller inter-strand interfaces II and III
In contrast to ASC, NLRP3-PYDs form a heterogeneous mixture of filaments and disk-like structures [46]. ASC oligomers analyzed by atomic force microscopy have been shown to form disk-like particles—rather than filaments—with a diameter of 12 nm and a height of 1 nm [33]. Of note, the pro-apoptotic protein apoptotic protease-activating factor 1 (Apaf-1) forms a structurally related ring-like assembly, termed apoptosome, which triggers cell death [47]. Whether the disk-like structures observed during ASC oligomerization are intermediate forms, or whether they represent artifacts remains uninvestigated.
Inflammasome sensors form seeds for ASC filaments
When ASC specks were first described in apoptotic cells in 1999 [6], it had not been described that they are built up by cross-linked fibrils. The concept of inflammasome sensor-induced speck formation was first established in 2001 in overexpression experiments with ASC and pyrin, which interacts via its PYD with ASC [48]. The ASC speck can be formed by endogenous or over-expressed full-length ASC [6].
In non-activated cells, ASC is found in its soluble form, as a high thermodynamic energy barrier prevents spontaneous oligomerization [49]. However, inflammasome sensors are capable of forming an oligomeric PYD cluster, which acts as a seed and recruits the first ASC molecules to form a helical filament [46] (Fig. 2d). It is thought that activation of the inflammasome sensor molecules leads to conformational changes (for example, in the case of NLRs), or brings several inflammasome sensor molecules closely together by binding to a common ligand (as with AIM2-like inflammasome sensors binding to nucleic acids), leading to PYD self-interaction [46]. This induced protein seed is then thought to lower the high energy barrier of ASC oligomerization, and causes unlimited ASC polymerization by prion-like propagation [49]. Although the conformational changes of monomeric to fibrillar ASC are only minor [45, 46], highly sub-stoichiometric amounts of fibrillar ASC are sufficient to induce further oligomerization of monomeric ASC, a hallmark of prion-like behavior [49].
It was shown that both the PYDs of the inflammasome sensor AIM2 and the PYD of ASC are capable of forming filaments on their own; however, AIM2 prefers interactions with ASC, whereas ASC prefers self-interactions [46]. A possible explanation for this phenomenon may be that AIM2-PYD forms a one-start helix, whereas ASC forms a three-start helix [50] (Fig. 2e). Similar to actin and tubulin, which also allow for different filament symmetries [51, 52], ASC is expected to tolerate minimal changed interfaces between subunits [50].
A different strategy to form a seed for ASC polymerization is achieved by the NLR family apoptosis inhibitory protein (NAIP)/NLRC4 inflammasome, which does not depend on ASC, but is strongly enhanced by its recruitment [53, 54]. Upon activation, the NAIP/NLRC4 complex can recruit linker ASC molecules via homotypic CARD–CARD interaction, which cluster and form a PYD seed to initiate ASC filament formation [25, 49, 55]. In the absence of ASC, NLRC4 recruits pro-caspase-1 via CARD–CARD interactions, which is sufficient to induce caspase-1/gasdermin-D-dependent pyroptosis, but not IL-1β/IL-18 cytokine maturation [25].
Charge-based interactions define interfaces for PYD interaction
The ASC–PYD is highly polar, containing positive charges in the helices α2 and α3, as well as in the connecting loop between these helices. Negative charges are found in the helices α1 and α4 [38]. Proof of a more specific interaction instead of non-specific hydrophobic interactions was proposed by bioinformatics and mutational screenings of ASC [56, 57]. A role for complementary charges as driving force in filament assembly was further supported by studies of Lu et al., who showed that, in a orthogonal cross section of the ASC filament, one side is essentially positively charged, while its opposite is mainly negatively charged [46]. That charges are indeed responsible for the ASC–PYD interaction is further supported by evolutionary conservation of the charged residues in the helices α1, α2, α3, and α4 of the pyrin-only protein (POP) ASC2, which can interact via its PYD with ASC [38, 58]. Originally, the interactions between ASC–PYDs were thought to be formed only by one specific interface between two PYDs [38, 57]. However, both targeted mutagenesis and structural analysis showed that PYD self-interaction is stabilized by several interfaces [46, 56, 59] (Fig. 2f).
Within the right-handed, three-start helix, each PYD interacts via three asymmetric interfaces (types I to III) with six adjacent PYDs [45]. The type I interface is formed between PYDs within one of the three helical strands and consists of a surface area of 880 Å2. The type I interface is not only the largest, but also the most conserved interface. The type II and type III interfaces are formed between different strands of the helix and provide an interaction surface of 540 and 360 Å2, respectively [46] (Fig. 2e, f). Each interface is defined via the interaction of two opposing surfaces. Residues of the α1 and α4 helices, which are mainly negatively charged, form the surface Ia. This surface interacts with the α2 and α3 helices on the opposing Ib site that provide an overall positively charge [38, 46]. Notably, the involvement of α3 is minimized in PYDs compared to other DD family members. The inter-strand type II interaction is determined by the corner of the α4–α5 helices (IIa) and the corner of the α5–α6 helices (IIb). Finally, the corner of α2–α3 (IIIa) and α1–α2 (IIIb) forms the type III interface.
Although PYDs are part of the DD superfamily, the ASC–PYD–PYD self-interaction interfaces are strikingly different from the interfaces formed in other DD complex structures. This can be particularly observed when comparing the ASC PYD type III interface with the type III interface in MyD88/MyD88 self-interactions [46].
When transformed from the monomeric to the filamentous state, ASC molecules retain their secondary structure and the position of all six α-helices. However, a minimal packaging effect takes place, resulting in conformational changes of some residues located in the subunit interfaces [45], especially in the region of the variable loop between α2 and α3 [46]. The highly variable loop between α2 and α3, which is unique to PYDs, is involved in all three interfaces [46], which might explain the conformational changes upon polymerization in that region.
ASC–CARD is required for speck formation and downstream signaling
In the ASC fibril, the self-interaction of ASC is PYD driven, while the CARD recruits pro-caspase-1 [10] (Fig. 2d). ASC–PYD and ASC full length form the same filamentous backbone structure, while the CARD is neither essential nor does it influence the structure of the PYD in the filament [34, 45]. Remarkably, ASC–PYD-only and ASC–CARD-only protein fragments both form similar fibrils when expressed separately [48]. In the early studies, it was assumed that the ASC–PYD and ASC–CARD not only form homotypic interaction but also heterotypic interaction when expressed as single-domain fragments [60]. However, more recent studies found that they form mutually exclusive filaments of only one domain fragment [56]. This phenomenon supports the current model of speck formation [45, 46] (Fig. 2c, d).
The ASC speck is composed of multiple filamentous structures which cluster together and build up a high molecular globular complex that resembles a bird’s nest with filaments in the periphery [48] (Fig. 2b, c). More detailed studies in inflammasome-competent THP-1 cells found single filaments next to condensed globular specks [46]. These findings led to the assumption that the ASC speck consists of cross-linked ASC filaments, which could be achieved via inter-strand ASC–CARD–CARD interactions [45, 46, 55]. This explains why ASC full-length filaments branch more often and form a three-dimensional globular speck, in contrast to the one-dimensional ASC–PYD-only filaments [45]. Filaments of full-length ASC have much rougher surface compared to the smooth surfaces of the PYD-only filament [45], indicating that the CARDs are located on the outside of the filaments and that the two domains of ASC are not only independent of each other in the monomeric state [33], but, due to the flexible linker, as well in filaments [34, 45]. Thereby, inter- and intra-strand CARD–CARD interactions would be possible, explaining why the flexible linker between PYD and CARD is necessary for the formation of globular specks [56] (Fig. 2c).
Indeed, the CARDs of ASC are responsible for the condensed speck formation instead of filaments [25, 55]. Schmidt et al. analyzed for the first time ASC speck formation on a molecular level in cells that express endogenous ASC. Instead of genetic tools to perturb the system, these investigators used an intracellular heavy chain-only alpaca antibody that binds specifically to the interface II of ASC–CARD. This interface is required for CARD–CARD interactions between two adjacent ASC molecules, but also between ASC and pro-caspase-1. The authors could show that the formation of the ASC filaments is not inhibited in cells expressing the antibody, but these filaments are no longer branched and do not form specks [55]. Dick et al. used a mutagenesis-based approach to show that the CARD domain is crucial to link individual ASC filaments [25]. Although it had already been shown that the ASC–CARD is needed to form condensed speck foci [61], previous studies had only analyzed ASC speck formation in vitro in overexpression systems, with protein fragments of ASC, or with ASC fused to large protein tags. Since most inflammasome components tend to become activated and oligomerize upon overexpression [14, 18, 46, 48, 62, 63], the use of small intracellular antibodies in cells that endogenously express the protein of interest opens a new range of possibilities for studying inflammasomes. Nevertheless, the use of intracellular antibodies raises other issues, as antibody binding might interfere with other interaction partners of ASC. For example, Schmidt et al. cannot exclude the possibility that the CARD of ASC is needed to bring together the ASC filaments mediated by an active transport mechanism.
Homotypic ASC–CARD interactions can form seeds to facilitate the formation of filaments by the CARD of pro-caspase-1, which emanate from the ASC filaments [49, 55, 64] (Fig. 2d). The conformation of the CARD of pro-caspase-1 was shown to be thermodynamically unstable [65], and the CARD of ASC was discovered to be as well at least partially unfolded [45]. Most likely, this could prevent the ASC filaments from being too densely cross-linked via CARD–CARD interactions, leaving enough free CARDs to interact with pro-caspase-1. In a full ternary inflammasome complex, pro-caspase-1 is overstoichiometric to ASC, and the ASC filaments are not as large as in binary complexes of ASC and NLRP3/AIM2 [46]. The structure of a full ternary endogenous inflammasome complex has not yet been determined. However, endogenous specks were shown to contain a multitude of caspase-1 compared to ASC, and in vitro assays show that caspase-1 filaments branch from the ASC fibers [46]. Taken together, this suggests a model in which every single ASC filament forms the center of a star-shaped complex (when seen from above), nucleating the formation of long CARD-mediated pro-caspase-1 filaments to every side [46]. The local concentration of pro-caspase-1 is considerably increased by this polymerization process and allows for caspase-1 auto-proteolysis and activation [10, 66–68]. A similar polymerization mechanism for pro-caspase-1 was shown for the ASC-independent NAIP/NLRC4 inflammasome by direct recruitment via the CARDs of NLRC4 [64, 69].
ASC speck formation is a prion-like event
On a cellular level, the process of ASC speck assembly was described to take no longer than 100 s in HeLa and THP-1 cells expressing fluorescently labeled ASC [10, 70]. To form the ASC speck, ASC is homogenously depleted from the whole cytoplasm independently of the distance to the speck. Only ASC from the nucleus is recruited tenfold slower, although speck formation takes place close to the nucleus [70] (Fig. 2a). It is worth mentioning that ASC location in the cell is a controversial issue. In contrast to the before stated, a primarily nuclear localization was described for ASC in homeostatic macrophages and monocytes, followed by a redistribution to the cytoplasmic, perinuclear region upon inflammasome activation and speck formation [12]. However, Bryan et al. do not differentiate between a priming and an inflammasome activation effect. However, it might be possible that priming does not only affect the expression level of ASC but as well its localization. In a more recent publication, it was reported that ASC relocates in complex with IKKα from the nucleus to the perinuclear region after priming, and it is released from IKKα after NLRP3 activation [11].
As soon as the PYDs of inflammasome sensors are clustered and form a seed for filamentous polymerization of ASC, the reaction is irreversible, no longer dependent on the original initiating signal, and follows an energetic gradient until all the ASC molecules of an activated cell are recruited [49]. Consequently, cells are either activated and contain a speck, or they are inactive and do not contain any speck. A gradual response is not likely, as the aggregation proceeds in a rapid prionoid process. In contrast, other filament forming proteins, such as actin or tubulin, display a non-prionoid filament formation [71]. For both of them, every monomer is regulated by itself via nucleotide hydrolysis, and therefore, the soluble and filamentous forms are in an equilibrium in every cell [49].
Over the last years, the idea of higher order signaling platforms within the innate immune system became increasingly popular [72]. Prionoid filament formation results in a potent signal amplification cascade, which guarantees extreme sensitivity of the system [49]. Indeed, prion-like amplification cascades are evolutionary conserved systems of signal transduction in innate immunity [73–75]. Similar to ASC, the adaptor protein mitochondrial antiviral-signaling (MAVS) can form prion-like filament downstream of viral RNA-sensing pathways. Although ASC and MAVS fulfill many criteria of prions, they differ from bona fide prions in that they are neither enriched in glutamine and asparagine nor intrinsically disordered [49].
In contrast to the before mentioned, the recently suggested alternative inflammasome in monocytes, which senses LPS and subsequently produces mature IL-1β, does not induce ASC speck formation. However, its activation is still dependent on NLRP3, ASC, and caspase-1 [76]. Whether the signal transduction from NLRP3 via ASC to caspase-1 occurs based on single-molecule interactions without a polymerization step or a whole new theory is necessary to explain this mechanism remains elusive.
Regulation of speck formation
Inflammasome activation followed by caspase-1 activation, pyroptosis, and IL-1β release represents a very potent pro-inflammatory signal of the innate immune system [5, 77, 78]. To prevent undesirable tissue damage due to inflammasome overactivation, its function needs to be tightly controlled. The activation and assembly of inflammasomes are regulated by a multitude of different mechanisms at the level of every involved component. The plurality of all signals orchestrates the complex fine-tuning which is necessary to keep the inflammasome under control [5, 79]. Here, we focus on the regulatory mechanisms that directly affect ASC.
ASC splicing
Splicing represents one of the mechanisms that regulate ASC speck formation. Indeed, PYCARD consists of three exons: exon 1 encodes the PYD, exon 2 encodes the flexible linker region, and exon 3 encodes the CARD of ASC (Fig. 3). Hence, the genomic organization of PYCARD is ideally suited to produce splice forms that could have regulatory functions similar to the POPs and CARD-only proteins (COPs) (see below).

ASC mRNA is alternatively spliced into four different isoforms. PYCARD consists of three exons. Exon 1 encodes the PYD, exon 2 encodes the flexible linker, and exon 3 encodes the CARD. ASC-a represents the canonical sequence, and contains all three exons, ASC-b lacks exon 2, ASC-c lacks 60 aa within the PYD and is the product of cryptic splice sides within exon 1, ASC-d is derived by a frame shift after the first 35 aa of the PYD, which results in a completely new exon 2 (exon 2′)
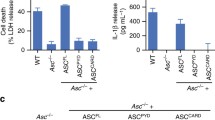
An alternative splice variant missing exon 2 was already mentioned in 2000 shortly after the initial description of ASC [6, 8]. Several years later, the protein product of the alternative spliced ASC mRNA was identified in pyroptosome purifications as a protein species of 19 kDa size, slightly smaller than full-length ASC [10]. This shorter ASC protein form was analyzed by mass spectrometry and was confirmed as being ASC missing the flexible linker between PYD and CARD, encoded by exon 2 [80]. At the same time, this and two additional splice variants of ASC were discovered by Bryan et al., who combined antibody mapping and in silico analysis of expressed sequence tags. Thus, a nomenclature for ASC splice variants was established. The full-length isoform was called ASC-a (named ASC in the rest of the manuscript), and represents the canonical sequence. The names ASC-b, ASC-c, and ASC-d were given to the shorter isoforms [81]. ASC-b lacks the flexible linker encoded by exon 2 and is derived by canonical alternative splicing [81] (Fig. 3). Taniguchi et al. showed that co-expression of either ASC-a or ASC-b together with pro-caspase-1 and pro-IL-1β in COS-7 cells led to caspase-1 and IL-1β maturation. Of note, the secreted amount of IL-1β was significantly higher in cells expressing ASC-b [80]. In view of this finding, it was speculated that the flexible linker would act as a hinge that changes the structure of ASC to an inactive conformation. Thereby, ASC-b, which lacks that hinge, would be constitutively active. However, in light of the recent structural findings on inflammasome assembly [45, 46] (see above), one would expect the flexible linker to be necessary for speck formation. The data published by Bryan et al. show the expected opposite of the older, before mentioned results of Taniguchi et al. They showed that over-expressed ASC-b cannot form aggregates on its own. Co-expressed with NLRP3, ASC-b formed specks, yet these were not as compact and the IL-1β maturation was less efficient. When co-expressed with ASC-a, ASC-b interfered with the formation of normal condensed specks, but rather induced larger irregular-shaped specks (Fig. 4a) [81].

Regulation of ASC speck assembly and function. a Pyrin only proteins (POPs) act as decoy-binding partners for PYD domains. They can either block ASC–PYD–PYD interactions or interactions between ASC and specific inflammasome sensors. The splice variants ASC-b and ASC-c as well as the CARD-only proteins (COPs) block different CARD–CARD interactions. b Post-translational modifications that regulate ASC function. Phosphorylation (P) and ubiquitination (Ub) that activate or inhibit ASC function are depicted. The respective kinase or the E3 ubiquitin ligase is indicated for every modification, as well as the inflammasome sensors involved in the process
ASC-c lacks 60 aa within exon 1, responsible for the assembly of the α-helices 3–6 of the PYD, and is produced by activation of cryptic splice sites within exon 1 [81] (Fig. 3). In overexpression models, ASC-c shows an exclusive cytoplasmic localization and self-polymerizes into long filaments. It co-localizes with caspase-1 and reduces the efficiency of ASC-a speck formation (Fig. 4a). IL-1β secretion upon inflammasome activation decreases in a dose-dependent manner with the overexpression of ASC-c, which suggests a regulatory role similar to COPs [81]. Endogenous-expressed ASC-c is hardly detectable at the protein level in human THP-1 cells and not at all in human peripheral blood macrophages, while it is highly expressed in mouse J774A.1 cells. This led to the conclusion that ASC-c might compensate for the lack of COPs in mice [81].
ASC-d contains the first 35 aa of ASC (helices 1 and 2 of the PYD) fused to a completely unrelated 69 aa peptide, and results from a frame shift which might be caused by RNA editing (Fig. 3). The potential role of ASC-d remains elusive, and seems not to be inflammasome related [81].
Endogenous expression at the protein level could only be demonstrated for ASC-a, ASC-b, and ASC-c. ASC-b and ASC-c are LPS inducible and detectable in human macrophages [81]. These studies suggest that splicing of PYCARD can result in various forms of the protein, which may modulate the effectiveness of ASC speck formation. It should be kept in mind that the stoichiometry of the alternative spliced forms to the full-length protein form might be critical for the regulation of ASC function. Hence, experiments using overexpression of splice variants could lead to misleading results, especially if further post-translational mechanisms are also involved in regulating ASC function.
PYD-only proteins
The process of ASC speck assembly represents another target of regulation. In humans, POPs and COPs can behave as decoy interaction partners for the PYD of ASC and other inflammasome proteins, or for the CARD domain of caspase-1, respectively.
Four different POPs have been described in humans (POP1–4), with no murine orthologs. However, two murine POPs have been predicted [Pyrin domain containing (Pydc) 3 and 4], which share less than 15 % sequence similarity to any human POP [82, 83]. A possible regulatory role for the murine POPs has not yet been described.
POP1
POP1, originally named ASC2, was first discovered (but not characterized) by an in silico approach in 2001 [84]. It consists of 89 aa and shares 88 % sequence similarity (64 % identity) with the PYD of ASC, from which it was probably derived by gene duplication [58]. Two similar but not identical NMR structures of POP1 were published at the same time by Espejo et al. and Natarajan et al. [85, 86]. The structure proposed by Espejo et al. features a slightly different orientation and an unstructured region between the helices α2 and α4 compared to the classical DD fold [86]. According to Natarajan et al., POP1 structurally resembles the ASC–PYD and folds into a classical DD bundle of six antiparallel α-helices, including the additional loop between α2 and α3 that is present in the ASC–PYD [85].
Endogenous expression of POP1 is found in monocytes, macrophages, dendritic cells, and granulocytes. It shows a diffuse distribution mostly throughout the cytosol, but it has been detected in the nucleus as well [58].
When ASC and POP1 are co-expressed, they interact and co-localize within a speck, while solitary overexpression of POP1 does not lead to POP1 aggregation [58]. POP1 uses its positive electrostatic potential surface patch (EPSP), which is formed by its helices α2 and α3, to bind to a negative EPSP formed by the helices α1 and α4 of ASC–PYD [87]. Despite its high similarity to ASC–PYD, POP1 does not interact with NLRP3-PYD, suggesting that POP1 disturbs efficient ASC fibrillar assembly rather than ASC binding to NLRP3 [87] (Fig. 4a).
The role of POP1 in inflammasome regulation is highly controversial. Counter-intuitively, POP1 enhanced IL-1β processing in a dose-dependent manner in overexpression experiments [58]. Another report confirmed that POP1 does not impair ASC and NLRP3-dependent IL-1β processing [88]. However, recent studies showed that POP1 inhibits inflammasome activation and cytokine release by impairing the inflammasome nucleation [89]. The observed differences might be explained by a more complex regulation of inflammasomes by POP1, or by additional regulatory steps that act in a context or cell specific manner. TNF-α stimulation, for example, does not only lead to phosphorylation of ASC–PYD but also of POP1 [58]. Moreover, POP1 influences the NF-κB signaling pathway by interacting with the IKK complex and suppressing the kinase activity of IKKα and IKKβ [58], which promote NF-κB translocation to the nucleus upon pro-inflammatory stimuli [90]. Since NF-κB signaling is also required for the expression of NLRP3 and pro-IL-1β, it is difficult to define whether POP1–ASC or POP1–IKK interactions are responsible for the observed effect on inflammasomes. The fact that stable expression of POP1 in THP-1 cells does not alter IκBα phosphorylation makes it more intricate to decipher the role of POP1 in inflammasome regulation [89].
POP2
POP2 was discovered by two groups in parallel [91, 92]. POP2 is a 97 aa protein encoded by a gene located on the human chromosome 3. It is not obviously derived from a gene duplication event, as no similar PYD-containing protein is encoded nearby in the genome. The NLRP2-PYD displays the closest similarity to POP2 (69 %), while the ASC- and NLRP3–PYD show similarities of only around 40 % [91, 92]. POP2 is an evolutionarily young gene only present in humans and closely related primates [93]. It is expressed in a wide range of hematopoietic cell lines, where it can be found in the cytoplasm and the nucleus [92]. Its expression is upregulated by diverse inflammatory stimuli, including LPS, TNF-α, and phorbol myristate acetate [88].
POP2 interacts with NLRP2 and ASC, and co-localizes with ASC specks [91, 92] (Fig. 4a). POP2 is accepted as a negative regulator of the inflammasome, inhibiting speck formation, caspase-1 activation, and IL-1β maturation [91, 92]. Yet, its mechanism of action is controversial. Some reports point towards a binding to the ASC–PYD, whereas others indicate interaction with the PYD of the inflammasome sensor [83, 91, 92]. Although POP2 induces speck formation when co-transfected with ASC, it blocks speck assembly when expressed with ASC and NLRP3-PYD, suggesting an interaction between POP2 and NLRP3-PYD. Similar effects were observed for NLRP1 and NLRP12 (Fig. 4a) [92]. The helix α1 of POP2 is essential for inflammasome inhibition. Especially, the acidic residues 6, 8, and 16 in α1 are necessary for PYD–PYD interaction and inflammasome inhibition [88]. The necessity of the negative charges in α1 together with the corresponding negative charges in α4 suggests an interaction with the positive EPSP of the helices α2 and α3 of the ASC–PYD, the same interface which was suggested for the interaction with NLRP3. All together, this led to the proposal of POP2 being a competitive inhibitor of NLRP3 [83, 87]. Besides these direct activities, similar to POP1, POP2 is also an inhibitor of NF-κB [88, 92]. However, in contrast to POP1, it does not act at the level of IKKα, but rather inhibits p65 transactivation via its helix α1 [88].
POP3 and 4
POP3 is located within an interferon-inducible gene cluster on chromosome 1 that contains AIM2 and several other genes associated with host defense [94]. POP3 is a protein of 113 aa built up by only five instead of six α-helices, as the region of the fifth α-helix seems to be unstructured [94]. The same non-canonical DD fold is found in NLRP1 [43]. Most likely, POP3 is derived from a partial gene duplication of AIM2, with which it shares a highly similar sequence (61 % identical), whereas it bears faint homology with the PYDs of ASC or NLRP3 [94]. POP3 does not interact with ASC, but inhibits AIM2-induced ASC speck formation and inflammasome assembly by competing with ASC for AIM2-PYD binding (Fig. 4a). It does not interfere with NLRP3-induced ASC speck formation and subsequent cytokine maturation [94].
The most recently described POP4 is the protein product of the pseudogene NLRP2P on the X chromosome. It shares 80 % identity with POP2, is LPS inducible, and has a regulatory effect on the NF-κB signaling pathway. However, it does not seem to interfere with ASC-dependent inflammasome formation (Fig. 4a) [95].
vPOPs
Besides the human POPs, a number of viruses contain homologous open reading frames for virus-encoded POPs (vPOPs): the vPOPs encoded by the rabbit myxoma virus (M013L), the Shope fibroma virus (gp013L), the swine poxvirus (SPV14L), the Yaba-like disease virus (18L), and the mule deer poxvirus (DPV83gp024) display significant similarities to POP1 and ASC–PYD [96, 97].
Both the vPOPs M013L and gp013L interact with ASC, co-localize with ASC specks (Fig. 4a), and inhibit caspase-1 activation and subsequent IL-1β/IL-18 cytokine maturation upon diverse stimuli [96, 97]. M013L was shown to be crucial for the viral immune evasion. M013L deficient myxoma viruses are not able to infect lymphocytes and monocytes and are sensed by NLRP3 and TLRs, which activate the inflammasome and trigger NF-κB signaling pathways in human myeloid cells [97, 98].
CARD-only proteins
Upon inflammasome sensor activation and ASC polymerization, several units of pro-caspase-1 are recruited to the ASC speck structure. Since caspase-1 represents the ultimate effector that catalyzes the production of mature IL-1β, it seems reasonable that its activity is regulated. This is believed to happen, at least in humans, by the COPs. COPs were predicted to function analogously to POPs and regulate inflammasome activation by modulating CARD–CARD interactions (Fig. 4a).
CARD16 (also known as pseudo-ICE), CARD17 (INCA), and CARD18 (ICEBERG) constitute the most studied COPs. All three consist of only a CARD domain and share a highly conserved amino acidic sequence with the CARD of pro-caspase-1. Due to this, and the fact that all three genes are located in close proximity to CASP1 on chromosome 11q22.3, it is believed that CARD16, 17, and 18 originated from gene duplication of CASP1. Of note, COPs are restricted to the genome of primates [83].
CARD17
CARD17 represents the best-characterized inflammasome inhibitor within the COP family. Expression of CARD17 at low nanomolar levels inhibits IL-1β secretion upon NLRP3 inflammasome activation [99, 100]. CARD17 has been shown to bind to pro-caspase-1 and prevents the polymerization of caspase-1-CARD filaments [99, 100]. Electron microscopy analysis localized CARD17 exclusively at the tip of caspase-1-CARD filaments, suggesting that CARD17 binds to the caspase-1-CARD oligomer and blocks the elongation of these filaments [99]. CARD17 is 81 % identical to the CARD of pro-caspase-1, and 83 % identical to CARD16 [100]. A structural model of CARD17, based on the molecular structure of CARD16, suggests that CARD17 possesses defective type Ib and type IIb interfaces. This would explain the mechanism used by CARD17 to impede pro-caspase-1 filament elongation [99]. Whether CARD17 binds to the CARD of ASC remains unanswered. However, CARD17 overexpression does not disrupt the formation of fibers made of CARD-only fragments of ASC [99, 101].
CARD18
CARD18, which is 52 % identical to the CARD of pro-caspase-1 [102], does not interact with ASC, but can nucleate the formation of caspase-1 filaments and is also incorporated into them [99]. However, and in contrast to CARD17, CARD18 overexpression in a THP-1 system does not affect the IL-1β release elicited by LPS and nigericin (a well-known activator of the classical pathway of NLRP3 inflammasome) [99]. Whether CARD18 plays a role in dampening IL-1β release elicited by an inflammasome-independent pathway remains controversial. On that note, CARD18 has been shown to inhibit caspase-1 activation and IL-1β secretion induced upon 6 h of LPS treatment at a nanomolar range. In this scenario, CARD18 could interfere with other proteins that interact with caspase-1 and enhance its autocatalysis, such as the receptor-interacting serine/threonine-protein kinase 2 (RIPK2) [102].
CARD16
CARD16 shares 97 % identity with the CARD domain of pro-caspase-1 and can interact with it [103, 104]. CARD16 overexpression experiments have shown that CARD16 blocks the secretion of IL-1β upon a long-term treatment with micromolar concentrations of LPS [103, 104]. In contrast to these data, transient expression of CARD16 in HeLa cells that also over-expressed caspase-1 and IL-1β showed an increase in IL-1β secretion [101]. Under these conditions, over-expressed CARD16 co-localized with caspase-1-CARD filaments and with ASC specks [101]. Both the IL-1β release and the co-localization with caspase-1 CARD filaments were reduced in CARD16 mutants that presented defective type I and type III interfaces [101, 105]. Further research is needed to understand the contradictory results obtained for CARD16 function, as well as to elicit the potential role of CARD16 under conditions of inflammasome activation.
Post-translational modifications
Post-translational modifications (PTMs) of proteins are involved in signal transduction and play an essential role in the control of the innate immune response [106–108]. The components of inflammasomes are also subjected to a variety of PTMs that regulate their function [4, 79]. In this section, we will focus on how PTMs of ASC regulate inflammasome signaling. The role of PTMs on other components of the inflammasome is reviewed elsewhere [4, 79].
Ubiquitination
Ubiquitination refers to the post-translational modification process in which one or several ubiquitin proteins are attached to a target protein [109]. It represents a three-step divergent process executed by three different enzymes: ubiquitin-activating enzymes (E1s), ubiquitin-conjugating enzymes (E2s), and ubiquitin-ligating enzymes (E3s). E1 activates ubiquitin in an ATP-dependent manner by catalyzing the formation of a thioester bond between the thiol group of the E1’s cysteine itself and the carboxyl group of the C-terminal glycine of ubiquitin. Activated ubiquitin is next transferred to E2, and subsequently, E3 catalyzes its ligation to the ε-amino group of a lysine of the target protein [110].
Ubiquitination can occur once (monoubiquitination) or repeatedly at the same site (polyubiquitination) [109]. In the latter case, the ubiquitin chains can be either linked to the ε-amino group of any of the seven lysines of the previous ubiquitin (K6, K11, K27, K29, K33, K48, or K63), or to the amino group of the N-terminal Met of the previous ubiquitin (linear ubiquitination) [111, 112]. Different ubiquitination patterns result in diverse outcomes. Whereas K48 polyubiquitination targets proteins for proteasomal degradation, other linkage types, such as K63 or linear ubiquitination, can regulate, for example, protein–protein interactions [109]. ASC has been proposed as a target for both K63 and linear ubiquitination, suggesting that ASC ubiquitination regulates signaling rather than proteasomal degradation of the protein.
Rodgers et al. demonstrated that ASC linear ubiquitination is required for NLRP3 and AIM2 inflammasome assembly [113] (Fig. 4b). This process is dependent on the expression of the linear ubiquitin chain assembly complex (LUBAC), which consists of heme-oxidized IRP2 ubiquitin ligase-1 (HOIL-1L), HOIL-1-interacting protein (HOIP), and SHANK-associated RH domain interactor (SHARPIN) [112, 113]. The E3 ligase activity of both HOIL-1L and HOIP is required for the efficient linear ubiquitination of ASC, cleavage of pro-caspase-1, and secretion of IL-1β upon NLRP3 activation [113].
Shi et al. reported increased K63-linked ubiquitin on ASC after AIM2 inflammasome activation in THP-1 cells [114]. The authors suggest an inhibitory role for ASC ubiquitination and propose that K63 polyubiquitination of ASC would promote p62-dependent inflammasome degradation by targeting the complex to autophagosomes (Fig. 4b). However, no direct proof is provided that ASC K63 polyubiquitination is required for degradation; and the exact site of ubiquitination on ASC has not yet been identified. Since Shi et al. could not identify the E3 ubiquitin ligase responsible for the K63 polyubiquitination [114], it has been debated that this could be a misinterpreted linear ubiquitination, due to cross reactivity of K63-ubiquitination antibodies against linear ubiquitination sites [115]. However, this fact would fail to explain why the alleged K63 polyubiquitination upon AIM2 stimulation results in decreased inflammasome activity by targeting it into autophagosomes, whereas linear ubiquitination of ASC boosts AIM2 inflammasome activity. Shi et al. showed that ASC becomes ubiquitinated after AIM2 inflammasome activation [114], but ASC ubiquitination patterns may vary depending on which inflammasome is activated. Although NLRP3 stimuli also induce autophagy [114], it has not been assessed whether ASC is polyubiquitinated after NLRP3 challenge, and whether this leads to autophagosomal degradation of the inflammasome.
In contrast to the inhibitory role of K63 polyubiquitination described above, another study showed a stimulatory effect of ASC K63 polyubiquitination in the context of a virus infection. Infection of cells with the vesicular stomatitis virus (VSV), an RNA virus that activates NLRP3, promoted K63 polyubiquitination on Lys174 of ASC [116] (Fig. 4b). The authors proposed that K63 polyubiquitination of ASC is conducted by the E3 ubiquitin ligase TRAF3 (TNF receptor-associated factor 3), and this process facilitates ASC speck formation and IL-1β secretion. The adaptor protein MAVS, which is known to interact with TRAF3 during antiviral response [117], was suggested to recruit TRAF3 to promote ASC K63 polyubiquitination [116]. However, the axis MAVS-TRAF3-ASC seems to be required only for RNA-dependent activation of NLRP3, since MAVS deficiency does not affect stimulation with either crystalline NLRP3 activators, the NLRC4 activator flagellin, or the AIM2 activator poly(dA:dT) [116].
Phosphorylation
Phosphorylation plays a key role in controlling the inflammatory response [118–120], and several studies have demonstrated that phosphorylation of ASC influences inflammasome function.
The c-Jun N-terminal kinase (Jnk) and the spleen tyrosine kinase (Syk) signaling pathways have been reported to regulate ASC function. Both Syk and Jnk become phosphorylated and activated after treatment with NLRP3 or AIM2 stimuli and interact as well with the inflammasome complex [121, 122]. The absence or inhibition of either Syk or Jnk leads to defects in ASC oligomerization, but does not affect NLRP3–ASC interaction [122]. Syk directly phosphorylates human ASC at the residue Tyr146 (Tyr144 in murine ASC) [121, 122] and Tyr187 [121] (Fig. 4b). Expression of ASC mutants that cannot be phosphorylated at one or both of these residues leads to reduced ASC oligomerization and IL-1β secretion in a reconstituted inflammasome system in HEK cells [121]. In contrast to NLRP3 and AIM2 inflammasomes, Jnk and Syk do not play any role in NLRC4 inflammasome activation [122]. This could be explained by the fact that NLRC4 can directly interact with pro-caspase-1 via CARD–CARD interactions and can form an inflammasome independently of ASC [4]. Syk activity is also not required for NLRP3 inflammasome function in dendritic cells [122, 123]. This cell-type specific pattern in the control of ASC function represents a challenge when trying to elucidate the molecular mechanisms that orchestrate inflammasome function. Of note, most of the experiments on mutants are carried out in artificial inflammasome-assembly systems, such as HEK cells, or in monocytic cell lines, instead of in primary cells.
As opposed to the proposed activating role of Syk in ASC aggregation, Lin et al. reported that LPS-primed Syk KO macrophages yield higher levels of mature IL-1β than WT macrophages when treated in vitro with NLRP3 activators. The authors found that LPS-primed Syk KO macrophages express higher levels of pro-IL-1β and NLRP3 [121]. Of note, Hara et al. measured mostly levels of IL-18—but not IL-1β—during the studies that demonstrate that Syk and Jnk are required for inflammasome function. This fact could have contributed to overlook any disorder in the IL-1β levels in the absence of Syk. Further investigations are necessary to understand and integrate the apparently contradictory negative role of Syk in LPS-induced NF-κB signaling and its activating role in inducing ASC aggregation.
Proteins that belong to the to the IKK family have also been described to play a role in ASC phosphorylation. Mice expressing non-functional mutants of IKKα manifest an aberrantly increased inflammatory response [124, 125] and present an enhanced inflammatory response upon challenge with NLRP3, AIM2, or NLRC4 activators [11] (Fig. 4b). Martin et al. additionally demonstrated that IKKα interacts with ASC in a phosphorylation-dependent manner and suggested that both IKKα’s kinase activity and phosphorylation of ASC at Ser193 and Ser16 are essential for this interaction [11] (Fig. 4b). In the same study, the IKK-related kinase IKKi, which usually promotes the activation of NF-κB and interferon regulatory factors [90], was identified as a critical regulator of inflammasome function [11]. The authors demonstrated that IKKi interacts with ASC after LPS treatment and that IKKi phosphorylates ASC at Ser58 (Fig. 4b). The non-phosphorylatable mutant S58A, which had previously been reported to lead to reduced inflammasome activation [122], did not shift from the nucleus to the perinuclear space after LPS challenge, and stayed bound to IKKα after LPS and ATP treatment [11]. Taken together, the study realized by Martin et al. suggests a model in which ASC is found phosphorylated and bound to IKKα in the nucleus during homeostasis. However, LPS priming induces Ser58 ASC phosphorylation by IKKi and release of the ASC–IKKα complex into the perinuclear area. Activation of NLRP3 or AIM2 leads to IKKα dephosphorylation, which further triggers ASC dephosphorylation and releases from IKKα, enabling inflammasome assembly. In the case of NLRP3, but not of AIM2, the protein phosphatase 2A is a promising candidate for IKKα dephosphorylation [11].
Extracellular specks
Balci-Peynircioglu et al. first observed that ASC specks could be released to the extracellular space and remain stable for long periods in a system of COS-7 cells that over-expressed ASC [126]. Six years later, release of ASC specks, including other components of the NLRP3 inflammasome, was confirmed in vivo, and its pro-inflammatory function was described [127, 128]. Both studies showed that ASC specks are released from the cell after the activation of NLRP3, NLRP1, or AIM2, but not of NLRC4.
The authors propose two different mechanisms by which extracellular ASC specks promote inflammation. On the one hand, extracellular ASC specks can directly activate pro-caspase-1, which is also released into the extracellular space. On the other hand, extracellular ASC specks can be engulfed by macrophages and induce phagolysosomal damage leading to NLRP3 inflammasome activation [129–131]. After phagolysosomal damage, the engulfed ASC specks are released into the cytoplasm, where they promote the oligomerization of soluble ASC. Both mechanisms lead to pro-IL-1β maturation and secretion of its active pro-inflammatory version [127, 128].
The nature of ASC specks as oligomers, their ability to induce oligomerization of soluble ASC molecules in other cells, and the potential to remain resistant to proteases fulfill all the characteristics of prionoids [132, 133]. Moreover, ASC specks are composed of fibrilar structures similar to the fibrils observed in amyloid structures [134, 135], even though no amyloid-fold was found in the ASC molecule [127, 128].
Both the reports of Baroja-Mazo et al. and our group proved that ASC specks are not only active in the cell that senses the threat, but also represent a way to spread the information throughout the organism, since ASC specks are found in biological fluids of challenged animals [127]. In humans, ASC particles appear in higher numbers in the serum of patients with clinically active CAPS [128]. ASC also concentrates in the cerebrospinal fluid of patients with traumatic brain injury [136]. In an animal model for this disease, the administration of anti-ASC neutralizing antibodies efficiently reduced inflammation [137]. Of note, our group reported the existence of anti-ASC autoantibodies in the serum of patients with autoimmune diseases, as well as in mice with induced experimental lupus. Moreover, pre-treatment with anti-ASC antibodies exacerbates the pro-inflammatory response by opsonizing and increasing engulfment of the extracellular ASC particles after crystal challenge [127]. Taking these two facts into account, targeting extracellular ASC specks to block their function—e.g., with anti-ASC antibodies that lack the Fc region—could potentially improve the outcome of not only CAPS patients, but also those ones suffering from autoimmune syndromes.
Another study reported that ASC expression in glomeruli of FMF patients positively correlates with the presence of amyloid deposits in the kidney, the main complication for FMF patients [126]. In these patient-derived samples, ASC expression was higher in the region immediately surrounding the amyloid deposits, which suggests that ASC may interact with the observed amyloid deposits in FMF patients [126].
ASC specks could also have a function in bridging innate and adaptive immunity. Since intracellular ASC specks co-aggregate with cytosolic ubiquitinated proteins by non-specific hydrophobic interactions, Sahilioglu et al. suggest that ASC speck formation could be implicated in antigen presentation [138]. They propose a similar mechanism to the one used by dendritic cells to aggregate ubiquitinated proteins in dendritic cell aggresome-like-induced structures [139]. Moreover, the fact that inflammasomes can be targeted to autophagosomes [114, 138] suggests another pathway by which proteins that co-aggregate with ASC specks can be cross-presented. Further work in this area is required to prove the veracity of these hypotheses.
Conclusion
Excessive inflammation is present in a multitude of diseases and results detrimental for the organism [5, 27]. Thus, it is not surprising that the function of every one of the inflammasome components remains tightly controlled. ASC represents no exception for this, as its function is regulated transcriptionally, post-transcriptionally, and through the binding of protein interaction partners. Alternative splicing of ASC transcripts can result in the expression of up to four ASC isoforms that show distinct properties. Changes in the expression levels of such isoforms could indeed affect inflammasome assembly and modulate the inflammatory response. Interaction of ASC, or other inflammasome components, with proteins, such as POPs and COPs, represents another level in the regulation of inflammasome function. Taking advantage of the effects of these interactions could be useful to develop novel therapeutic concepts based on the inhibition of ASC function. Both phosphorylation and ubiquitination have likewise been involved in regulating inflammasome function at the level of ASC. However, the sequential steps of PTMs that occur on ASC to promote its activation remain still elusive. Whether phosphorylation and ubiquitination, as well as the other mechanisms that regulate ASC function, can interact with each other and constitute a common pathway for ASC regulation has not yet been addressed. Further efforts are needed to integrate all the potential regulatory changes that can occur at every inflammasome component and to decode how these diverse mechanisms are synchronized to orchestrate inflammasome assembly.
The release of ASC specks by activated cells opens a new avenue in cell-to-cell communication and spreading of inflammation. The presence of ASC aggregates in samples of patients suffering from inflammatory diseases underlines the importance of blocking the potential pro-inflammatory functions that ASC can exert extracellularly. Targeting of ASC by antibodies that cannot elicit further immune responses might abolish the extracellular functions of ASC. In line with the characterization of extracellular ASC specks as prion-like aggregates [127, 128], the recently published mechanisms of ASC polymerization provided further evidence that ASC fibrillar assembly occurs in a prionoid manner [49]. Misfolded prion-like proteins and amyloid aggregates have been associated with an increasing number of human disorders [140]. In this respect, ASC has been found in the proximity of amyloid deposits in patients with autoinflammatory disease [126]. Taking into account that some misfolded proteins can induce heterologous seeding (also known as cross-seeding) of other proteins [141], it is tempting to speculate that extracellular ASC aggregation might play a role in favoring accumulation of other protein aggregates and promotion of disease. Thus, targeting ASC speck formation could ameliorate not only the inflammatory response after continuous inflammasome activation, but also other secondary effects caused by extracellular ASC specks accumulation.
Abbreviations
- aa:
-
Amino acid
- AIM2:
-
Absent in melanoma 2
- APAF-1:
-
Apoptotic protease-activating factor 1
- ASC:
-
Apoptosis-associated speck-like protein containing a CARD
- CAPS:
-
Cryopyrin-associated periodic syndrome
- CARD:
-
Caspase-recruitment domain
- COP:
-
CARD-only protein
- DAMP:
-
Damage-associated molecular pattern
- DD:
-
Death domain
- DED:
-
Death effector domain
- E1:
-
Ubiquitin-activating enzyme
- E2:
-
Ubiquitin-conjugating enzyme
- E3:
-
Ubiquitin-ligation enzyme
- EPSP:
-
Electrostatic potential surface patch
- FCAS:
-
Familial cold autoinflammatory syndrome
- FMF:
-
Familial Mediterranean fever
- HIN200:
-
Hematopoietic interferon-inducible nuclear antigens with 200 amino acid repeats
- HOIL-1:
-
Heme-oxidized IRP2 ubiquitin ligase-1
- HOIP:
-
HOIL-1 interacting protein
- IKK:
-
IκB kinase
- IL:
-
Interleukin
- Jnk:
-
c-Jun N-terminal kinase
- LPS:
-
Lipopolysaccharide
- LUBAC:
-
Linear ubiquitin chain assembly complex
- MAVS:
-
Mitochondrial antiviral-signaling
- MEFV:
-
Mediterranean fever
- MV:
-
Myxoma virus
- NAIP:
-
NLR family apoptosis inhibitory protein
- NF-κB:
-
Nuclear factor kappa-light-chain enhancer of activated B-cells
- NLR:
-
Nucleotide-binding oligomerization domain (NOD)-like receptor
- NLRC:
-
NOD-, LRR-, CARD-containing
- NLRP:
-
NOD-, leucine-rich repeat (LRR)-, PYD-containing
- NOD:
-
Nucleotide oligomerization domain
- NOMID:
-
Neonatal-onset multisystem inflammatory disorder
- PAMP:
-
Pathogen-associated molecular pattern
- POP:
-
Pyrin-only protein
- PRR:
-
Pattern-recognition receptor
- PTM:
-
Post-translational modification
- PYCARD:
-
PYD and CARD domain containing
- PYD:
-
Pyrin domain
- Pydc:
-
Pyrin domain containing
- RIPK2:
-
Receptor-interacting serine/threonine-protein kinase 2
- SHARPIN:
-
SHANK-associated RH domain interactor
- SPV:
-
Swine pox virus
- STED:
-
Stimulated emission depletion
- Syk:
-
Spleen tyrosine kinase
- TMS1:
-
Target of methylation-induced gene silencing 1
- TNF:
-
Tumor necrosis factor
- TRAF3:
-
TNF receptor-associated factor 3
- vPOP:
-
Virus-encoded POP
- VSV:
-
Vesicular stomatitis virus
References
Mogensen TH (2009) Pathogen recognition and inflammatory signaling in innate immune defenses. Clin Microbiol Rev 22:240–273. doi:10.1128/CMR.00046-08
Bianchi ME (2007) DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol 81:1–5. doi:10.1189/jlb.0306164
Razani B, Cheng G (2010) NF-kappaB: much learned, much to learn. Sci Signal. doi:10.1126/scisignal.3138pe29
Latz E, Xiao TS, Stutz A (2013) Activation and regulation of the inflammasomes. Nat Rev Immunol 13:397–411. doi:10.1038/nri3452
Broderick L, De Nardo D, Franklin BS et al (2015) The inflammasomes and autoinflammatory syndromes. Annu Rev Pathol Mech Dis. doi:10.1146/annurev-pathol-012414-040431
Masumoto J, Taniguchi S, Ayukawa K et al (1999) ASC, a novel 22-kDa protein, aggregates during apoptosis of human promyelocytic leukemia HL-60 cells. J Biol Chem 274:33835–33838
Masumoto J, Taniguchi S, Nakayama J et al (2001) Expression of apoptosis-associated speck-like protein containing a caspase recruitment domain, a pyrin N-terminal homology domain-containing protein, in normal human tissues. J Histochem Cytochem 49:1269–1275. doi:10.1177/002215540104901009
Conway KE, Mcconnell BB, Bowring CE et al (2000) TMS1, a novel proapoptotic caspase recruitment domain protein, is a target of methylation-induced gene silencing in human breast cancers. Cancer Res 60:6236–6242
Pelegrin P, Barroso-gutierrez C, Surprenant A (2008) P2X7 receptor differentially couples to distinct release pathways for IL-1 β in mouse macrophage. J Immunol 180:7147–7157. doi:10.4049/jimmunol.180.11.7147
Fernandes-Alnemri T, Wu J, Yu J-W et al (2007) The pyroptosome: a supramolecular assembly of ASC dimers mediating inflammatory cell death via caspase-1 activation. Cell Death Differ 14:1590–1604. doi:10.1038/sj.cdd.4402194
Martin BN, Wang C, Willette-Brown J et al (2014) IKKα negatively regulates ASC-dependent inflammasome activation. Nat Commun 5:4977. doi:10.1038/ncomms5977
Bryan NB, Dorfleutner A, Rojanasakul Y, Stehlik C (2009) Activation of inflammasomes requires intracellular redistribution of the apoptotic speck-like protein containing a caspase recruitment domain. J Immunol 182:3173–3182. doi:10.4049/jimmunol.0802367
Wang L, Manji GA, Grenier JM et al (2002) PYPAF7, a novel PYRIN-containing Apaf1-like protein that regulates activation of NF-κB and caspase-1-dependent cytokine processing. J Biol Chem 277:29874–29880. doi:10.1074/jbc.M203915200
Srinivasula SM, Poyet JL, Razmara M et al (2002) The PYRIN-CARD protein ASC is an activating adaptor for caspase-1. J Biol Chem 277:21119–21122. doi:10.1074/jbc.C200179200
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144:646–674. doi:10.1016/j.cell.2011.02.013
Salminen A, Kauppinen A, Hiltunen M, Kaarniranta K (2014) Epigenetic regulation of ASC/TMS1 expression: potential role in apoptosis and inflammasome function. Cell Mol Life Sci 71:1855–1864. doi:10.1007/s00018-013-1524-9
Shiohara M, Taniguchi S, Masumoto J et al (2002) ASC, which is composed of a PYD and a CARD, is up-regulated by inflammation and apoptosis in human neutrophils. Biochem Biophys Res Commun 293:1314–1318. doi:10.1016/S0006-291X(02)00384-4
Stehlik C, Lee SH, Dorfleutner A et al (2003) Apoptosis-associated speck-like protein containing a caspase recruitment domain is a regulator of procaspase-1 activation. J Immunol 171:6154–6163. doi:10.4049/jimmunol.171.11.6154
Drexler SK, Bonsignore L, Masin M et al (2012) Tissue-specific opposing functions of the inflammasome adaptor ASC in the regulation of epithelial skin carcinogenesis. Proc Natl Acad Sci USA 109:18384–18389. doi:10.1073/pnas.1209171109
Walter A, Schäfer M, Cecconi V et al (2013) Aldara activates TLR7-independent immune defence. Nat Commun 4:1560. doi:10.1038/ncomms2566
Feldmeyer L, Keller M, Niklaus G et al (2007) The inflammasome mediates UVB-induced activation and secretion of interleukin-1β by keratinocytes. Curr Biol 17:1140–1145. doi:10.1016/j.cub.2007.05.074
Dombrowski Y, Peric M, Koglin S et al (2011) Cytosolic DNA triggers inflammasome activation in keratinocytes in psoriatic lesions. Sci Transl Med. doi:10.1126/scitranslmed.3002001
Dunn JH, Liu W, Luo Y et al (2012) Dual role of apoptosis-associated speck-like protein containing a CARD (ASC) in tumorigenesis of human melanoma. J Invest Dermatol. doi:10.1038/jid.2012.317
Rathinam VAK, Fitzgerald KA (2016) Review inflammasome complexes: emerging mechanisms and effector functions. Cell 165:792–800. doi:10.1016/j.cell.2016.03.046
Dick MS, Sborgi L, Ru S et al (2016) ASC filament formation serves as a signal amplification mechanism for inflammasome. Nat Commun. doi:10.1038/ncomms11929
Broz P, Dixit VM (2016) Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol. doi:10.1038/nri.2016.58
Guo H, Callaway JB, Ting JP (2015) Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med 21:677–687. doi:10.1038/nm.3893
Kayagaki N, Stowe IB, Lee BL et al (2015) Caspase-11 cleaves gasdermin D for non-canonical inflammasome signaling. Nature. doi:10.1038/nature15541
Shi J, Zhao Y, Wang K et al (2015) Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. doi:10.1038/nature15514
Milhavet F, Cuisset L, Hoffman HM et al (2008) The infevers autoinflammatory mutation online registry: update with new genes and functions. Hum Mutat 29:803–808. doi:10.1002/humu.20720
Campbell L, Raheem I, Malemud CJ, Askari AD (2016) The relationship between NALP3 and autoinflammatory syndromes. Int J Mol Sci. doi:10.3390/ijms17050725
Ozen S, Bilginer Y (2014) A clinical guide to autoinflammatory diseases: familial Mediterranean fever and next-of-kin. Nat Rev Rheumatol 10:135–147. doi:10.1038/nrrheum.2013.174
de Alba E (2009) Structure and interdomain dynamics of apoptosis-associated speck-like protein containing a CARD (ASC). J Biol Chem 284:32932–32941. doi:10.1074/jbc.M109.024273
Ravotti F, Sborgi L, Cadalbert R et al (2016) Sequence-specific solid-state NMR assignments of the mouse ASC PYRIN domain in its filament form. Biomol NMR Assign 10:107–115
Park HH, Lo Y-C, Lin S-C et al (2007) The death domain superfamily in intracellular signaling of apoptosis and inflammation. Annu Rev Immunol 25:561–586. doi:10.1146/annurev.immunol.25.022106.141656
Huang B, Eberstadt M, Olejniczak ET et al (1996) NMR structure and mutagenesis of the Fas (APO-1/CD95) death domain. Nature 384:638–641
Tartaglia LA, Ayres TM, Wong GHW, Goeddel DV (1993) A novel domain within the 55 kd TNF receptor signals cell death. Cell 74:845–853
Liepinsh E, Barbals R, Dahl E et al (2003) The death-domain fold of the ASC PYRIN domain, presenting a basis for PYRIN/PYRIN recognition. J Mol Biol 332:1155–1163. doi:10.1016/j.jmb.2003.07.007
Bae JY, Park HH (2011) Crystal structure of NALP3 protein pyrin domain (PYD) and its implications in inflammasome assembly. J Biol Chem 286:39528–39536
Jin T, Perry A, Smith P et al (2013) Structure of the absent in melanoma 2 (AIM2) pyrin domain provides insights into the mechanisms of AIM2 autoinhibition and inflammasome assembly. J Biol Chem 288:13225–13235
Pinheiro AS, Proell M, Eibl C et al (2010) Three-dimensional structure of the NLRP7 pyrin domain: insight into pyrin-pyrin-mediated effector domain signaling in innate immunity. J Biol Chem 285:27402–27410
Pinheiro AS, Eibl C, Ekman-Vural Z et al (2011) The NLRP12 pyrin domain: structure, dynamics, and functional insights. J Mol Biol 413:790–803
Hiller S, Kohl A, Fiorito F et al (2003) NMR structure of the apoptosis- and inflammation-related NALP1 pyrin domain. Structure 11:1199–1205
Eibl C, Grigoriu S, Hessenberger M et al (2012) Structural and functional analysis of the NLRP4 pyrin domain. Biochemistry 51:7330–7341
Sborgi L, Ravotti F, Dandey VP et al (2015) Structure and assembly of the mouse ASC inflammasome by combined NMR spectroscopy and cryo-electron microscopy. Proc Natl Acad Sci USA. doi:10.1073/pnas.1507579112
Lu A, Magupalli VG, Ruan J et al (2014) Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell 156:1193–1206. doi:10.1016/j.cell.2014.02.008
Yuan S, Akey CW (2013) Apoptosome structure, assembly, and procaspase activation. Structure 21:501–515
Richards N, Schaner P, Diaz A et al (2001) Interaction between pyrin and the apoptotic speck protein (ASC) modulates ASC-induced apoptosis. J Biol Chem 276:39320–39329. doi:10.1074/jbc.M104730200
Cai X, Chen J, Xu H et al (2014) Prion-like polymerization underlies signal transduction in antiviral immune defense and inflammasome activation. Cell 156:1207–1222. doi:10.1016/j.cell.2014.01.063
Lu A, Li Y, Yin Q et al (2015) Plasticity in PYD assembly revealed by cryo-EM structure of the PYD filament of AIM2. Cell Discov. doi:10.1038/celldisc.2015.13
Egelman EH, Francis N, DeRosier DJ (1982) F-actin is a helix with a random variable twist. Nature 298:131–135
Chrétien D, Metoz F, Verde F et al (1992) Lattice defects in microtubules: protofilament numbers vary within individual microtubules. J Cell Biol 117:1031–1040
Broz P, von Moltke J, Jones JW et al (2010) Differential requirement for Caspase-1 autoproteolysis in pathogen-induced cell death and cytokine processing. Cell Host Microbe 8:471–483. doi:10.1016/j.chom.2010.11.007
Broz P, Newton K, Lamkanfi M et al (2010) Redundant roles for inflammasome receptors NLRP3 and NLRC4 in host defense against Salmonella. J Exp Med 207:1745–1755
Schmidt FI, Lu A, Chen JW et al (2016) A single domain antibody fragment that recognizes the adaptor ASC defines the role of ASC domains in inflammasome assembly. J Exp Med. doi:10.1084/jem.20151790
Sahillioglu AC, Sumbul F, Ozoren N, Haliloglu T (2014) Structural and dynamics aspects of ASC speck assembly. Structure 22:1722–1734. doi:10.1016/j.str.2014.09.011
Vajjhala PR, Mirams RE, Hill JM (2012) Multiple binding sites on the pyrin domain of ASC protein allow self-association and interaction with NLRP3 protein. J Biol Chem 287:41732–41743
Stehlik C, Krajewska M, Welsh K et al (2003) The PAAD/PYRIN-only protein POP1/ASC2 is a modulator of ASC-mediated nuclear-factor-kappa B and pro-caspase-1 regulation. Biochem J 373:101–113. doi:10.1042/BJ20030304
Vajjhala PR, Kaiser S, Smith SJ et al (2014) Identification of multifaceted binding modes for pyrin and ASC pyrin domains gives insights into pyrin inflammasome assembly. J Biol Chem 289:23504–23519
Masumoto J, Taniguchi S, Sagara J (2001) Pyrin N-terminal homology domain- and caspase recruitment domain-dependent oligomerization of ASC. Biochem Biophys Res Commun 280:652–655. doi:10.1006/bbrc.2000.4190
Proell M, Gerlic M, Mace PD et al (2013) The CARD plays a critical role in ASC foci formation and inflammasome signalling. Biochem J 449:613–621
Masumoto J, Taniguchi S, Nakayama K et al (2001) Murine ortholog of ASC, a CARD-containing protein, self-associates and exhibits restricted distribution in developing mouse embryos. Exp Cell Res 133:128–133. doi:10.1006/excr.2000.5078
Sanders MG, Parsons MJ, Howard AG et al (2015) Single-cell imaging of inflammatory caspase dimerization reveals differential recruitment to inflammasomes. Cell Death Dis 6:e1813. doi:10.1038/cddis.2015.186
Zhang L, Chen S, Ruan J et al (2015) Cryo-EM structure of the activated NAIP2- NLRC4 inflammasome reveals nucleated polymerization. Sci Express 4:12–14. doi:10.1126/science.aac5789
Chen Y-R, Clark AC (2004) Kinetic traps in the folding/unfolding of procaspase-1 CARD domain. Protein Sci 13:2196–2206. doi:10.1110/ps.03521504
Thornberry NA, Bull HG, Calaycay JR et al (1992) A novel heterodimeric cysteine protease is required for interleukin-1 beta processing in monocytes. Nature 356:768–774
Martinon F, Burns K, Tschopp J (2002) The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-β. Mol Cell 10:417–426. doi:10.1016/S1097-2765(02)00599-3
Lu A, Wu H (2015) Structural mechanisms of inflammasome assembly. FEBS J 5:435–444. doi:10.1111/febs.13133
Hu Z, Zhou Q, Zhang C et al (2015) Structural and biochemical basis for induced self-propagation of NLRC4. Sci Express 4:1–11. doi:10.1126/science.aac5489
Cheng J, Waite AL, Tkaczyk ER et al (2010) Kinetic properties of ASC protein aggregation in epithelial cells. J Cell Physiol 222:738–747. doi:10.1002/jcp.22005
Kueh HY, Mitchison TJ (2009) Structural plasticity in actin and tubulin polymer dynamics. Science 325:960–963
Wu H (2013) Higher-order assemblies in a new paradigm of signal transduction. Cell 153:287–292
Daskalov A, Paoletti M, Ness F, Saupe SJ (2012) Genomic clustering and homology between HET-S and the NWD2 STAND protein in various fungal genomes. PLoS One 7:e34854. doi:10.1371/journal.pone.0034854
Qiao Q, Yang C, Zheng C et al (2013) Structural architecture of the CARMA1/Bcl10/MALT1 signalosome: nucleation-induced filamentous assembly. Mol Cell 51:766–779. doi:10.1016/j.molcel.2013.08.032
Li J, McQuade T, Siemer AB et al (2012) The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell 150:339–350. doi:10.1016/j.cell.2012.06.019
Gaidt MM, Ebert TS, Chauhan D et al (2016) Human monocytes engage an alternative inflammasome pathway. Immunity. doi:10.1016/j.immuni.2016.01.012
Garlanda C, Dinarello CA, Mantovani A (2013) The interleukin-1 family: back to the future. Immunity 39:1003–1018. doi:10.1016/j.immuni.2013.11.010
de Vasconcelos NM, van Opdenbosch N, Lamkanfi M (2016) Inflammasomes as polyvalent cell death platforms. Cell Mol Life Sci 73:2335–2347. doi:10.1007/s00018-016-2204-3
Man SM, Kanneganti T-D (2015) Regulation of inflammasome activation. Immunol Rev 265:6–21
Taniguchi S, Matsushita K, Takeoka M et al (2009) A splice variant of ASC regulates IL-1beta release and aggregates differently from intact ASC. Mediators Inflamm. doi:10.1155/2009/287387
Bryan NB, Dorfleutner A, Kramer SJ et al (2010) Differential splicing of the apoptosis-associated speck like protein containing a caspase recruitment domain (ASC) regulates inflammasomes. J Inflamm (Lond) 7:23. doi:10.1186/1476-9255-7-23
Dorfleutner A, Chu L, Stehlik C (2015) Inhibiting the inflammasome: one domain at a time. Immunol Rev 265:205–216
Le HT, Harton JA (2013) Pyrin- and CARD-only proteins as regulators of NLR functions. Front Immunol 4:1–10. doi:10.3389/fimmu.2013.00275
Pawlowski K, Pio F, Chu ZL et al (2001) PAAD—a new protein domain associated with apoptosis, cancer and autoimmune diseases. Trends Biochem Sci 26:85–87. doi:10.1016/S0968-0004(00)01729-1
Natarajan A, Ghose R, Hill JM (2006) Structure and dynamics of ASC2, a pyrin domain-only protein that regulates inflammatory signaling. J Biol Chem 281:31863–31875. doi:10.1074/jbc.M605458200
Espejo F, Patarroyo ME (2006) Determining the 3D structure of human ASC2 protein involved in apoptosis and inflammation. Biochem Biophys Res Commun 340:860–864. doi:10.1016/j.bbrc.2005.12.087
Srimathi T, Robbins SL, Dubas RL et al (2008) Mapping of POP1-binding site on pyrin domain of ASC. J Biol Chem 283:15390–15398
Atianand MK, Harton JA (2011) Uncoupling of Pyrin-only protein 2 (POP2)-mediated dual regulation of NF-κB and the inflammasome. J Biol Chem 286:40536–40547. doi:10.1074/jbc.M111.274290
de Almeida L, Khare S, Misharin AV et al (2015) The PYRIN domain-only protein POP1 inhibits inflammasome assembly and ameliorates inflammatory disease. Immunity 43:264–276
Verhelst K, Verstrepen L, Carpentier I, Beyaert R (2013) IkB kinase e (IKKe): a therapeutic target in inflammation and cancer. Biochem Pharmacol 85:873–880. doi:10.1016/j.bcp.2013.01.007
Dorfleutner A, Bryan NB, Talbott SJ et al (2007) Cellular pyrin domain-only protein 2 is a candidate regulator of inflammasome activation. Infect Immun 75:1484–1492
Bedoya F, Sandler LL, Harton JA (2007) Pyrin-only protein 2 modulates NF-κB and disrupts ASC:CLR interactions. J Immunol 178:3837–3845. doi:10.4049/jimmunol.178.6.3837
Atianand MK, Fuchs T, Harton JA (2011) Recent evolution of the NF-κB and inflammasome regulating protein POP2 in primates. BMC Evol Biol 11:56. doi:10.1186/1471-2148-11-56
Khare S, Ratsimandresy RA, de Almeida L et al (2014) The PYRIN domain-only protein POP3 inhibits ALR inflammasomes and regulates responses to infection with DNA viruses. Nat Immunol 15:343–353. doi:10.1038/ni.2829
Porter KA, Duffy EB, Nyland P et al (2014) The CLRX.1/NOD24 (NLRP2P) pseudogene codes a functional negative regulator of NF-κB, pyrin-only protein 4. Genes Immun 15:392–403
Dorfleutner A, Talbott SJ, Bryan NB et al (2007) A Shope Fibroma virus PYRIN-only protein modulates the host immune response. Virus Genes 35:685–694
Johnston JB, Barrett JW, Nazarian SH et al (2005) A poxvirus-encoded pyrin domain protein interacts with ASC-1 to inhibit host inflammatory and apoptotic responses to infection. Immunity 23:587–598. doi:10.1016/j.immuni.2005.10.003
Rahman MM, McFadden G (2011) Myxoma virus lacking the pyrin-like protein M013 is sensed in human myeloid cells by both NLRP3 and multiple Toll-like receptors, which independently activate the inflammasome and NF-κB innate response pathways. J Virol 85:12505–12517
Lu A, Li Y, Schmidt FI et al (2016) Molecular basis of caspase-1 polymerization and its inhibition by a new capping mechanism. Nat Struct Mol Biol 23:1–12. doi:10.1038/nsmb.3199
Lamkanfi M, Denecker G, Kalai M et al (2004) INCA, a novel human caspase recruitment domain protein that inhibits interleukin-1β generation. J Biol Chem 279:51729–51738. doi:10.1074/jbc.M407891200
Karasawa T, Kawashima A, Usui F et al (2015) Oligomerized CARD16 promotes caspase-1 assembly and IL-1β processing. FEBS Open Bio 5:348–356. doi:10.1016/j.fob.2015.04.011
Humke EW, Shriver SK, Starovasnik MA et al (2000) ICEBERG: a novel inhibitor of interleukin-1β Generation. Cell 103:99–111. doi:10.1016/S0092-8674(00)00108-2
Lee SH, Stehlik C, Reed JC (2001) COP, a caspase recruitment domain-containing protein and inhibitor of caspase-1 activation processing. J Biol Chem 276:34495–34500. doi:10.1074/jbc.M101415200
Druilhe A, Srinivasula SM, Razmara M et al (2001) Regulation of IL-1beta generation by pseudo-ICE and ICEBERG, two dominant negative caspase recruitment domain proteins. Cell Death Differ 8:649–657. doi:10.1038/sj.cdd.4400881
Kersse K, Lamkanfi M, Bertrand MJM et al (2011) Interaction patches of procaspase-1 caspase recruitment domains (CARDs) are differently involved in procaspase-1 activation and receptor-interacting protein 2 (RIP2)-dependent nuclear factor κB signaling. J Biol Chem 286:35874–35882. doi:10.1074/jbc.M111.242321
Hu H, Sun S-C (2016) Ubiquitin signaling in immune responses. Cell Res 26:457–483. doi:10.1038/cr.2016.40
Cohen P (2014) Immune diseases caused by mutations in kinases and components of the ubiquitin system. Nat Immunol 15:521–529. doi:10.1038/ni.2892
Heaton SM, Borg NA, Dixit VM (2015) Ubiquitin in the activation and attenuation of innate antiviral immunity. J Exp Med 213:1–13. doi:10.1084/jem.20151531
Komander D, Rape M (2012) The ubiquitin code. Annu Rev Biochem 81:203–229. doi:10.1146/annurev-biochem-060310-170328
Hochstrasser M (2009) Origin and function of ubiquitin-like proteins. Nature. doi:10.1038/nature07958
Shimizu Y, Taraborrelli L, Walczak H (2015) Linear ubiquitination in immunity. Immunol Rev 266:190–207. doi:10.1111/imr.12309
Rieser E, Cordier SM, Walczak H (2013) Linear ubiquitination: a newly discovered regulator of cell signalling. Trends Biochem Sci 38:94–102. doi:10.1016/j.tibs.2012.11.007
Rodgers MA, Bowman JW, Fujita H et al (2014) The linear ubiquitin assembly complex (LUBAC) is essential for NLRP3 inflammasome activation. J Exp Med 211:1333–1347. doi:10.1084/jem.20132486
Shi C-S, Shenderov K, Huang N-N et al (2012) Activation of autophagy by inflammatory signals limits IL-1β production by targeting ubiquitinated inflammasomes for destruction. Nat Immunol 13:255–263. doi:10.1038/ni.2215
Matsumoto ML, Dong KC, Yu C et al (2012) Engineering and structural characterization of a linear polyubiquitin-specific antibody. J Mol Biol 418:134–144. doi:10.1016/j.jmb.2011.12.053
Guan K, Wei C, Zheng Z et al (2015) MAVS promotes inflammasome activation by targeting ASC for K63-linked ubiquitination via the E3 ligase TRAF3. J Immunol 194:4880–4890. doi:10.4049/jimmunol.1402851
Saha SK, Pietras EM, He JQ et al (2006) Regulation of antiviral responses by a direct and specific interaction between TRAF3 and Cardif. EMBO J 25:3257–3263. doi:10.1038/sj.emboj.7601220
Park H, Ishihara D, Cox D (2011) Regulation of tyrosine phosphorylation in macrophage phagocytosis and chemotaxis. Arch Biochem Biophys 510:101–111
Clark K (2014) Protein kinase networks that limit TLR signalling. Biochem Soc Trans 42:11–24
Christian F, Smith EL, Carmody RJ (2016) The regulation of NF-κB subunits by phosphorylation. Cells. doi:10.3390/cells5010012
Lin Y-C, Huang D-Y, Wang J-S et al (2015) Syk is involved in NLRP3 inflammasome-mediated caspase-1 activation through adaptor ASC phosphorylation and enhanced oligomerization. J Leukoc Biol 97:1–11. doi:10.1189/jlb.3HI0814-371RR
Hara H, Tsuchiya K, Kawamura I et al (2013) Phosphorylation of the adaptor ASC acts as a molecular switch that controls the formation of speck-like aggregates and inflammasome activity. Nat Immunol 14:1247–1255. doi:10.1038/ni.2749
Gross O, Poeck H, Bscheider M et al (2009) Syk kinase signalling couples to the Nlrp3 inflammasome for anti-fungal host defence. Nature. doi:10.1038/nature07965
Zhu F, Xia X, Liu B et al (2007) IKKa shields 14-3-3s, aG2/M cell cycle checkpoint gene, from hypermethylation, preventing its silencing. Mol Cell 27:214–227. doi:10.1016/j.molcel.2007.05.042
Lawrence T, Bebien M, Liu GY et al (2005) IKKalpha limits macrophage NF-kappaB activation and contributes to the resolution of inflammation. Nature 434:1138–1143. doi:10.1038/nature03491
Balci-Peynircioglu B, Waite AL, Schaner P et al (2008) Expression of ASC in renal tissues of familial mediterranean fever patients with amyloidosis: postulating a role for ASC in AA type amyloid deposition. Exp Biol Med (Maywood) 233:1324–1333. doi:10.3181/0803-RM-106
Franklin BS, Bossaller L, De Nardo D et al (2014) The adaptor ASC has extracellular and “prionoid” activities that propagate inflammation. Nat Immunol. doi:10.1038/ni.2913
Baroja-Mazo A, Martín-Sánchez F, Gomez AI et al (2014) The NLRP3 inflammasome is released as a particulate danger signal that amplifies the inflammatory response. Nat Immunol 15:1–5. doi:10.1038/ni.2919
Dostert C, Pétrilli V, Van Bruggen R et al (2008) Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science 320:674–677. doi:10.1126/science.1156995
Halle A, Hornung V, Petzold GC et al (2008) The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol 9:857–865. doi:10.1038/ni.1636
Hornung V, Bauernfeind F, Halle A et al (2008) Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol 9:847–856. doi:10.1038/ni.1631
Aguzzi A, Nuvolone M, Zhu C (2013) The immunobiology of prion diseases. Nat Rev Immunol 13:888–902. doi:10.1038/nri3553
Holmes BB, Diamond MI (2012) Cellular mechanisms of protein aggregate propagation. Curr Opin Neurol 25:721–726. doi:10.1097/WCO.0b013e32835a3ee0
Serpell LC (2000) Alzheimer’s amyloid fibrils: structure and assembly. Biochim Biophys Acta 1502:16–30. doi:10.1016/S0925-4439(00)00029-6
Greenwald J, Riek R (2010) Biology of amyloid: structure, function, and regulation. Structure 18:1244–1260. doi:10.1016/j.str.2010.08.009
Adamczak S, Dale G, de Rivero Vaccari JP et al (2012) Inflammasome proteins in cerebrospinal fluid of brain-injured patients as biomarkers of functional outcome: clinical article. J Neurosurg 117:1119–1125. doi:10.3171/2012.9.JNS12815
de Rivero Vaccari JP, Lotocki G, Alonso OF et al (2009) Therapeutic neutralization of the NLRP1 inflammasome reduces the innate immune response and improves histopathology after traumatic brain injury. J Cereb Blood Flow Metab 29:1251–1261. doi:10.1038/jcbfm.2009.46
Sahillioğlu AC, Özören N (2015) Artificial loading of ASC specks with cytosolic antigens. PLoS One 10(8):e0134912. doi:10.1371/journal.pone.0134912
Lelouard H, Gatti E, Cappello F et al (2002) Transient aggregation of ubiquitinated proteins during dendritic cell maturation. Nature 417:177–182. doi:10.1038/417177a
Knowles TPJ, Vendruscolo M, Dobson CM (2014) The amyloid state and its association with protein misfolding diseases. Nat Rev Mol Cell Biol 15:384–396. doi:10.1038/nrm3810
Morales R, Moreno-Gonzalez I, Soto C (2013) Cross-seeding of misfolded proteins: implications for etiology and pathogenesis of protein misfolding diseases. PLoS Pathog 9:1–4. doi:10.1371/journal.ppat.1003537
Acknowledgments
The ASC speck STED image showed in Fig. 2 was recorded with the help of Bjørnar Sporsheim at the Cellular and Molecular Imaging Core Facility (CMIC), Norwegian University of Science and Technology (NTNU). We thank Andrea Stutz for critical reading of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
F. Hoss and J. F. Rodriguez-Alcazar contributed equally.
Rights and permissions
About this article

Cite this article
Hoss, F., Rodriguez-Alcazar, J.F. & Latz, E. Assembly and regulation of ASC specks. Cell. Mol. Life Sci. 74, 1211–1229 (2017). https://doi.org/10.1007/s00018-016-2396-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-016-2396-6