
Abstract
The central nervous system (CNS) is the most delicate system in human body, with the most complex structure and function. It is vulnerable to trauma, infection, neurodegeneration and autoimmune diseases, and activates the immune system. An appropriate inflammatory response contributes to defence against invading microbes, whereas an excessive inflammatory response can aggravate tissue damage. The NLRP3 inflammasome was the first one studied in the brain. Once primed and activated, it completes the assembly of inflammasome (sensor NLRP3, adaptor ASC, and effector caspase-1), leading to caspase-1 activation and increased release of downstream inflammatory cytokines, as well as to pyroptosis. Cumulative studies have confirmed that NLRP3 plays an important role in regulating innate immunity and autoimmune diseases, and its inhibitors have shown good efficacy in animal models of various inflammatory diseases. In this review, we will briefly discuss the biological characteristics of NLRP3 inflammasome, summarize the recent advances and clinical impact of the NLRP3 inflammasome in infectious, inflammatory, immune, degenerative, genetic, and vascular diseases of CNS, and discuss the potential and challenges of NLRP3 as a therapeutic target for CNS diseases.
Similar content being viewed by others


NLRP3 is a protein consisting of an amino-terminal Pyrin domain (PYD), a central Nacht domain, and a C-terminal leucine-rich-repeats domain (LRR domain). Once primed and activated, it can complete the assembly of inflammasome (sensor NLRP3, adaptor ASC, and effector Caspase1), resulting in caspase-1 activation, IL-1β and IL-18 release enzyme cleavage and activity increase, as well as to pyroptosis, thus playing an important role in regulating innate immunity and autoimmune diseases [1]. In contrast to the attention paid to autoimmune and infectious diseases of the CNS, the role of immune mechanisms in degenerative and epilepsy and other CNS diseases has long been neglected. In recent years, the booming experiment and inspection technology have provided a large number of evidences for immune mechanisms are involved in CNS diseases, including but not limited to autoimmune and infectious diseases, and have shown potential therapeutic effects in modulating immune signaling mechanisms. The NLRP3 inflammasome was the first one studied in the brain, with impressive results. Numerous studies have demonstrated that the NLRP3 inhibitor MCC950, the cystic fibrosis transmembrane conductance regulator inhibitor C172 and its analogs CY-09,and oridonin show good curative effect in various preclinical immunopathological models such as Alzheimer’s disease, traumatic brain injury, atherosclerosis, diabetic encephalopathy, human immunodeficiency virus (HIV)-associated neurocognitive disorders (HANDs), cerebral edema, cerebral ischemia-reperfusion injury, hypoxic ischemic encephalopathy, etc [1,2,3,4,5,6]. The application of NLRP3 inflammasome inhibitors in CNS diseases will be eagerly awaited. In this review, we summarize the latest biological characteristics of NLRP3 inflammasome, how the inflammasome is primed and activated in CNS infectious, inflammatory immune, degenerative, vascular, genetic, metabolic, tumor and epilepsy diseases and its role in the occurrence and development of diseases after activation, explore the potential and challenges of NLRP3 as a therapeutic target for CNS diseases.
Overview of NLRP3 inflammasome
NLRP3 inflammasome priming and activation
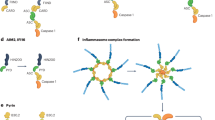
NLRP3 inflammasome, consisting of the sensor (NLRP3), adapter (ASC; also known as PYCARD) and effector (caspase-1) (Fig. 1), has been shown to play an important role in diseases such as arteriosclerosis, type 2 diabetes mellitus, neurodegenerative diseases, gout, arthritis and silicosis [1]. Priming of the NLRP3 inflammasome is a prerequisite for its activation. Priming can upregulate NLRP3 、pro-caspase-1、pro-IL-1β and pro-IL-18 expression, and induce post-translational modifications of NLRP3(such as ubiquitination, phosphorylation, and SUMOylation) [7,8,9]. The priming is achieved through the identification of pathogen-associated molecular patterns and damage-associated molecular patterns. Bauernfeind et al. confirmed that TNF, IL-1, HMGB1, IFN, TGF-1β and LPS could upregulate NLRP3 expression by inducing nuclear factor-κB (NF-κB) activation [10, 11]. Primed NLRP3 induces inflammasome assembly and full activation upon recognition of appropriate activator. The activators include K+ efflux, Cl− efflux, Ca2+ mobilization, lysosome destruction, anti-Golgi dissociation, mitochondrial dysfunction, mitochondrial ROS production and mitochondrial DNA release into the cytosol [12, 13]. There is mounting evidence that mitochondria are not only involved in the regulation of NLRP3 inflammasome activation, but also the docking site of inflammasome assembly. Once activated, NLRP3 associates with both mitochondria and mitochondria associated membranes. Cardiolipin, mitochondrial antiviral signaling protein and mitomycin 2 are considered to be the connection points between NLRP3 and mitochondria, playing important roles in NLRP3 localization and activation [14, 15]. These processes are called Canonical activation of the NLRP3 inflammasome pathways (Fig. 2).

Schematic representation of NLRP3 inflammasome structure.NLRP3 inflammasome, consisting of the sensor (NLRP3), adapter (ASC) and effector (caspase-1)

Canonical activation of the NLRP3 inflammasome pathways. NLRP3 canonical activation requires two steps, a priming step and an activation step.The priming is achieved through the identification of pathogen-associated molecular patterns and damage-associated molecular patterns. TNF, IL-1, HMGB1, IFN, TGF-1β and LPS could upregulate NLRP3 expression by inducing nuclear NF-κB activation.Primed NLRP3 induces inflammasome assembly and full activation upon recognition of K + efflux, Cl − efflux, Ca2 + mobilization, lysosome destruction, anti-Golgi dissociation, mitochondrial dysfunction, mitochondrial ROS production and mitochondrial DNA release into the cytosol.NLRP3 inflammasome formation includes binding of NLRP3 with NEK7, oligomerization of NLRP3, assembly of ASCs into fibrils, and recruitment and activation of Caspase 1. Actived NLRP3 inflammasome can induce caspase-1-dependent pyroptosis in host cells. Active GSDMD, which bind to phosphatidylinositol phosphates and phosphatidyl serine in the membrane leaflets, aggregate and insert into the plasma membrane to form a pore. Inflammatory cytokines such as IL-1β and IL-18 are released into the interstitial space and activate corresponding receptors in adjacent cells, causing an immune cascade and amplifying the injury effect
However, findings in recent years have revealed that NLRP3 inflammasome activation does not always follow a two-step activation pattern. The LPS released by Gram-negative bacteria can activate human caspase-4/5 or mouse caspase-11 to lyse GSDMD and induce pyroptosis.Activated caspase-11 activates pannexin-1 to release ATP and induce K + efflux, thus mediating the activation and oligomerization of NLRP3.And caspase-4 is highly expressed in human cells, so its activation does not need priming [16, 17]. This process is called Noncanonical activation of the NLRP3 inflammasome pathways (Fig. 1).Hornung et al. found another NLRP3 inflammasome activation pathway in human monocytes: mediating TLR4-TRIF-RIPK1-FADD-CASP8 signaling could activeNLRP3 inflammasome and release mature IL-1β, but did not lead to pyroptosis [18]. Lin et al. observed that simultaneous stimulation of TLR and NLRP3 by pathogenic microorganisms could lead to rapid assembly of NLRP3 inflammasome and pyroptosis in mice macrophages, which depends on the TLR signaling molecule IL-1 receptor-associated kinase and its kinase activity [19]. This process is called alternative activation of the NLRP3 inflammasome pathways (Fig. 3).

Noncanonical and alternative activation of the NLRP3 inflammasome pathways.Noncanonical activation of NLRP3 inflammasome is induced by gram-negative bacteria. Release of LPS from engulfed bacteria into the cytoplasm activates human Casp4/5 or mouse Casp11, which cleaves GSDMD to induce pyroptosis and indirectly activates the NLRP3 inflammasome to activate Casp1 and IL-1b and IL-18. Noncanonical activation does not require priming as Casp4 is present at a high level. The alternative pathway of activation is elicited by TLR4 agonists like LPS, which activates the TLR4-TRIF-RIPK1-FADD-Casp8 signaling. Casp8 activates the NLRP3 inflammasome but does not require K1 efflux, ASC speck formation, or pyroptosis
In addition, NLRP3 works with other sensors to co-activate the inflammasome. Freeman et al. found that lysophosphatidylcholine could induce NLRC4 and NLRP3 to form different inflammasomes in microglia and astrocytes, synergistically promote the mature IL-1β release, and play an important role in its hyperplasia [20]. Kalantari et al. demonstrated that Plasmodium and Aspergillus could activate NLRP3 and AIM2 inflammasomes, forming a dual cytoplasmic surveillance system [21, 22]. Davis et al. confirmed that NLRC5 bound to NLRP3 in an NBD-dependent but LRR-inhibitory manner in the presence of pathogens, and the two co-activated the inflammasome [23]. NIMA-associated kinase 7 is demonstrated not to interact with other inflammasome sensors (such as NLRC4 and AIM2), but to specifically interact with NLRP3 by polymerizing into complexes essential for ASC spot formation and caspase-1 activation, suggesting that NEK7 is a core component unique to the NLRP3 inflammasome [24,25,26]. In addition, Dos observed strong colocalization of NLRP3 and vimentin in activated alveolar macrophages, and the colocalization between NLRP3 and ASC was absent after vimentin knockdown, suggesting that vimentin is associated with components of the inflammasome [27]. Lang et al. reported that the activated macrophage migration inhibitory factor could not only regulate the interaction of NLRP3- vimentin, but also interact with NLRP3 and directly participated in the NLRP3 inflammasome assembly and activation, indicating that migration inhibitory factor’s role in inflammasome activation is independent of its role as a cytokine [28]. Recently, the proteins which interact with NLRP3 inflammasome complex but are not its components have been successively reported. Such as Pyrin-only proteins (POPs, also known as PYDC proteins) and Card-only proteins (COPs). POPs include POP1-POP4. Of these, POP1 and POP2 combine with ASC, thus inhibiting the interaction between NLRP3 and ASC and preventing the over-activation of NLRP3 [29, 30]. In human COPS, COP1, ICOBERG and INCA can bind to full-length caspase-1, thus preventing its own activation and limiting inflammasome activation [29] (Fig. 2).
NLRP3 inflammasome induces pyroptosis and its inhibitors
Activation of NLRP3 inflammasome can induce caspase-1-dependent pyroptosis in host cells. Gasdermin D is a mediator of pyroptosis. After interacting with caspase-1, it can bind to phosphatidylinositol phosphates and phosphatidyl serine in the membrane leaflets, aggregate and insert into the plasma membrane to form a pore. Inflammatory cytokines such as IL-1β and IL-18 are released into the interstitial space and activate corresponding receptors in adjacent cells, causing an immune cascade and amplifying the injury effect [31] (Fig. 2).There are more and more reports on NLRP3 as a therapeutic target for a variety of diseases in recent years.MCC950, a sulfonylurea compound, is the most effective and specific inhibitor of NLRP3, and has shown promising therapeutic effects in various preclinical immunopathological models [1], including CAPS, experimental autoimmune encephalomyelitis, Alzheimer’s disease, traumatic brain injury, arrhythmias, myocardial infarction, diabetic encephalopathy, HANDs, acute pancreatitis, spinal cord injury, brain edema, cerebral ischemia-reperfusion injury, brain injury following intracranial haemorrhage, age-related osteoporosis, neonatal hypoxic-ischemic encephalopathy, post-stroke cognitive impairment and other diseases [5, 32]. Furthermore, the inhibitor of cystic fibrosis transmembrane conductance regulator C172 and its analogues CY-09, oridonin have also been shown to specifically inhibit NLRP3 inflammasome [2, 4, 33] (Table 1; Fig. 4).

NLRP3 inflammasome signal path inhibitors in experimental models of central nervous system diseases. MCC950 inhibited NLRP3 activation and ATPase activity. JC-124 prevented the oligomerization in the interaction between NLRP3 and ASC. Baicalin inhibited the upregulated expression of TLR4, suppressed the phosphorylation and degradation of IκBα, and inhibited the subsequent nuclear translocation of NF-κB p65. Parthenolide curbed the phosphorylation of NF-κB. Ginsenoside Rh2 inhibited the NLRP3 activation. CY-09 prevented NLRP3 oligomerization. OLT1177 inhibited ATPase activity. Oridonin blocked the interaction between NLRP3 and NEK7, thereby inhibiting NLRP3 inflammasome assembly and activation. Bay 11-7082 curbed the phosphorylation of NF-κB. Neferine inhibited NLRP3 activation. Docosahexaenoic acid reduced the expression of NLRP3.Propofol depressed the TXNIP expression, oxidative stress, and NLRP3 inflammasome activation. Mangiferin reduced the expression of TXNIP, depress the oxidative stress and enhance the endogenous antioxidants. ACT001 restrained NF-κB nuclear translocation in microglia cells through inhibiting AKT phosphorylation. HET0016 inhibited the phosphorylation of p38 MAPK. Prussian Blue Nanozyme inhibited the assembly and activation of NLRP3 inflammasome.UNC9995 inhanced β-arrestin2 interacting with NLRP3 to interfere inflammasome assembly. Glibenclamide inhibition of NLRP3 activation. Andrographolide promoted the parkin-dependent autophagic flux formation in microglia; resulting in the removal of defective mitochondria which in turn inhibit NLRP3 inflammasome activation
The role of NLRP3 inflammasome in CNS diseases
CNS infection
Viral infections of the CNS
NLRP3 inflammasome can recognize various pathogen-related molecular patterns and danger-related molecular patterns during viral replication, and is a key player in the organism’s antiviral response. Currently, NLRP3 has been found to be localized mostly in microglia, and also expressed in other brain cells such as oligodendrocytes, brain endothelial cells and astrocytes [34,35,36]. Zika virus (ZIKV) is a neurotropic virus, which can not only break through the BBB and invade the CNS, but also damage peripheral nerves. It can also be transmitted vertically from mother to her fetus, and pose a serious threat to fetal health. Gim et al. found that ZIKV NS5 protein combined with Nacht and LRR domains of NLRP3 in cytoplasm to form a NS5-NLRP3-ASC globular structure, which promoted NLRP3 inflammasome activation and IL-1β release, and induced aggressive inflammation response [37]. Knockdown of NLRP3 can inhibit ZIKV-mediated IL-1β secretion in mice, indicating that negative regulation of NLRP3 inflammasome or preventing NS5 binding to NLRP3 and ASC can reduce the damage caused by ZIKV infection-induced aggressive inflammatory reaction. Since 2007, large-scale outbreaks of hand, foot and mouth disease caused by enterovirus have occurred continuously in China. The dominant prevalence of enterovirus 71(EV71) has led to severe CNS complications and death. A study has demonstrated that EV71 3D RNA polymerase in macrophages and peripheral blood monocytes can directly interact with NLRP3 to form 3D-NLRP3-asc circular structure, promote NLRP3 inflammasome assembly and activate IL-1β [38]. XIAO et al. confirmed that VIM mediated NLRP3 inflammasome activation in EV71-infected mice and promoted brain inflammation and neuronal injury, but failed to verify whether 3D-NLRP3-asc ring structure was formed in the CNS [39]. Japanese encephalitis virus is a common cause of acute epidemic viral encephalitis. ROS production and K+ efflux mediated by Japanese encephalitis virus infection can induce NLRP3 inflammasome assembly, Caspase-1 activation and IL-1β release [40]. Taken together, negative regulation of NLRP3 inflammasome can reduce the damage caused by CNS virus infection-induced aggressive inflammatory response.
Bacterial infections of the CNS
Streptococcus pneumoniae (S. pneumoniae) activates NLRP3 through its virulence factor pneumolysin [41]. Knockdown of NLRP3 and ASC attenuates brain inflammation in the mice model of S. pneumoniae meningitis [42]. Similarly, Kim et al. found that S. pneumoniae could induce caspase-1 activation and downstream inflammatory cytokines maturation in a mice meningitis model, suggesting that NLRP3 inflammasome plays a key role in this process, and inhibition of inflammasome activation is a practical strategy for the treatment of S. pneumoniae meningitis [43].
Other pathogen infections of the CNS
Kalantari et al. [21, 22] demonstrated that Plasmodium and Aspergillus could activate NLRP3 and AIM2 inflammasomes to form a dual-cytoplasmic surveillance system.Lee et al. found that Mycobacterium tuberculosis can stimulate microglia-leukocyte interaction, and act as a priming signal to activate NLRP3 inflammasome of microglia and induce IL-1β maturation in patients with tuberculous meningitis. Jin et al.found that Ginsenoside Rh2 can inhibit the TLR4/NF-κ b signaling pathway, block the activation of NLRP3 inflammasome, and improve the neuronal damage induced by Toxoplasma gondii infection [44].
Traumatic brain injury
Traumatic brain injury (TBI) is a change in brain function caused by external mechanical forces on the head or neck, and it is one of the most common causes of death and disability [45]. Peripheral edema is the main mechanism of secondary brain injury after craniocerebral injury. Yi et al. found that NLRP3 inflammasome expression increased in mice after TBI, and NLRP3 gene knockdown could alleviate brain edema, suggesting that NLRP3 inflammasome plays an important role in regulating brain edema and secondary inflammation after TBI [46]. Xu et al. detected that NLRP3 inflammasome expression was up-regulated in the cortex surrounding brain injury, especially in microglia. MCC950 could alleviate brain edema, reduce lesion volume, and decreases cell death in TBI mice, thereby reducing the severity of brain injury and showing a good neuroprotective effect [47]. However, this effect disappeared in mice with depleted microglia, indicating that NLRP3 inflammasome is involved in the inflammatory response to TBI, and MCC950’ s specific inhibition of NLRP3 inflammasome may be a promising therapeutic approach for TBI.In addition, JC124, HET0016, ACT001, pathenolide, mangiferin, propofol and other drugs have been reported to reduce the activation of NLRP3 inflammasome and caspase-1 around injury in TBI mice, and alleviate neurological deficits and neuronal damage [48,49,50,51,52].
Immunoinflammatory diseases of the CNS
Anti-NMDAR encephalitis
Autoimmune encephalitis has broadly come to mean a kind of nerve dysfunction is triggered by auto-specific antibodies attacking the CNS and mediating inflammatory brain parenchyma lesions. Its prevalence is increasing year by year. Anti-N-methyl-d-aspartate receptor (NMDAR) encephalitis is the main type of autoimmune encephalitis, which progresses rapidly. Some patients still have serious neurological deficits or mental disorders even after standardized treatment, seriously affecting the health and quality of life of patients [53]. Peng et al. observed that NLRP3 and IL-1β in cerebrospinal fluid were increased significantly in patients with anti-NMDAR encephalitis, and they were positively correlated with each other. Further follow-up showed that the decrease of maximum modified Rankin scale was positively correlated with the decrease of NLRP3 inflammasome in cerebrospinal fluid in patients with anti-NMDAR encephalitis [54]. It is suggested that NLRP3 inflammasome and its downstream inflammatory cytokines are involved in the neuroinflammatory process of anti-NMDAR encephalitis. its level reflects the severity of anti-NMDAR encephalitis patients, and can help judge the severity and prognosis of anti-NMDAR encephalitis.
NLRP3 autoimmune GFAP astrocytopathy
Autoimmune glial fibers acidic protein (GFAP) astrocytopathy is an autoimmune meningitis/encephalomyelitis mediated by GFAP antibody. At present, there are no recognized diagnostic criteria and treatment guidelines for it, and it is difficult to diagnose and treat. Luo et al. observed that the levels of NLRP3 inflammasome and its downstream cytokines IL-1β, IL-6 and IL-17 were significantly increased in cerebrospinal fluid of patients with GFAP astrocytic disease, and NLRP3 expression was positively correlated with cytokines and disease severity. Moreover, they found that NLRP3 and inflammatory cytokines levels were significantly positively correlated with anti-GFAP antibody titers and disease severity, indicating that NLRP3 inflammasome is activated significantly in patients with autoimmune GFAP astrocytopathy, triggers downstream inflammatory response, and participates in the occurrence of the disease [55]. These results suggest that NLRP3 inflammasome may be a new target for evaluating the severity and treatment of autoimmune GFAP astrocytopathy.
NLRP3 neuromyelitis optica spectrum disorders
Neuromyelitis optica spectrum disorders (NMOSDs) are a group of CNS inflammatory demyelinating diseases mediated by antigen-antibody. Patients who do not receive immunosuppressive therapy have poor prognosis and high mortality [56]. Peng et al. found that the levels of NLRP3, mtDNA, IL-1β, IL-6, and IL-17 in cerebrospinal fluid of NMOSDs patients were higher than those of the control group, and the EDSS scores of NMOSDs patients at the relapse stage was positively correlated with NLRP3 and mtDNA in CSF [57], suggesting that NLRP3 inflammasome-mediated pyroptosis after mitochondrial injury may play an important role in the pathogenesis of NMOSDs.
Multiple sclerosis
Multiple sclerosis (MS) is an autoimmune inflammatory demyelinating disease of CNS known as the most common CNS disease which leads to the disability of young and middle-aged people [58]. The expression of NLRP3 and IL-1β was increased in microglia of MS patients, and the percentage of NLRP3 and IL-1β positive CNS cells was positively correlated with demyelinating activity and imaging severity [59]. The activated NLRP3 inflammasome promotes the release of a large amount of mature IL-1β into the extracellular space, causing a cascade of inflammatory reactions and aggravating the progression of MS [60]. Using specific NLRP3 inflammasome inhibitors can alleviate axonal injury and disease severity in experimental autoimmune encephalomyelitis mice [59]. Using interferon-β can inhibit IL-1β release and reduce NLRP3 inflammasome activity, thus shortening the course of MS [61]. Additionally, Voet et al. found that deficiency of NF-κB regulatory protein A20 in microglia aggravated MS-like disease. The reason for this was that A20 deficiency caused NLRP3 inflammasome hyperactivation, leading to increased secretion of IL-1β [62]. OLT1177, a selective NLRP3 inflammasome inhibitor, ameliorates neurological decline and nerve tissue damage in EAE mice, demonstrating its beneficial role in the treatment of MS [63]. The aforementioned studies indicate that NLRP3 inflammasome and IL-1β are potential prognostic biomarkers and potential therapeutic targets for MS. Inhibiting the activities of NLRP3 and downstream inflammatory cytokines such as IL-1β may help attenuate persistent inflammatory response in CNS and s slow disease progression.
Stroke
Stroke is a kind of disorder of cerebrovascular circulation caused by various factors, which leads to ischemic and hypoxic necrosis of brain tissue. It is usually divided into hemorrhagic stroke and ischemic stroke [64]. With the rapid development of global population aging and the rising prevalence of risk factors such as obesity, hypertension, diabetes and hyperlipidemia, the stroke incidence is increasing year by year and the age of onset is younger, which seriously threatens human health [65]. Under the existing medical technology and conditions, its morbidity, disability and mortality are extremely high。 And it is the second largest cause of death and the third largest cause of disability among adults in the world [66]. Therefore, it is a major clinical challenge to find new treatment targets and formulate new treatment strategies to reduce its disability and mortality .
Ischemic stroke and cerebral ischemia-reperfusion injury
Ischemic stroke accounts for more than 80% of all stroke patients, which poses a serious threat to human health [67]. The key to treatment is to restore cerebral blood flow perfusion in ischemic area as soon as possible. However, cerebral blood flow reperfusion may aggravate ischemic tissue injury and inflammation, resulting in some ischemic brain tissue injury or dysfunction (i.e., cerebral ischemia-reperfusion injury) [68]. It has been confirmed that knockdown of NLRP3 gene can reduce cerebral infarct volume, attenuate brain edema, maintain BBB permeability, and decrease nerve cell death in mice with ischemic stroke [69]. Using MCC950 can improve the neurological deficit and enhance the long-term survival rate but not reduce the cerebral infarct volume of diabetic mice with transient middle cerebral artery occlusion, which may be due to the inhibitory effect of MCC950 is not as complete as NLRP3 knockdown [68]. Currently, the important roles of NLRP3 inflammasome assembly, caspase-1 activation, IL-β and other downstream inflammatory cytokines release have been confirmed in cerebral ischemia/reperfusion injury after ischemic stroke. Meanwhile, the effect of NLRP3 inflammasome related signaling pathways on this disease has also drawn more and more attention. Hou et al. found that up-regulating Nrf2 could lead to decreased expression of TXNIP, NLRP3 inflammasome, Caspase-1, IL-18, and IL-1β, and the protective effect of Nrf2 was basically eliminated after knockdown of Trx1 in the mice model of middle cerebral artery occlusion, suggesting that Nrf2 can inhibit NLRP3 inflammasome activation by regulating Trx1/TXNIP complex [70]. In addition, An et al. reported that histidine play a neuroprotective role by inhibiting NLRP3-mediated pyroptosis by modulating AMPK/GSK3β signaling pathway [71]. He et al. found that ATF4 gene overexpression can up-regulate Parkin expression, enhance phagocytic activity, and inhibit inflammatory response mediated by NLRP3 inflammasome. Knockdown of Parkin gene effectively reversed the above process, confirming that ATF4 can reduce cerebral ischemia-reperfusion injury by inhibiting NLRP3 inflammasome activation, and the mechanism may correlate with the inhibition of Parkin-dependent mitotic activity [72]. There are some studies have confirmed that meisoindigo and anthocyanin from myrica rubra also can inhibit NLRP3 inflammasome activation and regulate microglia/macrophage polarization by regulating TLR4/NF-κB signaling pathway, thus playing a neuroprotective role in ischemic stroke and cerebral ischemia-reperfusion injury [73, 74].
Hemorrhagic stroke
Although the incidence of hemorrhagic stroke is lower than that of ischemic stroke, its symptoms are more severe, the rate of disability and mortality are higher. Intracerebral hemorrhage(ICH) is the most common subtype of hemorrhagic stroke, with a 28-day mortality rate as high as 47%, which is much higher than that of ischemic stroke (3%) [67]. The catastrophic outcome of intracerebral hemorrhage is the result of local brain parenchyma destruction caused by hematoma and the secondary brain injury caused by BBB dysfunction, brain edema around hematoma, nerve inflammation and so on [75]. It was found that NLRP3, caspase-1, and IL-1β levels in CD1mice microgliaa increased significantly at 3 h after ICH. Knocking down NLRP3 by siRNA could reduce IL-1 β level and neutrophil infiltration around the hematoma [76], indicating that NLRP3 is an important participant in neuroinflammation after ICH. Overactivated NLRP3 in choroid plexus epithelium after ICH can regulate cerebrospinal fluid secretion through ion cotransporter NKCC1 and participate in the occurrence and development of hydrocephalus after ICH. Knocking out NLRP3 or applying MCC950 can reduce the secretion of cerebrospinal fluid and alleviate the neurological deficit and hydrocephalus [77]. Activated M2 microglia can help hematoma clearance by phagocytosis of red blood cells and tissue fragments. MitoQ can inhibit NLRP3 activation by reducing the production of mitochondrial ROS, and promote the transformation of microglia to M2 phenotype, thus reducing brain edema and neuroinflammation after ICH, promoting hematoma clearance, and improving the prognosis of ICH experimental animals [78]. By inhibiting the activation of NLRP3 and partially maintaining the integrity of the BBB after ICH, OLT1177 can reduce vascular leakage, reduce neuronal loss and brain edema in the damaged hemisphere, and effectively alleviate neurological impairment [79]. Didymin can attenuate microglial apoptosis, neutrophil infiltration, brain edema and BBB damage after ICH by up-regulating Rkip to inhibit the assembly of NLRP3 inflammasome, and improve neurological impairment [80]. Ursolic acid can down-regulate NLRP3 expression by inhibiting NF- κ B phosphorylation, thus reducing BBB destruction, perihematoma edema and neuronal loss after ICH [81]. At the same time, some studies have noted that the number of Helicobacter pylori in the gut of mice increased after ICH. After intraperitoneal injection of MCC950, the proportion of bacteroides, bifidobacteria and Bacteroides in the gut of mice increased, while the Helicobacter pylori decreased, and the brain edema and neurological impairment around hematoma were improved [82]. Unfortunately, this study did not deeply explore the bidirectional regulation and causal relationship between intestinal microflora, NLRP3 inflammasome and cerebral hemorrhage after ICH. In addition, silymarin, atorvastatin, baicalein, mst4, h2s, a68930-induced activation of drd1, fimasartan, p2y6 receptor activation, histone deacetylation 10, verbascoside, verapamil, cordycepin, dexmedetomidine, andrographolide and others can directly or indirectly inhibit NLRP3 activation to reduce nerve inflammation, improve secondary brain injury after ICH, and play a neuroprotective role [83–96].
-
Subarachnoid hemorrhage (SAH) is the second most common subtype of hemorrhagic stroke. Early brain injury characterized by brain edema, delayed cerebral ischemia caused by cerebral vasospasm, and persistent neuroinflammation after SAH are considered to be associated with adverse outcomes [83]. It has been found that the expressions of NLRP3, ASC and active caspase-1 in monocytes of aneurysmal subarachnoid hemorrhage patients are significantly increased, and MCC950 can effectively block the release of IL-1 β, IL-18 and tissue factor mediated by excessive activation of NLRP3 inflammasome [84]. A number of experiments on animal models of SAH further confirmed that red blood cells dissolved in the subarachnoid space after SAH event, oxidative stress and endoplasmic reticulum stress triggered by hemolytic products, potassium efflux and extracellular accumulation of ATP mediated the assembly of NLRP3 inflammasome. Inhibition of NLRP3 activation can effectively improve early brain injury and delayed cerebral ischemia after SAH [85,86,87]. The relationship between the overactivation of NLRP3 and the adverse outcome of SAH has been fully verified. NLRP3 is undoubtedly a potential target for SAH therapy. Studies have confirmed that MCC950 can effectively improve brain edema, BBB destruction, neutrophil infiltration, microthrombosis, neurological dysfunction and delayed cerebral vasospasm after SAH by inhibiting NLRP3 activation to reduce the polarization of microglia [87]. Low-dose fimasartan can significantly reduce the activation of NLRP3/ASC/caspase-1/NF- κ B pathway after ICH, thus alleviating the cerebral edema and improving neurological function [88]. INT-777, a TGR5 agonist, can inhibit the NLRP3 activation through TGR5/cAMP/PKA signaling pathway, alleviate acute cerebral edema and neuroinflammation, and hippocampal neuronal degeneration 28 days after SAH [89]. Inhibition of NLRP3 activation by PHLDA1 can induce M2 polarization of microglia after SAH and improve neuroinflammation and neuronal apoptosis [90]. Takinib can reduce oxidative damage, neuroinflammation, brain edema and neuronal apoptosis after SAH by inhibiting TAK1-ROS-NLRP3 signaling pathway, and improve neurological deficit [91]. In addition, resveratrol, schisandrin B, fluoxetine, melatonin and pterostilbene have also been reported to play a protective role in SAH by inhibiting the activation of NLRP3 [92,93,94,95]. To sum up, targeting NLRP3 is a potential therapeutic strategy to improve the adverse outcome of SAH.
Degenerative diseases of the CNS
Alzheimer’s disease
Alzheimer’s disease (AD) is a neurodegenerative disease manifested by neurological degeneration and cognitive decline, resulting from the amyloid-β (Aβ) protein accumulation, Neurofibrillary tangles formed by hyperphosphorylated tau, and long-term persistent aseptic neuroinflammation, which is the first cause of dementia [96]. There is still no widely recognized drug for preventing or treating AD on the market, despite a great deal of effort has been put in it. It was found that amyloid-β protein, Aβ oligomers, and hyperphosphorylated tau can activate NLRP3 inflammasome [97, 98]. The activated NLRP3 inflammasome in microglia can promote IL-1β maturation, induce pyroptosis, and increase amyloid-β protein accumulation and tau protein phosphorylation [97, 99]. In conclusion, NLRP3 inflammasome plays an important role in the occurrence and development of AD. Blocking NLRP3 inflammasome assembly or specifically inhibiting its downstream inflammatory cytokine IL-1β is an effective strategy to reduce the deterioration of AD neuronal function. At present, NLRP3 inhibitors MCC590, JC124, OLT1177 ,CY-09, andbaicalin have shown therapeutic effects in AD animal models, effectively improving neurological degeneration and cognitive decline in AD mice, but their safety and therapeutic effect in AD patients have not been validated [100,101,102,103,104].
Parkinson’s disease
Parkinson’s disease (PD) is a neurodegenerative disease manifested by movement disorders, tremor and balance disorders. Its main pathological features are progressive loss of dopaminergic neurons in the substantia nigra pars compacta, Lewy bodies and Alpha-synuclein (α-Syn) accumulation, and excessive neuroinflammation [105]. Several studies have shown that accumulated α-Syn can activate the NLRP3 inflammasome. Zhou et al. found that the intracellular ROS production and lysosomal cathepsin B release increased after microglia ingested α-Syn, thereby activating NLRP3 inflammasome and promoting IL-1β and IL-18 maturation [106]. Panicker et al. found that α-Syn could not only activate NLRP3 but also induce NF-κB-p65 nuclear translocations to promote NLRP3 priming. In addition, they confirmed that α-Syn uptake by microglia was co-regulated by Fyn kinase and CD36, and α-Syn mediated NLRP3 priming and activation was also regulated by Fyn kinase. NLRP3 inflammasome assembly activates caspase-1, and the activated caspase-1 directly cleaves a-Syn to increase its aggregation tendency. syn-induced inflammasome priming and activation and inflammasome-mediated a-Syn truncation and aggregation form a vicious circle, thus eventually leading to neuronal death [107]. Therefore, inhibition of NLRP3 inflammasome assembly, caspase-1 activation and the release of downstream inflammatory cytokines are important means to protect neural function. In recent years, several studies have found that baicalein, andrographolide, berberine, Genkwanin, Dl-3-n-Butylphthalide, glibenclamide, UNC9995(Drd2 biased agonist), Dopamine, Prussian Blue Nanozyme, KPT-8602 (XPO1 Inhibitor), Cordycepin, long noncoding RNA (HOTTIP), Long noncoding RNA GAS5, Long-Noncoding RNA LncZFAS1 and others can improve the neurobehavioral function of Parkinson’s disease mice by inhibiting NLPR3 inflammasome activation, showing good efficacy [108,109,110,111,112,113,114,115,116,117,118,119]. However, there is still a long way from animal experiments to clinical applications.
Amyotrophic lateral sclerosis
Amyotrophic lateral sclerosis (ALS) is a kind of progressive motor neuron degenerative disease characterized by Skeletal muscle weakness and atrophy from degeneration of motor neurons in cortex, brainstem and spinal cord [120]. Its etiology is still unknown. About 10% of ALS cases can be attributed to genetic mutations, among which copper-zinc superoxide dismutase (SOD1) gene mutation is the first discovered and the most studied mutation [121]. Misfolding of specific proteins is at the heart of ALS. And the SOD1 mutant protein, a major component of some familial and sporadic ALS protein deposits, spreads its misfolded conformation like a prion [122, 123]. Johann et al. found that the expression of NLRP3 and ASC increased in astrocytes of SOD1G93AALS model mice and postmortem spinal cord tissues of ALS patients [35]. Deora et al. further discovered that endocytosis of aggregated or soluble SOD1G93A mutant protein in microglia of SOD1G93AALS model mice could increase ROS production, promote Caspase-1 and IL-1β cleavage, ASC spot formation and IL-1β secretion. Of these the aggregation SOD1 protein had a more significant promoting effect. The specific NLRP3 inhibitor MCC950 can block IL-1β secretion, proving that NLRP3 inflammasome plays a key role in SOD1 mutant protein-induced IL-1β secretion in microglia [124]. The existing treatments have not been effective in changing the trajectory of ALS and have only modestly improved survival. Therefore, inhibition of NLRP3 activation may be a potential way to slow down ALS proliferative neuroinflammatory cells death and ameliorate disease progression. Currently, It has been found that anti-inflammatory cyclic dipeptide His-pro and diphenyl diselenide can reduce protein nitrification and inhibit NLRP3 inflammasome activation by reducing NO and ROS levels, thus playing a neuroprotective role in ALS model [125, 126].
HIV associated neurocognitive disorders
Combination antiretroviral therapy is the main anti-HIV treatment at present. Nearly 50% of infected people in the world have received cART. Although cART is able to prolong survival. Nearly 50% of those infected continue to suffer from human immunodeficiency virus (HIV)-associated neurocognitive disorders (HANDs) such as learning, memory and executive dysfunction [127, 128]. Antiretroviral drug-related neurotoxicity, persistent low levels of viral replication in the CNS, viral proteins such as HIV-1envelope protein gp120 and proinflammatory cytokines are involved in HIV-1-mediated neurodegeneration [129,130,131]. Walsh et al. found ASC ectopia, caspase1 cleavage, and IL-1β release in human microglia exposed to HIV-1 gp120 increased. NLRP3 deficiency and elevated extracellular K+ inhibited HIV-1-mediated IL-1β release [132]. Similar to the aforementioned experimental result, Chivero et al. found that HIV-1 trans-activator of transcription protein can promote caspase1 cleavage and IL-1β release in rat glial cells, and blocking NLRP3 can reduce IL-1β secretion. This result was verified in brain tissue from SIV-infected monkeys [133]. Further studies have confirmed that gp120LAV could bind to CXCR4 of microglia after shedding from the virus surface during HIV-1 infection, thus promoting nuclear translocation of NF-κB P65 and K+ efflux. NF-κB activation could induce NLRP3priming, and K+ efflux could promote NLRP3 inflammasome assembly, pyroptosis and IL-1β release, ultimately leading to neuronal death. And MCC950 showed a good neuroprotective effect on gp120LAV-induced neuronal death and dysfunction [134]. Therefore, NLRP3 inflammasome activation may be a potential target for HIV/AIDS therapeutic intervention, and NLRP3 inhibitors, IL-1 and other downstream inflammatory cytokine receptor antagonists hold the promise of being therapeutic drugs to improve HANDs.
X-linked adrenoleukodystrophy
X-linked adrenoleukodystrophy (X-ALD) is a hereditary metabolic disease composed of the accumulation of very long-chain fatty acids in adrenal cortex, CNS and plasma induced by ATP- binding cassette subfamily D member 1 (ABCD1) gene mutation. Its pathogenesis is not yet clear [135]. Jang et al. found increased expression of cholesterol 25-hydroxylase in fibroblasts of X-ALD patients with ABCD1 dysfunction and brain homogenates of ABCD1-deficient mice, and further confirmed that 25-HC could promote the NLRP3 inflammasome and caspase-1 activation and IL-1β production through potassium efflux, mtROS production, mitochondrial damage, and liver X receptor, thus inducing microglia recruitment and oligodendrocyte death [136]. Knockdown of NLRP3 attenuates 25-HC-mediated neuroinflammation and plays a neuroprotective role. Therefore, NLRP3 may be a potential therapeutic target for X-ALD.
Glioma of the CNS
Glioma is the most common and fatal primary CNS tumor in adults. Surgery, chemoradiotherapy and other regimens commonly used at present can not significantly improve patient prognosis, quality of life and survival time [137, 138]. It was found that NLRP3, ASC, caspase-1 and IL-1β were over-expressed in human glioma tissues and significantly correlated with the World Health Organization classification. Down-regulation of NLRP3 could reduce the expression of ASC, caspase-1 and IL-1β, inhibit epithelial-mesenchymal transition and PTEN/AKT signaling pathway, thus inhibiting the proliferation, migration and invasion of glioma cell lines [139]. Xue et al. found that knockdown of NLRP3 inhibited the growth and invasion of glioma cells, and also decreased the expression of IL-1β and NF-κB p65. Silencing NLRP3 inhibited the proliferation and invasion of tumor cells promoted by exogenous IL-1β.Eliminating IL-1β attenuated the pro-proliferative effects of NLRP3 overexpression. Blocking NF-κB inhibited the overexpression of IL-1β and NLRP3 [140]. These results indicate that NLRP3 can regulate the IL-1β/NF-κB p65 signaling pathway to promote the growth and invasion of glioma. To sum up, inhibiting NLRP3 inflammasome activity and its signaling pathway can inhibit the growth and invasion of glioma cells. Ding et al. found that miR-223-3p expression was decreased, while NLRP3 was increased in glioma tissues. MiR-223-3p mimics could inhibit the proliferation and migration of tumor cells by down-regulating the expression of various inflammatory cytokines such as IL-1β, monocyte chemoattractant protein-1, IL-8 and IL-18. And up-regulating NLRP3 expression could attenuate the effect of mir-223-3p mimics [141]. Role of mir-223-3p in negative regulation of NLRP3-mediated inflammatory response makes it likely to be an inhibitory factor of glioma, slow down the disease process and prolong the survival time of patients.β-hydroxybutyrate was reported to inhibit the activation of NLRP3 inflammasome and the expression of caspase-1 and IL-1β in C6 glioma cells, thus inhibiting the migration of C6 cells [142]. IP-Se-06, a selenylated imidazo [1,2-a] pyridine, was also found to reduce NLRP3 and caspase-1 levels in A172 glioblastoma cells [143].
Epilepsy
Epilepsy is a common end-point of many brain disorders such as tumor, infection, immune abnormality, metabolic disorder and traumatic brain injury. It can be the result of a single gene mutation (hereditary epilepsy) or a component of neurodevelopmental disorder [144]. Epilepsy affects approximately 1% of the population of all ages worldwide, refractory epilepsy accounts for more than 30%. Patients with epilepsy often require long-term and regular medication, but refractory epilepsy cannot be well controlled by existing drugs [145]. Therefore, it is urgent to develop new drugs to treat epilepsy. At present, the cellular and molecular mechanisms of epilepsy are still unclear, but a large amount of clinical and experimental evidence indicates that CNS or systemic inflammatory disorders and their mediated neuronal damage, gliosis cell proliferation, abnormal neural connections and hyperexcitable neural networks play an important role in the development of epilepsy [146, 147]. Meng et al. demonstrated for the first time that NLRP3 and IL-1β levels increase after status epilepticus in rat hippocampus, blocking NLRP3 inflammasome assembly or Caspase-1 activation could reduce the serum IL-1β and IL-18 levels, the number and severity of seizures after SE, and slow down the disease progression. knocking out NLRP3 or caspase-1 at 6 weeks after SE could maintain neuronal activity and reduce neuronal loss in hippocampal CA1 and CA3 regions [148]. Wu et al. found that the NLRP3 level in the epileptogenic brain tissue of the children with intractable temporal lobe epilepsy was higher than that in the normal brain tissue adjacent to the epileptogenic zone, and the IL-1β level in peripheral blood was higher than that in healthy children, which was positively correlated with the onset time of single seizure [149]. Qin et al. demonstrated that GPR120 overexpression reduced neuronal death after SE through down-regulating NLRP3 inflammasome expression, and knocking down GPR120 showed the opposite effect [150]. Therefore, inhibiting the activity of NLRP3 inflammasome and its downstream inflammatory cytokines is essential to reduce the occurrence of epilepsy and control its progression. Rong et al. found that amentoflavone could inhibit pentylenetetrazol-induced NLRP3 inflammasome activation and the release of IL-18, IL-1β and TNF-α in mice hippocampus, thus reducing seizures and playing a neuroprotective role [151]. He et al. demonstrated that curcumin could inhibit NLPR3 inflammasome activation and reduce hippocampal neuron loss during epilepsy [152]. Moreover, ibuprofen, CY-09, MCC950, neferine, eicosapentaenoic acid and docosahexaenoic acid have been reported to inhibit NLRP3 inflammasome activation to reduce neuroinflammation, exert anti-epileptic and neuroprotective effects [153,154,155,156,157]. Traditional Chinese medicine such as Chaihu-Longgu-Muli decoction was also found to exert anti-epileptic effect by inhibiting nlrp3 activity [158].
The applied perspectives of NLRP3 inhibitors
To sum up, inhibition of NLRP3 inflammasome signaling pathway has shown good protective effect in a variety of animal models of CNS disease. NLRP3 is a potential therapeutic target for CNS disease. The development and clinical application of NLRP3 inflammasome and its downstream inflammatory cytokine inhibitors are of great value. Unfortunately, several inhibitors has become available show adverse reactions in clinical trials. The phase II clinical trials of MCC950 (specific NLRP3 inflammasome inhibitor) in the treatment of rheumatoid arthritis was suspended due to liver toxicity. The furan structure of MCC950 is considered to be one of the causes of drug-induced liver damage [159]. GDC-2394 is an oral selective NLRP3 inhibitor, which was first published in 2022. Because it removed furan structure extensively compared with MCC950, introduced basic amine into the scaffold to increase solubility and reduce nephrotoxicity, and successfully passed the safety study of cynomolgus monkeys. So researchers place high hopes that it will be able to complete clinical trials [160]. However, in a phase 1 clinical trial that evaluated the safety, pharmacokinetics and pharmacodynamics of GDC-2394 in healthy volunteers, two volunteers developed grade 4 drug-induced liver damage during the GDC-2394-midazolam interaction phase, so the trial was suspended. Indoleamine, a metabolite of GDC-2394, is considered to be associated with hepatotoxicity, and the concentration of indoleamine after GDC-2394 alone is the same as that after GDC-2394 combined with midazolam, so midazolam is not considered to be the perpetrator of liver damage [161]. However, this does not mean that NLRP3 inhibitors have no future. At present, more and more new inhibitors have been developed or gradually passed clinical trials.Elnoflast (RO7486967) is a reversible small molecule selective NLRP3 inhibitor. A randomized, double-blind phase 1b clinical trial confirmed that Selnoflast at a dose of 450 mg/day for 7 days was well tolerated without serious adverse reactions, and successfully reached the expected level of plasma and tissue exposure [162]. DFV890 is an oral selective NLRP3 inhibitor that can inhibit the activity of NLRP3 by directly binding to NLRP3 and locking it in an inactive conformation. A human safety, tolerance and pharmacokinetic experiment showed that 100 mg once a day or 25 mg twice a day of DFV890 was sufficient to stably inhibit 90% of IL-1 β release.10% of the subjects developed mild to moderate intensity of papules and / or itching rash at the dose of 100 mg QD or 30 mg bid.And the rash subsided spontaneously after discontinuation, without involving the epidermis, and no serious adverse reactions occurred. However, no subjects developed a rash at the dose of 25 mg bid, suggesting that the low dose DFV890 was well tolerated by healthy subjects and had no safety problems [163]. Dapansutrile (OLT1177) is an oral small molecule selective NLRP3 inhibitor, which can directly target NLRP3 by inhibiting ATPase activity. In the phase 1 study of OLT1177 in healthy subjects, no serious adverse reactions occurred, showing good safety and tolerance [164]. In a phase 2a clinical trial of gout patients, low\medium and high doses of OLT1177 significantly reduced the joint swelling and pain of the subjects, and almost no adverse events related to OLT1177 occurred [165]. In the phase 1B safety and pharmacodynamics study of NYHAII-III systolic heart failure patients, OLT1177 at a dose of 2000 mg/ day increased subjects’ the left ventricular ejection fraction from 31.5% (27.5–39) to 36.5% (27.5–45) (P = 0.039). And there were no drug-related water and sodium retention, renal function damage, heart rate and blood pressure abnormalities during the treatment [166]. In a word, the prospect of developing and applying NLRP3 inhibitors to treat CNS diseases is still bright.
Conclusion
NLRP3 inflammasome has long been a research hotspot, which has been proved to play an important role in the occurrence and development of infectious, inflammatory immune, vascular, genetic metabolic, tumor, epilepsy and other diseases in the CNS. Although there is mounting evidence that a variety of NLRP3 inhibitors show good efficacy in preclinical immunopathological models such as AD, TBI, atherosclerosis, diabetic encephalopathy, HANDs, cerebral edema, cerebral ischemia-reperfusion injury and ischemic hypoxic encephalopathy [1,2,3,4,5,6]. But the activation of inflammasome is crucial to the defense of pathogens, and the loss of IL-1β and other inflammatory cytokines may adversely affect the immune defense of the body, only specific NLRP3 inflammasome inhibitors have a chance of being used clinically. The side effects of MCC950 and others in clinical trials cast a shadow on its clinical application, but OLT1177 and others have successively completed phase 1 clinical trials, which makes us firmly believe that the prospect of targeting NLRP3 inflammasome in the treatment of CNS diseases is bright.With the deepening of research and the rapid development of molecular biology, the molecular principles of inflammasome assembly will be uncovered. NLRP3 is expected to be a potential target for the treatment of CNS diseases, and will play a greater role in the treatment of CNS diseases.
Data availability
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- CNS:
-
central nervous system
- PYD:
-
pyrin domain
- LRR domain:
-
leucine-rich-repeats domain
- HIV:
-
human immunodeficiency virus
- HANDs:
-
HIV-associated neurocognitive disorders
- NF:
-
κB: factor-κB
- POPs:
-
pyrin-only proteins
- COPs:
-
card-only proteins
- ZIKV:
-
Zika virus
- EV71:
-
enterovirus 71
- S. pneumoniae:
-
Streptococcus pneumoniae
- TBI:
-
traumatic brain injury
- NMDAR:
-
Anti-N-methyl-d-aspartate receptor
- GFAP:
-
autoimmune glial fibers acidic protein
- NMOSDs:
-
neuromyelitis optica spectrum disorders
- MS:
-
multiple sclerosis
- AD:
-
alzheimer’s disease
- Aβ:
-
amyloid-β
- PD:
-
parkinson’s disease
- ALS:
-
amyotrophic lateral sclerosis
- SOD1:
-
superoxide dismutase
- X-ALD:
-
X-linked adrenoleukodystrophy
- ABCD1:
-
ATP- binding cassette subfamily D member 1
- ICH:
-
Intracerebral hemorrhage
- SAH:
-
Subarachnoid hemorrhage
References
Swanson KV, Deng M, Ting JP. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol. 2019;19(8):477–89.
He H, Jiang H, Chen Y, et al. Oridonin is a covalent NLRP3 inhibitor with strong anti-inflammasome activity. Nat Commun. 2018;9(1):2550.
Ismael S, Nasoohi S, Ishrat T. MCC950, the selective inhibitor of Nucleotide Oligomerization Domain-Like receptor Protein-3 Inflammasome, protects mice against traumatic brain Injury. J Neurotrauma. 2018;35(11):1294–303.
Huang Y, Jiang H, Chen Y et al. Tranilast directly targets NLRP3 to treat inflammasome-driven diseases. EMBO Mol Med. 2018;10(4).
Zhai Y, Meng X, Ye T et al. Inhibiting the NLRP3 inflammasome activation with MCC950 ameliorates Diabetic Encephalopathy in db/db mice. Molecules. 2018;23(3).
Lahooti B, Chhibber T, Bagchi S, Varahachalam SP, Jayant RD. Therapeutic role of inflammasome inhibitors in neurodegenerative disorders. Brain Behav Immun. 2021;91:771–83.
Barry R, John SW, Liccardi G, et al. SUMO-mediated regulation of NLRP3 modulates inflammasome activity. Nat Commun. 2018;9(1):3001.
Niu T, De Rosny C, Chautard S, et al. NLRP3 phosphorylation in its LRR domain critically regulates inflammasome assembly. Nat Commun. 2021;12(1):5862.
Akther M, Haque ME, Park J, Kang TB, Lee KH. NLRP3 Ubiquitination-A New Approach to target NLRP3 inflammasome activation. Int J Mol Sci. 2021;22(16).
Bauernfeind FG, Horvath G, Stutz A, et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol. 2009;183(2):787–91.
Paik S, Kim JK, Silwal P, Sasakawa C, Jo EK. An update on the regulatory mechanisms of NLRP3 inflammasome activation. Cell Mol Immunol. 2021;18(5):1141–60.
Guo C, Chi Z, Jiang D, et al. Cholesterol homeostatic Regulator SCAP-SREBP2 integrates NLRP3 inflammasome activation and cholesterol Biosynthetic Signaling in macrophages. Immunity. 2018;49(5):842–e856847.
Zhong Z, Liang S, Sanchez-Lopez E, et al. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature. 2018;560(7717):198–203.
Subramanian N, Natarajan K, Clatworthy MR, Wang Z, Germain RN. The adaptor MAVS promotes NLRP3 mitochondrial localization and inflammasome activation. Cell. 2013;153(2):348–61.
Iyer SS, He Q, Janczy JR, et al. Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity. 2013;39(2):311–23.
Kayagaki N, Warming S, Lamkanfi M, et al. Non-canonical inflammasome activation targets caspase-11. Nature. 2011;479(7371):117–21.
Shi J, Zhao Y, Wang Y, et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature. 2014;514(7521):187–92.
Gaidt MM, Ebert TS, Chauhan D, et al. Hum Monocytes Engage Altern Inflammasome Pathw Immun. 2016;44(4):833–46.
Lin KM, Hu W, Troutman TD, et al. IRAK-1 bypasses priming and directly links TLRs to rapid NLRP3 inflammasome activation. Proc Natl Acad Sci U S A. 2014;111(2):775–80.
Freeman L, Guo H. NLR members NLRC4 and NLRP3 mediate sterile inflammasome activation in microglia and astrocytes. J Exp Med. 2017;214(5):1351–70.
Kalantari P, DeOliveira RB, Chan J, et al. Dual engagement of the NLRP3 and AIM2 inflammasomes by plasmodium-derived hemozoin and DNA during malaria. Cell Rep. 2014;6(1):196–210.
Karki R, Man SM, Malireddi RKS, et al. Concerted activation of the AIM2 and NLRP3 inflammasomes orchestrates host protection against aspergillus infection. Cell Host Microbe. 2015;17(3):357–68.
Davis BK, Roberts RA, Huang MT, et al. Cutting edge: NLRC5-dependent activation of the inflammasome. J Immunol. 2011;186(3):1333–7.
Schmid-Burgk JL, Chauhan D, Schmidt T, et al. A genome-wide CRISPR (clustered regularly interspaced short palindromic repeats) screen identifies NEK7 as an essential component of NLRP3 inflammasome activation. J Biol Chem. 2016;291(1):103–9.
He Y, Zeng MY, Yang D, Motro B, Núñez G. NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature. 2016;530(7590):354–7.
Shi H, Wang Y, Li X, et al. NLRP3 activation and mitosis are mutually exclusive events coordinated by NEK7, a new inflammasome component. Nat Immunol. 2016;17(3):250–8.
dos Santos G, Rogel MR, Baker MA, et al. Vimentin regulates activation of the NLRP3 inflammasome. Nat Commun. 2015;6:6574.
Lang T, Lee JPW, Elgass K, et al. Macrophage migration inhibitory factor is required for NLRP3 inflammasome activation. Nat Commun. 2018;9(1):2223.
Indramohan M, Stehlik C, Dorfleutner A. COPs and POPs Patrol Inflammasome activation. J Mol Biol. 2018;430(2):153–73.
Bedoya F, Sandler LL, Harton JA. Pyrin-only protein 2 modulates NF-kappaB and disrupts ASC:CLR interactions. J Immunol. 2007;178(6):3837–45.
Shi J, Zhao Y, Wang K, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526(7575):660–5.
Corcoran SE, Halai R, Cooper MA. Pharmacological inhibition of the nod-like receptor family pyrin domain containing 3 inflammasome with MCC950. Pharmacol Rev. 2021;73(3):968–1000.
Jiang H, He H, Chen Y, et al. Identification of a selective and direct NLRP3 inhibitor to treat inflammatory disorders. J Exp Med. 2017;214(11):3219–38.
Nagyoszi P, Nyul-Toth A, Fazakas C, et al. Regulation of NOD-like receptors and inflammasome activation in cerebral endothelial cells. J Neurochem. 2015;135(3):551–64.
Johann S, Heitzer M, Kanagaratnam M, et al. NLRP3 inflammasome is expressed by astrocytes in the SOD1 mouse model of ALS and in human sporadic ALS patients. Glia. 2015;63(12):2260–73.
Liu HD, Li W, Chen ZR, et al. Expression of the NLRP3 inflammasome in cerebral cortex after traumatic brain injury in a rat model. Neurochem Res. 2013;38(10):2072–83.
Gim E, Shim DW, Hwang I, Shin OS, Yu JW. Zika Virus impairs host NLRP3-mediated inflammasome activation in an NS3-dependent manner. Immune Netw. 2019;19(6):e40.
Wang W, Xiao F, Wan P, et al. EV71 3D protein binds with NLRP3 and enhances the Assembly of Inflammasome Complex. PLoS Pathog. 2017;13(1):e1006123.
Xiao HS, Xie Q, Zhong JY, et al. [Effect of vimentin on activation of NLRP3 inflammasome in the brain of mice with EV71 infection]. Nan Fang Yi Ke Da Xue Xue Bao. 2018;38(6):704–10.
Kaushik DK, Gupta M, Kumawat KL, Basu A. NLRP3 inflammasome: key mediator of neuroinflammation in murine Japanese encephalitis. PLoS ONE. 2012;7(2):e32270.
McNeela EA, Burke A, Neill DR, et al. Pneumolysin activates the NLRP3 inflammasome and promotes proinflammatory cytokines independently of TLR4. PLoS Pathog. 2010;6(11):e1001191.
Hoegen T, Tremel N, Klein M, et al. The NLRP3 inflammasome contributes to brain injury in pneumococcal meningitis and is activated through ATP-dependent lysosomal cathepsin B release. J Immunol. 2011;187(10):5440–51.
Kim JY, Paton JC, Briles DE, Rhee DK, Pyo S. Streptococcus pneumoniae induces pyroptosis through the regulation of autophagy in murine microglia. Oncotarget. 2015;6(42):44161–78.
Jin GN, Lu JM, Lan HW, et al. Protective effect of ginsenoside Rh2 against Toxoplasma gondii infection-induced neuronal injury through binding TgCDPK1 and NLRP3 to inhibit microglial NLRP3 inflammasome signaling pathway. Int Immunopharmacol. 2022;112:109176.
Menon DK, Schwab K, Wright DW, Maas AI. Position statement: definition of traumatic brain injury. Arch Phys Med Rehabil. 2010;91(11):1637–40.
Yi HJ, Lee JE, Lee DH, et al. The role of NLRP3 in traumatic brain injury and its regulation by pioglitazone. J Neurosurg. 2019;133(4):1083–91.
Xu X, Yin D, Ren H, et al. Selective NLRP3 inflammasome inhibitor reduces neuroinflammation and improves long-term neurological outcomes in a murine model of traumatic brain injury. Neurobiol Dis. 2018;117:15–27.
Ding W, Cai C, Zhu X, Wang J, Jiang Q. Parthenolide ameliorates neurological deficits and neuroinflammation in mice with traumatic brain injury by suppressing STAT3/NF-κB and inflammasome activation. Int Immunopharmacol. 2022;108:108913.
Ma J, Xiao W, Wang J, et al. Propofol inhibits NLRP3 inflammasome and attenuates Blast-Induced Traumatic Brain Injury in rats. Inflammation. 2016;39(6):2094–103.
Fan K, Ma J, Xiao W, et al. Mangiferin attenuates blast-induced traumatic brain injury via inhibiting NLRP3 inflammasome. Chem Biol Interact. 2017;271:15–23.
Cai L, Gong Q, Qi L, et al. ACT001 attenuates microglia-mediated neuroinflammation after traumatic brain injury via inhibiting AKT/NFκB/NLRP3 pathway. Cell Commun Signal. 2022;20(1):56.
Chen X, Ning Y, Wang B, et al. HET0016 inhibits neuronal pyroptosis in the immature brain post-TBI via the p38 MAPK signaling pathway. Neuropharmacology. 2023;239:109687.
Titulaer MJ, McCracken L, Gabilondo I, et al. Treatment and prognostic factors for long-term outcome in patients with anti-NMDA receptor encephalitis: an observational cohort study. Lancet Neurol. 2013;12(2):157–65.
Peng Y, Liu B, Pei S, et al. Higher CSF levels of NLRP3 inflammasome is Associated with poor prognosis of Anti-N-methyl-D-Aspartate receptor encephalitis. Front Immunol. 2019;10:905.
Luo Y, Yan W, Zhou Z, et al. Elevated levels of NLRP3 in Cerebrospinal Fluid of patients with autoimmune GFAP astrocytopathy. Front Neurol. 2019;10:1019.
Jarius S, Wildemann B, Paul F. Neuromyelitis Optica: clinical features, immunopathogenesis and treatment. Clin Exp Immunol. 2014;176(2):149–64.
Peng Y, Chen J, Dai Y, et al. NLRP3 level in cerebrospinal fluid of patients with neuromyelitis optica spectrum disorders: increased levels and association with disease severity. Mult Scler Relat Disord. 2019;39:101888.
Noseworthy JH, Lucchinetti C, Rodriguez M. Weinshenker B.G. multiple sclerosis. N Engl J Med. 2000;343(13):938–52.
Malhotra S, Costa C, Eixarch H, et al. NLRP3 inflammasome as prognostic factor and therapeutic target in primary progressive multiple sclerosis patients. Brain. 2020;143(5):1414–30.
Olcum M, Tastan B, Kiser C, Genc S, Genc K. Microglial NLRP3 inflammasome activation in multiple sclerosis. Adv Protein Chem Struct Biol. 2020;119:247–308.
Malhotra S, Río J, Urcelay E, et al. NLRP3 inflammasome is associated with the response to IFN-β in patients with multiple sclerosis. Brain. 2015;138(Pt 3):644–52.
Voet S, Mc Guire C, Hagemeyer N, et al. A20 critically controls microglia activation and inhibits inflammasome-dependent neuroinflammation. Nat Commun. 2018;9(1):2036.
Sánchez-Fernández A, Skouras DB, Dinarello CA, López-Vales R. OLT1177 (Dapansutrile), a selective NLRP3 inflammasome inhibitor, ameliorates experimental autoimmune Encephalomyelitis Pathogenesis. Front Immunol. 2019;10:2578.
Campbell BCV, Khatri P, Stroke. Lancet. 2020;396(10244):129–42.
Elmaleh-Sachs A, Schwartz JL, Bramante CT, et al. Obes Manage Adults: Rev Jama. 2023;330(20):2000–15.
Feigin VL, Nguyen G, Cercy K, et al. Global, Regional, and Country-Specific Lifetime risks of Stroke, 1990 and 2016. N Engl J Med. 2018;379(25):2429–37.
Tu WJ, Wang LD. China stroke surveillance report 2021. Mil Med Res. 2023;10(1):33.
Hong P, Li FX, Gu RN, et al. Inhibition of NLRP3 inflammasome ameliorates cerebral ischemia-reperfusion Injury in Diabetic mice. Neural Plast. 2018;2018:9163521.
Yang F, Wang Z, Wei X, et al. NLRP3 deficiency ameliorates neurovascular damage in experimental ischemic stroke. J Cereb Blood Flow Metab. 2014;34(4):660–7.
Hou Y, Wang Y, He Q, et al. Nrf2 inhibits NLRP3 inflammasome activation through regulating Trx1/TXNIP complex in cerebral ischemia reperfusion injury. Behav Brain Res. 2018;336:32–9.
An P, Xie J, Qiu S, et al. Hispidulin exhibits neuroprotective activities against cerebral ischemia reperfusion injury through suppressing NLRP3-mediated pyroptosis. Life Sci. 2019;232:116599.
He Q, Li Z, Meng C et al. Parkin-dependent Mitophagy is required for the inhibition of ATF4 on NLRP3 inflammasome activation in Cerebral Ischemia-Reperfusion Injury in rats. Cells. 2019;8(8).
Ye Y, Jin T, Zhang X, et al. Meisoindigo Protects against Focal Cerebral Ischemia-Reperfusion Injury by inhibiting NLRP3 inflammasome activation and regulating Microglia/Macrophage polarization via TLR4/NF-κB signaling pathway. Front Cell Neurosci. 2019;13:553.
Cui HX, Chen JH, Li JW, Cheng FR, Yuan K. Protection of Anthocyanin from Myrica rubra against cerebral ischemia-reperfusion Injury via Modulation of the TLR4/NF-κB and NLRP3 pathways. Molecules. 2018;23(7).
Puy L, Parry-Jones AR, Sandset EC, et al. Intracerebral haemorrhage. Nat Rev Dis Primers. 2023;9(1):14.
Ma Q, Chen S, Hu Q, et al. NLRP3 inflammasome contributes to inflammation after intracerebral hemorrhage. Ann Neurol. 2014;75(2):209–19.
Zhang Z, Tan Q, Guo P, et al. NLRP3 inflammasome-mediated choroid plexus hypersecretion contributes to hydrocephalus after intraventricular hemorrhage via phosphorylated NKCC1 channels. J Neuroinflammation. 2022;19(1):163.
Chen W, Guo C, Huang S, et al. MitoQ attenuates brain damage by polarizing microglia towards the M2 phenotype through inhibition of the NLRP3 inflammasome after ICH. Pharmacol Res. 2020;161:105122.
Fang M, Xia F, Wang J, et al. The NLRP3 inhibitor, OLT1177 attenuates brain injury in experimental intracerebral hemorrhage. Int Immunopharmacol. 2024;131:111869.
Gu L, Sun M, Li R, et al. Didymin suppresses Microglia pyroptosis and Neuroinflammation through the Asc/Caspase-1/GSDMD pathway following experimental Intracerebral Hemorrhage. Front Immunol. 2022;13:810582.
Lei P, Li Z, Hua Q et al. Ursolic Acid alleviates Neuroinflammation after Intracerebral Hemorrhage by Mediating Microglial pyroptosis via the NF-κB/NLRP3/GSDMD pathway. Int J Mol Sci. 2023;24(19).
Xiao L, Zheng H, Li J, et al. Targeting NLRP3 inflammasome modulates gut microbiota, attenuates corticospinal tract injury and ameliorates neurobehavioral deficits after intracerebral hemorrhage in mice. Biomed Pharmacother. 2022;149:112797.
Claassen J, Park S. Spontaneous subarachnoid haemorrhage. Lancet. 2022;400(10355):846–62.
Díaz-García E, Nanwani-Nanwani K, García-Tovar S, et al. NLRP3 Inflammasome Overactivation in patients with Aneurysmal Subarachnoid Hemorrhage. Transl Stroke Res. 2023;14(3):334–46.
Luo Y, Lu J, Ruan W, Guo X, Chen S. MCC950 attenuated early brain injury by suppressing NLRP3 inflammasome after experimental SAH in rats. Brain Res Bull. 2019;146:320–6.
Liu C, Yao K, Tian Q, et al. CXCR4-BTK axis mediate pyroptosis and lipid peroxidation in early brain injury after subarachnoid hemorrhage via NLRP3 inflammasome and NF-κB pathway. Redox Biol. 2023;68:102960.
Dodd WS, Noda I, Martinez M, Hosaka K, Hoh BL. NLRP3 inhibition attenuates early brain injury and delayed cerebral vasospasm after subarachnoid hemorrhage. J Neuroinflammation. 2021;18(1):163.
Yang X, Sun J, Kim TJ, et al. Pretreatment with low-dose fimasartan ameliorates NLRP3 inflammasome-mediated neuroinflammation and brain injury after intracerebral hemorrhage. Exp Neurol. 2018;310:22–32.
Hu X, Yan J, Huang L, et al. INT-777 attenuates NLRP3-ASC inflammasome-mediated neuroinflammation via TGR5/cAMP/PKA signaling pathway after subarachnoid hemorrhage in rats. Brain Behav Immun. 2021;91:587–600.
Lai J, Chen G, Wu Z, et al. PHLDA1 modulates microglial response and NLRP3 inflammasome signaling following experimental subarachnoid hemorrhage. Front Immunol. 2023;14:1105973.
Wang W, Pang C, Zhang J, et al. Takinib inhibits microglial M1 polarization and oxidative damage after subarachnoid hemorrhage by targeting TAK1-dependent NLRP3 inflammasome signaling pathway. Front Immunol. 2023;14:1266315.
Liu H, Zhao L, Yue L, et al. Pterostilbene attenuates early brain Injury following subarachnoid hemorrhage via inhibition of the NLRP3 inflammasome and Nox2-Related oxidative stress. Mol Neurobiol. 2017;54(8):5928–40.
Li JR, Xu HZ, Nie S, et al. Fluoxetine-enhanced autophagy ameliorates early brain injury via inhibition of NLRP3 inflammasome activation following subrachnoid hemorrhage in rats. J Neuroinflammation. 2017;14(1):186.
Chen S, Ding YH, Shi SS, Tu XK. Schisandrin B inhibits NLRP3 inflammasome pathway and attenuates early Brain Injury in rats of subarachnoid hemorrhage. Chin J Integr Med. 2022;28(7):594–602.
Zhang X, Wu Q, Zhang Q, et al. Resveratrol attenuates early Brain Injury after experimental subarachnoid hemorrhage via inhibition of NLRP3 inflammasome activation. Front Neurosci. 2017;11:611.
Ising C, Heneka MT. Functional and structural damage of neurons by innate immune mechanisms during neurodegeneration. Cell Death Dis. 2018;9(2):120.
Stancu IC, Cremers N, Vanrusselt H, et al. Aggregated tau activates NLRP3-ASC inflammasome exacerbating exogenously seeded and non-exogenously seeded tau pathology in vivo. Acta Neuropathol. 2019;137(4):599–617.
Halle A, Hornung V, Petzold GC, et al. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol. 2008;9(8):857–65.
Heneka MT, McManus RM, Latz E. Inflammasome signalling in brain function and neurodegenerative disease. Nat Rev Neurosci. 2018;19(10):610–21.
Kuwar R, Rolfe A, Di L, et al. A novel inhibitor targeting NLRP3 inflammasome reduces neuropathology and improves cognitive function in Alzheimer’s Disease Transgenic mice. J Alzheimers Dis. 2021;82(4):1769–83.
Li JM, Hu T, Zhou XN, et al. The involvement of NLRP3 inflammasome in CUMS-induced AD-like pathological changes and related cognitive decline in mice. J Neuroinflammation. 2023;20(1):112.
Naeem A, Prakash R, Kumari N, et al. MCC950 reduces autophagy and improves cognitive function by inhibiting NLRP3-dependent neuroinflammation in a rat model of Alzheimer’s disease. Brain Behav Immun. 2024;116:70–84.
Jin X, Liu MY, Zhang DF, et al. Baicalin mitigates cognitive impairment and protects neurons from microglia-mediated neuroinflammation via suppressing NLRP3 inflammasomes and TLR4/NF-κB signaling pathway. CNS Neurosci Ther. 2019;25(5):575–90.
Han S, He Z, Hu X et al. Inhibiting NLRP3 inflammasome activation by CY-09 helps to restore cerebral glucose metabolism in 3×Tg-AD mice. Antioxid (Basel). 2023;12(3).
Gasser T. Molecular pathogenesis of Parkinson disease: insights from genetic studies. Expert Rev Mol Med. 2009;11:e22.
Zhou Y, Lu M, Du RH, et al. MicroRNA-7 targets nod-like receptor protein 3 inflammasome to modulate neuroinflammation in the pathogenesis of Parkinson’s disease. Mol Neurodegener. 2016;11:28.
Wang W, Nguyen LT, Burlak C, et al. Caspase-1 causes truncation and aggregation of the Parkinson’s disease-associated protein α-synuclein. Proc Natl Acad Sci U S A. 2016;113(34):9587–92.
Xu W, Zhang L, Geng Y, Liu Y, Zhang N. Long noncoding RNA GAS5 promotes microglial inflammatory response in Parkinson’s disease by regulating NLRP3 pathway through sponging miR-223-3p. Int Immunopharmacol. 2020;85:106614.
Lun P, Ji T, Wan DH, et al. HOTTIP downregulation reduces neuronal damage and microglial activation in Parkinson’s disease cell and mouse models. Neural Regen Res. 2022;17(4):887–97.
Liu S, Wang S, Gu R, et al. The XPO1 inhibitor KPT-8602 ameliorates Parkinson’s disease by inhibiting the NF-κB/NLRP3 pathway. Front Pharmacol. 2022;13:847605.
Ma X, Hao J, Wu J, et al. Prussian Blue Nanozyme as a pyroptosis inhibitor alleviates neurodegeneration. Adv Mater. 2022;34(15):e2106723.
Pike AF, Longhena F, Faustini G, et al. Dopamine signaling modulates microglial NLRP3 inflammasome activation: implications for Parkinson’s disease. J Neuroinflammation. 2022;19(1):50.
Zhu J, Sun T, Zhang J, et al. Drd2 biased agonist prevents neurodegeneration against NLRP3 inflammasome in Parkinson’s disease model via a β-arrestin2-biased mechanism. Brain Behav Immun. 2020;90:259–71.
Qiu X, Wang Q, Hou L, et al. Inhibition of NLRP3 inflammasome by glibenclamide attenuated dopaminergic neurodegeneration and motor deficits in paraquat and maneb-induced mouse Parkinson’s disease model. Toxicol Lett. 2021;349:1–11.
Rui W, Li S, Xiao H, Xiao M, Shi J. Baicalein attenuates neuroinflammation by inhibiting NLRP3/caspase-1/GSDMD pathway in MPTP Induced mice Model of Parkinson’s Disease. Int J Neuropsychopharmacol. 2020;23(11):762–73.
Ahmed S, Kwatra M, Ranjan Panda S, Murty USN, Naidu VGM. Andrographolide suppresses NLRP3 inflammasome activation in microglia through induction of parkin-mediated mitophagy in in-vitro and in-vivo models of Parkinson disease. Brain Behav Immun. 2021;91:142–58.
Huang S, Liu H, Lin Y, et al. Berberine protects against NLRP3 inflammasome via ameliorating autophagic impairment in MPTP-Induced Parkinson’s Disease Model. Front Pharmacol. 2020;11:618787.
Li Q, Zhang P, Cai Y. Genkwanin suppresses MPP(+)-induced cytotoxicity by inhibiting TLR4/MyD88/NLRP3 inflammasome pathway in a cellular model of Parkinson’s disease. Neurotoxicology. 2021;87:62–9.
Que R, Zheng J, Chang Z, et al. Dl-3-n-Butylphthalide rescues dopaminergic neurons in Parkinson’s Disease models by inhibiting the NLRP3 inflammasome and ameliorating mitochondrial impairment. Front Immunol. 2021;12:794770.
Paez-Colasante X, Figueroa-Romero C, Sakowski SA, Goutman SA, Feldman EL. Amyotrophic lateral sclerosis: mechanisms and therapeutics in the epigenomic era. Nat Rev Neurol. 2015;11(5):266–79.
Rosen DR, Siddique T, Patterson D, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362(6415):59–62.
Sibilla C, Bertolotti A. Prion properties of SOD1 in amyotrophic lateral sclerosis and potential therapy. Cold Spring Harb Perspect Biol. 2017;9(10).
Wang J, Farr GW, Zeiss CJ, et al. Progressive aggregation despite chaperone associations of a mutant SOD1-YFP in transgenic mice that develop ALS. Proc Natl Acad Sci U S A. 2009;106(5):1392–7.
Deora V, Lee JD, Albornoz EA, et al. The microglial NLRP3 inflammasome is activated by amyotrophic lateral sclerosis proteins. Glia. 2020;68(2):407–21.
Grottelli S, Mezzasoma L, Scarpelli P, et al. Cyclo(His-Pro) inhibits NLRP3 inflammasome cascade in ALS microglial cells. Mol Cell Neurosci. 2019;94:23–31.
Zhang C, Wang H, Liang W, et al. Diphenyl diselenide protects motor neurons through inhibition of microglia-mediated inflammatory injury in amyotrophic lateral sclerosis. Pharmacol Res. 2021;165:105457.
McArthur JC, Steiner J, Sacktor N, Nath A. Human immunodeficiency virus-associated neurocognitive disorders: mind the gap. Ann Neurol. 2010;67(6):699–714.
Saylor D, Dickens AM, Sacktor N, et al. HIV-associated neurocognitive disorder–pathogenesis and prospects for treatment. Nat Rev Neurol. 2016;12(4):234–48.
Carroll A, Brew B. HIV-associated neurocognitive disorders: recent advances in pathogenesis, biomarkers, and treatment. F1000Res. 2017;6:312.
Jayadev S, Garden GA. Host and viral factors influencing the pathogenesis of HIV-associated neurocognitive disorders. J Neuroimmune Pharmacol. 2009;4(2):175–89.
Vassallo M, Dunais B, Durant J, et al. Relevance of lipopolysaccharide levels in HIV-associated neurocognitive impairment: the neuradapt study. J Neurovirol. 2013;19(4):376–82.
Walsh JG, Reinke SN, Mamik MK, et al. Rapid inflammasome activation in microglia contributes to brain disease in HIV/AIDS. Retrovirology. 2014;11:35.
Chivero ET, Guo ML, Periyasamy P, et al. HIV-1 Tat Primes and activates microglial NLRP3 inflammasome-mediated neuroinflammation. J Neurosci. 2017;37(13):3599–609.
He X, Yang W, Zeng Z, et al. NLRP3-dependent pyroptosis is required for HIV-1 gp120-induced neuropathology. Cell Mol Immunol. 2020;17(3):283–99.
Mosser J, Douar AM, Sarde CO, et al. Putative X-linked adrenoleukodystrophy gene shares unexpected homology with ABC transporters. Nature. 1993;361(6414):726–30.
Jang J, Park S, Jin Hur H, et al. 25-hydroxycholesterol contributes to cerebral inflammation of X-linked adrenoleukodystrophy through activation of the NLRP3 inflammasome. Nat Commun. 2016;7:13129.
Wen PY, Reardon DA. Neuro-oncology in 2015: progress in glioma diagnosis, classification and treatment. Nat Rev Neurol. 2016;12(2):69–70.
Mauer M, Stupp R, Taphoorn MJ, et al. The prognostic value of health-related quality-of-life data in predicting survival in glioblastoma cancer patients: results from an international randomised phase III EORTC brain Tumour and Radiation Oncology groups, and NCIC Clinical Trials Group study. Br J Cancer. 2007;97(3):302–7.
Yin XF, Zhang Q, Chen ZY, et al. NLRP3 in human glioma is correlated with increased WHO grade, and regulates cellular proliferation, apoptosis and metastasis via epithelial-mesenchymal transition and the PTEN/AKT signaling pathway. Int J Oncol. 2018;53(3):973–86.
Xue L, Lu B, Gao B, et al. NLRP3 promotes Glioma Cell Proliferation and Invasion via the Interleukin-1β/NF-κB p65 signals. Oncol Res. 2019;27(5):557–64.
Ding Q, Shen L, Nie X, et al. MiR-223-3p overexpression inhibits cell proliferation and migration by regulating inflammation-associated cytokines in glioblastomas. Pathol Res Pract. 2018;214(9):1330–9.
Shang S, Wang L, Zhang Y, Lu H, Lu X. The Beta-hydroxybutyrate suppresses the Migration of Glioma Cells by inhibition of NLRP3 inflammasome. Cell Mol Neurobiol. 2018;38(8):1479–89.
Dos Santos DC, Rafique J, Saba S, et al. IP-Se-06, a Selenylated Imidazo[1,2-a]pyridine, Modulates Intracellular Redox State and causes Akt/mTOR/HIF-1α and MAPK Signaling Inhibition, promoting Antiproliferative Effect and apoptosis in Glioblastoma Cells. Oxid Med Cell Longev. 2022;2022:3710449.
Goldberg EM, Coulter DA. Mechanisms of epileptogenesis: a convergence on neural circuit dysfunction. Nat Rev Neurosci. 2013;14(5):337–49.
Hauser RM, Henshall DC, Lubin FD. The epigenetics of Epilepsy and its progression. Neuroscientist. 2018;24(2):186–200.
Rana A, Musto AE. The role of inflammation in the development of epilepsy. J Neuroinflammation. 2018;15(1):144.
Vezzani A, Fujinami RS, White HS, et al. Infections, inflammation and epilepsy. Acta Neuropathol. 2016;131(2):211–34.
Meng XF, Tan L, Tan MS, et al. Inhibition of the NLRP3 inflammasome provides neuroprotection in rats following amygdala kindling-induced status epilepticus. J Neuroinflammation. 2014;11:212.
Wu C, Zhang G, Chen L, et al. The role of NLRP3 and IL-1β in Refractory Epilepsy Brain Injury. Front Neurol. 2019;10:1418.
Qin Z, Song J, Lin A, et al. GPR120 modulates epileptic seizure and neuroinflammation mediated by NLRP3 inflammasome. J Neuroinflammation. 2022;19(1):121.
Rong S, Wan D, Fan Y, et al. Amentoflavone affects epileptogenesis and exerts neuroprotective effects by inhibiting NLRP3 inflammasome. Front Pharmacol. 2019;10:856.
He Q, Jiang L, Man S, et al. Curcumin reduces neuronal loss and inhibits the NLRP3 inflammasome activation in an epileptic rat model. Curr Neurovasc Res. 2018;15(3):186–92.
Liu R, Wu S, Guo C, et al. Ibuprofen exerts antiepileptic and neuroprotective effects in the rat model of Pentylenetetrazol-Induced Epilepsy via the COX-2/NLRP3/IL-18 pathway. Neurochem Res. 2020;45(10):2516–26.
Lin TY, Hung CY, Chiu KM et al. Neferine, an Alkaloid from Lotus seed embryos, exerts antiseizure and neuroprotective effects in a Kainic Acid-Induced Seizure Model in rats. Int J Mol Sci. 2022;23(8).
Wang X, Xiao A, Yang Y, et al. DHA and EPA prevent seizure and Depression-Like Behavior by inhibiting Ferroptosis and neuroinflammation via different Mode-of-actions in a Pentylenetetrazole-Induced Kindling Model in mice. Mol Nutr Food Res. 2022. https://doi.org/10.1002/mnfr.202200275. e2200275.
Hong Y, Wei C, Fu M, et al. MCC950 alleviates seizure severity and angiogenesis by inhibiting NLRP3/ IL-1β signaling pathway-mediated pyroptosis in mouse model of epilepsy. Int Immunopharmacol. 2024;126:111236.
Shen K, Jiang W, Zhang C, et al. Molecular mechanism of a specific NLRP3 inhibitor to Alleviate Seizure Severity Induced by Pentylenetetrazole. Curr Mol Pharmacol. 2021;14(4):579–86.
Xia S, Yang P, Li F, et al. Chaihu-Longgu-Muli Decoction exerts an antiepileptic effect in rats by improving pyroptosis in hippocampal neurons. J Ethnopharmacol. 2021;270:113794.
Mangan MSJ, Olhava EJ, Roush WR, et al. Targeting the NLRP3 inflammasome in inflammatory diseases. Nat Rev Drug Discov. 2018;17(8):588–606.
McBride C, Trzoss L, Povero D, et al. Overcoming Preclinical Safety obstacles to Discover (S)-N-((1,2,3,5,6,7-Hexahydro-s-indacen-4-yl)carbamoyl)-6-(methylamino)-6,7-dihydro-5H-pyrazolo[5,1-b][1,3]oxazine-3-sulfonamide (GDC-2394): a potent and selective NLRP3 inhibitor. J Med Chem. 2022;65(21):14721–39.
Tang F, Kunder R, Chu T, et al. First-in-human phase 1 trial evaluating safety, pharmacokinetics, and pharmacodynamics of NLRP3 inflammasome inhibitor, GDC-2394, in healthy volunteers. Clin Transl Sci. 2023;16(9):1653–66.
Klughammer B, Piali L, Nica A, et al. A randomized, double-blind phase 1b study evaluating the safety, tolerability, pharmacokinetics and pharmacodynamics of the NLRP3 inhibitor selnoflast in patients with moderate to severe active ulcerative colitis. Clin Transl Med. 2023;13(11):e1471.
Gatlik E, Mehes B, Voltz E, et al. First-in-human safety, tolerability, and pharmacokinetic results of DFV890, an oral low-molecular-weight NLRP3 inhibitor. Clin Transl Sci. 2024;17(5):e13789.
Marchetti C, Swartzwelter B, Gamboni F, et al. OLT1177, a β-sulfonyl nitrile compound, safe in humans, inhibits the NLRP3 inflammasome and reverses the metabolic cost of inflammation. Proc Natl Acad Sci U S A. 2018;115(7):E1530–9.
Klück V, Jansen T, Janssen M, et al. Dapansutrile, an oral selective NLRP3 inflammasome inhibitor, for treatment of gout flares: an open-label, dose-adaptive, proof-of-concept, phase 2a trial. Lancet Rheumatol. 2020;2(5):e270–80.
Wohlford GF, Van Tassell BW, Billingsley HE, et al. Phase 1B, Randomized, Double-Blinded, dose escalation, Single-Center, repeat Dose Safety and Pharmacodynamics Study of the oral NLRP3 inhibitor dapansutrile in subjects with NYHA II-III systolic heart failure. J Cardiovasc Pharmacol. 2020;77(1):49–60.
Amo-Aparicio J, Daly J, Højen JF, Dinarello CA. Pharmacologic inhibition of NLRP3 reduces the levels of α-synuclein and protects dopaminergic neurons in a model of Parkinson’s disease. J Neuroinflammation. 2023;20(1):147.
Zhao XJ, Zhu HY, Wang XL, et al. Oridonin ameliorates traumatic Brain Injury-Induced neurological damage by improving mitochondrial function and antioxidant capacity and suppressing neuroinflammation through the Nrf2 pathway. J Neurotrauma. 2022;39(7–8):530–43.
Wang X, Xiao A, Yang Y, et al. DHA and EPA prevent seizure and Depression-Like Behavior by inhibiting Ferroptosis and neuroinflammation via different Mode-of-actions in a Pentylenetetrazole-Induced Kindling Model in mice. Mol Nutr Food Res. 2022;66(22):e2200275.
Acknowledgements
We sincerely appreciate the financial support provided by the China NationalNatural Scientific Foundation grants (No: 81873762), Youth Fund Project of Hunan Natural Science Foundation(2022JJ40699), Science and Technology Department of Hunan Province Funds (No: 2022SK2032, 2018SK2069 and 2023SK4018).
Funding
This project was supported by National Natural Science Foundation of China (No: 81873762), Science and Technology Department of Hunan Province Funds (No: 2022SK2032 and 2023SK4018), Provincial Natural Science Foundation of Hunan (2022JJ40699 and 2024JJ8249).
Author information
Authors and Affiliations
Contributions
LQL and LZ conceived the idea of this review. LZ wrote and edited this manuscript and created figures and Tables. YFT, PH, SLL, ZS, HP, YQC, JWL and WXD reviewed and revised the manuscript. LQL, JX and LJL provided direction and guidance throughout the preparation of the manuscript. All authors critically reviewed and edited the content of this manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhang, L., Tang, Y., Huang, P. et al. Role of NLRP3 inflammasome in central nervous system diseases. Cell Biosci 14, 75 (2024). https://doi.org/10.1186/s13578-024-01256-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13578-024-01256-y