
Abstract
Unwarranted exposure due to liberal use of metals for maintaining the lavish life and to achieve the food demand for escalating population along with an incredible boost in the average human life span owing to orchestrated progress in rejuvenation therapy have gradually increased the occurrence of Parkinson’s disease (PD). Etiology is albeit elusive; association of PD with metal accumulation has never been overlooked due to noteworthy similitude between metal-exposure symptoms and a few cardinal features of disease. Even though metals are entailed in the vital functions, a hysterical shift, primarily augmentation, escorts the stern nigrostriatal dopaminergic neurodegeneration. An increase in the passage of metals through the blood brain barrier and impaired metabolic activity and elimination system could lead to metal accumulation in the brain, which eventually makes dopaminergic neurons quite susceptible. In the present article, an update on implication of metal accumulation in PD/Parkinsonism has been provided. Moreover, encouraging and paradoxical facts and fictions associated with metal accumulation in PD/Parkinsonism have also been compiled. Systematic literature survey of PD is performed to describe updated information if metal accumulation is an epicenter or merely an outcome. Finally, a perspective on the association of metal accumulation with pesticide-induced Parkinsonism has been explained to unveil the likely impact of the former in the latter.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Metals are indispensable component of several biologically active proteins, which play decisive roles in the metabolic activity of the central nervous system [1]. Although deficiency of metals is associated with a few behavioral and phenotypic abnormalities, accumulation is highly toxic to the central nervous system. Overexpression and aggregation of α-synuclein, oxidative stress, mitochondrial dysfunction, inflammation, and impaired class I and class II programmed cell death are major wrongdoers of Parkinson’s disease (PD) and metals are known to regulate such biological processes [1–6]. Accumulation of metals in the striatum and substantia nigra, two terribly affected areas of the brain in PD patients, is widely reported [1, 7]. Metals are reported to increase the defenselessness in experimental rodents and are associated with increased incidences of PD [8, 9, 20]. Imaging techniques have also demonstrated an accumulation of high level of metals in a toxin model of PD [10]. Like pesticides, natal metal exposure could exert lifelong effect in rodents owing to imprinting episode that intensifies the prevalence and severity of Parkinsonism upon adulthood [11, 12]. Oxidative stress is also contributed by metal accumulation. Naturally occurring agents encounter metal accumulation-mediated oxidative stress. Silymarin, resveratrol, melatonin, and their metabolites provide protection owing to their antioxidant, free radical scavenging and metal chelating properties [13–15] also showing the role of metal accumulation in PD.
Metal accumulation theory possibly came into the glare of publicity after emergence of the information showing incidences of gradual increase in the deposition of redox-reactive essential metals in the brain of PD patients [2, 11]. Perhaps, the theory got impetus after the development of rodent models employing individual, dual, or multiple metal combinations [1, 5]. Ample support to metal accumulation theory has come from adherence of inverse relationship between metal accumulation in PD brain and ameliorative strategies used to encounter it. Agents that encounter, scavenge, remediate, or reduce free radicals are found to lessen metal accumulation in the brain further strengthen the idea of metal accumulation theory [1, 8, 11, 16]. Furthermore, chelating agents and metal binding modulators, which reduce the severity of symptomatic disease features, also corroborate metal accumulation belief [17, 18].
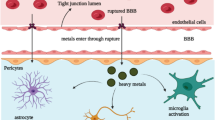
Unforeseen metal accumulation possibly happens when people are exposed to metal-rich pollutants, diets, and therapies [2, 11, 19, 20]. Buildup in dopaminergic neurons is also feasible, if there is disturbance in the metabolism, uptake, removal, and transport of metals or nutritional deprivation [11, 19, 21]. Despite a few confounding observations, epidemiological and experimental investigations unambiguously discourse that metal accumulation leads to phenotypic anomalies mimicking a few fundamental characteristics of PD [1, 2, 8, 9, 20]. Accumulation in the brain is regulated by the blood brain barrier (BBB) depending on the nature of metals and their ability to cross it. Once a metal enters the brain through specific transporters, it regulates the functional activity of pertinent enzyme. Therefore, metal accumulation is expected to alter the metabolic fate of dopaminergic neurons that are located in the substantia nigra and projected to the striatum. While timely elimination regulates the entry and deposition, unfortunately, the elimination system is not yet explicitly understood.
The growing evidence, which proves the role of metals in sporadic and rodent models of PD, impelled to hoard and discuss updated information in the current article. Three schools of thought prevail in the scientific arena about metal accumulation and PD. The followers of the first thought firmly accept that metal accumulation is an epicenter and believe that accumulation is a prerequisite of PD onset [9, 22]. The second camp deems that PD progression leads to metal accumulation, which is an outcome, and therefore, the concept of metal accumulation leading to PD could not be true [22]. Another thought, albeit with less supporters, disapproves any positive or negative impact of metal accumulation per se in PD. In the current article, we explicitly emphasized and scrutinized the major perspectives of metal accumulation and its relevance in PD pathogenesis. Literature testimonies on evaluated metal content in PD and toxin(s)-induced Parkinsonism have been discussed to pinpoint the merits of metal accumulation theory in the primary (sporadic/idiopathic) and secondary (causative factor(s)-induced) PD. Moreover, an attempt has been made to assess, if metal accumulation theory bears a clear-cut proof or an abstemious flaw or is simply a propagated myth. Finally, a perspective is presented to show how an evaluation of metal accumulation in the nigrostriatal region of the brain of pesticides-induced Parkinsonism could help in validating or ruling out the hypothesis “metal buildup is a prerequisite for PD pathogenesis”.
Iron Accumulation and PD
Iron deficiency is linked with diverse anomalies and clinical syndromes, such as anemia and respiratory problem. Unlike a few metals to be discussed in the latter part of the article, iron deficiency is not at all associated with the increased incidences of PD, but rather buildup is associated with neurodegeneration. Postmortem brain of patients and genetic and toxin models of PD have indicated noteworthy and selective accumulation of iron in the substantia nigra region suggesting a link of iron accumulation with PD risk [23–27]. Despite an age-dependent increase in the iron content in the adjacent tissues, buildup of iron in the substantia nigra is projected to be the main causative factor for the selective demise of dopaminergic neurons [24]. Multiple observations from in vivo iron imaging techniques, association studies of iron accumulation regulating genes, and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), rotenone, 6-hydroxydopamine (6-OHDA) and lipopolysaccharide-based models have supported the decisive role of iron in PD pathogenesis [10, 23, 25, 27–33]. Mutants of regulatory genes and their link with iron accumulation or iron supplementation and exposure-dependent animal experimentations and their association with disease susceptibility or increased vulnerability in adults, which were prior exposed to iron during critical period of development altogether narrate the magnitude of accumulation [11, 23, 28, 30–32, 34, 35]. Accumulation is also associated with non-depression-linked PD demonstrating the adverse effect of iron in all forms of this movement disorder [36].
Unquestionably, iron gets accumulated in the substantia nigra and other target tissues; its role is said to be elusive owing to lack of entire mechanism of disease pathogenesis [26]. However, iron buildup in the substantia nigra induces oxidative stress, formation of intracellular α-synuclein aggregates, motor impairment, disturbed iron homeostasis, cellular disintegration, intrinsic apoptosis, microglial activation, and neuronal death [27, 33, 35, 37]. Iron chelator reduces the extent of anomalies along with reduction in the nigral iron content confirming its role in PD pathogenesis [25, 37]. Moreover, iron chelator also shrinks the cellular iron pool and offers protection from 6-OHDA, MPTP, and rotenone-induced Parkinsonism, which are exemplified with iron accumulation [29, 33]. Involvement of ceruloplasmin in iron accumulation has come from a knockout study in which ferric ammonium citrate administration-mediated iron deposition in the brain of MPTP-treated rodents and its reversal by deferoxamine, a metal chelator, were found [27]. Iron accumulation is seen, if ceruloplasmin along with ferroportin 1, which helps in iron export, is reduced in 6-OHDA-induced degeneration showing that accumulation starts immediately after toxicant exposure [32]. Oxidation of ceruloplasmin in PD patients changes its chemical nature from basic to acidic and reduces ferroxidase activity that subsequently augments intracellular iron retention [30]. Elevated level of iron and overexpression of divalent metal transporter 1(DMT1) in affected tissue of Parkinsonian rats have shown the value of latter in the former process [34]. Moreover, reduction in Nedd4 family-interacting protein 1 (Ndfip1) is found to contribute to 6-OHDA-induced iron deposition through DMT1 degradation-dependent pathway [34]. P-type ATPase/ATP13A2 causes an enlargement of lysosome and late endosome and reduces iron-induced membrane permeabilization and thereby protects from iron-induced cell damage. However, defects in ATP13A2 gene leads to PD, which is characterized with the brain iron accumulation [31]. It indicates the role of iron in the regulation of clearance mechanism of defective proteins and organelles as well as its own through the action of a few selected proteins. Combination of a naturally occurring antioxidant and an iron-chelator is found be to rescued from impaired antioxidant defense system, α-synuclein accumulation, and aggregation, inhibits monoamine oxidase, and activates hypoxia-inducible factor-1 signaling pathway along with its downstream mediators viewing that iron buildup leads to oxidative stress and thereby PD [37]. Overexpression of human α-synuclein increases the intracellular iron content and its redistribution from the cytoplasm to the perinuclear region within α-synuclein-rich inclusions further indicate the role of iron in PD [38]. Microglial activation releases inflammatory cytokines under iron overload that subsequently leads to its accumulation by increasing the level of regulatory protein 1 and hepcidin through free radical production, which shows that microglia aggravates iron-induced PD pathogenesis [35].
Despite all supportive evidences, iron alone is not a culprit of sporadic PD. This is evident from a study in which patients were found to atypically possess iron accumulation with neurodegeneration along with dystonia and orofacial stereotypes. The study shows that symptoms of iron alone-induced Parkinsonism are different from sporadic PD [28]. Moreover, a link between age and iron accumulation in different brain regions is reported; heterogeneity of phase value of accumulation and overstressed propensity in the nigra indicate the need of further assessment for commenting upon the relationship between iron accumulation and PD [24].
Manganese Accumulation and PD
Manganism was diagnosed approximately two hundred years ago; epidemiological studies correlating manganese exposure with PD came into limelight during the last few decades [7]. Like other micronutrients, it is required for catalytic activity of a few enzymes and biological function of some proteins but excessive accumulation is reported to be extremely toxic [39]. Age, gender, genetics, environmental/endogenous exposure, and ethnicity altogether determine the fate of manganese-induced toxicity. In general, manganese exposure is less than the toxic level in the general population; but mostly, its accumulation is reported in PD patients of Chinese population [40, 41]. Augmented exposure or reduced excretion leads to its accumulation in the basal ganglion that could induce Parkinsonian symptom or manganism [42]. Manganese accumulation leads to aberrant dopaminergic neurotransmission by increasing oxidative stress, mitochondrial dysfunction, inflammation, and synaptodendritic degeneration, which are reduced by the specific remedial agents (such as antioxidants, anti-inflammatory agents, mitochondrial function regulators, neuroprotective agents, etc.) further showing its role in eliciting PD-like symptoms [43].
Manganese is accumulated in the Golgi apparatus where it gets detoxified. Moreover, manganese accumulation reduces total iron content that adds to neuronal injury [44]. Manganese activates the nuclear factor erythroid-2 related factor 2 and heme oxygenase-1 pathways through free radicals and ubiquitin-proteasome pathway-dependent mechanisms [45]. Sub-acute low-level manganese exposure disrupts the release and function of dopamine, gamma amino butyric acid, and glutamate neurotransmitter, which subsequently lead to neurobehavioral anomaly [46]. Rampant exposure to manganese has long-term impact on the regulation of extracellular dopamine release in the striatum [47]. Continuing exposure to manganese during critical periods of development causes oxidative stress and impaired motor coordination [48].
Hydrogen peroxide is the main culprit of oxidative stress in manganese-induced toxicity that is produced by the mitochondrial complex II [49]. Owing to distinct distribution in transportable state as compared with the rest of metals, it affects the mitochondrial electron transport chain of neurons located in the nigra and adjacent tissues [7]. Activity of the central histaminergic system is increased in PD and 6-OHDA-lesioned rats that is further increased by manganese exposure showing its contribution in sporadic as well as toxin(s)-induced PD [50]. While periodic/regular exposure to welding fume induces permanent change in dopaminergic neurons, it is ambiguous to establish whether exposure to manganese from welding electrodes, ferroalloy, and other means produces behavioral anomalies leading to manganism or not [41, 51, 52]. Exposure induces a few symptomatic features mimicking disease and some that are not at all associated with PD leading to the hypothesis that manganism is not identical to PD [46, 53]. Influence of manganese is reported to be quite high in the individuals possessing genetic predisposition as well [54]. Both manganism and sporadic PD are regulated by PARK genes in the similar way and oxidative stress, impaired mitochondrial function, α-synuclein aggregation, and aberrant ubiquitin proteasome system direct the nigrostriatal dopaminergic neurodegeneration in both the conditions [55]. Even very low concentration of manganese induces α-synuclein fibril formation [56]. Accumulation of welding fumes/manganese chloride impairs the mitochondrial function, degenerates tyrosine hydroxylase (TH) containing neurons, and alters the expression of PARK proteins showing that PARK genes play important role in manganism similar to sporadic PD [57]. Motor alterations and selective degeneration in the nigra induced by the inhalation of manganese mixtures are significantly reversed by levodopa indicating that manganism is similar to PD and manganese can be used to develop an appropriate disease model [58, 59]. Moreover, behavioral alterations and inflammation persist even after manganese gets cleared off from the cortical brain compartment [60] showing that the changes are irreversible similar to PD. Though the highest concentration of manganese is achieved in the basal ganglia, manganese-induced Parkinsonism is differentiated from sporadic PD owing to noticeable inhibition of dopamine release even in the absence of the loss of terminals of the nigrostriatal dopaminergic neurons [53]. Manganese-induced Parkinsonism does not involve the demise of midbrain dopaminergic neurons and levodopa is also not found to be effective according to a study [61], which further shows that manganism is different from PD. Dynamic mode of transport and detection and pharmacokinetic modeling of trafficking also indicate variability between manganese-induced Parkinsonism and sporadic PD [42]. Level of manganese is not altered in the substantia nigra but 20 % reduction in the striatum of PD patients is seen as compared with controls pointing out that accumulation of manganese is not associated with sporadic PD [62].
Copper Accumulation and PD
Long-term exposure or excessive accumulation increases disease occurrence owing to its free radical-generating property [63–65]. Contrary to this, copper rescues from PD symptoms in a study, which exhibited 34–45 % reduction in the substantia nigra of PD patients [62, 66, 67]. Moreover, no specific and straightforward relationship between copper intake and PD risk/protection has been observed in another study [68]. Despite conflicting reports, an increased level of copper-bound biologically active proteins has been consistently reported to be protective. It is supported by the verity that its supplementation increases the activity of copper bound proteins and ameliorates disease symptoms. Chelation reduces availability of copper to bound proteins and aggravates PD symptoms [69].
Neuroprotective properties of copper are partially contributed by its cofactor nature in antioxidant protein, superoxide dismutase (SOD). SOD scavenges superoxide radical and regulates electron transport chain [69]. A study performed in the cerebrospinal fluid of PD patients also indicated that free copper is toxic, while protein bound copper is protective in nature [70]. Neuroprotection is also mediated by its action against toxic effects of iron deposits [71]. Copper also prevents toxin-induced protein nitration and reduction in TH activity and its inactivation [72, 73]. Copper regulates the function of metallothioneins, ceruloplasmin, DJ-1, copper transporter 1, and P-type ATPase B proteins. Reduced expression of ceruloplasmin in the nigrostriatal pathway also shows that reduction of copper bound proteins is associated with PD [69, 74].
Copper induces the formation of oxidation products of catecholamine, which are regulated by chloride concentration and lead to DNA damage [75, 76]. Copper plays imperative role in oxidation kinetics responsible for dopaminergic neurodegeneration [76]. Copper ion directly regulates α-synuclein fibril formation and oligomerization [56, 77]. Copper-induced dopamine oxidation increases mitophagy and caspase-3-independent apoptotic degeneration [78]. While autophagy does not play any role in metal elimination, removal of damaged organelles and proteins could eliminate some metals. Nigral α-synuclein aggregation augments neuronal cell death if copper-dependent defense mechanism is impaired since its interaction with α-synuclein triggers modification and aggregation owing to the formation of reactive oxygen species [79, 80]. Although α-synuclein stimulates copper-mediated toxicity even without aggregation, aggregated forms are found to be more pathogenic [79, 81].
α-Synuclein increases the cellular sensitivity to copper showing that pathological role of α-synuclein aggregates depends on copper-binding capacity [82]. Copper also regulates an interaction of herbicide with α-synuclein [83]. Depending on pH, copper, and α-synuclein ratio, variable copper species have been reported. Binding affinity of copper (1+) with N-terminal and C-terminal regions of α-synuclein is found to be uneven and interaction leads to site-specific oxidation of the latter [84, 85]. Imbalance in the cellular copper homeostasis preferentially targets oxidation of N-terminal region while function and aggregation are regulated by C-terminal domain [86, 87]. Physiological form of α-synuclein that interacts with copper (1+) is found to be N-terminally acetylated, which subsequently abolishes binding of copper at high affinity [84, 88, 89]. Aggregation propensity and folding are regulated by remote histidine residue that regulates its binding with copper (2+) [84]. Copper also binds to histidine residue at position 50 of the carboxy terminal sequence that finally determines the fate of α-synuclein [69]. High-affinity form of α-synuclein undergoes fibrillation and partially folded conformation [69, 90]. Although α-synuclein exists in soluble and membrane-bound forms, copper exerts its effect mainly on the soluble form [63]. Reduced cellular copper clears off larger aggregates and oligomers that are intensely localized to the plasma membrane [82]. Copper (2+) regulates protein/vesicle coordination and extent of α-helix for the membrane-associated area [91]. Deletion of any terminal results in a loss of aggregation whereas deletion of C-terminal results in a loss of membrane association [82]. Both copper and dopamine interacts with both terminus and induce folding [87, 90]. Dopamine or dopamine/copper induces α-synuclein oligomerization and cross-linking more than that of free radical-mediated covalent modification [92].
DJ-1 interacts with copper and is regulated and/or stabilized by zinc [93]. DJ-1 changes the coordination geometry of copper leading to failure of metal transfer to SOD [94]. DJ-1 requires stable homodimer for mutation that weakens its formation and compromises competency [95]. DJ-1 enhances the cellular defense and mutations reduce its protective property [96]. Even a small genetic change or concomitant addition of dopamine sensitizes copper-induced cytotoxicity [96]. Aberrant expression of Parkin also substantiates the role of copper in PD [97].
Zinc Accumulation and PD
Maintenance of homeostatic relationship among essential metals is required for the normal functioning of the brain [98]. Zinc controls synaptic transmission along with iron and copper and regulates elevation in an impaired compartmentalization leading to deregulation of homeostasis [99]. Zinc acts as an antioxidant at the desired cellular concentration; depletion or excess induces free radical generation [65]. Antioxidant nature of zinc is not because of metal per se, but is rather due to zinc-containing proteins [100]. Alteration in the intracellular zinc content is associated with functional anomaly in neurons. Deficiency is implicated in growth problem, mental retardation, emotional disturbance, and physical and immunological aberrations in children while it is not associated with any specific problem in adults [101]. Deficiency also leads to excessive consumption of iron and copper, accumulation of manganese, ingestion difference of vitamin E/copper, and decreased consumption of vitamin B12 [102].
Higher concentration is even more toxic since metals oxidize macromolecules and reduce the cellular antioxidant defense system [103]. Zinc is profusely present in the hippocampus, cerebral cortex, thalamus, and olfactory cortex [104] and its accumulation in the nigrostriatal region leads to PD [62]. Although concentration of zinc in other brain regions of PD patients remains the same, it increases by 50–54 % in the substantia nigra and 18–35 % in the striatum indicating the role of its accumulation in neurodegeneration [62]. Presence of zinc-dependent matrix metalloproteinase-2 (MMP-2) in α-synuclein inclusions and selective augmentation in expression of MMP-2 in the striatum also indicate the same notion [105]. Since zinc is required for normal functioning of MMP-2, its accumulation increases the susceptibility of degenerative diseases that are characterized with α-synuclein aggregate formation, inflammation, BBB dysfunction, and myelin deterioration [105]. Zinc also increases an interaction of herbicide with α-synuclein [83]. Accumulation of zinc is associated with stern dopaminergic neuronal cell loss as evident from the overexpression of metallothionein, an indicator of metal homeostasis disturbance [74, 106]. A decrease in zinc-dependent SOD and ferroxidase activity in the cerebrospinal fluid of PD patients also indicates its relevance in the regulation of antioxidant defense system [70]. A chelator that crosses the BBB reduces the level of essential metals, such as zinc, and reduces neurotoxicity [18] confirming the role of zinc accumulation in PD.
Parkinsonian toxins that inhibit the mitochondrial complex I, such as N-methyl-4-phenylpyridinium (MPP+) and 6-OHDA, induce zinc accumulation in the substantia nigra pars compacta showing that cytosolic labile zinc accumulation could be an indicator of degenerating dopaminergic neurons [107, 108]. Similarly, 3-morpholinosydnonimine mediates the mitochondrial accumulation of metals, including zinc, and the induction of metallothionein gene protects from it; this further highlights the role of zinc in neurodegeneration [74, 109]. Zinc induces nicotinamide adenine dinucleotide phosphate-oxidase-dependent free radical generation, dopamine and glutathione depletion, apoptotic loss of TH-positive neurons, reduction in the expression of monoamine transporters, and microglial activation after long-term exposure to high doses showing its role in Parkinsonism [8]. Apocyanin, an antioxidant, and/or N-acetyl cysteine, an anti-inflammatory agent, are found to reduce zinc-induced alterations that validate the role of zinc in oxidative stress-induced PD in experimental models [16].
Zinc induces PD even in manganese-exposed population since manganese is competently and effectively transported by zinc transporters [110]. Two types of transporters are found to be responsible for manganese transport and/or storage in the brain; one is iron and another is zinc. But a stable relationship between manganese and zinc at the tissue and cellular levels suggests that zinc transport/storage is associated with manganese transport and accumulation [111]. Since zinc is known to induce its own transporters, little excess in its concentration could promote uncontrolled passage of manganese in the brain leading to PD-like symptoms in manganese exposed individuals. Moreover, zinc along with iron influences the effect of lead and affects dopaminergic neurons [112]. It is also supported by a study that has shown an increased expression of transporters and transferrin receptors owing to concurrent and altered level of metals [98].
Accumulation of zinc produces adverse effect on the BBB as elevated level is found to be associated with barrier dysfunction [35]. Defective BBB could allow an entry of unwanted molecules and radicals in the brain. Moreover, disturbance in zinc homeostasis leads to the lysosomal impairment, α-synuclein accumulation, and mitochondrial dysfunction through PARK 9-dependent mechanism [113]. Interestingly, higher intake of zinc is also reported to be protective against PD in a study [68]. The interaction of DJ-1, a neuroprotective and antioxidant protein, with copper is regulated and/or stabilized by zinc showing that zinc regulates neurodegeneration/protection even in familial PD [93].
Mercury Accumulation and PD
History of a possible association of mercury exposure with PD is quite old as dental amalgam fillings have been used from the ancient era and is projected to be associated with disease pathogenesis [114]. J.M. Charcot, who is appropriately referred to as the father of modern neurobiology, primarily emphasized the hereditary etiology of PD. But he never ruled out the possibility of mercury exposure as an etiological factor even in the absence of a clear cut association [115]. Mercury absorption occurs through the lungs; afterwards, it reaches to the bloodstream and subsequently enters and accumulates in the central nervous system [116]. Mercury exposure induces adverse effect on dopamine transporters leading to a dose-dependent depletion of the striatal dopamine [117]. Although an obvious monotonic dose–response association between PD and blood mercury is seen, scalp hair mercury is not found to be the first-rate disease predictor [118]. While no significant association between occupational mercury exposure and PD is seen in an epidemiological study, confidence interval of odds ratio did not ignore the likelihood [119]. In another epidemiological study, a rare clinical variant of mercury intoxication was found to be associated with Parkinsonism even in the absence of chronic exposure specific neuropsychiatric signs [120]. PD subjects are found to be associated with detectable blood mercury level, but barely a few controls are found to have the same, which substantiate that it plays a role in disease etiology [121]. High urinary excretion, which could be extrapolated as high mercury exposure, is also found to be linked with increased average tremor intensity (a hallmark of PD) within high-frequency window [122]. Higher mercury exposure is also related to abnormal facial expression [123] that is often reported to be a secondary characteristic of PD. However, inadequate longitudinal exposure assessment, negative confounding by better access to dental care in the elite groups, inadequate epidemiological studies, insufficient number of cases and controls for power statistics, and lack of animal experimentations have been the major limitations. Therefore, better designed studies are still needed to confirm its association with PD [114].
Magnesium Accumulation and PD
Magnesium is the second most abundant divalent intracellular metal cation involved in the intracellular processes [124]. Association of magnesium accumulation/dietary intake with PD is hard to define since both accumulation and low level are found to be associated with risk as well as protection in animal and epidemiological investigations. A multicenter hospital-based case–control study performed in Japan did not observe any relationship between magnesium intake and disease risk rather high intake was found to be neuroprotective [68]. Low amount owing to decreased function of dopaminergic neurons leads to catalepsy, which shows that low intake could be a contributory factor in PD [125]. On the other hand, magnesium (2+) acts as a calcium (2+) channel antagonist and reduces the damaging consequence of calcium (2+)-induced neuronal inflammation showing its protective efficacy [126]. Magnesium inhibits iron-induced α-synuclein aggregation that further confirms the notion [127]. Cytosolic content is regulated in the brain to equilibrate changes in rapidly available free energy and magnesium and to moderate cadmium/aluminum-mediated effects illustrating that low magnesium level could lead to neurodegeneration [128–130]. While magnesium prevents the length of dopaminergic neuritis, it can exacerbate MPTP-induced striatal dopamine depletion [131, 132]. Furthermore, low dose increases motor activity and latency to heat stimuli, but medium and high doses decrease the same and increase pole climbing time in MPTP-treated mice [132]. Magnesium exerts imprinting effect since low magnesium intake (1/5th of desired value) over generations is found to be associated with significant death of dopaminergic neurons in the substantia nigra [133].
Regardless of the fact that the mitochondria maintain magnesium homeostasis, its high level leads to dysfunction and formation of protein aggregates in the brain mitochondria [134]. Rotenone reduces magnesium-dependent block of N-methyl-D-aspartate current in dopaminergic neurons of the substantia nigra signifying that magnesium-mediated excitotoxic mechanism participates in rotenone-induced Parkinsonism [135]. Contrary to it, an increase in magnesium content is found to inhibit the cellular free radical generation, maintains energy production, and rescues from toxin-induced Parkinsonism [136]. A gradual decrease in the magnesium (2+) concentration in the mitochondria is seen in response to a neurotoxin in differentiated PC12 cells viewing that its specific concentration is needed to maintain the normal neuronal functions [137].
Association of magnesium deficiency or accumulation is also reflected from the studies, which have shown the presence/absence of association of ion channel protein variants with PD risk. Human solute carrier family 41 (magnesium transporter), member 1 (SLC41A1) gene encodes for sodium (1+)/magnesium (2+) exchanger, which is involved in magnesium (2+) efflux system [124]. SLC41A1 is found to be dysfunctional when magnesium (2+) efflux is impaired. Moreover, long-term and chronic intracellular magnesium (2+) deficiencies in PD patients are found to increase magnesium (2+) efflux by SLC41A1 variant p.A350 V [138, 139]. PD is also associated with reduced expression of transient receptor potential cation channel, subfamily M, member 2 and 7 channel proteins [140].
Studies available have shown no significant association of magnesium with PD or neuroprotection. Although low magnesium in the diet is associated with olfaction, magnesium along with calcium, iron, silicon, and zinc are not correlated with duration or severity of PD or anti-PD drugs [141, 142]. Mystifying observations across studies indicate that magnesium is associated with PD risk. Despite contradictory reports, it is believed that measurement of brain magnesium (2+) could help in differential disease diagnosis [128]. Overall, association of PD with magnesium accumulation/deficiency has been elusive and studies are still required to spell it out.
Lead Accumulation and PD
During recent years, chronic exposure to lead has been minimized owing to complete ban on gasoline, but it remains to be a major public health concern [143]. Limited studies are available showing an association between lead exposure and PD [144]. PD risk gets elevated by >2-fold in people in the highest quartile for lifetime lead exposure indicating that chronic exposure could be a risk factor [145]. While lead alone is not found to be associated with PD in a study, dual combination of lead, iron, or copper increases the risk [64]. An increase in the plasma lead level in urban PD subjects is found in a study [146] pointing out the accumulation of lead in the brain. A 10-fold increase in radioactive lead in the lipid fraction is also seen indicating that lead is primarily accumulated in the lipid fraction [147]. An increase in bone lead content is not found to be associated with risk but cumulative exposure augments PD risk in the typical patients [148]. Similarly, the tibia bone lead unlike the patella lead is seen to be associated with cognition deficit and cumulative exposure aggravates the condition [149]. Association of lead with PD is also evident from a study where a case was exposed to lead for 17 years in a car battery industry and was diagnosed with high level of lead in the blood. The person was initially characterized with the primary disease symptoms, followed by the secondary symptoms and later by the late stage disease symptoms. Moreover, the patient was also found to be levodopa responsive [150]. Lead exposure increases α-synuclein aggregation and aggresome formation and inhibits degradation and thereby supports the lead accumulation theory of PD [151]. Although studies have shown an association of lead accumulation/exposure, lead level is not found to be altered in the substantia nigra and striatum of patients as compared with other regions of the brain, which simply contradicts its adverse association with PD [62].
Aluminum Accumulation and PD
Except a few scattered contradictory observations, reports have shown an association of aluminum accumulation in the central nervous system and exposure to aluminum-containing antacids with increased incidence of neurodegenerative diseases [152–154]. Aluminum gets accumulated in the substantia nigra and/or striatum, particularly in the gray matter, and is linked with PD pathogenesis [130, 155]. Presence of aluminum in the Lewy bodies of the nigra of PD patients also indicates its significance [156]. Aluminum increases monoamine oxidase-B (MAO-B) enzyme activity and is associated with dopamine degradation and depletion thereby indicating its etiological worth in PD [157]. Augmentation in aluminum content in neuromelanin granules shows that it promotes oxidant formation, which accounts for the selective degeneration of neuromelanin-positive neurons [158, 159]. Accumulation of aluminum in hair and increase in urinary 8-hydroxy-2′-deoxyguanosine, an indicator of oxidative stress, in Mongolian patients further support the negative role of this metal in PD [160]. Aluminum enhances 6-OHDA-induced oxidative stress, reduces endogenous antioxidant defense enzymes, and increases nigrostriatal dopaminergic neurodegeneration, which additionally support the notion that this metal adds on PD hallmarks induced by a Parkinsonian toxin [161].
Aluminum increases interaction between pesticides and α-synuclein [83]. Exposure reduces TH-immunoreactivity, neurotransmitter content, and motor functions and induces fibril formation from aggregated α-synuclein leading to conformational change attributed to the development of a partially folded intermediate [56, 155]. While aggregation and structural changes are reported, newer and specific tools are expected to help in understanding the mechanism of aggregation [162]. It triggers phosphorus leading to homeostatic imbalance in the serum of patients showing that aluminum indirectly regulates PD pathogenesis [163]. Albeit most of the studies have shown an association of aluminum accumulation with PD, it is found to reduce lipid peroxidation and 6-OHDA-induced dopaminergic lesion in the striatum [100].
Calcium Accumulation and PD
Several direct and indirect studies have highlighted that calcium (2+) homeostasis in the endoplasmic reticulum is a decisive factor of neurodegeneration [164, 165]. Reticular calcium (2+) and activation of calcium–calmodulin–calcineurin cascade is regulated by α-synuclein showing that calcium homeostatic disturbance could be associated with disease pathogenesis [166]. Maintenance of calcium homeostasis owing to an accumulation of lactoferrin (an iron-binding protein) is required for protection against MPTP-induced neurodegeneration demonstrating its importance in the normal function of dopaminergic neurons [167]. Excessive concentration, deposition, or even slight changes in calcium content of dopaminergic neurons could lead to the onset of PD symptoms [154, 168]. Mitochondrial dysfunction and defective autophagy, two critical events of degeneration, are also regulated by the mitochondrial calcium influx [169, 170]. Attenuation in the mitochondrial calcium capacity or augmentation in oxidative stress lowers the threshold for opening of the mitochondrial permeability transition pore [171]. Moreover, dopamine transporter-1 receptor signal transduction pathway depends on L-type calcium (2+) channel in order to mediate cyclic adenosine monophosphate response element-binding protein phosphorylation [172]. The impaired mitochondrial calcium (2+) accumulation during agonist stimulation is a major consequence of human complex I deficiency [173]. Calcium-binding domain, which is located in the 15 amino acids at the acidic C-terminal end of α-synuclein, induces filament formation that continues through intermediate or proto-fibrillar species leading to PD [174].
Cadmium Accumulation and PD
Indisputably, the central nervous system disorders are the second most health risk associated with metal exposure [175]. Although cadmium accumulation is less studied as compared with iron, manganese, and copper, a trend of increased disease risk with its exposure is reported [176]. High cadmium level along with manganese, iron, lead, and aluminum are also seen in Mongolian PD subjects as compared with Japanese [160]. An old person, who was acutely exposed to cadmium, was found to possess PD-like features showing that cadmium exposure damages the basal ganglion, the most affected site in PD [177]. Effects of cadmium on cognition, behavior, learning deficits, and altered dopaminergic function are also reported [112] showing that cadmium accumulation could lead to PD. Cadmium alters the interaction of Parkinsonian toxins with α-synuclein that further shows its role in disease pathogenesis [83]. Cadmium exposure also modulates ubiquitin proteasome pathway, antioxidant enzymes, phase II enzymes, and cell cycle regulators implicated in PD pathogenesis [178]. Besides, an auto-transplantation study has shown lack of any change in cadmium along with a few other metals after operation showing that cadmium accumulation is significantly associated with PD pathogenesis [179]. Even though an association between PD risk among nurses and exposure to cadmium is reported, no explicit and unambiguous association between adulthood ambient exposure to cadmium and PD risk is seen [144].
Arsenic Accumulation and PD
It is not yet clear if arsenic accumulation induces the nigrostriatal dopaminergic neurodegeneration or not. Undeniably, arsenic (3+) induces oxidative stress leading to the activation of early transcription factors [180]. Clear cut evidence is still not available that could have explicitly explained its role in PD. Arsenic (3+) synergistically enhances dopamine toxicity in differentiated dopaminergic neurons in culture [181] indicating that it aggravates dopaminergic neuronal cell death. Moreover, glutathione transferase-ω E155 deletion linked with abnormal arsenic excretion and age-at-onset of PD, which further indicates that arsenic could be a critical player [182]. Despite a few supports, arsenic accumulation theory of PD gets jolted owing to the presence of its low concentration in PD patients in comparison with controls [183].
Cobalt Accumulation and PD
Association of cobalt accumulation is appraised; however, no study has yet confirmed its substantial association with PD [106]. While cobalt (2+) induces rapid formation of discrete annular α-synuclein oligomeric species, cobalt (3+) causes significant acceleration in α-synuclein fibril formation [56, 174]. Since fibril and oligomer formation are very much associated with PD, therefore, the role of cobalt in PD pathogenesis needs to be explored further.
Is Metal Accumulation an Epicenter or Outcome of PD?
Metals get accumulated owing to lack of fully operational excretion machinery irrespective of their route of entry [184–186]. Accumulation in the brain also happens due to metal-mediated increase in the BBB permeability. Accumulation leads to oxidative damage, metal–metal interaction, estrogen-like effects, and epigenetic modifications [187]. Metal accumulation is contributed by both innate and acquired factors, which are evident from the studies that have shown accumulation of a specific metal in a fussy population [184–186]. For instance, iron gets accumulated in the brain of the people of Irish, Scottish, British and Scandinavian ancestry much more in comparison with the people of rest of the world. It shows that hereditary/innate factors regulate metal accumulation process [186]. Presence of higher accumulation in males as compared with females indicates implicit role of acquired factors in metal buildup [186].
Exposure to manganese, copper, lead, iron, mercury, aluminum, zinc, and cadmium appears to be the main environmental risk factors (Table 1) along with pesticides [22, 100, 188, 189]. However, it is still a Demigod subject if metal accumulation is an epicenter or merely an outcome [9] (Table 2). Although a trend of increase in disease risk with metal exposure at work place is reported, limited studies exist in this direction [144, 176]. One school of thought believes that metal accumulation is neither an epicenter nor an outcome, but rather it is an indispensable process, which has zilch contribution to PD rather to keep healthy. The notion is reliant on the plethora of information that demonstrate the protective effect of metals or their essentiality for the catalytic activity of a few enzymes opposite to metal accumulation theory that deems accumulation as a causative or contributory factor. For example, metal supplementation is needed to maintain the normal physiology of dopaminergic neurons of the substantia nigra, one of the most badly affected tissues, during metal deficiency [22]. Furthermore, with the best of our knowledge, no scientific evidence is available in the literature that could have explicitly demonstrated an increased prevalence of PD in populations, which are more prone to metal accumulation [186] as compared with populations in which accumulation is found to be less. Such proofs validate the hypothesis that metal accumulation is not associated with PD or at least it is not a causative factor. Moreover, a few population studies have also shown lack of relationship between the two [22].
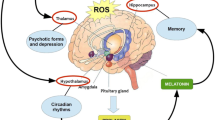
Campaigners of the jingle “causative factor theory” contradict the tune of “no impact theory” or “outcome/consequence theory”. Supporters of “causative factor theory” believe that protection offered by metals is not because of metal per se rather it is an antioxidant property of metal bound proteins that provides defense (Fig. 1). Elevated level of metals in the substantia nigra of patients in comparison with the rest of the brain indicates that metal accumulation is a contributory factor [104, 107, 108]. Accumulation theory explains that higher oxidation state of metals induces oxidation of macromolecules and aggregation of α-synuclein [22]. High metal conjugates lead to glutathione depletion in the substantia nigra of PD patients showing that oxidative stress occurs owing to metal accumulation and metal accumulation does not occur because of oxidative stress. Higher level of metals is seen in Mongolian PD patients (population that is not genetically prone to metal accumulation like Irish) as compared with Japanese further shows that accumulation leads to oxidative stress and could be a causative factor [160]. Excess dopamine, the main neurotransmitter, and levodopa, the main PD therapy, are also known to increase oxidative stress [188, 189] showing that stress could be a secondary event. Moreover, presence of high nitrate content in the cerebrospinal fluid in PD patients treated with levodopa or with dopamine agonist vis-à-vis their respective controls further supports it. However, nitrite (an end product of nitric oxide metabolism that is an indicator of nitrosative stress/oxidative stress) is apparently unrelated with PD risk in another study [190] showing that stress could be secondary to metal accumulation. Activated microglial cells are reported to contain high level of free metals, such as iron [22]. Since two major events in PD pathogenesis, i.e., oxidative stress and microglial activation occur in the nigrostriatal tissues owing to metal accumulation but not vice versa show that metal accounts for the initiation of neurodegeneration.

Metals in PD pathogenesis: accumulation of free form of metals, such as copper, iron, or zinc, and metals per se, such as aluminum, cadmium, or arsenic, in the brain cause/protect dopaminergic neurodegeneration. Metal, if bound to an antioxidant and free form magnesium, per se rescue from PD
Campaigners of the view, which considers metal accumulation as an outcome of PD pathogenesis, provide substantial evidences in their support. They believe that low level of natural antioxidants, such as glutathione and SOD, and impaired activity of metal bound antioxidants are the major roots of disease pathogenesis. Reduced antioxidant defense system directs metal accumulation or availability of free metals in the substantia nigra [22] leading to the hypothesis that accumulation is an outcome and not the starting point. This theory is also supported by the fact that dopamine and levodopa form conjugates with glutathione in PD patients leading to oxidative stress that ultimately leads to PD [188, 189, 191]. Moreover, believers of this theory simply disagree with “causative factor hypothesis” with a convincing logic “if metal accumulation is the main cause why low level of a few metals is also associated with PD”. Moreover, neurotoxins mainly inhibit the mitochondrial complex I and lead to oxidative stress and provoke the elimination of metals from metal bound proteins that subsequently increase free metal content [107–109]. Therefore, availability of excessive free metals could be a consequence and not a cause. However, exposure to metals could induce hydrogen peroxide production by the inhibition of the mitochondrial complex II suggesting that metal accumulation leads to oxidative stress and could be the primary event [49]. Moreover, population studies correlating metal exposure and disease pathogenesis are still limited. With inadequate studies, it is hard to pin down a hypothesis that could be near to reality. Animal experimentations performed till date is either one metal centric or one parameter centric; therefore, interpretation biasness could not be excluded. Moreover, most of the experimental animals do not develop PD naturally and disease needs to be induced by specific neurotoxin; therefore, animal results, even if positive, could not be completely extrapolated to humans and relied upon. Overall, the study seems to be inconsistent and inconclusive owing to lack of meta-analysis of genes that regulate the level of metals in the brain of exposed populations and their association with PD across the globe. Such studies need to be performed extensively along with multiple contributory factor-based investigations in order to reach up to a definite conclusion, if metal accumulation is an epicenter or an outcome or none of the above.
Does Exposure to Pesticides also Induce Metal Accumulation in the Brain?
A few direct and indirect studies have shown a possibility of metal accumulation in the substantia nigra of pesticides and non-pesticides, such as MPP+, 6-OHDA, lipopolysaccharide, and rotenone, models of Parkinsonism [10, 23, 25, 28–33, 107, 108]. Extent of exposure time to pesticides and heavy metals has also been linked with the age at onset in non-familial PD [192]. A number of studies have shown an implication of metals and pesticides either alone or in combination of two in PD pathogenesis [1, 193]. However, in combinational exposure, the level of metals is rarely measured to counter an uncertainty “if pesticide or Parkinsonian toxin induces metal accumulation in the nigrostriatal tissues of the brain”. Moreover, metal containing pesticides are also found to induce an additional degree of oxidative stress in dopaminergic system of rodents that is exposed to a non-metal containing Parkinsonian pesticide. Such observation directly indicates the existence of a possible crosstalk between metal and Parkinsonian pesticide in order to exert sternness of disease features [194]. Cypermethrin-induced changes in the non-neuronal system of lower animals are found to be induced by cadmium showing that metals could induce toxic effects of the pesticides [195]. The similar likelihood in non-human primates and rodents could not be ignored until disapproved in the pre-clinical studies. Presence of even low level of pesticide in the diet is found to induce cadmium accumulation in the kidney irrespective of the dietary content of zinc and copper [196] showing that pesticide exposure leads to metal accumulation. All these three metals are individually known to induce the symptomatic features of Parkinsonism at some or the other concentrations. Moreover, dietary deficiency of one metal is also reported to lead an accumulation of other in the brain [20]. Such postulations indicate a possibility that during nutritional deprivation, pesticide exposure leads to metal accumulation.
Despite extensive research employing various models and research tools, it has still remained an unrequited Demigod issue “if metal accumulation is an epicenter or an outcome”. Pesticides are found to induce α-synuclein aggregation and bound to metal-induced partially folded α-synuclein [83]. High iron content in the brain of toxin-induced rodent model of Parkinsonism [29] has also indicated that toxins induce metal accumulation. Parkinsonian toxins that inhibit the mitochondrial complex I also induce metal accumulation in the substantia nigra indicating that metal accumulation could be an indicator of degenerating dopaminergic neurons [107, 108]. But detailed studies are not yet performed to assess the accumulation level of all suspected metals in the substantia nigra employing all pesticide and toxin models of PD. Experimental evidences would be expected to narrate the appropriateness of suggested theories more precisely. Therefore, such studies need to be performed and a correlation between metal accumulation and pesticide exposure needs to be established. Once such correlation is established and is found to be affirmative, a clear cut wrapping up can be drawn whether metal accumulation is an epicenter or an outcome. The Demigod question that exists today in PD biology could be answered agreeably if time- and dose-dependent accumulations of all suspected metals could be measured in the target tissues of control and Parkinsonian toxin/pesticide-treated animals and subsequently correlated with behavioral and supplementary phenotypic disease symptoms.
Conclusion
Despite a strong association between metal accumulation and PD, it is not yet clear whether metal accumulation leads to PD or PD leads to metal accumulation.
References
Singh C, Ahmad I, Kumar A (2007) Pesticides and metals induced Parkinson’s disease: involvement of free radicals and oxidative stress. Cell Mol Biol (Noisy-le-Grand) 53(5):19–28
Andruska KM, Racette AB (2015) Neuromythology of manganism. Curr Epidemiol Rep 2:143–148
Dell’Acqua S, Pirota V, Anzani C, Rocco MM, Nicolis S, Valensin D, Monzani E, Casella L (2015) Reactivity of copper-α-synuclein peptide complexes relevant to Parkinson’s disease. Metallomics 7:1091–1102
Myhre O, Utkilen H, Duale N, Brunborg G, Hofer T (2013) Metal dyshomeostasis and inflammation in Alzheimer’s and Parkinson’s diseases: possible impact of environmental exposures. Oxidative Med Cell Longev. doi:10.1155/2013/726954
Prakash A, Bharti K, Majeed AB (2015) Zinc: indications in brain disorders. Fundam Clin Pharmacol 29:131–149
Rivera-Mancía S, Ríos C, Montes S (2011) Manganese accumulation in the CNS and associated pathologies. Biometals 24:811–825
Robison G, Zakharova T, Fu S, Jiang W, Fulper R, Barrea R, Marcus MA, Zheng W, et al. (2012) X-rayfluorescence imaging: a new tool for studying manganese neurotoxicity. PLoS One 7(11):e48899. doi:10.1371/journal.pone.0048899
Kumar V, Singh BK, Chauhan AK, Singh D, Patel DK, Singh C (2015) Minocycline rescues from zinc-induced nigrostriatal dopaminergic neurodegeneration: biochemical and molecular interventions. Mol Neurobiol. doi:10.1007/s12035-015-9137-y
Singh N, Haldar S, Tripathi AK, McElwee MK, Horback K, Beserra A (2014) Iron in neurodegenerative disorders of protein misfolding: a case of prion disorders and Parkinson’s disease. Antioxid Redox Signal 21:471–484
Oh ES, Heo C, Kim JS, Suh M, Lee YH, Kim JM (2014) Hyperspectral fluorescence imaging for cellular iron mapping in the in vitro model of Parkinson’s disease. J Biomed Opt 19(5):051207. doi:10.1117/1.JBO.19.5.051207
Hare DJ, Arora M, Jenkins NL, Finkelstein DI, Doble PA, Bush AI (2015) Is early-life iron exposure critical in neurodegeneration? Nat Rev Neurol 11:536–544
Singh AK, Tiwari MN, Upadhyay G, Patel DK, Singh D, Prakash O, Singh MP (2012) Long-term exposure to cypermethrin induces the nigrostriatal dopaminergic neurodegeneration in adult rats: postnatal exposure enhances the susceptibility during adulthood. Neurobiol Aging 33:404–415
Galano A, Medina ME, Tan DX, Reiter RJ (2015) Melatonin and its metabolites as copper chelating agents and their role in inhibiting oxidative stress: a physicochemical analysis. J Pineal Res 58(1):107–116
Singhal NK, Srivastava G, Patel DK, Jain SK, Singh MP (2011) Melatonin or silymarin reduces maneb- and paraquat-induced Parkinson’s disease phenotype in the mouse. J Pineal Res 50:97–109
Ur Rasheed MS, Tripathi MK, Mishra AK, Shukla S, Singh MP (2015) Resveratrol protects from toxin-induced parkinsonism: plethora of proofs hitherto petty translational value. Mol Neurobiol. doi:10.1007/s12035-015-9124-3
Kumar A, Singh BK, Ahmad I, Shukla S, Patel DK, Srivastava G, Kumar V, Pandey HP, et al. (2012) Involvement of NADPH oxidase and glutathione in zinc-induced dopaminergic neurodegeneration in rats: similarity with paraquat neurotoxicity. Brain Res 1438:48–64
Grubman A, Lidgerwood GE, Duncan C, Bica L, Tan JL, Parker SJ, Caragounis A, Meyerowitz J, et al. (2014b) Deregulation of subcellular biometal homeostasis through loss of the metal transporter, Zip7, in a childhood neurodegenerative disorder. Acta Neuropathol Commun 2:25
Ward RJ, Dexter DT, Crichton RR (2012) Chelating agents for neurodegenerative diseases. Curr Med Chem 19:2760–2772
Aschner M (2000) Manganese: brain transport and emerging research needs. Environ Health Perspect 108S:429–432
Cheng P, Yu J, Huang W, Bai S, Zhu X, Qi Z, Shao W, Xie P (2015) Dietary intake of iron, zinc, copper, and risk of Parkinson’s disease: a meta-analysis. Neurol Sci 36:2269–2275
Bishop GM, Dang TN, Dringen R, Robinson SR (2011) Accumulation of non-transferrin-bound iron by neurons, astrocytes, and microglia. Neurotox Res 19:443–451
Agim ZS, Cannon JR (2015) Dietary factors in the etiology of Parkinson’s disease. BioMed Res Int 2015:672838. doi:10.1155/2015/672838
Ayton S, Lei P (2014) Nigral iron elevation is an invariable feature of Parkinson’s disease and is a sufficient cause of neurodegeneration. BioMed Res Int 2014:581256. doi:10.1155/2014/581256
Dashtipour K, Liu M, Kani C, Dalaie P, Obenaus A, Simmons D, Gatto NM, Zarifi M (2015) Iron accumulation is not homogenous among patients with Parkinson’s disease. Parkinsons Dis 2015:324843. doi:10.1155/2015/324843
Grolez G, Moreau C, Sablonnière B, Garçon G, Devedjian JC, Meguig S, Gelé P, Delmaire C, et al. (2015) Ceruloplasmin activity and iron chelation treatment of patients with Parkinson’s disease. BMC Neurol 15:74
Mochizuki H, Yasuda T (2012) Iron accumulation in Parkinson’s disease. J Neural Transm 119:1511–1514
You LH, Li F, Wang L, Zhao SE, Wang SM, Zhang LL, Zhang LH, Duan XL, et al. (2015) Brain iron accumulation exacerbates the pathogenesis of MPTP-induced Parkinson’s disease. Neuroscience 284:234–246
Fekete R (2012) Late onset neurodegeneration with brain-iron accumulation presenting as parkinsonism. Case Rep Neurol Med 2012:387095. doi:10.1155/2012/387095
Mena NP, García-Beltran O, Lourido F, Urrutia PJ, Mena R, Castro-Castillo V, Cassels BK, Nunez MT (2015) The novel mitochondrial iron chelator 5-((methylamino)methyl)-8-hydroxyquinoline protects against mitochondrial-induced oxidative damage and neuronal death. Biochem Biophys Res Commun 463:787–792
Olivieri S, Conti A, Iannaccone S, Cannistraci CV, Campanella A, Barbariga M, Codazzi F, Pelizzoni I, et al. (2011) Ceruloplasmin oxidation, a feature of Parkinson’s disease CSF, inhibits ferroxidase activity and promotes cellular iron retention. J Neurosci 31:18568–18518
Rinaldi DE, Corradi GR, Cuesta LM, Adamo HP, de Tezanos Pinto F (2015) The Parkinson-associated human P5B-ATPase ATP13A2 protects against the iron-induced cytotoxicity. Biochim Biophys Acta 1848:1646–1655
Wang J, Bi M, Xie J (2015) Ceruloplasmin is involved in the nigral iron accumulation of 6-OHDA-lesioned rats. Cell Mol Neurobiol 35:661–668
Workman DG, Tsatsanis A, Lewis FW, Boyle JP, Mousadoust M, Hettiarachchi NT, Hunter M, Peers CS, et al. (2015) Protection from neurodegeneration in the 6-hydroxydopamine (6-OHDA) model of Parkinson’s with novel 1-hydroxypyridin-2-one metal chelators. Metallomics 7:867–876
Jia W, Xu H, Du X, Jiang H, Xie J (2015) Ndfip1 attenuated 6-OHDA-induced iron accumulation via regulating the degradation of DMT1. Neurobiol Aging 36:1183–1193
Wang J, Song N, Jiang H, Wang J, Xie J (2013) Pro-inflammatory cytokines modulate iron regulatory protein 1 expression and iron transportation through reactive oxygen/nitrogen species production in ventral mesencephalic neurons. Biochim Biophys Acta 1832:618–625
Fukushima T, Tan X, Luo Y, Wang P, Song J, Kanda H, Hayakawa T, Kumagai T, et al. (2013) Heavy metals in blood and urine and its relation to depressive symptoms in Parkinson’s disease patients. Fukushima J Med Sci 59:76–80
Weinreb O, Mandel S, Youdim MB, Amit T (2013) Targeting dysregulation of brain iron homeostasis in Parkinson’s disease by iron chelators. Free Radic Biol Med 62:52–64
Ortega R, Carmona A, Roudeau S, Perrin L, Dučić T, Carboni E, Bohic S, Cloetens P, et al. (2015) α-synuclein over-expression induces increased iron accumulation and redistribution in iron-exposed neurons. Mol Neurobiol. doi:10.1007/s12035-015-9146-x
Horning KJ, Caito SW, Tipps KG, Bowman AB, Aschner M (2015) Manganese is essential for neuronal health. Annu Rev Nutr 35:71–108
Fukushima T, Tan X, Luo Y, Kanda H (2010) Relationship between blood levels of heavy metals and Parkinson’s disease in China. Neuroepidemiology 34:18–24
Park RM (2013) Neurobehavioral deficits and parkinsonism in occupations with manganese exposure: a review of methodological issues in the epidemiological literature. Saf Health Work 4:123–135
Kwakye GF, Paoliello MM, Mukhopadhyay S, Bowman AB, Aschner M (2015) Manganese-induced parkinsonism and Parkinson’s disease: shared and distinguishable features. Int J Environ Res Public Health 12:7519–7540
Milatovic D, Gupta RC, Yu Y, Zaja-Milatovic S, Aschner M (2011) Protective effects of antioxidants and anti-inflammatory agents against manganese-induced oxidative damage and neuronal injury. Toxicol Appl Pharmacol 256:219–226
Carmona A, Devès G, Roudeau S, Cloetens P, Bohic S, Ortega R (2010) Manganese accumulates within golgi apparatus in dopaminergic cells as revealed by synchrotron X-ray fluorescence nanoimaging. ACS Chem Neurosci 1:194–203
Li H, Wu S, Shi N, Lian S, Lin W (2011) Nrf2/HO-1 pathway activation by manganese is associated with reactive oxygen species and ubiquitin-proteasome pathway, not MAPKs signaling. J Appl Toxicol 31:690–697
O’Neal SL, Lee JW, Zheng W, Cannon JR (2014) Subacute manganese exposure in rats is a neurochemical model of early manganese toxicity. Neurotoxicology 44:303–313
Khalid M, Aoun RA, Mathews TA (2011) Altered striatal dopamine release following a sub-acute exposure to manganese. J Neurosci Methods 202:182–191
Cordova FM, Aguiar AS Jr, Peres TV, Lopes MW, Gonçalves FM, Pedro DZ, Lopes SC, Pilati C, et al. (2013) Manganese-exposed developing rats display motor deficits and striatal oxidative stress that are reversed by Trolox. Arch Toxicol 87:1231–1244
Liu Y, Barber DS, Zhang P, Liu B (2013) Complex II of the mitochondrial respiratory chain is the key mediator of divalent manganese-induced hydrogen peroxide production in microglia. Toxicol Sci 132:298–306
Brus R, Jochem J, Nowak P, Adwent M, Boroń D, Brus H, Kostrzewa RM (2012) Effect of pre- and postnatal manganese exposure on brain histamine content in a rodent model of Parkinson’s disease. Neurotox Res 21:143–148
Sriram K, Lin GX, Jefferson AM, Stone S, Afshari A, Keane MJ, McKinney W, Jackson M, et al. (2015) Modifying welding process parameters can reduce the neurotoxic potential of manganese-containing welding fumes. Toxicology 328:168–178
Sriram K, Lin GX, Jefferson AM, Roberts JR, Wirth O, Hayashi Y, Krajnak KM, Soukup JM, et al. (2010b) Mitochondrial dysfunction and loss of Parkinson’s disease-linked proteins contribute to neurotoxicity of manganese-containing welding fumes. FASEB J 24:4989–5002
Guilarte TR, Gonzales KK (2015) Manganese-induced parkinsonism is not idiopathic Parkinson’s disease: environmental and genetic evidence. Toxicol Sci 146:204–212
Aboud AA, Tidball AM, Kumar KK, Neely MD, Ess KC, Erikson KM, Bowman AB (2012) Genetic risk for Parkinson’s diseasecorrelates with alterations in neuronal manganese sensitivity between two human subjects. Neurotoxicology 33:1443–1449
Roth JA (2014) Correlation between the biochemical pathways altered by mutated Parkinson-related genes and chronic exposure to manganese. Neurotoxicology 44:314–325
Uversky VN, Li J, Fink AL (2011) Metal-triggered structural transformations, aggregation, and fibrillation of human alpha-synuclein: a possible molecular NK between Parkinson’s disease and heavy metal exposure. J Biol Chem 276:44284–44296
Sriram K, Lin GX, Jefferson AM, Roberts JR, Chapman RS, Chen BT, Soukup JM, Ghio AJ, et al. (2010a) Dopaminergic neurotoxicity following pulmonary exposure to manganese-containing welding fumes. Arch Toxicol 84:521–540
Ordoñez-Librado JL, Anaya-Martinez V, Gutierrez-Valdez AL, Montiel-Flores E, Corona DR, Martinez-Fong D, Avila-Costa MR (2010) L-DOPA treatment reverses the motor alterations induced by manganese exposure as a Parkinson disease experimental model. Neurosci Lett 471:79–82
Sanchez-Betancourt J, Anaya-Martínez V, Gutierrez-Valdez AL, Ordoñez-Librado JL, Montiel-Flores E, Espinosa-Villanueva J, Reynoso-Erazo L, Avila-Costa MR (2012) Manganese mixture inhalation is a reliable Parkinson disease model in rats. Neurotoxicology 33:1346–1355
Santos D, Batoréu MC, Tavares de Almeida I, Davis Randall L, Mateus ML, Andrade V, Ramos R, Torres E, et al. (2013) Evaluation of neurobehavioral and neuroinflammatory end-points in the post-exposure period in rats sub-acutely exposed to manganese. Toxicology 314:95–99
Guilarte TR (2010) Manganese and Parkinson’s disease: a critical review and new findings. Environ Health Perspect 118:1071–1080
Dexter DT, Wells FR, Lees AJ, Agid F, Agid Y, Jenner P, Marsden CD (1989) Increased nigral iron content and alterations in other metal ions occurring in brain in Parkinson’s disease. J Neurochem 52:1830–1836
Dudzik CG, Walter ED, Abrams BS, Jurica MS, Millhauser GL (2013) Coordination of copper to the membrane-bound form of α-synuclein. Biochemistry 52:53–60
Gorell JM, Johnson CC, Rybicki BA, Peterson EL, Kortsha GX, Brown GG, Richardson RJ (1999) Occupational exposure to manganese, copper, lead, iron, mercury and zinc and the risk of Parkinson’s disease. Neurotoxicology 20:239–247
Stelmashook EV, Isaev NK, Genrikhs EE, Amelkina GA, Khaspekov LG, Skrebitsky VG, Illarioshkin SN (2014) Role of zinc and copper ions in the pathogenetic mechanisms of Alzheimer’s and Parkinson’s diseases. Biochemistry (Mosc) 79:391–396
Hung LW, Villemagne VL, Cheng L, Sherratt NA, Ayton S, White AR, Crouch PJ, Lim S, et al. (2012) The hypoxia imaging agent CuII(atsm) is neuroprotective and improves motor and cognitive functions in multiple animal models of Parkinson’s disease. J Exp Med 209:837–854
Mariani S, Ventriglia M, Simonelli I, Donno S, Bucossi S, Vernieri F, Melgari JM, Pasqualetti P, et al. (2013) Fe and Cu do not differ in Parkinson’s disease: a replication study plus meta-analysis. Neurobiol Aging 34:632–633
Miyake Y, Tanaka K, Fukushima W, Sasaki S, Kiyohara C, Tsuboi Y, Yamada T, Oeda T, et al. (2011) Dietary intake of metals and risk of Parkinson’s disease: a case-control study in Japan. J Neurol Sci 306:98–102
Montes S, Rivera-Mancia S, Diaz-Ruiz A, Tristan-Lopez L, Rios C (2014) Copper and copper proteins in Parkinson’s disease. Oxidative Med Cell Longev. doi:10.1155/2014/147251
Boll MC, Alcaraz-Zubeldia M, Montes S, Rios C (2008) Free copper, ferroxidase and SOD1 activities, lipid peroxidation and NO(x) content in the CSF: a different marker profile in four neurodegenerative diseases. Neurochem Res 33:1717–1723
Rubio-Osornio M, Montes S, Heras-Romero Y, Guevara J, Rubio C, Aguilera P, Rivera-Mancia S, Floriano-Sánchez E, et al. (2013) Induction of ferroxidase enzymatic activity by copper reduces MPP + −evoked neurotoxicity in rats. Neurosci Res 75:250–255
Alcaraz-Zubeldia M, Boll-Woehrlen MC, Montes-López S, Pérez-Severiano F, Martínez-Lazcano JC, Díaz-Ruiz A, Ríos C (2009) Copper sulfate prevents tyrosine hydroxylase reduced activity and motor deficits in a Parkinson’s disease model in mice. Rev Investig Clin 61:405–411
Rubio-Osornio M, Montes S, Pérez-Severiano F, Aguilera P, Floriano-Sánchez E, Monroy-Noyola A, Rubio C, Ríos C (2009) Copper reduces striatal protein nitration and tyrosine hydroxylase inactivation induced by MPP+ in rats. Neurochem Int 54:447–451
Hozumi I (2013) Roles and therapeutic potential of metallothioneins in neurodegenerative diseases. Curr Pharm Biotechnol 14:408–413
Nishino Y, Ando M, Makino R, Ueda K, Okamoto Y, Kojima N (2011) Different mechanisms between copper and iron in catecholamines-mediated oxidative DNA damage and disruption of gene expression in vitro. Neurotox Res 20:84–92
Pham AN, Waite TD (2014) Cu(II)-catalyzed oxidation of dopamine in aqueous solutions: mechanism and kinetics. J Inorg Biochem 137:74–84
Jinsmaa Y, Sullivan P, Gross D, Cooney A, Sharabi Y, Goldstein DS (2014) Divalent metal ions enhance DOPAL-induced oligomerization of alpha-synuclein. Neurosci Lett 569:27–32
Paris I, Perez-Pastene C, Couve E, Caviedes P, Ledoux S, Segura-Aguilar J (2009) Copper dopamine complex induces mitochondrial autophagy preceding caspase-independent apoptotic cell death. J Biol Chem 284:13306–13315
Carboni E, Lingor P (2015) Insights on the interaction of alpha-synuclein and metals in the pathophysiology of Parkinson’s disease. Metallomics 7:395–404
Davies KM, Bohic S, Carmona A, Ortega R, Cottam V, Hare DJ, Finberg JP, Reyes S, et al. (2014) Copper pathology in vulnerable brain regions in Parkinson’s disease. Neurobiol Aging 35:858–866
Anandhan A, Rodriguez-Rocha H, Bohovych I, Griggs AM, Zavala-Flores L, Reyes-Reyes EM, Seravalli J, Stanciu LA, et al. (2015) Overexpression of alpha-synuclein at non-toxic levels increases dopaminergic cell death induced by copper exposure via modulation of protein degradation pathways. Neurobiol Dis 81:76–92
Wang X, Moualla D, Wright JA, Brown DR (2010) Copper binding regulates intracellular alpha-synuclein localisation, aggregation and toxicity. J Neurochem 113:704–714
André C, Truong TT, Robert JF, Guillaume YC (2005) Effect of metals on herbicides-alpha-synuclein association: a possible factor in neurodegenerative disease studied by capillary electrophoresis. Electrophoresis 26:3256–3264
De Ricco R, Valensin D, Dell’Acqua S, Casella L, Dorlet P, Faller P, Hureau C (2015) Remote His50 acts as a coordination switch in the high-affinity N-terminal centered copper(II) site of α-synuclein. Inorg Chem 54:4744–4751
Miotto MC, Rodriguez EE, Valiente-Gabioud AA, Torres-Monserrat V, Binolfi A, Quintanar L, Zweckstetter M, Griesinger C, et al. (2014b) Site-specific copper-catalyzed oxidation of α-synuclein: tightening the link between metal binding and protein oxidative damage in Parkinson’s disease. Inorg Chem 53:4350–4358
Lu Y, Prudent M, Fauvet B, Lashuel HA, Girault HH (2011) Phosphorylation of α-synuclein at Y125 and S129 alters its metal binding properties: implications for understanding the role of α-synuclein in the pathogenesis of Parkinson’s disease and related disorders. ACS Chem Neurosci 2:667–675
Miotto MC, Binolfi A, Zweckstetter M, Griesinger C, Fernández CO (2014a) Bioinorganic chemistry of synucleinopathies: deciphering the binding features of met motifs and his-50 in AS-Cu(I) interactions. J Inorg Biochem 141:208–211
Miotto MC, Valiente-Gabioud AA, Rossetti G, Zweckstetter M, Carloni P, Selenko P, Griesinger C, Binolfi A, et al. (2015) Copper binding to the N-terminally acetylated, naturally occurring form of alpha-synuclein induces local helical folding. J Am Chem Soc 137:6444–6447
Moriarty GM, Minetti CA, Remeta DP, Baum J (2014) A revised picture of the Cu(II)-α-synuclein complex: the role of N-terminal acetylation. Biochemistry 53:2815–2817
Tavassoly O, Nokhrin S, Dmitriev OY, Lee JS (2014) Cu(II) and dopamine bind to α-synuclein and cause large conformational changes. FEBS J 281:2738–2753
Lucas HR, Lee JC (2011) Copper(II) enhances membrane-bound α-synuclein helix formation. Metallomics 3:280–283
Ha Y, Yang A, Lee S, Kim K, Liew H, Lee SH, Lee JE, Lee HI, et al. (2014) Dopamine and Cu+/2+ can induce oligomerization of α-synuclein in the absence of oxygen: two types of oligomerization mechanisms for α-synuclein and related cell toxicity studies. J Neurosci Res 92:359–368
Tashiro S, Caaveiro JM, Wu CX, Hoang QQ, Tsumoto K (2014) Thermodynamic and structural characterization of the specific binding of Zn(II) to human protein DJ-1. Biochemistry 53:2218–2220
Girotto S, Cendron L, Bisaglia M, Tessari I, Mammi S, Zanotti G, Bubacco L (2014) DJ-1 is a copper chaperone acting on SOD1 activation. J Biol Chem 289:10887–10899
Puno MR, Patel NA, Møller SG, Robinson CV, Moody PC, Odell M (2013) Structure of Cu(I)-bound DJ-1 reveals a biscysteinate metal binding site at the homodimer interface: insights into mutational inactivation of DJ-1 in parkinsonism. J Am Chem Soc 135:15974–15977
Björkblom B, Adilbayeva A, Maple-Grødem J, Piston D, Ökvist M, Xu XM, Brede C, Larsen JP, et al. (2013) Parkinson disease protein DJ-1 binds metals and protects against metal-induced cytotoxicity. J Biol Chem 288:22809–22820
Aboud AA, Tidball AM, Kumar KK, Neely MD, Han B, Ess KC, Hong CC, Erikson KM, et al. (2015) PARK2 patient neuroprogenitors show increased mitochondrial sensitivity to copper. Neurobiol Dis 73:204–212
Garcia SJ, Gellein K, Syversen T, Aschner M (2007) Iron deficient and manganese supplemented diets alter metals and transporters in the developing rat brain. Toxicol Sci 95:205–214
Barnham KJ, Bush AI (2008) Metals in Alzheimer’s and Parkinson’s diseases. Curr Opin Chem Biol 12:222–228
Méndez-Alvarez E, Soto-Otero R, Hermida-Ameijeiras A, López-Real AM, Labandeira-García JL (2002) Effects of aluminum and zinc on the oxidative stress caused by 6-hydroxydopamine autoxidation: relevance for the pathogenesis of Parkinson’s disease. Biochim Biophys Acta 1586:155–168
Prasad AS (2009) Impact of the discovery of human zinc deficiency on health. J Am Coll Nutr 28:257–265
Fukushima T, Tan X, Luo Y, Kanda H (2011) Serum vitamins and heavy metals in blood and urine, and the correlations among them in Parkinson’s disease patients in China. Neuroepidemiology 36:240–244
Jomova K, Vondrakova D, Lawson M, Valko M (2010) Metals, oxidative stress and neurodegenerative disorders. Mol Cell Biochem 345:91–104
Frederickson CJ, Suh SW, Silva D, Frederickson CJ, Thompson RB (2000) Importance of zinc in the central nervous system: the zinc-containing neuron. J Nutr 130:1471S–1483S
Bassil F, Monvoisin A, Canron MH, Vital A, Meissner WG, Tison F, Fernagut PO (2015) Region-specific alterations of matrix metalloproteinase activity in multiple system atrophy. Mov Disord. doi:10.1016/j.nbd.2014.11.018
Grubman A, Pollari E, Duncan C, Caragounis A, Blom T, Volitakis I, Wong A, Cooper J, et al. (2014a) Deregulation of biometal homeostasis: the missing link for neuronal ceroid lipofuscinoses? Metallomics 6:932–943
Lee JY, Son HJ, Choi JH, Cho E, Kim J, Chung SJ, Hwang O, Koh JY (2009) Cytosolic labile zinc accumulation in degenerating dopaminergic neurons of mouse brain after MPTP treatment. Brain Res 1286:208–214
Sheline CT, Zhu J, Zhang W, Shi C, Cai AL (2013) Mitochondrial inhibitor models of Huntington’s disease and Parkinson’s disease induce zinc accumulation and are attenuated by inhibition of zinc neurotoxicity in vitro or in vivo. Neurodegener Dis 11:49–58
Sharma SK, Ebadi M (2003) Metallothionein attenuates (SIN-1)-induced oxidative stress in dopaminergic neurons. Antioxid Redox Signal 5:251–264
Fujishiro H, Yoshida M, Nakano Y, Himeno S (2014) Interleukin-6 enhances manganese accumulation in SH-SY5Y cells: implications of the up-regulation of ZIP14 and the down-regulation of ZnT10. Metallomics 6:944–949
Robison G, Zakharova T, Fu S, Jiang W, Fulper R, Barrea R, Zheng W, Pushkar Y (2013) X-ray fluorescence imaging of the hippocampal formation after manganese exposure. Metallomics 5:1554–1565
Hubbs-Tait L, Nation JR, Krebs NF, Bellinger DC (2005) Neurotoxicants, micronutrients, and social environments: individual and combined effects on children’s development. Psychol Sci Public Interest 6:57–121
Park JS, Blair NF, Sue CM (2015) The role of ATP13A2 in Parkinson’s disease: clinical phenotypes and molecular mechanisms. Mov Disord 30:770–779
Bates MN (2006) Mercury amalgam dental fillings: an epidemiologic assessment. Int J Hyg Environ Health 209:309–316
Goetz CG (2010) Shaking up the Salpetriere: Jean-Martin Charcot and mercury-induced tremor. Neurology 74:1739–1742
Reinhardt JW (1992) Side-effects: mercury contribution to body burden from dental amalgam. Adv Dent Res 6:110–113
Lin CY, Liou SH, Hsiech CM, Ku MC, Tsai SY (2011) Dose-response relationship between cumulative mercury exposure index and specific uptake ratio in the striatum on Tc-99 m TRODAT SPECT. Clin Nucl Med 36:689–693
Ngim CH, Devathasan G (1989) Epidemiologic study on the association between body burden mercury level and idiopathic Parkinson’s disease. Neuroepidemiology 8:128–141
Ohlson CG, Hogstedt C (1981) Parkinson’s disease and occupational exposure to organic solvents, agricultural chemicals and mercury--a case-referent study. Scand J Work Environ Health 7:252–256
Finkelstein Y, Vardi J, Kesten MM, Hod I (1996) The enigma of parkinsonism in chronic borderline mercury intoxication, resolved by challenge with penicillamine. Neurotoxicology 17:291–295
Dantzig PI (2006) Parkinson’s disease, macular degeneration and cutaneous signs of mercury toxicity. J Occup Environ Med 48:656
Biernat H, Ellias SA, Wermuth L, Cleary D, de Oliveira Santos EC, Jorgensen PJ, Feldman RG, Grandjean P (1999) Tremor frequency patterns in mercury vapor exposure, compared with early Parkinson’s disease and essential tremor. Neurotoxicology 20:945–952
Charles LE, Burchfiel CM, Fekedulegn D, Kashon ML, Ross GW, Petrovitch H, Sanderson WT (2006) Occupational exposures and movement abnormalities among Japanese-American men: the Honolulu-Asia aging study. Neuroepidemiology 26:130–139
Kolisek M, Nestler A, Vormann J, Schweigel-Röntgen M (2012) Human gene SLC41A1 encodes for the Na+/Mg2 + exchanger. Am J Phys Cell Phys 302:C318–C326
Taniguchi R, Nakagawasai O, Tan-no K, Yamadera F, Nemoto W, Sato S, Yaoita F, Tadano T (2013) Combined low calcium and lack magnesium is a risk factor for motor deficit in mice. Biosci Biotechnol Biochem 77:266–270
Lee M, Jantaratnotai N, McGeer E, McLarnon JG, McGeer PL (2011) Mg2+ ions reduce microglial and THP-1 cell neurotoxicity by inhibiting Ca2+ entry through purinergic channels. Brain Res 1369:21–35
Golts N, Snyder H, Frasier M, Theisler C, Choi P, Wolozin B (2002) Magnesium inhibits spontaneous and iron-induced aggregation of alpha-synuclein. J Biol Chem 277:16116–16123
Iotti S, Malucelli E (2008) In vivo assessment of Mg2+ in human brain and skeletal muscle by 31P-MRS. Magnes Res 21:157–162
Kiss SA, Dombovári J, Oncsik M (1991) Magnesium inhibits the harmful effects on plants of some toxic elements. Magnes Res 4:3–7
Yasui M, Kihira T, Ota K (1992) Calcium, magnesium and aluminum concentrations in Parkinson’s disease. Neurotoxicology 13:593–600
Hashimoto T, Nishi K, Nagasao J, Tsuji S, Oyanagi K (2008) Magnesium exerts both preventive and ameliorating effects in an in vitro rat Parkinson disease model involving 1-methyl-4-phenylpyridinium (MPP+) toxicity in dopaminergic neurons. Brain Res 1197:143–151
Tariq M, Khan HA, al Moutaery K, al Deeb SM (1998) Effect of chronic administration of magnesium sulfate on 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced neurotoxicity in mice. Pharmacol Toxicol 82:218–222
Oyanagi K, Kawakami E, Kikuchi-Horie K, Ohara K, Ogata K, Takahama S, Wada M, Kihira T, et al. (2006) Magnesium deficiency over generations in rats with special references to the pathogenesis of the parkinsonism-dementia complex and amyotrophic lateral sclerosis of Guam. Neuropathology 26:115–128
Alleyne T, Mohan N, Adogwa A (2011) Elevated ferric, calcium and magnesium ions in the brain induce protein aggregation in brain mitochondria. West Indian Med J 61:122–127
Wu YN, Johnson SW (2009) Rotenone reduces Mg2 + −dependent block of NMDA currents in substantia nigra dopamine neurons. Neurotoxicology 30:320–325
Shindo Y, Yamanaka R, Suzuki K, Hotta K, Oka K (2015) Intracellular magnesium level determines cell viability in the MPP+ model of Parkinson’s disease. Biochim Biophys Acta. doi:10.1016/j.bbamcr.2015.08.013
Shindo Y, Fujii T, Komatsu H, Citterio D, Hotta K, Suzuki K, Oka K (2011) Newly developed Mg2 + −selective fluorescent probe enables visualization of Mg2+ dynamics in mitochondria. PLoS One 6(8):e23684. doi:10.1371/journal.pone.0023684
Kolisek M, Sponder G, Mastrototaro L, Smorodchenko A, Launay P, Vormann J, Schweigel-Röntgen M (2013) Substitution p.A350 V in Na2+/Mg2+ exchanger SLC41A1, potentially associated with Parkinson’s disease, is a gain-of-function mutation. PLoS One 8(8):e71096. doi:10.1371/journal.pone.0071096
Lin CH, Wu YR, Chen WL, Wang HC, Lee CM, Lee-Chen GJ, Chen CM (2014) Variant R244H in Na+/Mg2+ exchanger SLC41A1 in Taiwanese Parkinson’s disease is associated with loss of Mg2+ efflux function. Parkinsonism Relat Disord 20:600–603
Vink R, Cook NL, van den Heuvel C (2009) Magnesium in acute and chronic brain injury: an update. Magnes Res 22:158S–162S
Ådén E, Carlsson M, Poortvliet E, Stenlund H, Linder J, Edström M, Forsgren L, Håglin L (2011) Dietary intake and olfactory function in patients with newly diagnosed Parkinson’s disease: a case-control study. Nutr Neurosci 14(1):25–31
Forte G, Alimonti A, Violante N, Di Gregorio M, Senofonte O, Petrucci F, Sancesario G, Bocca B (2005) Calcium, copper, iron, magnesium, silicon and zinc content of hair in Parkinson’s disease. J Trace Elem Med Biol 19:195–201
Neal AP, Guilarte TR (2013) Mechanisms of lead and manganese neurotoxicity. Toxicol Res 2:99–114
Palacios N, Fitzgerald K, Roberts AL, Hart JE, Weisskopf MG, Schwarzschild MA, Ascherio A, Laden F (2014) A prospective analysis of airborne metal exposures and risk of Parkinson disease in the nurses’ health study cohort. Environ Health Perspect 122:933–938
Coon S, Stark A, Peterson E, Gloi A, Kortsha G, Pounds J, Chettle D, Gorell J (2006) Whole-body lifetime occupational lead exposure and risk of Parkinson’s disease. Environ Health Perspect 114:1872–1876
Kumudini N, Uma A, Devi YP, Naushad SM, Mridula R, Borgohain R, Kutala VK (2014) Association of Parkinson’s disease with altered serum levels of lead and transition metals among south Indian subjects. Indian J Biochem Biophys 51:121–126
Momcilovic B, Alkhatib HA, Duerre JA, Cooley M, Long WM, Harris TR, Lykken GI (2001) Environmental lead-210 and bismuth-210 accrue selectively in the brain proteins in Alzheimer disease and brain lipids in Parkinson disease. Alzheimer Dis Assoc Disord 15:106–115
Weisskopf MG, Weuve J, Nie H, Saint-Hilaire MH, Sudarsky L, Simon DK, Hersh B, Schwartz J, et al. (2010) Association of cumulative lead exposure with Parkinson’s disease. Environ Health Perspect 118:1609–16013
Weuve J, Press DZ, Grodstein F, Wright RO, Hu H, Weisskopf MG (2013) Cumulative exposure to lead and cognition in persons with Parkinson’s disease. Mov Disord 28:176–182
Sanz P, Nogué S, Vilchez D, Vilchez J, Casal A, Logroscino G (2007) Progressive supranuclear palsy-like parkinsonism resulting from occupational exposure to lead sulphate batteries. J Int Med Res 35:159–163
Grunberg-Etkovitz N, Lev N, Ickowicz D, Avital A, Offen D, Malik Z (2009) Accelerated proteasomal activity induced by Pb2+, Ga3+, or Cu2+ exposure does not induce degradation of alpha-synuclein. J Environ Pathol Toxicol Oncol 28:5–24
Altschuler E (1999) Aluminum-containing antacids as a cause of idiopathic Parkinson’s disease. Med Hypotheses 53:22–23
Singla N, Dhawan DK (2014) Influence of zinc on calcium-dependent signal transduction pathways during aluminium-induced neurodegeneration. Mol Neurobiol 50:613–625
Yanagihara R, Garruto RM, Gajdusek DC, Tomita A, Uchikawa T, Konagaya Y, Chen KM, Sobue I, et al. (1984) Calcium and vitamin D metabolism in Guamanian Chamorros with amyotrophic lateral sclerosis and parkinsonism-dementia. Ann Neurol 15:42–48
Erazi H, Ahboucha S, Gamrani H (2011) Chronic exposure to aluminum reduces tyrosine hydroxylase expression in the substantia nigra and locomotor performance in rats. Neurosci Lett 487:8–11
Hirsch EC, Brandel JP, Galle P, Javoy-Agid F, Agid Y (1991) Iron and aluminum increase in the substantia nigra of patients with Parkinson’s disease: an X-ray microanalysis. J Neurochem 56:446–451
Zatta P, Zambenedetti P, Milanese M (1999) Activation of monoamine oxidase type-B by aluminum in rat brain homogenate. Neuroreport 10:3645–3648
Good PF, Olanow CW, Perl DP (1992) Neuromelanin-containing neurons of the substantia nigra accumulate iron and aluminum in Parkinson’s disease: a LAMMA study. Brain Res 593:343–346
Meglio L, Oteiza PI (1999) Aluminum enhances melanin-induced lipid peroxidation. Neurochem Res 24:1001–1008
Komatsu F, Kagawa Y, Kawabata T, Kaneko Y, Chimedregzen U, Purvee B, Otgon J (2011) A high accumulation of hair minerals in Mongolian people: 2(nd) report; influence of manganese, iron, lead, cadmium and aluminum to oxidative stress, parkinsonism and arthritis. Curr Aging Sci 4:42–56
Sánchez-Iglesias S, Méndez-Alvarez E, Iglesias-González J, Muñoz-Patiño A, Sánchez-Sellero I, Labandeira-García JL, Soto-Otero R (2009) Brain oxidative stress and selective behaviour of aluminium in specific areas of rat brain: potential effects in a 6-OHDA-induced model of Parkinson’s disease. J Neurochem 109:879–888
Long X, Zhang C, Cheng J, Bi S (2008) A novel method for study of the aggregation of protein induced by metal ion aluminum(III) using resonance Rayleigh scattering technique. Spectrochim Acta A Mol Biomol Spectrosc 69:71–77
Ahmed SS, Santosh W (2010) Metallomic profiling and linkage map analysis of early Parkinson’s disease: a new insight to aluminum marker for the possible diagnosis. PLoS One 5(6):e11252. doi:10.1371/journal.pone.0011252
Belal C, Ameli NJ, El Kommos A, Bezalel S, Al’Khafaji AM, Mughal MR, Mattson MP, Kyriazis GA, et al. (2012) The homocysteine-inducible endoplasmic reticulum (ER) stress protein Herp counteracts mutant α-synuclein-induced ER stress via the homeostatic regulation of ER-resident calcium release channel proteins. Hum Mol Genet 21:963–977
Chigurupati S, Wei Z, Belal C, Vandermey M, Kyriazis GA, Arumugam TV, Chan SL (2009) The homocysteine-inducible endoplasmic reticulum stress protein counteracts calcium store depletion and induction of CCAAT enhancer-binding protein homologous protein in a neurotoxin model of Parkinson disease. J Biol Chem 284:18323–18333
Caraveo G, Auluck PK, Whitesell L, Chung CY, Baru V, Mosharov EV, Yan X, Ben-Johny M, et al. (2014) Calcineurin determines toxic versus beneficial responses to α-synuclein. Proc Natl Acad Sci U S A 111:E3544–E3552
Rousseau E, Michel PP, Hirsch EC (2013) The iron-binding protein lactoferrin protects vulnerable dopamine neurons from degeneration by preserving mitochondrial calcium homeostasis. Mol Pharmacol 84:888–898
Overk CR, Rockenstein E, Florio J, Cheng Q, Masliah E (2015) Differential calcium alterations in animal models of neurodegenerative disease: reversal by FK506. Neuroscience. doi:10.1016/j.neuroscience.2015.08.068
Cali T, Ottolini D, Negro A, Brini M (2012) α-synuclein controls mitochondrial calcium homeostasis by enhancing endoplasmic reticulum-mitochondria interactions. J Biol Chem 287:17914–17929
Marongiu R, Spencer B, Crews L, Adame A, Patrick C, Trejo M, Dallapiccola B, Valente EM, et al. (2009) Mutant Pink1 induces mitochondrial dysfunction in a neuronal cell model of Parkinson’s disease by disturbing calcium flux. J Neurochem 108:1561–1567
Gandhi S, Wood-Kaczmar A, Yao Z, Plun-Favreau H, Deas E, Klupsch K, Downward J, Latchman DS, et al. (2009) PINK1-associated Parkinson’s disease is caused by neuronal vulnerability to calcium-induced cell death. Mol Cell 33:627–638
Eaton ME, Macias W, Youngs RM, Rajadhyaksha A, Dudman JT, Konradi C (2004) L-type Ca2+ channel blockers promote Ca2+ accumulation when dopamine receptors are activated in striatal neurons. Brain Res Mol Brain Res 131:65–72
Visch HJ, Rutter GA, Koopman WJ, Koenderink JB, Verkaart S, de Groot T, Varadi A, Mitchell KJ, et al. (2004) Inhibition of mitochondrial Na + −Ca2+ exchange restores agonist-induced ATP production and Ca2+ handling in human complex I deficiency. J Biol Chem 279:40328–40336
Lowe R, Pountney DL, Jensen PH, Gai WP, Voelcker NH (2004) Calcium(II) selectively induces alpha-synuclein annular oligomers via interaction with the C-terminal domain. Protein Sci 13:3245–3252
Jumpponen M, Rönkkömäki H, Pasanen P, Laitinen J (2014) Occupational exposure to solid chemical agents in biomass-fired power plants and associated health effects. Chemosphere 104:25–31
Dhillon AS, Tarbutton GL, Levin JL, Plotkin GM, Lowry LK, Nalbone JT, Shepherd S (2008) Pesticide/environmental exposures and Parkinson’s disease in East Texas. J Agromed 13:37–48
Okuda B, Iwamoto Y, Tachibana H, Sugita M (1997) Parkinsonism after acute cadmium poisoning. Clin Neurol Neurosurg 99:263–265
Yu X, Robinson JF, Sidhu JS, Hong S, Faustman EM (2010) A system-based comparison of gene expression reveals alterations in oxidative stress, disruption of ubiquitin-proteasome system and altered cell cycle regulation after exposure to cadmium and methylmercury in mouse embryonic fibroblast. Toxicol Sci 114:356–377
Shi MT (1991) Determination of multiple chemical elements in CSF in Parkinson disease after intracerebral auto transplantation of the adrenal medulla. Zhonghua Wai Ke Za Zhi 29:129–132
Felix K, Manna SK, Wise K, Barr J, Ramesh GT (2005) Low levels of arsenite activates nuclear factor-kappaB and activator protein-1 in immortalized mesencephalic cells. J Biochem Mol Toxicol 19:67–77
Shavali S, Sens DA (2008) Synergistic neurotoxic effects of arsenic and dopamine in human dopaminergic neuroblastoma SH-SY5Y cells. Toxicol Sci 102:254–261
Schmuck EM, Board PG, Whitbread AK, Tetlow N, Cavanaugh JA, Blackburn AC, Masoumi A (2005) Characterization of the monomethylarsonate reductase and dehydroascorbate reductase activities of omega class glutathione transferase variants: implications for arsenic metabolism and the age-at-onset of Alzheimer’s and Parkinson’s diseases. Pharmacogenet Genomics 15:493–501
Larsen NA, Pakkenberg H, Damsgaard E, Heydorn K, Wold S (1981) Distribution of arsenic, manganese, and selenium in the human brain in chronic renal insufficiency, Parkinson’s disease, and amyotrophic lateral sclerosis. J Neurol Sci 51:437–446
Ala A, Walker AP, Ashkan K, Dooley JS, Schilsky ML (2007) Wilson’s disease. Lancet 369(9559):397–408
Crownover BK, Covey CJ (2013) Hereditary hemochromatosis. Am Fam Physician 87:183–190
Pietrangelo A (2003) Haemochromatosis. Gut 52 S(2):ii23–ii30. doi:10.1136/gut.52.suppl_2.ii23
Wang B, Du Y (2013) Cadmium and its neurotoxic effects. Oxidative Med Cell Longev 2013:898034
Olanow CW, Arendash GW (1994) Metals and free radicals in neurodegeneration. Curr Opin Neurol 7:548–558
Seidler A, Hellenbrand W, Robra BP, Vieregge P, Nischan P, Joerg J, Oertel WH, Ulm G, et al. (1996) Possible environmental, occupational, and other etiologic factors for Parkinson’s disease: a case-control study in Germany. Neurology 46:1275–1284
Molina JA, Jiménez-Jiménez FJ, Navarro JA, Vargas C, Gómez P, Benito-León J, Ortí-Pareja M, Cisneros E, et al. (1996) Cerebrospinal fluid nitrate levels in patients with Parkinson’s disease. Acta Neurol Scand 93:123–126
Jenner P (2003) Dopamine agonists, receptor selectivity and dyskinesia induction in Parkinson’s disease. Curr Opin Neurol 16:S3–S9
Ratner MH, Farb DH, Ozer J, Feldman RG, Durso R (2014) Younger age at onset of sporadic Parkinson’s disease among subjects occupationally exposed to metals and pesticides. Interdiscip Toxicol 7:123–133
Singh BK, Kumar A, Ahmad I, Kumar V, Patel DK, Jain SK,Singh C (2011) Oxidative stress in zinc-induced dopaminergic neurodegeneration: implications of superoxide dismutase and heme oxygenase-1. Free Radic Res 45:1207–1222
Fitsanakis VA, Amarnath V, Moore JT, Montine KS, Zhang J, Montine TJ (2002) Catalysis of catechol oxidation by metal-dithiocarbamate complexes in pesticides. Free Radic Biol Med 33:1714–1723
Yang Y, Ye X, He B, Liu J (2015) Cadmium potentiates toxicity of cypermethrin in zebrafish. Environ Toxicol Chem. doi:10.1002/etc.3200
Panemangalore M, Bebe FN (2005) Interaction between pesticides and essentialmetalcopperincreases the accumulation of copper in the kidneys of rats. Biol Trace Elem Res 108:169–184
Acknowledgments
Department of Science and Technology, India, Indian Council of Medical Research, India, and University Grants Commission, India are deeply acknowledged for rendering research fellowship to Mohd Sami ur Rasheed, Sonam Tripathi, and Saumya Mishra, respectively. The work was monetarily supported by the CSIR through a network project entitled, “Neurodegenerative Diseases: Causes and Corrections (BSC0115/Molecules in Neurodegenerative Diseases/miND)”. The CSIR-IITR communication number of this article is 3357.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors state no conflicts of interest.
Additional information
IITR Communication Number: 3357
Mohd Sami ur Rasheed and Sonam Tripathi contributed equally to this work.
Rights and permissions
About this article

Cite this article
Rasheed, M.S.u., Tripathi, S., Mishra, S. et al. Coherent and Contradictory Facts, Feats and Fictions Associated with Metal Accumulation in Parkinson’s Disease: Epicenter or Outcome, Yet a Demigod Question. Mol Neurobiol 54, 4738–4755 (2017). https://doi.org/10.1007/s12035-016-0016-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-016-0016-y