
Abstract
Antibiotic and herbicide resistance genes have been used in transgene technology as powerful selection tools. Nonetheless, once transgenic events have been obtained their presence is no longer needed and can even be undesirable. In this work, we have developed a system to excise the selectable marker and the cre recombinase genes from transgenic banana cv. ‘Grande Naine’ (Musa AAA). To achieve this, the embryo specific REG-2 promoter was isolated from rice and its expression pattern in banana cell clumps, somatic embryos and regenerated plantlets was characterized by using a pREG2::uidA fusion construct. Subsequently, the REG-2 promoter was placed upstream of the cre gene, conferring Cre functionality in somatic embryos and recombination of lox sites resulting in excision of the selectable marker and cre genes. PCR analysis revealed that 41.7 % of the analysed transgenic plants were completely marker free, results that were thereafter confirmed by Southern blot hybridization. These results demonstrate the feasibility of using developmentally controlled promoters to mediate marker excision in banana. This system does not require any extra handling compared to the conventional transformation procedure and might be useful in other species regenerating through somatic embryogenesis.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Plant genetic engineering is a technology that is critical for future food, feed, energy, and even pharmaceutical needs (Castle et al. 2008; Eckardt et al. 2009; Edgerton 2009; Farré et al. 2010; Varshney et al. 2011). It has a notable potential to engage important socioeconomic problems, especially in the developing world (Farré et al. 2010). In order to produce genetically engineered plants, firstly a recombinant DNA has to be introduced in the plant cell and secondly, cells that have integrated the DNA into the appropriate plant genome (nuclear or plastid) should be differentiated from the cells that do not contain the recombinant DNA. This distinction is possible by the use of selectable marker genes (SMGs), which in most cases are genes that encode proteins conferring resistance to antibiotics or herbicides (Miki and McHugh 2004). Thus, SMGs are considered indispensable for transgene technologies. However, the use of SMGs is one of the major issues of public concern regarding biotech crops. In addition, the continued presence of SMGs after selection is not necessary and on the contrary, may raise technological problems. For example, it precludes the retransformation with the same selectable marker gene. In addition, their regulatory sequences (promoter and terminator) can influence the expression of other transgenes and/or endogenous genes. Moreover, some SMGs may induce pleiotropic effects under certain conditions (Abdeen and Miki 2009; Miki et al. 2009). Thus, as new technologies become available, recommendations have been made to avoid or eliminate marker genes from genetically modified plants (Corrado and Karali 2009).
Several approaches have been developed to obtain marker-free transgenic plants (reviewed by Hare and Chua 2002; Puchta 2003; Darbani et al. 2007), but some of these are difficult or impossible to apply in crops that have long generation times or are vegetatively propagated. For these plant species the most suitable way to obtain marker-free transgenic plants is the use of site-specific recombination systems like Cre/lox from bacteriophage P1 (Hoess et al. 1982; Hoess and Abremski 1985), FLP/frt from Saccharomyces cerevisiae (Cox 1983; Senecoff et al. 1985) or R/RS from Zygosaccharomyces rouxii (Araki et al. 1985), combined with an auto-excision strategy. In this strategy, the recombinase gene (cre, FLP or R) is placed under the control of an inducible or tissue specific promoter. Then, on the same vector, the SMG and recombinase expression cassettes are placed between two directly repeated recombination sites (lox, frt or RS). Once selection is achieved, recombinase expression can be activated. The activation of the recombinase can be induced either chemically (Sugita et al. 2000; Zuo et al. 2001; Schaart et al. 2004; Sreekala et al. 2005; Zhang et al. 2006), by heat shock (Kilby et al. 1995; Hoff et al. 2001; Zhang et al. 2003; Wang et al. 2005; Cuellar et al. 2006; Luo et al. 2008; Fladung et al. 2010; Rao et al. 2010; Akbudak and Srivastava, 2011; Chong-Pérez et al. 2012a) or by tissue specific promoters (Mlynárová et al. 2006; Luo et al. 2007, Li et al. 2007; Verweire et al. 2007; Bai et al. 2008; Moravčíková et al. 2008; Kopertekh et al. 2009; 2010). The latter system to induce the recombinase activity has never been tested in vegetatively propagated species like banana (Musa spp.).
Bananas and plantain (Musa spp.) are important crops in the context of food production and income commodities for many countries in the developing world. However their production is seriously affected by some important constraints, like pathogenic fungi, bacteria, viruses, nematodes and abiotic stress factors. Research has been focused on improving some economically important cultivars like those that belong to the Cavendish subgroup (Musa AAA). However, classical breeding efforts have been limited because of the polyploidy, parthenocarpy and long life cycle of the commercial varieties. As an alternative, genetic transformation methods have been developed in the last two decades to transfer foreign DNA to the banana genome. These include protoplast electroporation (Sági et al. 1994), particle bombardment (Sági et al. 1995; Becker et al. 2000; Vishnevetsky et al. 2011) and A. tumefaciens (May et al. 1995; Ganapathi et al. 2001; Khanna et al. 2004; Acereto-Escoffié et al. 2005; Pei et al. 2005; Tripathi et al. 2005; Pérez-Hernández et al. 2006; Ghosh et al. 2009; Subramanyam et al. 2011). In these methods only three SMGs have been used, the antibiotic resistance genes nptII and hpt, and the herbicide resistance gene als. Besides reducing public concerns and technical problems described above, the release of marker-free transgenic banana might also reduce the need for time-consuming and expensive safety evaluations, which at the same time means reduction in cost in developing and marketing new GM products (Hare and Chua 2002).
In the present work we evaluated the promoter of the rice embryo globulin gene (REG-2) (Sun et al. 1996; Qu and Takaiwa 2004) to control cre gene expression as the basis for a marker gene auto-excision system. The activity of the REG-2 promoter in rice was reported as localized exclusively in seed embryo and aleurone with no expression in the endosperm (Qu and Takaiwa 2004). We verified the expression pattern conferred by this promoter in the course of a banana transformation/regeneration procedure. We next tested whether the REG-2 promoter is able to drive excision of the antibiotic resistance gene hpt and the cre gene during development of transgenic somatic embryos in banana.
Materials and methods
Cloning of the rice embryo globulin gene (REG-2) promoter in destination vector pGW-A and pCAMBIA1301
A 1.252 kb (upstream of the ATG) promoter fragment of the REG-2 gene was amplified from genomic DNA of rice (Oryza sativa, cv. Nipponbare, japonica group) using primers containing the attB1 and attB2 sites for Gateway™ cloning and EcoRI and NcoI (underlined letters) for classical cloning. Gateway primers used to clone the REG-2 promoter (bold is the sequence-specific part) were as follows: REG-2 forward (EcoRI), 5′-GGG GAC AAG TTT GTA CAA AAA AGC AGG CTG AGA ATT C GT CGA CGA GCG AGT CAT TAG-3′ and REG-2 reverse (NcoI), R: 5′-GGG GAC CAC TTT GTA CAA GAA AGC TGG GT CCA TGG AGG TGT TCG ATC GAT CCT AGC-3′. PCR was carried out using PrimeSTAR™HS DNA polymerase (Takara, Japan) in 50 μl reaction mixture containing 1 μl of total DNA (200 ng), 10 μl of 5X PCR Prime STAR™ buffer, 400 μM of each dNTP, 5 pM of each primer and 0.625 u of DNA polymerase. The REG-2 promoter fragment was amplified in a Mastercycler (Eppendorf, Germany) with the following conditions: 98 °C for 1 min, followed by 35 cycles of denaturation at 98 °C for 10 s, annealing at 55 °C for 10 s, and synthesis at 72 °C for 90 s, and by a final extension at 72 °C for 10 min. The PCR product was introduced in pDONR-Zeo™ via BP reaction (overnight), following the manufacturer’s instructions (Invitrogen, USA), resulting in the REG-2 Entry clone™.
Subsequently, the EcoRI-NcoI fragment from the REG-2 Entry clone™ was used to substitute the 35S promoter region in the EcoRI-NcoI sites of pCAMBIA1301 (CAMBIA, Canberra, Australia), forming the pREG2::uidA plasmid (Fig. 1a). This vector allowed us to study the tissue specificity and expression pattern of the REG-2 promoter. Furthermore, plasmid pCAMBIA1301 was used as a control. The promoter was introduced in pGW-A (Verweire et al. 2007) to form the pREG2-A plasmid (Fig. 1b) via a gateway LR reaction. The LR product and pREG2::uidA were transformed in DH5α (Invitrogen) and grown on semisolid LB medium supplemented with 50 mg/l kanamycin at 37 °C, overnight. Ten to twenty colonies were selected and grown in LB liquid medium and DNA was isolated using a GenElute™ Plasmid DNA miniprep kit (Sigma, Germany) and checked by enzymatic restriction analysis, followed by gel electrophoresis. Both clones were verified by sequencing. Binary vector plasmids were transformed into Agrobacterium tumefaciens strain EHA105 (Hood et al. 1993) by the freeze–thaw method (Holsters et al. 1978).

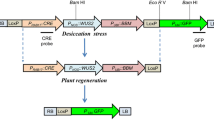
Diagrams of the T-DNA region of the plasmids used for genetic transformation experiments. a pREG2::uidA. A promoter testing plasmid derived from pCAMBIA1301 with the REG-2 promoter to drive expression of the reporter gene uidA. b pREG2-A. A self-activating gene excision construct (HCCN) as well as the expected product after Cre-mediated excision (N). REG-2: rice embryo globulin gene (REG-2) promoter; LB and RB, left border and right border of the T-DNA; uidA, β-glucuronidase gene; Tnos, polyadenylation signal of the nopaline synthase gene; lox, recombinase recognition sites; hpt, hygromycin phosphotransferase gene; Pnos, promoter of the nopaline synthase gene; T35S, CaMV 35S polyadenylation signal; codA, cytosine deaminase gene; nptII, neomycin phosphotransferase gene; cre-intron, cre recombinase gene containing an intron; P35S, CaMV 35S promoter. The REG-2 promoter was introduced via an LR reaction (Gateway™); this results in attB1 and attB2 sites upstream and downstream of the promoter sequence, respectively. Primers used for PCR analysis and restriction sites and probes for Southern analysis are indicated, respectively, above and below the constructs
Plant material, transformation and regeneration
Agrobacterium-mediated transformation of embryogenic cell suspensions (ECS) of the banana cv. ‘Grande Naine’ (Musa AAA) was done as described previously (Chong-Pérez et al. 2012a, b). Briefly, ECS were co-cultivated with the A. tumefaciens strain EHA105 harboring the plasmid pCAMBIA1301, pREG2::uidA or pREG2-A. Selection was applied on semi-solid ZZ medium containing timentin (200 mg/l) and hygromycin B (50 mg/l) or geneticin (G418, 50 mg/l). The cultures were maintained for two months in the dark at 27 ± 2 °C with bi-weekly subcultures. After eight weeks, small embryo colonies were selected separately, and transferred to non-selective RD1 medium (half strength MS medium, supplemented with 100 mg/l ascorbic acid, 100 mg/l myoinositol, 2.5 g/l Gelrite™ and 30 g/l sucrose, pH was adjusted at 5.8 before autoclaving) for four more weeks. Then, somatic embryos were relocated in 25 ml glass tubes containing 10 ml of germination medium (MS medium, 1.0 mg/l biotin, 0.5 mg/l 6-BAP, 2.0 mg/l IAA, 45 g/l sucrose, and 2.5 g/l Gelrite™, pH was adjusted to 5.8 before autoclaving). Cultures were done under natural light conditions at 27 ± 2 °C. After a month in these conditions, regenerated plants were subcultured to 25 ml glass tubes containing each 10 ml of MS medium supplemented with sucrose 3.0 % (w/v) (elongation medium). Colonies that did not germinate were transferred to fresh germination medium for another month.
Molecular analysis
PCR analysis
Genomic DNA was prepared from leaf material of plants transformed with pREG2-A by using the GenElute™ Plant Genomic DNA miniprep kit (Sigma, Germany). Around 50 ng of genomic plant DNA was used as template for the PCR reactions. TaKaRa Ex Taq™ polymerase was used (Takara, Japan). The efficiency of excision was calculated from the results of PCR with two combinations of three primers (Fig. 1b). The sequences of the three primers used to detect excision of the selectable marker gene cassette were C-3300-F, 5′-GCGGACGTTTTTAATGTACTGAATTAACG-3′; nptII-SR, 5′-CCGCATTGCATCAGCCATGATGG-3′; and codA-1, 5′-GTCGCCAACCCGCTGGTCAATATTC-3′. The PCR product expected to be amplified for the combination of C3300-F/nptII-SR primers is 724 bp if excision occurred, and otherwise 8.3 kb for pREG2-A. To avoid the possibility of false negative results due to the difficulty of amplifying the 8.3 kb fragment, PCR was also carried out with primers codA-1/nptII-SR, so that a 1441 bp fragment would be amplified if excision had not occurred. PCR conditions were as mentioned before (Chong-Pérez et al. 2012a) except that 0.1 % (w/v) BSA and 1.0 % (w/v) PVP was added to the PCR mixture (Xin et al. 2003).
Southern blot hybridization
Genomic DNA was prepared from leaves of in vitro plants by using the Nucleon Phytopure DNA extraction kit (RPN8511, GE Healthcare) according to the manufacturer’s instructions. Twenty micrograms of genomic DNA were digested with AseI/BstNI (Fig. 1b) during 2–3 h. Then, fragments were separated on a 0.8 % TAE agarose gel for 14 h at 25 V. The rest of the procedure (gel preparation, DNA transfer, hybridization and detection) was done as was mentioned before (Chong-Pérez et al. 2012a).
Histochemical GUS assays
A histochemical GUS assay was carried out in samples transformed with pREG2::uidA plasmid in several phases of the genetic transformation protocol. Embryogenic cells, pro-embryo masses, embryos and plantlets were stained with a modified assay solution containing 100 mM phosphate buffer (pH 7.0, 50 mM Na2HPO4 and 50 mM KH2PO4), 10 mM EDTA, 5 mM K-ferricyanide, 5 mM K-ferro-cyanide, 0.1 % Triton X-100, 1 mM X-Gluc (Jefferson et al. 1987) and incubated overnight at 37 °C.
Results
With the objective to develop an auto-excision system in banana that would not require any extra handling step, the promoter from the rice REG-2 gene was chosen to drive embryo-specific expression of the cre gene. REG-2 encodes a 7S-globulin type storage protein and is expressed in maturing embryos (Sun et al. 1996; Miyoshi et al. 1999; Qu and Takaiwa 2004). In rice seeds this gene shows temporally and spatially regulated expression, which increases during embryo development and is localized in the embryo and aleurone but not in the endosperm (Miyoshi et al. 1999; Qu and Takaiwa 2004). This gene expression profile would be suitable for tightly controlled expression of the cre gene in our transformation protocol. Indeed, the antibiotic selection is applied during cell multiplication and early stages of embryo formation. In the later stages of embryo formation and embryo maturation, selection can be omitted and we expected that the REG-2 promoter could induce Cre activity in this stage, resulting in excision of the selectable marker gene just before the germination.
The 35S promoter was replaced by the REG-2 promoter to drive the expression of the uidA reporter gene in the pCAMBIA1301 plasmid. The T-DNA region of the resulting plasmid pREG2::uidA is represented in Fig. 1a. The pattern of REG-2 promoter activity was visually tested through expression of β-glucuronidase at different time points after inoculation with A. tumefaciens (1, 4, 8 and 12 weeks). No GUS expression was detected after 1 and 4 weeks (Fig. 2a,b). After 8 weeks on selection medium, a few embryos showed GUS staining (Fig. 2c). However strong GUS activity was clearly detected after a month in RD1 embryo formation medium (Fig. 2d) and also on secondary embryos localized at the base of germinated plantlets, but not in the plantlets themselves (Fig. 2e). In contrast to the developmentally regulated expression pattern conferred by the REG-2 promoter, GUS expression under control of the 35S promoter was detected in all stages: cell clumps, embryos and plantlets derived from embryogenic cells (Fig. 2f–j).

Histochemical analysis of GUS expression directed by REG-2 embryo specific gene promoter (upper panel) and 35S promoter (lower panel) in transgenic somatic embryos of banana cv. ‘Grande Naine’ (Musa AAA). (a, f) one week; (b, g) 4 weeks; (c, h) 8 weeks and (d, i) 12 weeks after transformation with A. tumefaciens strain EHA105 containing pREG2::uidA (a–e) or pCAMBIA1301 (f–i)
The REG-2 promoter thus appeared suitable to drive recombinase expression in a Cre-lox based auto-excision strategy and was subsequently investigated during banana transformation with A. tumefaciens harboring the plasmid pREG2-A (Fig. 1b). pREG2-A is a derivative of the GSA vector (Verweire et al. 2007; Chong-Pérez et al. 2012a), which contains three transcriptional units between tandemly oriented lox sites. These are Pnos-hpt-Tnos, REG2-cre-i-T35S and P35S-codA-tnos. Outside the excision sequence, we used Pnos-nptII-Tnos as expression cassette of interest. The original non-recombined T-DNA is referred to as ‘‘HCCN’’ (contains the h pt, c odA, c re-i and n ptII genes), whereas the recombined T-DNA is referred to as ‘‘N’’ (because it has lost the hpt, codA, and cre-i genes and only contains the n ptII gene).
Transformed cells were cultured for eight weeks on ZZ medium containing hygromycin (resistance provided by the hpt gene located between the lox sites) or G418 (resistance provided by nptII, located outside the lox sites). The selection efficiency with either of these antibiotics was compared in order to verify whether premature excision of the hpt gene had occurred. Indeed, if activation of the cre gene and concomitant excision of the hpt gene would occur during the 8 weeks on selection medium, a substantially lower transformation frequency would be expected in the hygromycin selection. In a first experiment transformation efficiency on hygromycin (H effic ) was 76.6 ± 24.0 embryo colonies per plate whereas on G418 (G effic ) it was 76.3 ± 18.0 colonies per plate. In a second experiment H effic was 30.6 ± 6.0 and G effic 28.0 ± 5.3. In both experiments the ratio H effic /G effic was 1.0, which indicates that there was no premature recombination of the lox sites.
Thereafter, hygromycin resistant colonies were transferred to RD1 antibiotic-free medium, where the complete development of somatic embryos and their subsequent maturation occurred in a normal way. Then, it is in this period that we expected the activation of the cre gene by the REG-2 promoter, and hence the auto-excision of the selectable marker genes and Cre expression cassettes. Although the counter-selectable marker gene codA is present in the transformation vector, we did not attempt to use 5-fluorocytosine selection, as we previously found this selection system to be problematic in our banana transformation protocol (Chong-Pérez et al. 2012a).
To investigate whether gene excision took place, genomic DNA from regenerated lines was analyzed by PCR with the combination of three primers. Primers C3300-F/nptII-SR should amplify a 724 bp band (“N” fragment, Fig. 1b) if the excision occurred or, an 8.3 kb band, if no excision occurred (the original “HCCN” T-DNA fragment, Fig. 1b). To confirm these results, a second PCR is carried out with the primer set codA-1/nptII-SR which should amplify a 1441 bp fragment corresponding to the non-recombined “HCCN” T-DNA (Fig. 1b), whereas no amplification is expected if excision took place. Based on this principle, 44 lines were arbitrarily selected for PCR analysis with primer set C3300-F/nptII-SR (PCR 1) and primer set codA-1/nptII-SR (PCR 2). As is shown in Fig. 3a, PCR1 yielded only the recombined “N” fragment of 724 bp in 22 lines (lines 14, 15, 21, 23, 24, 25, 27, 29, 31, 33, 40, 42, 44, 50, 59, 71, 82, 90, 100, 203, 205 and 109) whereas four lines showed only the unrecombined “HCCN” fragment of 8.3 kb (lines 5, 13, 70 and 105), six lines showed both bands (lines 8, 32, 72, 73, 113 and 200) and 12 lines did not show any signal (lines 4, 20, 22, 28, 30, 39, 41, 52, 53, 97, 119 and 131). PCR2 results showed that a 1441 bp “HCCN” T-DNA fragment was present in 21 lines, which includes the six lines that showed two bands in PCR1 (8, 32, 72, 73, 113 and 200), the four lines that showed only the 8.3 kb fragment (5, 13, 70 and 105), seven lines that yielded only the excision “N” fragment (21, 25, 31, 40, 71, 100 and 205) and four lines that did not show any signal in PCR1 (22, 39, 41 and 52) (Fig. 3a). These results confirmed the PCR1 results and also the strategy to use the second set of primers to detect possible masked results. Indeed, from the 22 lines that were positive for the excision fragment, the PCR2 revealed that 15 lines (14, 15, 23, 24, 27, 29, 33, 42, 44, 50, 59, 82, 90, 201 and 109) were completely marker-free, corresponding to 34 % of the analyzed lines (15/44). Moreover, 13 lines showed both “N” and “HCCN” fragments (8, 21, 25, 31, 32, 40, 71, 72, 73, 100, 113, 200 and 205), which could be explained by multiple insertions of the T-DNA in their genome or because of the coexistence of two types of cells due to incomplete excision, resulting in a chimeric plant. On the other hand, eight lines (5, 13, 22, 39, 41, 52, 70 and 105) showed no excision and surprisingly, eight lines (4, 20, 28, 30, 53, 97, 119 and 131) did not show any signal, which indicates that there were escapes in the selection process (Fig. 3a). Then, excluding the escape events, from the 36 transgenic lines analysed, the total gene excision rate at the somatic embryo development stage in this experiment, including both complete and chimeric excisions, was (15 + 13)/36 = 77.8 % based on the PCR analysis.

PCR and Southern blot analysis to verify excision events in banana cv. ‘Grande Naine’ (Musa AAA) regenerants derived from somatic embryos. a PCR results. PCR1, primers C-3300-F/nptII-SR yield a 724 bp fragment specific for the excision allele N and/or an 8.3 kb fragment indicative for the original HCCN allele; PCR2, primers codA-1/nptII-SR yield a 1441 bp fragment specific for the original allele HCCN. Genomic DNA was extracted from pREG2-A regenerants derived from somatic embryos; Co−, untransformed plant; C+, plasmid control; H2O, water was added instead of template DNA. MWM, Molecular weight marker Gene Ruler™ DNA Ladder Mix (Fermentas). b Southern blot hybridization analysis of transgenic banana plants of pREG2-A lines. Twenty micrograms total DNA were digested with AseI and BstNI, and separated fragments were hybridized with a digoxigenin-labeled nptII probe (488 bp). Co−, untransformed control plant; Co+ one copy reconstruction of linearized plasmid; MWM, digoxigenin-labeled DNA molecular weight marker VII (Roche)
Additionally, in order to confirm the results obtained in PCR analyses, 19 lines were randomly selected for Southern blot hybridization. Genomic DNA from these lines was digested with BstNI/AseI and hybridized with a probe corresponding to a 488 bp fragment of the nptII gene. Plants in which excision occurred are foreseen to display an 842 bp band, while a 1717 bp band is indicative for the original non-recombined T-DNA sequence (Fig. 1b). As is shown in Fig. 3b, nine lines showed successful recombination of the lox sites and displayed only the 842 bp band (14, 23, 24, 25, 44, 15, 50, 82 and 109). This confirms the PCR results, except for line 25, which displayed a 1441 bp band in PCR2. On the other hand seven lines displayed only the 1717 bp band indicating that excision did not occur in these lines (13, 39, 5, 70, 71, 105 and 113). Interestingly, in lines 71 and 113 the excision signal (842 bp) did not appear, as was expected from the results in PCR1 (Fig. 3); this is probably due to the chimeric nature of these lines. Only line 8 showed the bands at 1717 and 842 bp in the Southern blot hybridization, as expected from the PCR analysis. Perhaps this line has multiple insertions of the T-DNA and in some or one of these the recombination of lox sites yielded the “N” fragment while in the other T-DNA(s) no recombination occurred (Fig. 3b).
Furthermore to confirm the excision of the marker gene from the banana genome, the 724 bp PCR products amplified in PCR1 from six marker-free lines were sequenced and for all of them the sequencing results were as expected for a precise recombination (Fig. 4).

Sequence confirmation of marker gene excision in the genome of transgenic banana line 14 cv.‘Grande Naine’ (Musa AAA). The 724 bp fragment was amplified in PCR1 from genomic DNA using the primers (C3300-F, 5′-GCGGACGTTTTTAATGTACTGAATTAACG-3′ and nptII-SR, 5′-CCGCATTGCATCAGCCATGATGG-3′). Here, part of the sequence is shown which is exactly as predicted after site-specific recombination of the lox sites
Discussion
Several approaches have been proposed to remove selectable marker genes after the transformation-selection process in order to create marker-free transgenic plants. Site-specific recombination is a highly versatile system that can be applied to any plant species in several ways. One of these is the auto-excision strategy, in which the conditional expression of the recombinase can be tightly controlled by inducible promoters. This system is of capital importance for vegetatively propagated species and species with a long generation time, where it is impossible or highly impractical to use systems that require segregation and/or crossing.
In this work we take advantage of a developmentally regulated system based on Cre/lox and an embryo specific REG-2 promoter to create marker-free banana plants. The success of the system was depending on appropriate temporal and spatial expression of this rice promoter in banana cell clumps and somatic embryos. In rice, the specificity and expression strength of the REG-2 promoter was studied by Qu and Takaiwa (2004); these authors showed that REG-2 promoter activity was strictly in the mature embryo and aleurone layer, with no expression in the endosperm. To our knowledge, the activity of this promoter has not been tested in somatic embryos. The activation of this promoter was not observed at the early transformation stage during banana cell clump multiplication, whereas at 8 weeks after co-cultivation some activity was detected, which can likely be ascribed to the asynchronous nature of somatic embryo development. This low level promoter activity did not prevent effective use of the hpt selectable marker located between the lox sites in the auto-excision construct, as no drop in transformation efficiency was observed on hygromycin containing medium. The REG-2 promoter was strongly induced in somatic embryos at 12 weeks after co-cultivation, however GUS expression was not detected in regenerated plants, indicating that REG-2 promoter activity is also tightly regulated during banana somatic embryogenesis. This expression pattern appears very suitable for selectable marker gene excision in transformation systems where plants regenerate via a similar somatic embryogenesis pathway like grapevine (Vidal et al. 2009), forage grasses (Bettany et al. 2003), rice (Wagiran et al. 2010) and trees, like conifers (Klimaszewska et al. 2005). The functionality of this and/or other developmentally regulated promoters should however be tested in each particular case.
In this work the potential of the REG-2 promoter was tested to drive cre gene expression, aiming to remove floxed sequences during somatic embryo development. If escape events are not considered, in total 36 transgenic lines were analysed, from which PCR revealed 41.7 % (15/36) plants with complete excision, as well as 36.1 % (13/36) of chimeric plants and/or plants that keep an unrecombined T-DNA copy. Southern blot hybridization confirmed PCR results and demonstrated complete excision of the selectable marker and cre genes from the banana genome. However, PCR results and Southern blot hybridization analysis were contradictory in some lines (25, 71 and 113); this discrepancy may partly be due to the higher sensitivity of the PCR assay, but could also indicate that (some of) these events are chimeric. The chimerism could be associated with insufficient expression of the cre gene during embryo development. Indeed, this is a phenomenon inherent to transgene technology and can be connected to many factors like copy number, complex integration patterns and genomic position (Hobbs et al. 1993; Jorgensen et al. 1996; De Wilde et al. 2000). Relation between cre expressions levels and recombination efficiency has been observed in higher plants (Russell et al. 1992; Marjanac et al. 2008). However, other results have indicated that cre expression is less susceptible to variation when this gene is driven by tissue specific or developmentally regulated promoter (Van Ex et al. 2009). An embryo multiplication step could be introduced in the transformation protocol in order to reduce the incidence of chimeric plants, based on the unicellular origin of the secondary embryos (Maximova et al. 2002).
In the auto-excision system presented here the selectable marker gene is present for about eight weeks. This time apparently was not enough to kill all non-transgenic cells in the culture resulting in the appearance of escapes in the selection process. Probably an increase of the selection pressure (e.g. by applying an antibiotic gradient) combined with a synchronization in the somatic embryogenesis process could be a solution for this problem. Additionally, a treatment with abscisic acid (ABA) in the maturation step could be included in the transformation protocol. In fact ABA has been found to induce the expression of some genes in zygotic and somatic embryos, like Late-Embryogenic Abundant (LEA) genes (Ikeda et al. 2006). Likewise, REG-2 is induced in rice zygotic embryos in the presence of ABA (Sun et al. 1996) and the same effect could be expected in somatic embryos. Moreover, a recent report indicates that when ABA was included in the maturation medium, the germination of plantain somatic embryos was increased around fivefold (Sholi et al. 2009), which makes this treatment more attractive.
Although the proposed method has some limitations with respect to the occurrence of chimeric plants and selection escapes, it allows obtaining completely marker-free banana plants with an efficiency of 41.7 %. The total rate of excision was 77.8 % counting complete and partial excision. This efficiency is comparable with previously obtained results when heat-shock inducible promoters were used to excise selectable marker genes in the same banana cultivar (Chong-Pérez et al. 2012a). Other approaches have combined the use of developmentally inducible promoters and auto-excision systems. The excision efficiencies obtained in these systems are variable. The highest excision frequencies (100 %) were obtained for systems based on the tobacco NTM 19 microspore-specific promoter (Mlynárová et al. 2006), the Arabidopsis thaliana SDS and AP1 germline promoters (Verweire et al. 2007), the pollen-specific BGP1, PAB5 promoters (Luo et al. 2007) in the model plant species A. thaliana and/or tobacco. Other systems have used seed specific promoters. For example, Moravčíková et al. (2008) showed that the promoter of the Arabidopsis cruciferin C (CRUC) gene allowed the production of marker-free transgenic tobacco plants; the seed-specific napin promoter from Brassica napus was used to drive Cre expression and produce marker-free transgenic plants of B. napus and N. benthamiana (Kopertekh et al. 2009; 2010). Recently, the tomato-derived LAT52 pollen-specific promoter coupled with the CinH-RS2 recombination system was successfully used to excise selectable marker genes from tobacco pollen (Moon et al. 2011). Likewise, Bai et al. (2008) have reported the excision of a selectable marker gene in rice in a system where the cre gene was controlled by a floral specific promoter OsMADS45, which is the only report for monocotyledonous species. Moreover, in soybean 5–30 % excision efficiency was observed when the cre gene was driven by the embryo specific gene promoter app1 from Arabidopsis thaliana (Li et al. 2007). This system, showed for the first time the possibility to drive Cre activation with a developmentally regulated promoter under in vitro conditions. In all the other methodologies mentioned above marker gene excision occurs during sexual reproduction, thus these systems are not applicable to species that are vegetatively propagated and/or have a long life cycle.
In this work we demonstrate the feasibility of the use of the embryo-specific REG-2 promoter to drive the expression of the recombinase gene in the late stage of development of somatic embryos to create marker-free transgenic banana plants. To our understanding, this is the first report of the application of this technology in a vegetatively propagated species.
References
Abdeen A, Miki B (2009) The pleiotropic effects of the BAR gene and glufosinate on the Arabidopsis transcriptome. Plant Biotechnol J 7:266–282
Acereto-Escoffié POM, Chi-Manzanero BH, Echeverría-Echeverría S, Grijalva R, James KA et al (2005) Agrobacterium-mediated transformation of Musa acuminata cv. ‘Grand Nain’ scalps by vacuum infiltration. Sci Hortic 105:359–371
Akbudak MA, Srivastava V (2011) Improved FLP recombinase, FLPe, efficiently removes marker gene from transgene locus developed by Cre–lox mediated site-specific gene integration in rice. Mol Biotechnol 49:82–89
Araki H, Jearnpipatkul A, Tatsumi H, Sakurai T, Ushio K, Muta T, Oshima Y (1985) Molecular and functional organization of yeast plasmid pSR1. J Mol Biol 182:191–203
Bai X, Wang Q, Chengcai C (2008) Excision of a selective marker in transgenic rice using a novel Cre/loxP system controlled by a floral specific promoter. Transgenic Res 17:1035–1043
Becker DK, Dugdale B, Smith MK, Harding RM, Dale JL (2000) Genetic transformation of Cavendish banana (Musa spp. AAA group) cv. ‘Grand Nain’ via microprojectile bombardment. Plant Cell Rep 19:229–234
Bettany AJE, Dalton SJ, Timms E, Manderyck B, Dhanoa MS, Morris P (2003) Agrobacterium tumefaciens-mediated transformation of Festuca arundinacea (Schreb.) and Lolium multiflorum (Lam.). Plant Cell Rep 21:437–444
Castle LA, Wu G, McElroy D (2008) Agricultural input traits: past, present and future. Curr Opin Biotechnol 17:105–112
Chong-Pérez B, Kosky RG, Reyes M, Rojas L, Ocaña B, Tejeda M, Pérez B, Angenon G (2012a) Heat shock induced excision of selectable marker genes in transgenic banana by the Cre-lox site-specific recombination system. J Biotechnol 15:265–273
Chong-Pérez B, Reyes M, Rojas L, Ocaña B, Pérez B, Kosky RG, Angenon G (2012b) Establishment of embryogenic cell suspension cultures and Agrobacterium-mediated transformation in banana cv. ‘Dwarf Cavendish’ (Musa AAA): effect of spermidine on transformation efficiency. Plant Cell Tissue Organ Cult 111:79–90
Corrado G, Karali M (2009) Inducible gene expression systems and plant biotechnology. Biotechnol Adv 27:733–743
Cox MM (1983) The FLP protein of the yeast 2 mm plasmid: expression of a eukaryotic genetic recombination system in Escherichia coli. Proc Natl Acad Sci USA 80:4223–4227
Cuellar W, Gaudin A, Solorzano D, Casas A, Nopo L, Chudalayandi P, Medrano G, Kreuze J, Ghislain M (2006) Self-excision of the antibiotic resistance gene nptII using a heat inducible Cre-loxP system from transgenic potato. Plant Mol Biol 62:71–82
Darbani B, Eimanifar A, Stewart CN Jr, Camargo WN (2007) Methods to produce marker-free transgenic plants. Biotechnol J 2:3–90
De Wilde C, Van Houdt H, De Buck S, Angenon G, De Jaeger G, Depicker A (2000) Plants as bioreactors for protein production: avoiding the problem of transgene silencing. Plant Mol Biol 43:347–359
Eckardt NA, Cominelli E, Galbiati M, Tonelli C (2009) The future of science: food and water for life. Plant Cell 21:368–372
Edgerton MD (2009) Increasing crop productivity to meet global needs for feed, food, and fuel. Plant Physiol 149:7–13
Farré G, Ramessar K, Twyman RM, Capell T, Christou P (2010) The humanitarian impact of plant biotechnology: recent breakthroughs vs bottlenecks for adoption. Curr Opin Plant Biol 13:219–225
Fladung M, Schenk TMH, Polak O, Becker D (2010) Elimination of marker genes and targeted integration via FLP/FRT recombination system from yeast in hybrid aspen (Populus tremula L. × P. tremuloides Michx.). Tree Genet Gen 6:205–217
Ganapathi TR, Higgs NS, Balint-Kurti PJ, Arntzen CJ, May GD, van Eck JM (2001) Agrobacterium –mediated transformation of embryogenic cell suspensions of the banana cultivar Rasthali (AAB). Plant Cell Rep 20:157–162
Ghosh A, Ganapathi TR, Nath P, Bapat VA (2009) Establishment of embryogenic cell suspension cultures and Agrobacterium-mediated transformation in an important Cavendish banana cv. Robusta (AAA). Plant Cell, Tissue Organ Cult 97:131–139
Hare PD, Chua NH (2002) Excision of selectable marker genes from transgenic plants. Nat Biotechnol 20:575–580
Hobbs SL, Warkentin TD, DeLong CM (1993) Transgene copy number can be positively or negatively associated with transgene expression. Plant Mol Biol 21:17–26
Hoess RH, Abremski K (1985) Mechanism of strand cleavage and exchange in the Cre-lox site-specific recombination system. J Mol Biol 181:351–362
Hoess RH, Ziese M, Sternberg N (1982) P1 site-specific recombination: nucleotide sequence of the recombining sites. Proc Natl Acad Sci USA 79:3398–3402
Hoff T, Schnorr K-M, Mundy J (2001) A recombinase-mediated transcriptional induction system in transgenic plants. Plant Mol Biol 45:41–49
Holsters M, De Waele D, Depicker A, Messens E, Van Montagu M, Schell J (1978) Transfection and transformation of A. tumefaciens. Mol Gen Genet 163:181–187
Hood EE, Gelvin SB, Melchers LS, Hoekema A (1993) New Agrobacterium helper plasmids for gene-transfer to plants. Transgenic Res 2:208–218
Ikeda M, Umehara M, Kamada H (2006) Embryogenesis-related genes: its expression and roles during somatic and zygotic embryogenesis in carrot and Arabidopsis. Plant Biotechnol 23:153–161
Jefferson RA, Kavanagh TA, Bevan MW (1987) GUS fusions: beta-glucuronidase as a sensitive and versatile gene fusion marker in higher plants. EMBO J 6:3901–3907
Jorgensen RA, Cluster PD, English J, Que QD, Napoli CA (1996) Chalcone synthase cosuppression phenotypes in petunia flowers: comparison of sense vs antisense constructs and single-copy vs complex T-DNA sequences. Plant Mol Biol 31:957–973
Khanna H, Becker D, Kleidon J, Dale J (2004) Centrifugation assisted Agrobacterium tumefaciens-mediated transformation (CAAT) of embryogenic cell suspensions of banana (Musa spp. Cavendish AAA and Lady Finger AAB). Mol Breed 14:239–252
Kilby NJ, Davies GJ, Snaith MR (1995) FLP recombinase in transgenic plants: constitutive activity in stably transformed tobacco and generation of marked cell clones in Arabidopsis. Plant J 8:637–652
Klimaszewska K, Rutledge RG, Seguin A (2005) Genetic transformation of conifers utilizing somatic embryogenesis. Methods Mol Biol 286:151–164
Kopertekh L, Broer I, Schiemann J (2009) Developmentally regulated site-specific marker gene excision in transgenic B. napus plants. Plant Cell Rep 28:1075–1083
Kopertekh L, Schulze K, Frolov A, Strack D, Broer I, Schiemann J (2010) Cre-mediated seed-specific transgene excision in tobacco. Plant Mol Biol 72:597–605
Li Z, Xing A, Moon BP, Burgoyne SA, Guida AD, Liang H, Lee C, Caster CS, Barton JE, Klein TM, Falco SC (2007) A Cre/loxP- mediated self-activating gene excision system to produce marker gene free transgenic soybean plants. Plant Mol Biol 65:329–341
Luo K, Duan H, Zhao D, Zheng X, Deng W, Chen Y, Stewart CN Jr, McAvoy R, Jiang X, Wu Y, He A, Pei Y, Li Y (2007) ‘GM-gene-deletor’: fused loxP-FRT recognition sequences dramatically improve the efficiency of FLP or CRE recombinase on transgene excision from pollen and seed of tobacco plants. Plant Biotechnol J 5:263–274
Luo K, Sun M, Deng W, Xu S (2008) Excision of selectable marker gene from transgenic tobacco using the GM-gene-deletor system regulated by a heat-inducible promoter. Biotechnol Lett 30:1295–1302
Marjanac G, De Paepe A, Peck I, Jacobs A, De Buck S, Depicker A (2008) Evaluation of CRE-mediated excision approaches in Arabidopsis thaliana. Transgenic Res 17:239–250
Maximova SN, Alemanno L, Young A, Ferriere N, Traore A, Guiltinan MJ (2002) Efficiency, genotypic variability, and cellular origin of primary and secondary somatic embryogenesis of Theobroma cacao L. In Vitro Cell Dev Biol Plant 38:252–259
May G, Afza R, Mason H, Wiecko A, Novak F, Arntzen C (1995) Generation of transgenic banana (Musa acuminata) plants via Agrobacterium-mediated transformation. Nat Biotechnol 13:486–492
Miki B, McHugh S (2004) Selectable marker genes in transgenic plants: applications, alternatives and biosafety. J Biotechnol 107:193–232
Miki B, Abdeen A, Manabe Y, MacDonald P (2009) Selectable marker genes and unintended changes to the plant transcriptome. Plant Biotechnol J 7:211–218
Miyoshi K, Nakata E, Nagato Y, Hattori T (1999) Differential in situ expression of three ABA-regulated genes of rice, RAB16A, REG2 and OSBZ8, during seed development. Plant Cell Physiol 40:443–447
Mlynárová L, Conner AJ, Nap JP (2006) Directed microspore-specific recombination of transgenic alleles to prevent pollen-mediated transmission of transgenes. Plant Biotechnol J 4:445–452
Moon HS, Abercrombie LL, Eda S, Blanvillain R, Thomson JG, Ow D, Stewart CN (2011) Transgene excision in pollen using a codon optimized serine resolvase CinH-RS2 site-specific recombination system. Plant Mol Biol 75:621–631
Moravčíková J, Vaculková E, Bauer M, Libantová J (2008) Feasibility of the seed specific cruciferin C promoter in the self-excision Cre/loxP strategy focused on generation of marker-free transgenic plants. Theor Appl Genet 117:1325–1334
Pei XW, Chen SK, Wen RM, Ye S, Huang JQ, Zhang YQ, Wang BQ, Wang ZX, Jia SR (2005) Creation of transgenic bananas expressing human lysozyme gene for Panama Wilt resistance. J Integr Plant Biol 47:971–977
Pérez-Hernández JB, Remy S, Swennen R, Sági L (2006) Banana (Musa spp.). In: Wang K (ed) Methods in molecular biology, 344: Agrobacterium protocols, vol 2. Humana, Totowa, pp 167–175
Puchta H (2003) Marker-free transgenic plants. Plant Cell, Tissue Organ Cult 74:123–134
Qu LQ, Takaiwa F (2004) Evaluation of tissue specificity and expression strength of rice seed component gene promoters in transgenic rice. Plant Biotech J 2:113–125
Rao MR, Moon HS, Schenk TMH, Becker D, Mazarei M, Stewart CN Jr (2010) FLP/FRT recombination from yeast: application of a two gene cassette scheme as an inducible system in plants. Sensors 10:8526–8535
Russell SH, Hoopes JL, Odell JT (1992) Directed excision of a transgene from the plant genome. Mol Gen Genet 234:49–59
Sági L, Remy S, Panis B, Swennen R, Volckaert G (1994) Transient gene expression in electroporated banana (Musa spp., cv. Bluggoe, ABB group) protoplasts isolated from regenerable embryogenic cell suspensions. Plant Cell Rep 13:262–266
Sági L, Panis B, Remy S, Schoofs H, De Smit K, Swennen R, Cammue BP (1995) Genetic transformation of banana and plantain (Musa spp.) by particle bombardment. Biotechnology 13:481–485
Schaart JG, Krens FA, Pelgrom KTB, Mendes O, Rouwendal GJA (2004) Effective production of marker-free transgenic strawberry plants using inducible site-specific recombination and a bifunctional selectable marker gene. Plant Biotechnol J 2:233–240
Senecoff JF, Bruckner RC, Cox MM (1985) The FLP recombinase of the yeast 2-mm plasmid: characterization of its recombination site. Proc Natl Acad Sci USA 82:7270–7274
Sholi NJY, Chaurasia A, Agrawal A, Sarin NB (2009) ABA enhances plant regeneration of somatic embryos derived from cell suspension cultures of plantain cv. Spambia (Musa sp.). Plant Cell, Tissue Organ Cult 99:133–140
Sreekala C, Wu L, Gu K, Wang D, Tian D (2005) Excision of a selectable marker in transgenic rice (Oryza sativa L.) using a chemically regulated CRE/loxP system. Plant Cell Rep 24:86–94
Subramanyam K, Subramanyam K, Sailaja K, Srinivasulu M, Lakshmidevi K (2011) Highly efficient Agrobacterium-mediated transformation of banana cv. Rasthali (AAB) via sonication and vacuum infiltration. Plant Cell Rep 30:425–436
Sugita K, Kasahara T, Matsunaga E, Ebinuma H (2000) A transformation vector for the production of marker-free transgenic plants containing a single copy transgene at high frequency. Plant J 22:461–469
Sun JL, Nakagawa H, Karita S, Ohyama K, Hattori T (1996) Rice embryo globulins: amino-terminal amino acid sequences, cDNA cloning and expression. Plant Cell Physiol 37:612–620
Tripathi L, Tripathi JN, Hughes Jd’A (2005) Agrobacterium-mediated transformation of plantain (Musa spp.) cultivar Agbagba. Afr J Biotechnol 4:1378–1383
Van Ex F, Verweire D, Claeys M, Depicker A, Angenon G (2009) Evaluation of seven promoters to achieve germline directed Cre-lox recombination in Arabidopsis thaliana. Plant Cell Rep 28:1509–1520
Varshney RK, Bansal KC, Aggarwal PK, Datta K, Craufurd PQ (2011) Agricultural biotechnology for crop improvement in a variable climate: hope or hype? Trends Plant Sci 16:363–371
Verweire D, Verleyen K, De Buck S, Claeys M, Angenon G (2007) Marker-free transgenic plants through genetically programmed auto-excision. Plant Physiol 145:1220–1231
Vidal JR, Rama J, Taboada L, Martin C, Ibañez M, Segura A, González-Benito ME (2009) Improved somatic embryogenesis of grapevine (Vitis vinifera) with focus on induction parameters and efficient plant regeneration. Plant Cell, Tissue Organ Cult 96:85–94
Vishnevetsky J, White TL, Palmateer AJ, Flaishman M, Cohen Y, Elad Y, Melcheva M, Hanania U, Sahar N, Dgani O, Perl A (2011) Improved tolerance toward fungal diseases in transgenic Cavendish banana (Musa spp. AAA group) cv. Grand Nain. Transgenic Res 20:61–72
Wagiran A, Ismail I, Zain CRCM, Abdullah R (2010) Agrobacterium tumefaciens-mediated transformation of the isopentenyltransferase gene in japonica rice suspension cell culture. Aust J Crop Sci 4:421–429
Wang Y, Chen B, Hu Y, Li J, Lin Z (2005) Inducible excision of selectable marker gene from transgenic plants by the Cre/lox site-specific recombination system. Transgenic Res 14:605–614
Xin Z, Velten JP, Oliver MJ, Burke J (2003) High-throughput DNA extraction method suitable for PCR. Biotechniques 34:820–826
Zhang W, Subbarao S, Addae P, Shen A, Armstrong C, Peschke V, Gilbertson L (2003) Cre/lox-mediated marker gene excision in transgenic maize (Zea mays L.) plants. Theor Appl Genet 107:1157–1168
Zhang Y, Li H, Ouyang B, Lu Y, Ye Z (2006) Chemical-induced autoexcision of selectable markers in elite tomato plants transformed with a gene conferring resistance to lepidopteran insects. Biotechnol Lett 28:1247–1253
Zuo J, Niu QW, Moller SG, Chua NH (2001) Chemical-regulated, site-specific DNA excision in transgenic plants. Nat Biotechnol 19:157–161
Acknowledgments
This work was supported by the Institutional University Collaboration programme with Universidad Central “Marta Abreu” de Las Villas from the Flemish Interuniversity Council (VLIR IUC UCLV). Borys Chong-Pérez was partially supported by the International Foundation for Science (grant C/4976-1).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Chong-Pérez, B., Reyes, M., Rojas, L. et al. Excision of a selectable marker gene in transgenic banana using a Cre/lox system controlled by an embryo specific promoter. Plant Mol Biol 83, 143–152 (2013). https://doi.org/10.1007/s11103-013-0058-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11103-013-0058-8