
Abstract
This work is focused on the generation of selectable marker-free transgenic tobacco plants using the self excision Cre/loxP system that is controlled by a strong seed specific Arabidopsis cruciferin C (CRUC) promoter. It involves Agrobacterium-mediated transformation using a binary vector containing the gus reporter gene and one pair of the loxP sites flanking the cre recombinase and selectable nptII marker genes (floxed DNA). Surprisingly, an ectopic activation of CRUC resulting in partial excision of floxed DNA was observed during regeneration of transformed cells already in calli. The regenerated T0 plants were chimeric, but no ongoing ectopic expression was observed in these one-year-long invitro maintained plants. The process of the nptII removal was expected in the seeds; however, none of the analysed T0 transgenic lines generated whole progeny sensitive to kanamycin. Detailed analyses of progeny of selected T0-30 line showed that 10.2% GUS positive plants had completely removed nptII gene while the remaining 86.4% were still chimeras. Repeated activation of the cre gene in T2 seeds resulted in increased rate of marker-free plants, whereas four out of ten analysed chimeric T1 plants generated completely marker-free progenies. This work points out the feasibility as well as limits of the CRUC promoter in the Cre/loxP strategy.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Selectable marker genes encoding antibiotics or herbicide resistance are essential for identifying those rare plant cells that have taken up foreign DNA upon transformation of plants (Bevan et al. 1983). However, many consumers and environmental groups have voiced their concern over the release of transformed plants with such genes into environment. One of the approaches developed for the removal of marker genes is the Cre/loxP system that consists of the cre recombinase gene and two 34 bp loxP sites. The Cre recombinase mediates recombination events and causes the excision of a DNA segment between two directly oriented adjacent loxP sites (floxed DNA). In the Cre/loxP system termed as “self excision”, the floxed DNA comprises both the cre and selectable marker genes as a part of the same T-DNA. Upon activation, the Cre recombinase removes its own gene sequence as well as the marker gene. The expression of the cre is induced by either heat shock, or spraying with chemicals (Hoff et al. 2001; Cuellar et al. 2006; Zhang et al. 2006). The requirements for external induction and intrinsic problems with efficacy and costs have, however, hampered widespread field applications of such an advanced technology. A more sophisticated approach includes self excision controlled by an endogenous stimulus that is an integral part of plant biology. Recently Mlynárová et al. (2006), Luo et al. (2007), Li et al. (2007) and Verweire et al. (2007) reported that the excision of floxed DNA was more efficient when the cre recombinase was driven by tissue-specific promoters. Out of these promoters, only a limited number was embryo specific (APP1, PAB5) (Li et al. 2007; Luo et al. 2007). Although the latter authors succeeded in producing marker-free transgenic plants, the efficiency of excision events concerning the number of transgenic embryogenic cells or progeny plants with a completely removed floxed DNA remains limiting. For example, the embryogenic APP1 promoter driving an excision in transformed soybean embryogenic culture resulted in 13% events with complete excision, 31% events yielded chimeras and in 56% events the excision failed (Li et al. 2007). These authors, therefore, suggested an application of another, more robust embryo-specific promoter with uniform expression in a small time window after globular stage of soybean somatic embryo development. For example, the cruciferin C (CRUC) promoter from Arabidopsis thaliana exerts 4- and 19-fold higher transcriptional activity than the PAB5 and APP1 promoters, respectively (Schmid et al. 2005). The expression from CRUC starts between mid-globular and early heart embryo stages (Becerra et al. 2006), gradually increases and accumulates in cotyledons and hypocotyls (Höglund et al. 1992). According to the Arabidopsis Microarray Database (AMD) (Zimmermann et al. 2004) this promoter is active in the seeds and silique but not in any other tissue type including callus.
This work is geared towards studying the feasibility of the CRUC promoter in the self excision Cre/loxP-based generation of marker-free plants. This promoter is expected to control the excision of the antibiotic resistance gene during development of transgenic seeds. In our system, the T-DNA of plant transformation vector harbours the loxP cassette with both the cre recombinase and nptII genes and the gus gene outside of the loxP sites. The efficiency of the nptII gene removal was investigated in T1 and T2 progenies of selected self-pollinated transformants.
Material and methods
Vector construction
The CRUC promoter sequence was isolated from Arabidopsis thaliana (Stuitje et al. 2003). Subsequently, it was fused to the DsRFP (rfp) gene (pEV1) to confirm its tissue-specificity, and to the plant intron-containing variant of the cre (cre INT) recombinase terminated by nosT (Mlynárová and Nap 2003) yielding pEV2 to study the Cre/loxP-controlled excision events.
To obtain the binary vector pEV1 (Fig. 1a), first the CRUC promoter was cloned as a SalI-NcoI fragment from pEV2 together with the rfp gene as an NcoI-XbaI fragment from pFLUAR101 (Stuitje et al. 2003) into pBluescript SK + . The CRUC/rfp/nosT fusion was subsequently cloned as a XbaI-Asp718 fragment into pBinPlus (van Engelen et al. 1995).

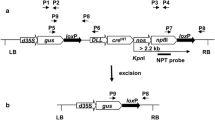
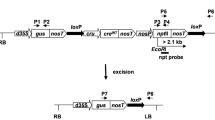
Plant binary vectors used in transformation experiments. a T-DNA structure of plant binary vector pEV1. The T-DNA consists of the DsRed (rfp) gene under control of the cruciferin (CRU) promoter and the neomycin phosphotransferase (nptII) gene regulated by the NOS promoter. Both genes were terminated by nosT. b T-DNA configuration of plant binary vector pEV2. The T-DNA consists of the β-glucuronidase (gus) gene driven by the double CaMV 35S promoter (d35S); the intron-containing cre recombinase gene (cre INT) under control of the cruciferin (CRU) promoter and the neomycin phosphotransferase (nptII) gene driven by the NOS promoter. All three genes were terminated by nosT. Black arrows indicate the presence and orientation of the loxP sites. The restriction sites used for Southern blot analyses are indicated as well as predicted fragment size. The primers used for PCR analysis are indicated as P1/P2, P3/P4 and P5/P6. c The T-DNA configuration generated after excision of the loxP embedded DNA. The primers used for PCR analysis are indicated as P7/P6
The pEV2 construct (Fig. 1b) was prepared by cloning NOS/nptII/nosT-loxP and dCaMV35S/gus/nosT-loxP-CRUC/cre INT /nosT fragments into the binary vector pUN as described by Vaculková et al. (2007). The plasmid pEV2 was evaluated for stability and recombination events in Escherichia coli DH5αF′ as described by Mlynárová and Nap (2003).
Both binary vectors pEV1 and pEV2 were introduced into A. tumefaciens LBA 4404 (separately) and their stability was verified by restriction analyses after retransformation into E. coli.
Plant material and transformation
Tobacco (Nicotiana tabacum cv. Petit Havana SR1) was transformed with A. tumefaciens LBA 4404 carrying binary vectors pEV1 or pEV2 using leaf disc transformation protocol described previously (Mlynárová et al. 1994). The transformed tissue was selected on the medium with 50 mg/l kanamycin. Regenerated shoots that showed difficulties with rooting were transferred and rooted without the presence of antibiotic. Transgenic plants (confirmed by PCR) were transferred to the soil, cultivated in the greenhouse conditions and allowed to set the seed after self-pollination.
Segregation assays
Two different segregation assays were used to test for the kanamycin resistance in progenies of the transgenic plants. In the germination assay, surface-sterilised seeds (approximately 100 seeds per plant) were germinated on MS medium containing 1% sucrose, 0.8% agar and 50 mg/l kanamycin. Three weeks later, the seedlings were evaluated for the kanamycin resistance. The green seedlings were considered to be kanamycin-resistant (KmR) and the yellow or pale green as kanamycin-sensitive (KmS).
In the rooting assay, surface-sterilised seeds germinated on MS medium containing 1% sucrose, 0.8% agar under non-selective conditions. Two weeks later, the seedlings were transferred into 100 ml glass culture vessels onto the fresh MS medium and grown for 6 weeks. Next, the plants devoid of roots were allowed to re-root in the presence of 50 mg/l kanamycin. Those that formed roots in 3 weeks were evaluated as kanamycin-resistant (R+), whereas non-rooted plantlets as sensitive (R−).
For GUS segregation assays, the GUS activity was detected on the seedlings/plants germinated/grown in both presence and absence of 50 mg/l kanamycin.
β-glucuronidase assays
A histochemical GUS assay was carried out according to the method of Jefferson et al. (1987). Leaf explants were incubated in a solution of 2 mM 5-bromo-4-chloro-3-indolyl glucuronide (X-gluc, Duchefa, The Netherlands), 50 mM phosphate (pH 7) at 37°C in the dark overnight.
Fluorimetric GUS assays were performed as described by Mlynárová et al. (1994). The GUS activity was expressed in picomoles of methylumbelliferone released per min per µg of soluble protein. The concentration of proteins was determined according to Bradford (1976).
RFP fluorescence
An assay for RFP fluorescence was conducted on the embryos isolated from the transgenic tobacco seeds (EV1). The activity of RFP was screened with fluorescent Axioplan 2 microscope (Carl Zeiss, Germany) using filter sets BP 546 and LP 590, and were photographed by Sony DXC-S500 Digital Camera System.
DNA analyses
Genomic DNA was isolated from tobacco leaves using the DNeasy Plant Mini Kit (Qiagen, UK). Internal PCR primers for detection of the gus gene were P1 (5′-GAT AAC GTG CTG ATG GTG CAC GAC-3′) and P2 (5′-GGC AAT ACT CCA CAT CAC CAC GCT-3′), for detection of the nptII gene were P3 (5′-GAT GGA TTG CAC GCA GGT TCT-3′) and P4 (5′-ATG GGT CAC GAC GAG ATC ATC-3′). The PCR reactions were carried out in 50 µl mixture containing 100-200 ng of DNA template, 15 pmol of each primer, 200 µM dNTPs, 1× PCR buffer and 1 U of Taq DNA polymerase (Finnzymes, Finland). The first PCR step of 94°C for 4 min was followed by 30 cycles of 94°C for 45 s, 64°C for 45 s and 72°C for 2 min. The last step was performed at 72°C for 7 min.
For Southern blot analysis, 10 μg of total DNA was digested with appropriate restriction enzymes (VspI or EcoRI), separated on a 1% agarose gel and blotted onto a Hybond N+ membrane (Amersham, UK). The GUS- and NPTII-specific probes were isolated as 2-kb and 1.4-kb restriction fragments (respectively) from an agarose gel and radioactively labelled using the Megaprime DNA labelling Kit (Amersham, UK). The hybridisation was performed in a hybridisation solution containing 10% dextran sulfate, 1% SDS, 1 M NaCl and 100 μg/l of salmon sperm DNA at 65°C. Hybridisation signals were visualised by autoradiography using a BAS2000 PhosphorImager (Fuji, Japan).
PCR screening for nptII excision
The presence or absence of the nptII gene was detected using two primer sets P5/P6 and P7/P6, respectively (Fig. 1b, c). The excision event (NPT−) was verified by amplification of a 0.59-kb fragment using the P7/P6 primers, whereas retaining of the nptII gene (NPT+) was detected by amplification of a 1.8-kb fragment using the P5/P6 primer set. The sequences of primers were: P5 (5′-ATG ACT GGG CAC AAC AGA CAA TCG-3′), P6 (5′-TCC GGC TCG TAT GTT GTG TGG AAT-3′), P7 (5′-ATA TGG CGC GTT GGC GGT AAC AAG-3′). The PCR reactions were carried out as described earlier.
Quantitative q-PCR was performed using the primers P1/P2 and P3/P4, Rotor-Gene 6000 real-time PCR system (Corbett Research, Australia), and ABsolute qPCR SYBR Green Mix (ABgene, UK) according to the manufacturer’s instructions. The corresponding reaction mixtures (20 μl) consisted of 1 × ABsolute qPCR SYBR Green Mix, 300 nM primers and 50–75 ng of template DNA. The cycling conditions consisted of 15 min of incubation at 95°C followed by 40 cycles of 95°C for 15 s, 64°C for 15 s and 72°C for 20 s. The standard curves for the gus and nptII genes were generated using serial dilutions of DNA from T1-80 (single-copy T-DNA plant without any detectable excision of nptII) as a template DNA. A melting curve analysis was performed at 72–95°C. The relative amount ratio of the nptII as a target gene was calculated based on the efficiency (E) and crossing point (CT) deviation of analysed samples versus control (T1-80) plant and expressed in comparison to the gus as a reference gene (Pfaffl 2001). The experiments were repeated three times.
Results
The Arabidopsis seed-specific promoter CRUC (Becerra et al. 2006) appears to be a good candidate for the systems in which the exact, developmentally regulated expression is required. Its specific activity was confirmed also in the transgenic tobacco seeds (EV1), where expression of the rfp gene driven by CRUC promoter was detected in the embryo, but not in the endosperm (Fig. 2).

Expression of rfp in transgenic tobacco seed (without testa). The seeds were obtained after self-pollination of the transgenic tobacco plants (EV1). a The seed before and b after extirpation of an embryo (arrow) from an endosperm (broken line). Bars 500 μm
Behaving as expected, the feasibility of this promoter in the self excision Cre/loxP strategy was investigated following the tobacco transformation with A. tumefaciens/pEV2.
Tobacco transformation and nptII excision in T0 plants
Leaf tissues transformed with pEV2 regenerated under selection pressure of kanamycin. However, the shoot formation delayed and the developed root system was weaker than the usual. For this reason, all histochemically GUS-positive plants were replaced and rooted under non-selective conditions. The presence of the gus and nptII genes was detected in the genomes of 74 transgenic plants by PCR using the primers P1/P2 and P3/P4 specific to the gus and nptII genes, respectively (data not shown). Randomly selected T0 transformants (34) were screened by Southern blot hybridisation using the GUS and NPTII probes for detection of single-copy T-DNA plants. An example of Southern blot analysis is shown in Electronic Supplementary Material S1. Based on the restriction map of pEV2 (Fig. 1b), the GUS probe was expected to hybridise to both internal gus as well as to border gus fragments. The latter indicated the number of integrated copies. In the case of the nptII gene the number of detected fragments corresponded to the number of transgene copies. The combined results of the GUS and NPTII hybridisations showed that 11 plants carried intact single copy T-DNA integration. Eight of them were used in further experiments.
As one of the possible reasons for poor rooting of the primary transformants, the loss of nptII gene due to the premature excision was investigated. PCR analyses with the P7/P6 (NPT−) and P5/P6 (NPT+) primers were conducted on eight single-copy transformants (Electronic Supplementary Material S2). Obtained data confirmed the occurrence of both (NPT+) and (NPT−) cells in each of them. In addition, the independent experiments showed a similar pattern already in the developing transgenic calli, 10 weeks after transformation (Electronic Supplementary Material S3). However, no ongoing excision of the nptII gene in the mature transgenic T0 plants over time was observed. This was confirmed in experiments, where DNAs isolated from the leaves of regenerated plants and the same plants maintained in invitro conditions for 1 year were analysed by qPCR (Fig. 3).

qPCR analyses of ongoing ectopic expression in mature T0 plants. Genomic DNAs isolated from the leaves of three regenerated T0 plants and the same plants maintained in invitro conditions for 1 year were analysed by qPCR using the P1/P2 and P3/P4 primers. The relative amount ratio of the nptII as a target gene was calculated based on E and ΔCT of an analysed sample versus the single-copy T1-80 plant (no nptII excision, group C) and expressed in comparison to the gus as a reference gene. Data are mean values and standard deviations based on three replicates. No significant differences in the nptII/gus relative amount ratios of analysed plants over period of 1 year were observed
Excision of the nptII gene in T1 plants
The eight single-copy T0 plants were grown to the maturity in the greenhouse to set the seeds after self-pollination. According to Mendelian rules, all single-copy T0 transformants are hemizygous for the transgene and their progeny segregate 3:1 for transgene-positive versus transgene-negative seedlings. Therefore, all seedlings should segregate in ratio 3:1 (GUS+:GUS−) for the gus gene. In the case of successful excision of the nptII gene (Fig. 1c), all seedlings are expected to segregate in ratio 0:1 (KmR:KmS) for the nptII gene. In contrast to GUS, the segregation ratio for the nptII gene in progenies of all analysed T0 plants was different than expected. In the germination assays, the number of kanamycin-sensitive seedlings ranged from 61.9% (T0-64) to 91.2% (T0-30) (Table 1). None of the eight T0 lines generated progeny that was completely sensitive to kanamycin.
In order to investigate the excision event in progeny in more detail, the seedlings of non-selectively germinated seeds of a line (T0-30) were subsequently analysed. PCR analyses with the primers pairs P7/P6 and P5/P6 were carried out to detect the recombination events in leaves of 82 intact T1 plants. Based on PCR analyses, the analysed plants fall into four groups (Fig. 4; Table 2).

Detection of the nptII gene excision in T1 plants. Photographs of ethidium bromide-stained 1% agarose gels carrying PCR products obtained on progeny of the single-copy T0-30 plant. a PCR results with the primers P1/P2 that amplified an internal 0.3 kb fragment of the gus gene (GUS +). b PCR results with the primers P7/P6 that amplified a 0.59 kb fragment indicating excision of the nptII gene (NPT-). c PCR results with the primers P5/P6 that amplified an 1.8 kb fragment corresponding to the nptII gene (NPT +). d PCR products obtained with the actin primers. The lane M contains 1 kb DNA ladder (Fermentas) as a size marker, the lanes A-D represent the PCR products on individual T1 plants distributed according to groups A, B, C and D, T0-30 parental T0-30 plant, NT non-transformed plant, EV2 plasmid pEV2 used for plant transformation
The first group (A) includes six plants that amplified the gus internal P1-P2 (GUS+) and P7-P6 (NPT−) fragments confirming presence of the gus gene and excision of the nptII gene, respectively. As was predicted (Fig. 1c), these plants carried gus (and showed GUS activity) but no nptII was detected. Therefore, these plants were considered as marker-free.
The second group (B) comprises 51 gus gene containing plants that showed amplification of both P7-P6 (NPT−) and P5-P6 (NPT+) fragments. In these plants, transgene excision apparently did not occur in all cells of developing embryos and therefore they can be considered as chimeric for the nptII gene.
The third group (C) includes two gus gene containing plants that showed only amplification of the P5-P6 (NPT+) fragment. The absence of the P7-P6 (NPT−) PCR product indicated no transgene excision as a result of failure of the Cre/loxP system at all.
The group (D) includes 23 plants, in which the presence of neither gus nor nptII genes was detected. They represent 25% of non-transgenic part of population based on Mendelian rules of heredity.
Taken together, the Cre-mediated excision occurred in 57 (groups A and B) out of 59 (groups A, B and C) GUS-positive transgenic T1 plants. However, only six plants (group A) can be considered as marker-free. In most plants (86.4%, group B) the incomplete excision of the nptII gene was observed. Marker free or nptII chimeric nature of selected T1 plants was also confirmed by Southern hybridisation (Fig. 5a). In addition, a variable extent of the nptII excision in analysed chimeric T1 plants was confirmed by qPCR (Fig. 5b).

Southern blot and qPCR analyses on T1 plants. a EcoRI-digested genomic DNA was probed with the NPTII specific probe. An absence of the corresponding signal indicates marker-free T1 plants. For each T1 plant, the corresponding GUS activity is given (in picomoles of methylumbelliferone released per min per µg of soluble protein). b The T1 plants were analysed by qPCR using the P1/P2 and P3/P4 primers. The relative amount ratio of nptII/gus was calculated based on E and ΔCT of an analysed sample versus the single-copy T1-80 plant (no nptII excision, group C). Data are mean values and standard deviations based on three replicates. The nptII/gus relative amount ratios between 0 (100% excision) and 1 (no excision) indicate the excision rate in chimeras
In plant transgenosis, the results of the germination assays are believed to be proof enough for the kanamycin resistance or sensitivity. To our surprise, the number of T1 plants evaluated as marker-free (6) by PCR (Table 2) was much lower than anticipated by the germination assay (58) (Table 1). In the attempt to find more reliable screening assay, all T1 plants (82) analysed by PCR were subjected to rooting in the presence of kanamycin. Data are summarised in Table 2. The results showed that 13 out of 59 GUS-positive T1 plants did not form any roots. As expected, six out of 13 (R−) were marker-free plants (group A). The remaining seven (R−) plants were chimeras (group B). In contrast to pale/yellow marker-free plants, they remained green. The rooting assay indicated less marker free-plants than the germination assay. At the same time, results of the rooting assay were more consistent with the data obtained by PCR. Based on the germination assay experiments we assume that among the KmS seedlings were also nptII chimeric ones with lower survival potential. We suppose that higher number of the KmS plants can be caused by higher tissue sensitivity of the germinating seedlings.
Excision of the nptII gene in T2 plants
To investigate whether the process of the excision was completed in T2 generation, ten randomly selected chimeric T1 plants (group B) and two marker-free plants (group A), were allowed to set the seeds after self-pollination. The progeny of T1 plants is expected to segregate in ratios 1:0 or 3:1 for the presence versus absence of the gus gene, depending on the homo- or hemizygous state of parental T1 plants, respectively. The segregation of GUS activity showed the hemizygous nature of two marker-free (group A) as well as five out of ten randomly selected T1 plants (group B). The germination and rooting assays in the presence of kanamycin revealed that progenies of four chimeric plants as well as two marker-free plants segregated in ratio 0:1 (KmR:KmS) indicating the absence of the nptII gene. This was also confirmed by PCR (data not shown). In contrast, none of the homozygous T1 plants generated completely kanamycin-sensitive progeny. The percentage of KmS plants ranged from 22.6% (T1-8) to 59.8% (T1-62) (Table 3). Hence, repeated activation of the cre gene in T2 seeds resulted in increased rate of the marker free plants.
Discussion
The self excision strategy of floxed DNA comprising the selectable marker gene and cre recombinase driven by a tissue-specific promoter seems to be very promising with respect to the release of selectable marker-free crops into the environment. The advantage of this approach is that expression of the cre recombinase was supposed to be limited to a relatively short period of time thus decreasing the possibility for negative effects of high levels of the Cre protein on plant morphology and development (Coppoolse et al. 2003). In our study, the tissue-specific CRUC promoter was applied to drive the cre gene expression while the removal of floxed sequence was expected during the seed development without any external stimulus. The activity of Arabidopsis CRUC promoter was clearly confirmed in the tobacco embryo (Fig. 2). Further, using this promoter we were able to generate the marker-free transgenic plants indicating its feasibility in the Cre/loxP strategy. However, we faced several unexpected difficulties. Poor rooting of T0 plants on the media in the presence of kanamycin made us study this phenomenon in detail. PCR analyses revealed chimeric nature (for the nptII gene) of these T0 plants. In addition, after an independent transformation experiment we found partial excision of floxed DNA already in the developing tobacco calli despite the fact that the CRUC promoter was shown not to be active in Arabidopsis callus (AMD database). This indicated an ectopic expression from the CRUC promoter in non-differentiated cells of tobacco callus and/or in developing adventive meristems from which the shoots developed. It seems that such ectopic excision did not continue in the tissue of mature T0 plants (Fig. 3). This raises the question: What could be the reason for premature activation of the CRUC promoter during the process of regeneration? Previously, it has been reported that the activity of many tissue-specific promoters might not exclusively be tightly tissue-specific and can be activated by different (a) biotic stimuli (Trindade et al. 2003), or by the presence of adjacent strong promoter sequences (Suzuki et al. 2001). In our system, both alternatives might be relevant. Since the pEV2 used for transformation experiments contained the gus gene fused to the double 35S promoter located near to the CRUC-cre expression unit, it is therefore possible that this promoter sequence affected the expression pattern of the cre gene. Similarly, Zheng et al. (2007) reported that the 35S promoter caused ectopic expression of pollen, ovule and early embryo-specific BAP5 promoter (Belostotsky and Meagher 1996) in calli and shoot primordia. However, the influence of the 35S promoter can vary at the organ level from one promoter to another; therefore, it is difficult to predict its effect on a particular gene promoter (Zheng et al. 2007). A possible effect of (a) biotic stress on an activity of the CRUC promoter could also be considered. Data in AMD database indicate that gene expression from the cruciferin promoter might be induced by agrobacteria surviving on the surface of the transformed tissue for longer time. Nevertheless, an ectopic activation of the cre-driving promoter (early nptII excision) interfered with regeneration of the transformants in our experiments. Such premature excision of a selectable marker gene could be a limiting factor in obtaining primary transformants and in the application of inducible promoters in the self excision Cre/loxP strategy.
Focusing on the nptII gene removal in the seeds we found that the process of DNA excision occurred in progenies of all T0 transgenic plants to a certain extent since both the kanamycin-sensitive as well as kanamycin-resistant seedlings were detected. A detailed analysis of T0-30 progeny showed that only six out of 59 transgenic T1 plants were marker-free and the excision failed in two plants while the remaining plants were chimeric. This chimerism could be caused by insufficient efficiency of the Cre/loxP system alone in higher plants and the timing of expression from the CRUC promoter in the developing embryo or by a combination of several factors. The differences in efficiency of the Cre-mediated recombination and variable cre expression in higher plants have also been observed by others (Keenan and Stemmer 2002; Marjanac et al. 2008). Recently Luo et al. (2007) studied the efficiency of nptII removal from tobacco pollen, seed or both using the phage Cre/loxP or yeast FLP/FRT systems. They observed 100% excision efficiency only in the case of combination of loxP-FRT fusion sequences as recognition sites for Cre or FLP. On the other hand, Mlynárová et al. (2006) using the Cre/loxP system in combination with pollen specific NTM19 promoter achieved high efficiency of the selectable marker gene removal from pollen of tobacco since only two KmR seedlings among about 16 800 KmS were observed. Such a high efficiency could coincide with activity of the NTM19 promoter in the early stage of pollen development in uninucleate microspore. In contrast, a multicellular embryo might represent a more complex structure for the complete removal of floxed sequences. Liu et al. (2005) achieved increased excision efficiency in the tissue of the transgenic plants when a nuclear localization signal (NLS) was fused to the cre recombinase driven by a heat shock-inducible promoter. This signal apparently enhanced the effect of an intrinsic, bipartite basic determinant of the cre gene which functions as a NLS. From this, it follows that if the process of excision has to occur in every cell of a plant tissue (or in a multicellular embryo as in our case), the transport of Cre through a nuclear membrane might be the bottleneck of the whole process.
Although the proposed approach showed some limitation regarding the excision efficiency in T1 tobacco plants, we were however able to select the hemizygous as well as homozygous (for desired gus gene) transgenic plants with stably removed selectable nptII gene in T2 generation. We also found that only the hemizygous T1 plants (4) produced marker-free progenies with 100% efficiency. Apparently, the hemizygous status was in the excision advantage comparing to the homozygous status. We suppose that this might coincide with the fact that recombination events have to occur on both alleles.
Although the CRUC promoter was expected to be an ideal candidate for controlling excision process in the seeds, we point out its ectopic activity during a regeneration of transgenic tobacco plants and the problems with a complete excision of the nptII gene in T1 progeny. It is not yet known whether these observations are the consequences of the plant species used for the transformation, properties of the Cre/loxP driving promoter and/or the vector construct. We hope that our work will evoke further discussions on the phenomenon of chimerism that is very likely to occur to a different extent in all systems where a recombination event has to take place in multicellular structures. An additional screening such as the rooting test might be useful for scientists to identify routinely and more effectively the chimeric plants.
References
Becerra C, Puigdomenech P, Vicient CM (2006) Computational and experimental analysis identifies Arabidopsis genes specifically expressed during early seed development. BMC Genomics 7:38. doi:10.1186/1471-2164-7-38
Belostotsky DA, Meagher RB (1996) A pollen-, ovule-, and early embryo-specific poly(A) binding protein from Arabidopsis complements essential functions in yeast. Plant Cell 8:1261–1275
Bevan MW, Flavell RB, Chilton MD (1983) A chimeric antibiotic resistance gene as a selection marker for plant cell transformation. Nature 304:184–187
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantise of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Coppoolse ER, deVroomen MJ, Roelofs D, Smit J, van Gennip F, Hersmus BJM, Nijkamp JJ, van Haaren MJ (2003) Cre recombinase expression can result in phenotypic aberrations in plants. Plant Mol Biol 51:263–279
Cuellar W, Gaudin A, Solórzano D, Casas A, Ñopo L, Chudalayandi P, Medrano G, Kreuze J, Ghislain M (2006) Self-excision of the antibiotic resistance gene nptII using a heat inducible Cre/loxP system from transgenic potato. Plant Mol Biol 62:71–82
Hoff T, Schnorr KM, Mundy J (2001) A recombinase-mediated transcriptional induction system in transgenic plants. Plant Mol Biol 45:41–49
Höglund AS, Rödin J, Larsson E, Rask L (1992) Distribution of napin and cruciferin in developing rape seed embryos. Plant Physiol 98:509–515
Jefferson RA, Kavanagh TA, Bevan MW (1987) GUS fusion: β-glucuronidase as sensitive and versatile gene fusion marker in higher plants. EMBO J 6:3901–3908
Keenan RJ, Stemmer WP (2002) Nontransgenic crops from transgenic plants. Nat Biotechnol 20:215–216
Li Z, Xing A, Moon BP, Burgoyne SA, Guida AD, Liang H, Lee C, Caster CS, Barton JE, Klein TM, Falco SC (2007) A Cre/loxP-mediated self-activating gene excision system to produce marker free transgenic soybean plants. Plant Mol Biol 65:329–341
Liu HK, Ch Yang, Wei ZM (2005) Heat shock-regulated site-specific excision of extraneous DNA in transgenic plants. Plant Sci 168:997–1003
Luo K, Duan H, Zhao D, Zheng X, Deng W, Chen Y, Stewart CN, McAvoy R, Jiang X, Wu Y, He A, Pei Y, Li Y (2007) ‘GM-gene-deletor’: fused loxP-FRT recognition sequences dramatically improve the efficiency of FLP or CRE recombinase on transgene excision from pollen and seed of tobacco plants. Plant Biotechnol J 5:263–274
Marjanac G, De Paepe A, Peck I, Jacobs A, De Buck S, Depicker A (2008) Evaluation of CRE-mediated excision approaches in Arabidopsis thaliana. Transgenic Res 17:239–250
Mlynárová L, Loonen A, Heldens J, Jansen RC, Keizer P, Stiekema WJ, Nap JP (1994) Reduced position effect in mature transgenic plants conferred by the chicken lysozyme matrix associated region. Plant Cell 6:417–426
Mlynárová L, Nap JP (2003) A self-excising Cre recombinase allows efficient recombination of multiple ectopic heterospecific lox sites in transgenic tobacco. Transgenic Res 12:45–57
Mlynárová L, Conner AJ, Nap JP (2006) Directed microspore-specific recombination of transgenic alleles to prevent pollen-mediated transmission of transgenes. Plant Biotechnol J 4:445–452
Pfaffl MW (2001) A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29:2002–2007
Schmid M, Davison TS, Henz SR, Pape UJ, Demar M, Vingron M, Schölkopf B, Weigel D, Lohmann J (2005) A gene expression map of Arabidopsis thaliana development. Nat Genet 37:501–506
Stuitje AR, Verbree EC, van der Linden KH, Mietkiewska E, Nap JP, Kneppers TJA (2003) Seed-expressed fluorescent proteins as versatile tools for easy (co)transformation and high-throughput functional genomics in Arabidopsis. Plant Biotechnol J 1:301–309
Suzuki M, Kao CY, Cocciolone S, McCarty DR (2001) Maize VP1 complements Arabidopsis abi3 and confers a novel ABA/auxin interaction in roots. Plant J 28:409–418
Trindade LM, Horvath B, Bachem C, Jacobsen E, Visser RGF (2003) Isolation and functional characterization of a stolon specific promoter from potato (Solanum tuberosum L.). Gene 303:77–87
Vaculková E, Moravčíková J, Matušíková I, Bauer M, Libantová J (2007) A modified low copy number binary vector pUN for Agrobacterium-mediated transformation. Biol Plant 51:538–540
van Engelen A, Molthoff JW, Conner AJ, Nap JP, Pereira A, Stiekema WJ (1995) pBINPLUS: an improved plant transformation vector based on pBIN19. Transgenic Res 4:288–290
Verweire D, Verleyen K, De Buck S, Claeys M, Angenon G (2007) Marker-free transgenic plants through genetically programmed auto-excision. Plant Physiol 145:1220–1231
Zhang Y, Li H, Ouyang B, Lu Y, Ye Z (2006) Chemical-induced auto-excision of selectable markers in elite tomato plants transformed with a gene conferring resistance to lepidopteran insects. Biotechnol Lett 28:1247–1253
Zheng X, Deng W, Luo K, Duan H, Chen Y, McAvoy R, Song S, Pei Y, Li Y (2007) The cauliflower mosaic virus (CaMV) 35S promoter sequence alters the level and patterns of activity of adjacent tissue- and organ-specific gene promoters. Plant Cell Rep 26:1195–1203
Zimmermann P, Hirsch-Hoffmann M, Hennig L, Gruissem W (2004) GENEVESTIGATOR. Arabidopsis microarray database and analysis toolbox. Plant Physiol 136:2621–2632
Acknowledgments
We thank Drs. Mlynárová and Nap for providing plasmids pFLUAR101, pLM91-containing CRUC-cre expression unit and for helpful discussion. We thank Dr. Salaj for help with microscopic techniques, and Dr. Matušíková for her kind proof reading of the manuscript. The authors thank Anna Fábelová for invitro plant care. The study was funded by Scientific Grant Agency of the Ministry of Education of Slovak Republic and Slovak Academy of Sciences 2/0011/08.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by C. F. Quiros.
Electronic supplementary material
Below is the link to the electronic supplementary material.
122_2008_866_MOESM1_ESM.tif
Electronic Supplementary Material S1 Examples of Southern blot analysis of parental T0 plants. a VspI-digested DNA probed with the GUS specific probe. The bands of a 2.9 kb correspond to the internal gus fragment. All additional bands (>1.5 kb) correspond to the border fragments and indicate the number of independent transgene copies. The absence of the internal 2.9 kb gus fragment in the plant T0-8 indicates an incomplete gus gene integration. b EcoRI-digested DNA probed with the NPTII specific probe. The bands (>2.1 kb) correspond to the number of independent transgene copies. NT – non-transgenic control plant, asterisks indicate single copy transformants used in further analyses. (TIFF 9924 kb)
122_2008_866_MOESM2_ESM.tif
Electronic Supplementary Material S2 PCR detection of premature excision in T0 plants. Photographs of ethidium bromide-stained 1% agarose gels carrying PCR products obtained from the single-copy T0 tobacco plants. a PCR with the primers P7/P6 that amplified a 0.59 kb fragment indicating excision of the nptII gene (NPT-). b PCR with the primers P5/P6 that amplified a 1.8 kb fragment corresponding to the nptII gene (NPT+). c PCR products obtained on the same plant set with the actin primers. The lane M contains 1 kb DNA ladder (Fermentas) as a size marker, next follow the PCR products of individual T0 plants, the last lane represents PCR product of plasmid pEV2 (EV2). (TIFF 6356 kb)
122_2008_866_MOESM3_ESM.tif
Electronic Supplementary Material S3 PCR analyses of premature excision in developing transgenic calli. Genomic DNA from transgenic calli was subjected to PCR analysis with the primers P7/P6 that amplified a 0.59 kb PCR fragment indicating the nptII gene excision (NPT-) and the primers P5/P6 that amplified a 1.8 kb fragment corresponding to the nptII gene (NPT+). The actin primers were used as a PCR control. The lane M contains 1 kb DNA ladder (Fermentas) as a size marker, C – transgenic calli, NT – non-transformed control, EV2 – plasmid pEV2. (TIFF 3590 kb)
Rights and permissions
About this article
Cite this article
Moravčíková, J., Vaculková, E., Bauer, M. et al. Feasibility of the seed specific cruciferin C promoter in the self excision Cre/loxP strategy focused on generation of marker-free transgenic plants. Theor Appl Genet 117, 1325–1334 (2008). https://doi.org/10.1007/s00122-008-0866-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-008-0866-4