
Abstract
The ability of the CRE recombinase to catalyze excision of a DNA fragment flanked by directly repeated lox sites has been exploited to modify gene expression and proved to function well in particular case studies. However, very often variability in CRE expression and differences in efficiency of CRE-mediated recombination are observed. Here, various approaches were investigated to reproducibly obtain optimal CRE activity. CRE recombination was analyzed either by transforming the CRE T-DNA into plants containing a lox-flanked fragment or by transforming a T-DNA harboring a lox-flanked fragment into plants producing the CRE recombinase. Although somatic CRE-mediated excision of a lox-flanked fragment was obtained in all transformants, a variable amount of germline-transmitted deletions was found among different independent transformants, irrespective of the orientation of transformation. Also, the efficiency of CRE-mediated excision correlated well with the CRE mRNA level. In addition, CRE-mediated fragment excision was compared after floral dip and after root tissue transformation when transforming in a CRE-expressing background. Importantly, less CRE activity was needed to excise the lox-flanked fragment from the transferred T-DNA after root tissue transformation than after floral dip transformation. We hypothesize that this is correlated with the lower T-DNA copy number inserted during root transformation as compared to floral dip transformation.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Several site-specific recombination systems from bacteriophages and yeast have been well characterized (Odell and Russell 1994; Ow 2002, Thomson and Ow 2006) and found to be functional in a wide range of organisms. The most widely used in plants is the bacteriophage P1 CRE/loxP site-specific recombination system (Gilbertson 2003). This CRE recombinase system has been exploited extensively to remove selectable marker genes (Dale and Ow 1990; Russell et al. 1992; Zuo et al. 2001; Hoa et al. 2002; Zhang et al. 2003; Sreekala et al. 2005). Another application is the resolution of complex transgene loci to single copies after direct gene transfer (Srivastava et al. 1999; Srivastava and Ow 2001). In addition, the CRE/lox recombination system for targeted DNA insertion proved to be efficient when DNA was introduced by particle bombardement (Srivastava et al. 2004; Chawla et al. 2006), but was ineffective either after polyethylene glycol-mediated protoplast transformation because of high rates of gene silencing or after Agrobacterium-mediated transformation because of low transformation frequencies (Vergunst and Hooykaas 1998; Vergunst et al. 1998; Day et al. 2000). In the above described examples, the lox constructs and the CRE gene were combined either by retransformation or by genetic crossings. The timing of gene expression can be highly controlled by using inducible promoters, such as promoters regulated by chemicals or heat shock for switching on CRE at the desired time point (Hoff et al. 2001; Joubès et al. 2004; Sreekala et al. 2005; Wang et al. 2005). Furthermore, strategies for transient expression of the CRE recombinase in plants have been successful either by cocultivation with recombinant Agrobacterium strains (Gleave et al. 1999), by use of an inducible self-excising CRE expression vector (Zuo et al. 2001), or by applying plant virus vector systems (Kopertekh et al. 2004; Jia et al. 2006).
Several reports have been published on variations in excision efficiency (Odell et al. 1990; Bayley et al. 1992; Russell et al. 1992; Hoa et al. 2002) that have been postulated to be caused by the chromosomal position of the transgenic loci and/or the difference in CRE recombinase activity in the lox target locus and CRE-expressing locus. The efficiency of recombination seemed to be related primarily to the CRE parent and has been hypothesized to be most probably due to different CRE expression levels in tobacco (Nicotiana tabacum) and Arabidopsis thaliana (Russell et al. 1992). In addition, the excision efficiency was much higher when the lox target plant was transformed with a CRE-containing T-DNA than when CRE-expressing plants were crossed with lox target plants, indicating that various degrees of excision efficiency are obtained with different combination methods for the lox-flanked gene and the CRE recombinase (Russell et al. 1992). The above examples illustrate that, although CRE-mediated excision can work reliably, many problems are observed with the reproducibility.
The objective of this work was to obtain a clear view on the effectiveness of CRE-mediated excision in A. thaliana. In particular, the question was asked whether CRE-mediated recombination is more efficient when the lox-flanked target is transformed into a CRE background or vice versa when a CRE-expressing T-DNA construct is transformed into a lox-flanked target fragment-containing plant. Therefore, firstly, we transformed a CRE-expressing T-DNA into plants containing a loxP-flanked ß-glucuronidase (GUS)-coding cassette and, secondly, a T-DNA with the loxP-flanked GUS cassette into two different homozygous CRE-expressing plants, either by floral dip transformation or by root explant cocultivation, allowing the CRE recombination activity to be compared in both series of transformants. By using different CRE-expressing plants, a possible direct correlation between the CRE recombination efficiency and the CRE expression level could be analyzed.
Results
Strategy to evaluate CRE-mediated excision of the GUS expression cassette
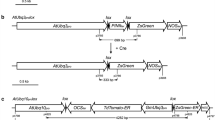
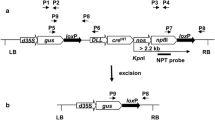
The presence or absence of the GUS gene between two tandemly repeated loxP sites was used as reporter system for CRE-mediated recombination (Fig. 1a, b) (De Buck et al. 2000). The K2L T-DNA contains a kanamycin resistance gene as selectable marker and a 35S-driven GUS gene flanked by two directly repeated loxP sites (Fig. 1a) (De Buck et al. 1998). The chosen CK2L6 line of Arabidopsis is homozygous for the transgenic locus containing a single K2L T-DNA copy inserted into chromosome 5 and displays stable and high GUS expression in the F2 generation (Theuns et al. 2002, De Buck et al. 2004). The excision of the GUS gene by introduction of CRE was analyzed by histochemical staining and by polymerase chain reaction (PCR) analysis (Fig. 1a, b; Materials and methods).

Strategy for CRE-mediated excision of the GUS expression cassette. (a) Schematic representation of the K2L T-DNA in the CK2L6 line. The K2L T-DNA consists of a kanamycin selectable marker and P35S-GUS-3′n expression cassette (not on scale). After digestion of the plant DNA with HindIII and SacI, a GUS fragment of 3199 bp will be observed after hybridization with the GUS probe (probe 1) when the GUS gene is present in the genome and a T-DNA/plant junction fragment of at least 3072 bp will be observed after hybridization with the NPTII probe (probe 2). (b) The derived K2L-Δ T-DNA after CRE-mediated recombination of the directly oriented loxP sites (L), resulting from the excision of the GUS expression cassette. (c) Schematic outline of the HSC T-DNA in pHsc. (d) Schematic outline of the CRE T-DNA in pCre. Abbreviations: 3′ocs, 3′ end of the octopine synthase gene; NPTII, neomycin phosphotransferase II gene; Pn, promoter of the nopaline synthase gene; L, loxP sequence recognition site of the CRE/loxP recombination system; P35S, cauliflower mosaic virus promoter; GUS, ß-glucuronidase-coding sequence; HPT, hygromycin phosphotransferase gene; 3′n, 3′ end of the nopaline synthase gene; 3′35S, 35S terminator; CRE, CRE recombinase gene; BAR, phosphinothricin transferase gene; PSSUARA, the small subunit promoter of Arabidopsis; TP, transit peptide; 3′g7, 3′ terminator of gene 7; attB1 and attB2, GATEWAY™-compatible recombination sites; H, HindIII; S, SacI; LB, left border; RB, right border; p3, p4, and p5, primers used for PCR analysis
Efficiency of CRE-mediated excision of the GUS expression cassette by transformation of a P35S-CRE T-DNA into CK2L6 Arabidopsis plants
In order to analyze the effectiveness of CRE recombinase to induce DNA recombination and excision of the GUS gene, CK2L6 Arabidopsis plants were retransformed by the floral dip method with the HSC T-DNA (Fig. 1c) that contains a P35S-driven CRE gene and a hygromycin-selectable marker gene (Materials and Methods). T2 progeny plants of ten independent hygromycin-resistant CK2L6-SC retransformants were stained for GUS activity (Table 1). All T2 seedlings derived from retransformant CK2L6-SC13 were GUS-negative (Fig. 2T) implying that the CRE recombinase excised the GUS cassette during the T1 generation. Among the progeny of the other nine CK2L6-SC retransformants, a variable number of GUS-negative and GUS-positive seedlings with a mosaic pattern were observed (Table 1). The mosaic pattern resulted from GUS gene excision in parts of the cells of the growing T2 progeny plants, implying inheritance of a non-excised form of the K2L T-DNA from the T1 CK2L6-SC retransformants. For example, eight of ten T2 seedlings of retransformant CK2L6-SC6 were GUS-negative and only two had a patchy GUS activity distribution, mainly in the hypocotyl and primary root cells (Fig. 2G, H, I). In retransformant CK2L6-SC3, seven T2 seedlings were GUS-negative and in the remaining three GUS was active in the root hairs of the primary root (Fig. 2B). Furthermore, in the CK2L6-SC5, CK2L6-SC7, CK2L6-SC9, CK2L6-SC10, and CK2L6-SC12 retransformants, four to six of the progeny plants displayed a patchy distribution of GUS activity in the cotyledons, hypocotyls, and the primary root cells (Fig. 2 CK2L6-SC5 (E, F); CK2L6-SC7 (J, K); CK2L6-SC9 (L, M); CK2L6-SC10 (N, O); CK2L6-SC12 (R, S)). Finally, in transformants CK2L6-SC4 and CK2L6-SC11, only two of the T2 seedlings were GUS-negative, while the remaining eight seedlings had large GUS-positive sectors in the cotyledons, hypocotyls, primary roots, and in some lateral roots (Fig. 2 CK2L6-SC4 (C, D); CK2L6-SC11 (P, Q)).

Histochemical GUS staining of 2-week-old seedlings. Histochemical analysis of T2 progeny from ten independent CK2L-SC retransformants. CK2L6 parental line (A); representative examples of T2 progeny plants from transformant CK2L6-SC3 (B), CK2L6-SC4 (C, D), CK2L6-SC5 (E, F), CK2L6-SC6 (G, H, I), CK2L6-SC7 (J, K), CK2L6-SC9 (L, M), CK2L6-SC10 (N, O), CK2L6-SC11 (P, Q), CK2L6-SC12 (R, S), and CK2L6-SC13 (T). Seedlings were grown for two weeks on K1 medium supplemented with 20 mg/l hygromycin
To rule out that silencing was the reason for the absence of GUS staining rather than CRE-mediated GUS excision, a molecular analysis was performed with the primer sets p3 + p4 and p3 + p5 (Fig. 1). For the T2 seedlings from the transformant CK2L6-SC13, only a 834 bp fragment was detected, indicating efficient CRE-mediated excision of the GUS gene in the analyzed leaves (Fig. 3). This result corresponds with the histochemical staining because no GUS activity was observed in any of the T2 progeny plants (Fig. 2T). The progeny from the other nine CK2L6-SC retransformants showed a signal for both the 834-bp fragment and the 1200-bp fragment, indicative for the presence of the GUS cassette, correlating with the mosaic GUS staining pattern (Fig. 3).

PCR analysis of genomic DNA prepared from T2 progeny from the ten independent CK2L6-SC retransformants. Primers 3, 4, and 5 were used as illustrated in Fig. 1. Seedlings were grown for two weeks on K1 medium supplemented with 20 mg/l hygromycin. The following controls were added: C1, CK2L plant; C2, KH15d plant (deletion line containing both the GUS expression cassette and the deletion [De Buck et al. 2001]), and C24 wild-type DNA
To investigate whether the observed differential CRE-mediated recombination efficiency could be caused by a difference in CRE expression levels, we performed a real-time quantitative PCR analysis. The retransformants CK2L6-SC11 and CK2L6-SC13 were chosen as representatives for low and high rates of GUS excision, respectively (Fig. 4). CRE mRNA levels in the CK2L6-SC13 retransformant were five-fold higher than those in the CK2L6-SC11 retransformant, corresponding with the GUS staining results of T2 progeny plants. This difference indicates that high CRE mRNA levels are required to obtain high rates of CRE-mediated recombination.

Expression analysis of P35S-CRE in the CK2L6-SC11, CK2L6-SC13, Cre1, and Cre13 lines. mRNA accumulation levels in pooled 2-week-old seedlings were determined by real-time PCR with ACTIN2 as constitutive control. The fold difference of expression in the analyzed lines compared to the untransformed control is shown. Error bars are standard deviation of two biological repeats. Seedlings were grown for two weeks on K1 medium supplemented with 20 mg/l hygromycin (CK2L6-SC11 and CK2L6-SC13) or non-selective K1 medium (Cre1 and Cre13)
In conclusion, although all ten CK2L6-SC retransformants showed functional CRE activity, the efficiency of CRE-mediated excision of the GUS expression cassette varied significantly between the different CK2L6-SC retransformants. Furthermore, we demonstrated a correlation between the CRE mRNA expression level and excision efficiency in the CK2L6-SC11 and the CK2L6-SC13 transformants.
Efficiency of CRE-mediated excision by transformation of the lox-GUS K2L T-DNA into CRE-expressing Arabidopsis plants
Two homozygous, single-locus CRE-expressing lines, Cre1 and Cre13 (Materials and Methods) containing the CRE T-DNA (De Buck et al. 2000) (Fig. 1d), were used to determine whether the CRE-mediated recombination can be obtained with a higher frequency when the target is transformed into CRE-expressing background (see above). Therefore, Cre1 and Cre13 plants were retransformed with the lox target-containing K2L T-DNA by floral dip transformation. Four out of six T2 progenies of the retransformants into Cre1 background (FC1K2L) were GUS-negative, but none of the T2 progenies of the ten analyzed retransformants into Cre13 background (FC13K2L) showed excision of the GUS cassette in all progeny plants (Table 2). Moreover, the T2 plants of FC1K2L-2-3, which still contained the GUS gene were only weakly stained, while seedlings of all FC13K2L T2 plants displayed a mosaic pattern ranging from minimally to completely GUS-stained (data not shown). Thus, the retransformed Cre1 and the Cre13 plants differed clearly in CRE efficiency for GUS excision from the K2L T-DNA. The PCR results fully corroborated the GUS staining pattern for all the transformants, meaning that a mosaic GUS pattern correlated with the amplification of the 1200-bp fragment that is diagnostic for the presence of the GUS cassette (Table 2). In the absence of GUS staining, only the diagnostic fragment of 834 bp was found indicative of the K2L T-DNA excision allele (Table 2).
Real-time PCR analysis revealed that the CRE mRNA level was approximately two-fold higher in the Cre1 plants than in the Cre13 plants, which is in agreement with the observed different rate of CRE-mediated recombination (Fig. 4).
The K2L T-DNA was also introduced into Cre1 and Cre13 plants via root explant transformation. T2 seedlings of nine retransformants in Cre1 (RC1K2L) and of eight retransformants in Cre13 (RC13K2L) were analyzed by GUS staining and PCR analysis (Table 3; Materials and Methods). Strikingly, none of the T2 progenies of the RC1K2L retransformants still contained the GUS gene. Also, the GUS cassette was excised in all of the T2 progeny plants of seven of the eight RC13K2L retransformants.
To explain the higher rate of GUS excision after root transformation than after floral dip transformation, GUS excision and T-DNA integration pattern for 1 FC1K2L, 5 FC13K2L, 5 RC1K2L, and 3 RC13K2L retransformants were determined by DNA gel blot analysis (Fig. 5). The HindIII/SacI digest was chosen to allow detection of the internal 3199-bp GUS fragment with the GUS probe and the visualization of the left T-DNA-plant junction fragment with the neomycin phosphotransferase II (NPTII) probe (Fig. 1). The obtained results with the GUS probe (Fig. 5a) fully confirmed the PCR analysis and GUS staining pattern. For retransformants FC1K2L3-3, no GUS fragment could be detected, while for all the tested FC13K2L retransformants the GUS fragment was present. In all RC1K2L and RC13K2L retransformants, the 3199-bp GUS fragment was absent, except in RC13K2L-27a where it could be visualized analogously to the PCR and GUS staining results. The presence of the other hybridization bands with the GUS probe is probably caused by partial digestion because of methylation of the HindIII-SacI restriction sites (Nelson et al. 1993). Moreover, inverted repeats of additional T-DNAs in the locus may cause inversion of the segments between two inverted loxP sites and result in new GUS fragments. Hybridization with the NPTII probe indicated that on average more T-DNA copies were integrated in floral dip than in root tissue transformants (Fig. 5b). On average, more than two LB/plant border junctions were observed for the floral dip retransformants, while retransformants obtained by root transformation contained on average one or two LB/plant border junctions.

DNA gel blot analysis on HindIII-SacI-digested DNA of FC1K2L, FC13K2L, RC1K2L and RC13K2L retransformants. (a) GUS excision analysis. A GUS fragment of 3199 bp was observed after hybridization with the GUS probe when the GUS gene is still present in the genome. When GUS excision occurred in all cells of the transformant, no GUS fragment was detected. The expected GUS fragment is indicated. (b) Integration pattern analysis. DNA gel blot analysis with the NPTII probe. On top of each lane, the name is given of the transformant from which DNA was prepared. Left panels, floral dip retransformants; right panels, root tissue retransformants. Seedlings were grown for two weeks on K1 medium supplemented with 50 mg/l kanamycin
All together, we demonstrated that when a loxP-containing T-DNA was transformed into CRE-expressing plants, the efficiency of CRE-mediated excision varied according to the CRE-expressing line, indicating that the CRE expression level is most probably the most discriminating factor to obtain high CRE activity. Moreover, our results convincingly show that GUS excision from the introduced K2L T-DNA was much more efficient after root explant than after floral dip transformation.
Discussion
We analyzed the CRE-mediated excision efficiency of a loxP-flanked GUS expression cassette in transgenic Arabidopsis plants after transformation of the CRE construct into the target plant and after transformation of the target into CRE-expressing plants. For this latter approach, both the floral dip and the more labour-intensive root explant transformation method were compared.
Upon floral dip transformation of CK2L6 plants with the P35S-CRE construct, a variable excision efficiency was observed among different independent transformants. One out of ten independent CK2L6-SC retransformants had a 100% GUS-negative progeny. As confirmed by molecular analysis, the absence of GUS staining was due to CRE-mediated excision of the GUS gene. In the other nine CK2L6-SC retransformants, the percentage of GUS-negative and GUS mosaic plants varied, but all transformants displayed CRE-mediated recombination. Different excision efficiencies are probably related to different expression levels of the CRE recombinase. Indeed, upon transformation, the T-DNA is often randomly integrated into multiple copies, resulting in silencing of the resident T-DNA transgenes and variability of transgene expression (Muskens et al. 2000; Van Houdt et al. 2000; De Buck et al. 2001). Thus, variable CRE activity levels could explain the complete or partial CRE-mediated excision of the GUS gene in different transformants. Real-time PCR analysis of transformants CK2L6-SC11 and CK2L6-SC13 demonstrated a correlation between CRE mRNA expression levels and observed CRE activity. In addition, transformation of the CK2L6 plants with the CRE recombinase, controlled either by the constitutive, but weaker, nopaline synthase promoter or the heat-shock-induced promoter Gmhsp 17.6L of soybean (Glycine max) (Severin and Schöffl 1990) did not result in efficient excision of the loxP-flanked GUS gene, because GUS-negative sectors were observed with a very low frequency (data not shown). This observation suggests that the nopaline synthase and the used heat-inducible promoter were too weak to obtain the required CRE recombinase levels.
Subsequently, two CRE-expressing plants were retransformed with the loxP-containing K2L T-DNA by floral dip and root tissue transformation. A difference with the previous approach is that upon transformation with the K2L T-DNA, multiple copies of the K2L T-DNA can integrate into the CRE-expressing plant. In addition, the K2L T-DNA can integrate into different loci in the genome, although mostly multiple T-DNA copies tend to integrate at the same locus (De Block and Debrouwer 1991; De Neve et al. 1997). Different CRE recombination rates were observed between the two CRE-expressing lines. Quantitative PCR revealed lower CRE mRNA levels in Cre13 plants than in Cre1 plants, explaining the less efficient GUS excision in the Cre13 than in the Cre1 retransformants. Because both CRE-expressing plants are single-locus transformants derived from transformation with the same P35S-CRE gene, the two-fold difference in CRE mRNA levels is most probably correlated with the transgene locus structure. Root explant transformation in the two CRE-expressing lines resulted in almost all GUS-negative T2 progenies. Thus, the CRE-mediated excision of the target was clearly better after root tissue transformation than after floral dip transformation. We postulate that this difference in excision rate is due to the fact that fewer T-DNA copies are integrated after root explant transformation than after floral dip transformation. As a consequence, a lower amount of CRE proteins would be needed in the RC1K2L and RC13K2L retransformants. Also, this hypothesis could explain that GUS excision efficiency is comparable in the CK2L6-SC13 and RC13K2L retransformants with only two allelic and on average one or two K2L T-DNA copies, respectively. The same CRE mRNA levels were not sufficient for efficient excision in the FC13K2L retransformants with complex T-DNA integration patterns. Moreover, also the used transformation method to introduce the K2L T-DNA9 in itself might affect the excision efficiency. Indeed, the expression of the P35S-CRE gene and/or the ability of different cell types to respond to CRE-mediated recombination can contribute to the differences noted between the two used transformation methods. The P35S-CRE expression might be higher in the root explant transformation-responsive cells than in the female gametocyte, or the chromatin might be less susceptible to CRE recombination by applying floral dip.
In conclusion, although the efficiency of CRE-mediated excision of the loxP-flanked GUS gene was variable, we obtained CRE-expressing Arabidopsis transformants in which a loxP-flanked GUS gene was fully deleted and the deleted locus transmitted to the next generation. In addition, the results demonstrate that a rather high CRE expression level is needed when constructs are transformed into the CRE-expressing background by floral dip, whereas lower CRE mRNA levels are required to obtain the same frequency of fragment excision after root explant transformation. Finally, our study revealed that CRE-mediated excision can be maximized by experimentally identifying a highly effective CRE-expressing line that can be preferentially used for further transformations with a lox-flanked fragment.
Materials and methods
Plasmid construction for HSC T-DNA, CRE T-DNA, and K2L T-DNA
For cloning of pHSC, the CRE-coding region was amplified by PCR from the pMM23 plasmid (Dale and Ow 1990) with primers including SacII and NcoI sites (forward CRE-specific primer 5′ tccccgcgggttgacatgtccaatttactgacc3′ and reverse CRE-specific primer 5′ catgccatgggaattcttactaatcgccatcttcc3′). The NcoI/SacII CRE fragment of 1049 bp was cloned upstream of the 35S terminator, resulting in pCreT35S. Subsequently, the SacI/ApaI CreT35S fragment of 1276 bp was cloned into the pPZP200 binary vector (Hajdukiewicz et al. 1994) with the hygromycin selectable marker, resulting in the pHCreT35S plasmid. To create a GATEWAY™-compatible vector pHGWC, the GW cassette rfA (Invitrogen, Carlsbad, CA, USA) for cloning different promoters was cloned upstream of the CRE recombinase. The P35S sequence was amplified by PCR with pXD610 DNA (De Loose et al. 1995) as a template. To obtain the P35S-entry clone, the PCR products flanked by attB sites were recombined into the pDONR201-KmR vector. The T-DNA vector pHSC was generated by an LR reaction in which the P35S entry clone was incubated with the pHGWC destination vector in the presence of LR clonase (Invitrogen).
The plasmid pK2L610, described by De Buck et al. (1998), carries the pK2L610 T-DNA, referred to as the K2L T-DNA throughout the text. For the construction of the CRE T-DNA, the reader is referred to De Buck et al. (2000).
Agrobacterium-mediated plant transformation and selection of transformants
The transformation vector pHSC was introduced by electroporation into Agrobacterium tumefaciens strain C58C1RifR, containing the pMP90 plasmid (Koncz and Schell 1986). The Cre1 and Cre13 lines were obtained after root explant transformation of Arabidopsis thaliana (L). Heynh. (ecotype C24) (Valvekens et al. 1988) and selection on phosphinothricin (10 mg/l) and those containing one locus were retained for further analysis. Transformation of CK2L6 plants (Theuns et al. 2002) with the HSC T-DNA was performed by the floral dip method (Clough and Bent 1998) to obtain transformants into the lox target background. Cre1 and Cre13 plants were transformed with the K2L T-DNA (De Buck et al. 1998) by floral dip (Clough and Bent 1998) and by root tissue transformation (Valvekens et al. 1988) to obtain transformants in the CRE background. Seeds of the dipped plants were harvested and sown on K1 medium supplemented with 20 mg/l hygromycin (for HSC T-DNA) or 50 mg/l kanamycin (for K2L T-DNA) for selection of primary transformants. The K1 medium used was germination medium supplemented with 0.5 g/l of N-morpholinoethanesulfonic acid. Selected primary transformants (T1) were self-fertilized and T2 seeds were collected.
Plant DNA preparation and DNA gel blot analysis
DNA of Arabidopsis seedlings was prepared from 10–30 mg frozen plant tissue with the Puregene™ DNA Purification system (Qiagen, Hilden, Germany). The NPTII and GUS probes (probe 2 and 1, respectively; Fig. 1) were prepared according to De Buck et al. (1999). In each lane of 1% agarose gels, 1 μg Arabidopsis DNA was loaded. A nonradioactive method (GeneImages random prime labeling module and GeneImages CDP star detection module; GE-Healthcare, Little Chalfont, UK) was used to label and detect the DNA.
Histochemical ß-glucuronidase assay
Two-week-old T2 seedlings were fixed in 90% cold acetone for 30 min with continuous shaking. The seedlings were washed with 0.1 M Na2HPO4/NaH2PO4 buffer (pH 7), incubated in 0.1 M Na2HPO4/NaH2PO4 buffer (pH 7) containing 10 mM EDTA, 0.5 mM K3[Fe(CN)6)], 0.5 mM K4[Fe(CN)6)], 1% dimethylsulfoxide, and 50 mg/ml 5-bromo-4-chloro-3-indolyl-ß-d-glucuronic acid overnight at 37°C, afterwards washed with 0.1 M Na2HPO4/NaH2PO4 buffer (pH 7), destained in 90% ethanol, and stored in 70% ethanol. Photographs were taken with a digital camera (AxioCam HRc; Zeiss, Jena, Germany) connected to a Stemi SV11 microscope (Zeiss).
PCR analysis
Genomic DNA was prepared from pooled leaf material of ten T2 plants per CK2L6-SC retransformants and from eight to ten full T2 seedlings of the Cre1 and Cre13 retransformants from 10–30 mg frozen plant tissue with the Puregene™ DNA Purification system (Qiagen). PCR analysis was performed with 50 ng of DNA and primers specific for the non-excised and excised fragments. The primers used were: p3, 5′tgatcctgtttcctgtgtgaaatt3′; p4, 5′ttgtaaggagatgcactgatttat3′; p5, 5′atttgcggccgctttaatagtaaattgtaatgttgt3′. The PCR conditions were as follows: initial denaturation at 94°C for 5 min, followed by 30 cycles of denaturation at 94°C for 45 s, annealing at 55°C for 1 min, and elongation at 72°C for 1 min; and a final elongation step at 72°C for 5 min. The efficiency of recombination was tested by PCR analysis with p3 + p4 and p3 + p5. When the CRE recombinase was active and the P35S-GUS cassette between the two loxP sites was deleted, a fragment of 834 bp was generated with p3 + p4. When no deletion of the P35S-GUS cassette in the K2L T-DNA occurred, a fragment of 1200 bp was amplified with p3 + p5.
Real-time PCR analysis
RNA was extracted from pooled 2-week-old Arabidopsis seedlings grown on K1 medium with TRIzol® reagent (MRC, Cincinnati, OH, USA) according to the manufacturer’s instructions. Poly d(T) cDNA was synthesized from 2 μg total RNA with Superscript II reverse transcriptase (Invitrogen). Of the obtained cDNA, 1 μl was used for a PCR with actin primers to check for contamination of residual DNA. The following primers were used: forward primer, 5′ccacctgaaaggaagt3′ and reverse primer, 5′aaaacaatgggactaaaacgca3′. CRE expression was quantified on an iCycler real-time PCR detection system (BioRad, Hercules, CA, USA) with the qPCR core kit for SYBR Green I (Eurogentec, Seraing, Belgium). PCRs were carried out in triplicate. Relative expression levels were first normalized to ACTIN2 expression and then to the respective untransformed controls with the 2−ΔΔC T method (Livak and Schmittgen 2001). Specific primer pairs were designed using Beacon Designer 4.0 (Premier Biosoft International, Palo Alto, CA, USA): At3g18780/ACTIN2, 5′gttgactacgagcaggagatgg3′ and 5′acaaacgagggctggaacaag3′; CRE, 5′tttcccgcagaacctgaagatg3′ and 5′atccgccgcataaccagtg3′.
References
Bayley CC, Morgan M, Dale EC, Ow DW (1992) Exchange of gene activity in transgenic plants catalyzed by the Cre-lox site-specific recombination system. Plant Mol Biol 18:353–361
Chawla R, Ariza-Nieto M, Wilson AJ, Moore SK, Srivastava V (2006) Transgene expression produced by biolistic-mediated, site-specific gene integration is consistently inherited by the subsequent generations. Plant Biotechnol J 4:209–218
Clough SJ, Bent AF (1998) Floral dip: a simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana. Plant J 16:735–743
Dale EC, Ow DW (1990) Intra- and intermolecular site-specific recombination in plant cells mediated by bacteriophage P1 recombinase. Gene 91:79–85
Day CD, Lee E, Kobayashi J, Holappa LD, Albert H, Ow DW (2000) Transgene integration into the same chromosome location can produce alleles that express at a predictable level, or alleles that are differentially silenced. Genes Dev 14:2869–2880
De Block M, Debrouwer D (1991) Two T-DNA’s co-transformed into Brassica napus by a double Agrobacterium tumefaciens infection are mainly integrated at the same locus. Theor Appl Genet 82:257–263
De Buck S, De Wilde C, Van Montagu M, Depicker A (2000) Determination of the T-DNA transfer and the T-DNA integration frequencies upon cocultivation of Arabidopsis thaliana root explants. Mol Plant-Microbe Interact 13:658–665
De Buck S, Jacobs A, Van Montagu M, Depicker A (1998) Agrobacterium tumefaciens transformation and cotransformation frequencies of Arabidopsis thaliana root explants and tobacco protoplasts. Mol Plant-Microbe Interact 11:449–457
De Buck S, Jacobs A, Van Montagu M, Depicker A (1999) The DNA sequences of T-DNA junctions suggest that complex T-DNA loci are formed by a recombination process resembling T-DNA integration. Plant J 20:295–304
De Buck S, Van Montagu M, Depicker A (2001) Transgene silencing of invertedly repeated transgenes is released upon deletion of one of the transgenes involved. Plant Mol Biol 46:433–445
De Buck S, Windels P, De Loose M, Depicker A (2004) Single-copy T-DNAs integrated at different positions in the Arabidopsis genome display uniform and comparable ß-glucuronidase accumulation levels. Cell Mol Life Sci 61:2632–2645
De Loose M, Danthinne X, Van Bockstaele E, Van Montagu M, Depicker A (1995) Different 5′ leader sequences modulate ß-glucuronidase accumulation levels in transgenic Nicotiana tabacum plants. Euphytica 85:209–216
De Neve M, De Buck S, Jacobs A, Van Montagu M, Depicker A (1997) T-DNA integration patterns in co-transformed plant cells suggest that T-DNA repeats originate from ligation of separate T-DNAs. Plant J 11:15–29
Gilbertson L (2003) Cre-lox recombination: cre-ative tools for plant biotechnology. Trends Biotechnol 21:550–555
Gleave AP, Mitra DS, Mudge SR, Morris BAM (1999) Selectable marker-free transgenic plants without sexual crossing: transient expression of cre recombinase and use of a conditional lethal dominant gene. Plant Mol Biol 40:223–235
Hajdukiewicz P, Svab Z, Maliga P (1994) The small, versatile pPZP family of Agrobacterium binary vectors for plant transformation. Plant Mol Biol 25:989–994
Hoa TTC, Bong BB, Huq E, Hodges TK (2002) Cre/lox site-specific recombination controls the excision of a transgene from the rice genome. Theor Appl Genet 104:518–525
Hoff T, Schnorr KM, Mundy J (2001) A recombinase-mediated transcriptional induction system in transgenic plants. Plant Mol Biol 45:41–49
Jia H, Pang Y, Chen X, Fang R (2006) Removal of the selectable marker gene from transgenic tobacco plants by expression of Cre recombinase from a Tobacco Mosaic Virus vector through agroinfection. Transgenic Res 15:375–384
Joubès J, De Schutter K, Verkest A, Inzé D, De Veylder L (2004) Conditional, recombinase-mediated, expression of genes in plant cell cultures. Plant J 37:889–896
Koncz C, Schell J (1986) The promoter of TL-DNA gene 5 controls the tissue-specific expression of chimaeric genes carried by a novel type of Agrobacterium binary vector. Mol Gen Genet 204:383–396
Kopertekh L, Jüttner G, Schiemann J (2004) PVX-Cre-mediated marker gene elimination from transgenic plants. Plant Mol Biol 55:491–500
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔC T method. Methods 25:402–408
Muskens MWM, Vissers APA, Mol JNM, Kooter JM (2000) Role of inverted DNA repeats in transcriptional and post-transcriptional gene silencing. Plant Mol Biol 43:243–260
Nelson M, Raschke E, McClelland M (1993) Effect of site-specific methylation on restriction endonucleases and DNA modification methyltransferases. Nucleic Acids Res 21:3139–3154
Odell J, Caimi P, Sauer B, Russell S (1990) Site-directed recombination in the genome of transgenic tobacco. Mol Gen Genet 223:369–378
Odell JT, Russell SH (1994) Use of site-specific recombination systems in plants. In: Paszkowski J (ed) Homologous recombination and gene silencing in plants. Kluwer Academic Publishers, Dordrecht, pp 219–270
Ow DW (2002) Recombinase-directed plant transformation for the post-genomic era. Plant Mol Biol 48:183–200
Russell SH, Hoopes JL, Odell JT (1992) Directed excision of a transgene from the plant genome. Mol Gen Genet 234:49–59
Severin K, Schöffl F (1990) Heat-inducible hygromycin resistance in transgenic tobacco. Plant Mol Biol 15:827–833
Sreekala C, Wu L, Gu K, Wang D, Tian D, Yin Z (2005) Excision of a selectable marker in transgenic rice (Oryza sativa L.) using a chemically regulated Cre/loxP system. Plant Cell Rep 24:86–94
Srivastava V, Anderson OD, Ow DW (1999) Single-copy transgenic wheat generated through the resolution of complex integration patterns. Proc Natl Acad Sci USA 96:11117–11121
Srivastava V, Ariza-Nieto M, Wilson AJ (2004) Cre-mediated site-specific gene integration for consistent transgene expression in rice. Plant Biotechnol J 2:169–179
Srivastava V, Ow DW (2001) Single-copy primary transformants of maize obtained through the co-introduction of a recombinase-expressing construct. Plant Mol Biol 46:561–566
Theuns I, Windels P, De Buck S, Depicker A, Van Bockstaele E, De Loose M (2002) Identification and characterization of T-DNA inserts by T-DNA fingerprinting. Euphytica 123:75–84
Thomson JG, Ow DW (2006) Site-specific recombination systems for the genetic manipulation of eukaryotic genomes. Genesis 44:465–476
Valvekens D, Van Montagu M, Van Lijsebettens M (1988) Agrobacterium tumefaciens-mediated transformation of Arabidopsis thaliana root explants by using kanamycin selection. Proc Natl Acad Sci USA 85:5536–5540
Van Houdt H, Kovařík A, Van Montagu M, Depicker A (2000) Cross-talk between posttranscriptionally silenced neomycin phosphotransferase II transgenes. FEBS Lett 467:41–46
Vergunst AC, Hooykaas PJJ (1998) Cre/lox-mediated site-specific integration of Agrobacterium T-DNA in Arabidopsis thaliana by transient expression of cre. Plant Mol Biol 38:393–406
Vergunst AC, Jansen LET, Hooykaas PJJ (1998) Site-specific integration of Agrobacterium T-DNA in Arabidopsis thaliana mediated by Cre recombinase. Nucleic Acids Res 26:2729–2734
Wang Y, Chen B, Hu Y, Li J, Lin Z (2005) Inducible excision of selectable marker gene from transgenic plants by the Cre/lox site-specific recombination system. Transgenic Res 14:605–614
Zhang W, Subbarao S, Addae P, Shen A, Armstrong C, Peschke V, Gilbertson L (2003) Cre/lox-mediated marker gene excision in transgenic maize (Zea mays L.) plants. Theor Appl Genet 107:1157–1168
Zuo J, Niu Q-W, Møller SG, Chua N-H (2001) Chemical-regulated, site-specific DNA excision in transgenic plants. Nat Biotechnol 19:157–161
Acknowledgements
The authors thank Geert Angenon and Dimitri Verweire for discussions, Katleen Hoorelbeke, Marieke Louwers, and Els Van Lerberge for practical assistance, and Martine De Cock for help in preparing the manuscript. This work was supported by grants from the 6th framework program of the European Union “GENINTEG” (LSHG-CT2003-503303), the “Bijzondere Onderzoeksfonds” of the Ghent University (BOF 01111400), and the European Union BIOTECH program (QLRT-2000-00078) with additional cofinancing from the Flemish Community.
Author information
Authors and Affiliations
Corresponding author
Additional information
Gordana Marjanac and Annelies De Paepe contributed equally to this work.
Rights and permissions
About this article
Cite this article
Marjanac, G., De Paepe, A., Peck, I. et al. Evaluation of CRE-mediated excision approaches in Arabidopsis thaliana . Transgenic Res 17, 239–250 (2008). https://doi.org/10.1007/s11248-007-9096-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11248-007-9096-9