
Abstract
Establishment of an efficient protocol for regeneration and genetic transformation is required in banana for the incorporation of useful traits. Therefore an efficient method has been developed for somatic embryogenesis, plant regeneration and transformation of Cavendish banana cultivar Robusta (AAA). Embryogenic cell suspension culture (ECS) was established using immature male flowers. Percentage appearance of embryogenic callus and distinct globular embryos was 10.3 and 11.1, respectively. ECS obtained was cocultivated under different cocultivation conditions with Agrobacterium tumefaciens strain EHA105 harboring pCAMBIA 1301 plant expression vector. Up to 30 transgenic plants/50 mg settled cell volume (SCV) was obtained with cocultivation in semisolid medium whereas no transgenics could be obtained with parallel experiments carried out in liquid medium. Histochemical GUS assay in different tissues of putatively transformed plants demonstrated expression of uidA gene. Among the putatively transformed plants obtained, a set of 4 were confirmed by PCR analysis and stable integration of the transgene by Southern analysis. GUS specific activity measured by a MUG (4-methylumbelliferyl-β-d-glucuronide) based flourometric assay revealed increase in transient GUS expression in semisolid as well as liquid cocultivation with centrifugation. This is the first report showing somatic embryogenesis and Agrobacterium tumefaciens mediated transformation using embryogenic cell suspension cultures in an important Cavendish banana cultivar Robusta. The present protocol will make possible agronomic improvement of this important commercially grown cultivar by introduction of disease resistance characteristics and antisense-mediated delayed fruit ripening strategies. Further, it will also assist in functional characterization of new gene or promoter elements isolated from this or other cultivars of banana.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Banana (Musa spp.) is one of the most important fruit crop in tropical and subtropical countries. There has been a 3-fold increase in world banana production since last three decades. Musa spp. makes an enormous contribution to food security and also provides export revenue in developing countries. Conventional breeding programmes have not been very successful for banana improvement because of impediments like long life cycle, triploidy and sterility in most of the edible cultivars. Genetic transformation offers a feasible means through which important economic traits could be introduced into these cultivars. In general, genetic transformation techniques and regeneration frequencies are reported to be highly genotype dependent. Thus optimization of transformation protocol for any particular type of cultivar becomes a prerequisite for agronomic improvement in that cultivar (Opabode 2006).
Plant regeneration has been carried out using different explants for a variety of banana cultivars. Embryogenic cell suspension cultures have been established using basal leaf sheaths and corm sections (Novak et al. 1989), highly proliferating shoot tip cultures (Dheda et al. 1991), zygotic embryos (Marroquin et al. 1993), and immature male flowers (Escalant et al. 1994; Cote et al. 1996; Navarro et al. 1997; Sagi et al. 1998; Becker et al. 2000). Among the Cavendish cultivars, embryogenic cell suspension culture has been established for cultivars Grand Naine and Gros Michael (Assani et al. 2002). There have been several reports on genetic transformation of banana. Transgenic banana plants have been obtained in different cultivars using biolistics (Sagi et al. 1995; Becker et al. 2000), Agrobacterium-mediated genetic transformation (May et al. 1995; Ganapathi et al. 2001), and electroporation (Sagi et al. 1994). Despite of variety independent nature of biolistic transformation, Agrobacterium-mediated genetic transformation remains the method of choice due to high transformation efficiency.
Herein, we report establishment of embryogenic cell suspension culture, Agrobacterium-mediated genetic transformation and regeneration of transgenic plants with high transformation efficiency. As we have used ECS for cocultivation, chances of getting chimeras are almost negligible. Also this makes the protocol less tedious and faster. To the best of our knowledge there is no report of somatic embryogenesis and genetic transformation in Dwarf Cavendish banana cultivar Robusta. Dwarf Cavendish forms the mainstay of Indian banana industry as it alone contributes 57% of total production. Robusta is the dominant Cavendish cultivar in India due to its high yield, ability to withstand strong winds; production of large bunches which satisfies the export standards, early maturity and fruits with consistent pulp having pleasant flavor and attractive color.
Materials and methods
Somatic embryogenesis and establishment of embryogenic cell suspension cultures
Embryogenic cell suspension culture of banana cultivar Robusta was initiated and maintained as described earlier (Cote et al. 1996). Floral apices were surface sterilized with 70% (v/v) alcohol followed by thorough rinsing in sterile distilled water for 3–4 times. Outer whorls were removed until the bud was reduced to approximately 1.5 cm in length. Individual flower whorls were isolated and cultured on MS medium (Murashige and Skoog 1962), supplemented with 2,4-dichlorophenoxy acetic acid (2,4-D, 4 mg/l), indole-3-acetic acid (IAA, 1 mg/l), naphthaleneacetic acid (NAA, 1 mg/l), biotin (1 mg/l) and 3% sucrose (hereafter referred as BCM). The pH of the medium was adjusted to 5.7 prior to autoclaving and gelled with 0.2% phytagel (Sigma, USA). The cultures were maintained in dark and a temperature of 25 ± 2°C for 3–4 months. The embryogenic callus developed was transferred to semisolid medium supplemented with 2,4-D (1 mg/ml), biotin (1 mg/ml), malt extract (100 mg/l), glutamine (100 mg/ml) and 4.5% sucrose (hereafter referred as M2 medium). The pH of the medium was adjusted to 5.3 prior to autoclaving. About 150 mg of friable embryogenic callus was transferred to 20 ml liquid M2 medium in 250 ml Erlenmeyer flask for initiation of ECS. These cultures were maintained on a gyratory shaker at 100 rpm under a photoperiod of 16 h (light)/8 h (dark) at 45 μmol photon m2 s−1 and temperature of 25 ± 2°C. Fine cell suspensions were obtained within 3 months of initiation. Embryogenic cells were subcultured every week by taking 0.5 ml packed cell volume (PCV) and 4.5 ml spent medium into 20 ml fresh liquid M2 medium.
Embryogenic cell suspension culture was aspirated on to glass fiber filters (Sartorius, Germany) and kept on semi-solid M2 medium for 6 days before transfer to semisolid SH medium (Schenk and Hildebrandt 1972), supplemented with picloram (1 mg/ml), malt extract (100 mg/l), glutamine (100 mg/l) and 4.5% sucrose (hereafter referred as BEM). The pH of this medium was adjusted to 5.3 prior to autoclaving. From this stage all subsequent cultures were maintained under a photoperiod of 16 h (light)/8 h (dark) at 45 μmol photon m2 s−1 and temperature of 25 ± 2°C. After 3 months of culture in this medium, mature somatic embryos were transferred to semisolid MS medium supplemented with benzylaminopurine (BAP, 1 mg/l) and 3% sucrose (hereafter referred as M4). The pH of this medium was adjusted to 5.7 prior to autoclaving. For development of complete plantlets, germinating somatic embryos were transferred to semisolid MS medium supplemented with NAA (1 mg/ml) and 3% sucrose (hereafter referred as RM). The pH was adjusted to 5.7 prior to autoclaving.
Agrobacterium strain, binary vector and preparation of Agrobacterium suspension
A. tumefaciens strain EHA105 (Hood et al. 1993), harboring the binary vector pCAMBIA 1301 (CAMBIA, Canberra, Australia) was used in the present study. The T-DNA of pCAMBIA 1301 contains CaMV 35S promoter driven gusA gene interrupted by a modified castor bean catalase intron and double enhanced 35S promoter driven hpt II gene conferring resistance to hygromycin.
A single Agrobacterium colony was grown overnight in liquid YENB (0.75% yeast extract, 0.8% nutrient broth) medium supplemented with 50 mg/l kanamycin at 26°C with an orbital shaking of 150 rpm. Next morning the culture was resuspended in the same medium such that OD600 nm is approximately 0.1. It was then grown for 4–5 h under same conditions until an OD600 nm of 0.8–1.0 was reached. The suspension was pelleted down at 6,000 g for 10 min. The pellet was subsequently resuspended in M2 medium supplemented with 100 μM acetosyringone (ACS) at a final OD600 nm of 0.1 or 0.2, to be used for infection.
Transformation of ECS
Seven days post sub cultured cells were sieved through an 85-μm sieve to remove larger cell clumps. Approximately 0.05 g SCV of sieved ECS was cocultivated with Agrobacterium in liquid M2 medium supplemented with 100 μM ACS. For semisolid cocultivation, ECS were aspirated onto glass fiber discs with a Buchner apparatus and transferred to semisolid M2 medium containing 100 μM ACS and maintained in dark for 3–7 days. Post-coculture ECS along with filters were transferred to solidified M2 medium containing carbenicillin (200 mg/l) and cefotaxime (200 mg/l). Three days further ECS and filters were transferred separately to BEM containing carbenicillin (200 mg/l), cefotaxime (200 mg/l) and hygromycin (5 mg/l). Regeneration was carried out in selective regeneration medium as described earlier. For liquid cocultivation, 10 ml M2 medium supplemented with 100 μM ACS was used. The cultures were incubated in dark under two conditions—at 100 rpm for 3–7 days (condition A) and initial 12 h at 40 rpm and thereafter at 100 rpm for 3–7 days (condition B). The remaining steps were similar to semisolid cocultivation. At all conditions, one set was centrifuged at 1,000 rpm for 5 min prior to cocultivation. Three replicates were used for each experiment.
GUS histochemical assay
Transformed tissues (cells from suspension, developed somatic embryos, leaves and roots from transgenic plants) were analyzed for β-glucuronidase expression by using X-GlcA (5-bromo-4-chloro-3-indolyl-β-d-glucuronide cyclohexylammonium salt) as the substrate (Jefferson 1987). The histochemical reaction was allowed to proceed at 37°C for 4 h to overnight. Subsequently, the tissues were cleared in 70% (v/v) ethanol. To serve as control against any background GUS staining, un-transformed tissues were included at all staining occasions.
Fluorometric GUS activity assay
β-glucuronidase specific activity was measured in transformed ECS and leaves using the fluorometric assay (Jefferson 1987). ECS were harvested 6 days post-cocultivation, from all cocultivation conditions and three independent transformation events. Leaves from plants showing positive GUS histochemical staining was also excised. Total protein extracts obtained using GUS extraction buffer (50 mM NaHPO4 pH 7.0, 10 mM β-mercaptoethanol, 10 mM EDTA, 0.1% (w/v) sodium lauryl sarcosine, 0.1% (w/v) Triton X-100) were used for GUS specific activity using 2 mM MUG assay buffer. Fluorescence was measured using a fluorimeter with emission and excitation filters set at 465 and 360 nm, respectively. Total protein concentration in samples was determined as described previously (Bradford 1976).
Polymerase chain reaction (PCR)
Genomic DNA was isolated from leaves of putatively transformed plants using GenElute Plant Genomic DNA Miniprep Kit (Sigma, USA). This DNA was fractionated on 0.8% agarose gel along with pCAMBIA 1301 plasmid loaded in one of the wells. High molecular weight genomic DNA, considerably larger than pCAMBIA 1301 plasmid was eluted from the gel and used for PCR. Following primers were synthesized based on the sequence of pCAMBIA 1301 Ti-plasmid.
-
Primer A 5′-AACAGGTATGGAATTTCGCCGATTT-3′
-
Primer B 5′-TTTATCCTAGTTTGCGCGCTATATTT-3′
-
Primer C 5′-CCTAGATCTCTTCGGTAATGAAAAA-3′
Primers A and B were used to amplify a 450 bp β-glucuronidase gene fragment which lies within the T-DNA borders. Primers A and C were used to amplify a 1,585 bp fragment, out of which 550 bp lies within the T-DNA borders and the remaining 1,035 bp is outside T-DNA. Eluted genomic DNA from one of the transgenic plants mixed with 10 pg pCAMBIA 1301 plasmid was also used as a template for both primer combinations. Genomic DNA isolated from untransformed Robusta leaves was treated as negative control whereas 1301 plasmid was taken as positive control. A 50 μl PCR mix contained 100 ng of each primer, 1 unit of Taq polymerase, 200 μM of each dNTP, 1× PCR buffer and 500 ng of eluted plant genomic DNA as template. PCR conditions used were 94°C for 5 min for initial melting followed by 35 cycles of amplification with each cycle consisting of the following steps: 94°C for 1 min, 54°C for 1½ min and 72°C for 2½ min with a final extension at 72°C for 15 min.
Southern analysis of transgenic plants
Southern analysis was carried out as previously described (Sunil Kumar et al. 2005). 10–20 micrograms of genomic DNA isolated from four independently transformed plants was digested with NcoI and Eco721 to release ~2 kb uidA gene from the T-DNA region of pCAMBIA 1301. The digested DNA was electrophoresed on 1% agarose gel and transferred to Hybond-N+ membrane (Amersham Pharamacia Biotech, UK) using standard protocols (Sambrook et al. 1989). These DNAs were hybridized against a random primed radiolabelled probe of 947 bp fragment obtained by PCR amplification of pCAMBIA1301 with uidA specific primers mentioned below.
-
Forward primer 5′-AAGCCAGACAGAGTCTGATATCTACC-3′
-
Reverse primer 5′-GAGTGAAGATCCCTTTCTTGTTACC-3′
Results were visualized by autoradiography.
Results
Somatic embryogenesis
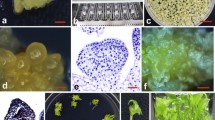
Flower buds swelled up within 1 month of culture with pronounced secretion of phenolics. Approximately 48.67% of the cultures showed the appearance of green nodular callus from the base of explants within 3–4 months of culture in BCM (Fig. 1a). At this stage the responding cultures were transferred to M2 medium in which around 10.3% formed white friable embryogenic callus (Fig. 1b). 11.1% appearance of distinct globular embryos (Fig. 1c) was observed in M2 medium. Suspension cultures were initiated in liquid M2 medium from friable embryogenic callus and globular embryos. Fine embryogenic cells with dense cytoplasm (Fig. 1d) were obtained after 3 months of initiation. These cultures were subcultured weekly. With every subculture, 2- to 3-fold increases in cell mass was observed.

Somatic embryogenesis and Agrobacterium mediated transformation in banana cv. Robusta. a Green nodular callus that developed on BCM. b White friable embryogenic callus that developed on transfer to M2 medium. c Distinct globular somatic embryos. d Embryogenic cells as observed in suspension cultures. e Germinating somatic embryos in M4 medium. f Development of somatic embryos under selection, note the necrosis of untransformed cells. g Development of somatic embryos in glass fiber filters kept on selection medium. h Transformed plants regenerated on selection medium. i GUS stained embryogenic mass with distinctly visible somatic embryos. j Leaf showing GUS expression. k GUS expression in roots
Embryogenic cells plated on BEM, showed the development of somatic embryos within 1 month of transfer. The somatic embryos showed conversion signs within 1 month of transfer to M4 medium (Fig. 1e). 100% of the germinating embryos formed complete plantlets on transfer to RM.
Agrobacterium mediated transformation
Distinct globular embryos developed within 1 month of transfer to selective BEM, whereas untransformed cells showed pronounced necrosis (Fig. 1f). Embryos developed on the filters kept under selection even after removing most of the ECS (Fig. 1g). These embryos were directly transferred to M4 for germination. This accelerated the entire process of regenerating transgenic plants by around 2 months. Coeloptile emergence was observed within 1–2 months of transfer to M4 medium with selection. Complete plantlets were obtained when germinating embryos were transferred to RM with selection (Fig. 1h). The entire transformation protocol was performed four times and for each transformation, 0.05 g SCV of ECS was taken. 30 independently transformed lines were obtained out of which a set of 4 was used for further molecular analysis. Characteristic blue GUS staining was observed in all tissues that survived in selective medium (Fig. 1i–k,). As the β-glucuronidase gene in pCAMBIA 1301 is interrupted by a modified castor bean catalase intron, all of this GUS activity was derived from plant protein synthesis machinery only.
Comparison of semi-solid and liquid cocultivation of embryogenic cell suspension cultures
Liquid cocultivation was carried out under two conditions that differed only in initial speed of shaking. Under both conditions of liquid cocultivation, ECS could survive the cocultivation phase but were confronted with bacterial overgrowth in later regeneration phases, which could not be controlled even on increasing the concentration of carbenicillin and cefotaxim. It increased as the cocultivation period was increased. On transfer to BEM, ECS failed to show any signs of embryogenesis instead necrosis was predominant. No transgenic plants were obtained with liquid cocultivation under both the above-mentioned conditions.
Maximum transient GUS expression of 6201.23 picomoles of 4-Methylumbellifrerone (4-MU) released/h/mg of total protein was noted with condition B of liquid cocultivation, whereas for condition A, maximum expression of 4355.92 pmoles 4-MU/h/mg total protein was recorded (Fig. 2a). In contrast when Agrobacterium inoculated cells were transferred to semi-solid cocultivation medium, no signs of necrosis was observed. The infected ECS showed vigorous growth. Most suitable cocultivation period was 72 h as necrosis and bacterial growth was predominant with increasing cocultivation period.

a Intracellular GUS specific activity in transformed cell suspension cultures of Robusta at different cocultivation conditions. S: solid cocultivation, A and B are two liquid cocultivation conditions, c—centrifuged samples, bacterial OD is written in brackets below. Note the expression of β-glucuronidase in transformed samples as compared to untransformed control. Data represents an average of three replicates. b GUS specific activity in leaves of transgenic plants. C: control plant, L1, 2, 3, and 4: independent transgenic in vitro plants. L1 (GH-5m): 5 months old copy of L1 and L1 (GH-1m): 1-month-old copy of L1 maintained in the green house
Maximum transient GUS expression of 6763.32 pmoles 4-MU/h/mg total protein was noted for semisolid cocultivation which was higher than both conditions of liquid cocultivation.
Effect of bacterial density on transformation
We have tested various bacterial densities for both solid and liquid cocultivation. From our earlier experiments (data not shown), we concluded that on increasing bacterial OD above 0.2, bacterial colonization and necrosis was prominent for this cultivar under both, semisolid and liquid cocultivation. Therefore further experiments were restricted to two OD’s 0.1 and 0.2. In semisolid cocultivation transgenic plants were obtained with both bacterial OD’s but the transformation frequency was higher for 0.2. Also maximum transient GUS expression of 6763.32 pmoles 4-MU/h/mg total proteins was recorded for 0.2 OD whereas for 0.1 OD, expression of only 2678.20 pmoles 4-MU/h/mg total protein was recorded (Fig. 2a).
Under both conditions of liquid cocultivation, transient GUS expression was higher when bacterial OD was 0.2. With 0.2 bacterial OD, transient expression level of 4355.92 pmoles 4-MU/h/mg total protein was recorded for condition A, whereas for condition B maximum transient expression level of 6201.23 pmoles 4-MU/h/mg total protein was recorded. With 0.1 OD, maximum transient expression level for condition A and B was 2489.21 pmoles 4-MU/h/mg total protein and 2456.07 pmoles 4-MU/h/mg total protein, respectively. Cells showed pronounced necrosis with both bacterial OD’s under liquid cocultivation and failed to show any signs of embryogenesis.
Effect of centrifugation on transient GUS expression with semi-solid and liquid cocultivation
A significant 2- to 3-fold increase was observed in transient GUS expression following application of centrifugal force during cocultivation in both semi-solid and liquid medium and at all bacterial OD’s (Fig. 2a).
GUS specific activity in leaves
High level of β-glucuronidase activity was noted in leaves of transgenic plants by quantitative MUG-based fluorometric assay (Fig. 2b). Among the various transgenic lines analyzed, maximum expression level of 52,494.88 pmoles 4-MU/min/mg total protein were recorded in L1. Expression was slightly higher for 5-month-old hardened L1 copy as compared to the 1-month-old copies maintained in green house.
PCR and Southern analysis
Stable insertion of transgene into banana genome was verified by a PCR based technique as described by Chen et al. (2006). Presence of Ti plasmid in the genomic DNA of putatively transformed plants will lead to false positive results thereby making conventional PCR unconvincing unless verifying the stable incorporation of transgene in the plant genome. To overcome this problem, a three-pronged approach was utilized. Firstly, genomic DNA was isolated from the transformed plants which were confirmed to be Agrobacterium free after being maintained on antibiotic-free medium (except hygromycin) for more than 6 months. Secondly, to further eradicate the possibility of Ti-plasmid contaminating genomic DNA samples, DNA isolated from transgenic plants was fractionated on agarose gel with pCAMBIA 1301 Ti-plasmid as reference. Subsequently only high molecular weight DNA was eluted from the gel (data not shown). This DNA was then used as template for PCR reaction. Finally, to confirm the absence of Ti-plasmid in genomic DNA samples, a control PCR reaction was carried out using primers A and C. As primer C is complementary to a region present outside T-DNA, no DNA fragment should be obtained if the genomic DNA samples are not contaminated with the Ti-plasmid. As shown in Fig. 3a, no amplification was observed when DNA isolated from wild type control plant was used as template with primers A and B (lane 2) and Primers A and C (lane 3). A 450 bp amplification product was observed when eluted DNA from transgenic plants was used as template with primers A and B (lanes 6, 12, 14 and 16), where as no product was detected with the same DNA templates when primers A and C were used (lanes 7, 13, 15 and 17). Also, if 10 pg pCAMBIA 1301 Ti-plasmid was added to one of the cleaned up genomic DNA samples, primers A and B gave a 450 bp product and primers A and C gave a 1,585 bp product (lanes 18 and 19). The results clearly rules out any chances of residual Agrobacterium in the transgenic plants.

Molecular analysis. a PCR based confirmation of stable transgene insertion into banana genome carried out with three primes (A, B and C) using 500 ng plant genomic DNA and/or 10 pg pCAMBIA1301 as template. Lanes 1, 11 and 20 1 kb DNA ladder. Lanes 2 and 3 genomic DNA derived from untransformed control plant with primers A, B and A, C, respectively. Lanes 4 and 5 pCAMBIA1301 plasmid as template with primers A, B and primers A, C, respectively. Lanes 6 and 7 eluted genomic DNA from transgenic plant 1 with primers A, B and A, C, respectively. Lanes 8–10 eluted genomic DNA from transgenic plant 1 with single primers A, B and C, respectively. Lanes 12 and 13 eluted genomic DNA from transgenic plant 2 with primers A, B and A, C, respectively. Lanes 14 and 15 eluted genomic DNA from transgenic plant 3 with primers A, B and A, C, respectively. Lanes 16 and 17 eluted genomic DNA from transgenic plant 4 with primers A, B and A, C, respectively. Lanes 18 and 19 eluted genomic DNA from transgenic plant 4 mixed with pCAMBIA 1301 plasmid as template. b Southern blot analysis for stable transgene integration into banana genome. Genomic DNA was digested with NcoI and Eco721 and hybridized with [α-32P]dCTP labeled uidA probe. Lane 1 1 kb ladder. Lane 2 untransformed control plant. Lane 3 linearized pCAMBIA 1301 plasmid DNA. Lanes 4–7 transgenic banana plants
Stable integration of the transgene in PCR positive plants was further confirmed by Southern analysis. Genomic DNA digested with NcoI and Eco721 released ~2 kb gusA gene internal to the T-DNA region and hybridized to a 947 bp radiolabelled probe specific for uidA gene from pCAMBIA 1301 (Fig. 3b).
Discussion
The main objective of the present investigation was to optimize a protocol that would aid in recovery of stably transformed transgenic plants with considerable transformation efficiency in an important banana cultivar Robusta. Morphologically normal transgenic plants obtained through induction of somatic embryos from transformed embryogenic cell suspension cultures, established the usefulness of the present protocol for introducing important economic traits, as improved fruit quality and disease resistance into banana genome. Expression of β-glucuronidase in banana leaf proves the efficacy of this system for expression of any other useful foreign protein in Robusta.
Agrobacterium mediated transformation has been successfully used to obtain transgenic banana plants in the past. Apical meristem of cultivar Grand Naine has been used as initial explants for transformation (May et al. 1995). The present study uses embryogenic cell suspension cultures for Agrobacterium infection. This cell suspension culture based method is less tedious and faster as repeated rounds of selection are not required. Chimerism can also be avoided as plants obtained from embryogenic cell suspension culture are of single cell origin (Roux et al. 2004). Other methods of transformation as electroporation (Sagi et al. 1994), and biolistics have been used for banana transformation (Sagi et al. 1995; Becker et al. 2000), but Agrobacterium remains to be the most preferred mode. Among the high demand Cavendish cultivars, transformation of Grand Naine alone have been reported so far. To the best of our knowledge, this is the first report of somatic embryogenesis and Agrobacterium mediated transformation of commercially important banana cultivar Robusta.
According to recent estimates (2006), 98% of banana production occurs in developing countries and India accounts for 19.71% of global production. Estimates say that worldwide production of banana in 2006 was 97.37 million tones and export value accounted to more than US $4 billion annually. Banana being a climacteric fruit, exhibits uncontrolled ripening behavior once initiated, thus loosing its market viability in a very short span of time. According to FAO reports, in 1997–1998, annual loss due to perished fruit may account up to US $20 billion in India of which banana contributes the most.
Several common practices have been employed in attempts to prolong the shelf life of banana. These include refrigeration and controlled atmosphere storage, the use of ethylene absorbent, surface coating and moist sawdust treatments (Abdullah et al. 1990), but the practices prove to be either too costly or ineffective. This shifted the focus of the researchers to the underlying molecular mechanism of ripening. Presently much knowledge is available in this context (Giovannoni 2001). Several genes associated with ripening in banana have been characterized, isolated and cloned (Clendennen et al. 1997; Medina-Suarez et al. 1997; Drury et al. 1999; Asif and Nath 2005). It has been reported that ripening could be delayed in banana by inhibiting fruit specific ethylene synthesis using sense suppression (Balint-Kurti et al. 2001). Similarly firmer fruits have been produced in tomato by antisense suppression of polygalacturonase gene (Smith et al. 1990). Establishment of β-glucuronidase positive Robusta plants opens up the possibility of introducing ripening related genes in desired orientation into Robusta genome. This would help in increasing the fruit shelf life of this edible cultivar. Since Robusta is one of the most dominant and highly priced banana cultivar, slow ripening Robusta fruits should prove to be a major breakthrough for exporting the fruits. Using this protocol we have already succeeded in transferring some of the ripening related genes into Robusta genome (data not shown).
The protocol described above can be implemented in genetic programs designed for creation of transgenic plants resistant to major diseases such as black sigatoka, fusarium wilt (fungal diseases) and banana bunchy top virus. Genes encoding antifungal activity (Broekaert et al. 1996), defensins (Remy et al. 1998) and lipid transfer protein (Sagi 2000) have been expressed in banana and transgenic plants have shown resistance.
In summary, we have demonstrated Agrobacterium mediated transformation of embryogenic cell suspension culture of an important banana cultivar, Robusta. We have reported a high transformation efficiency of 30 plants/0.05 g SCV. Centrifugation assisted Agrobacterium mediated transformation enhances transformation efficiency (Khanna et al. 2004). In this study also it is evident as high GUS specific activity was observed in ECS centrifuged prior to cocultivation. Transformation efficiency could have been further enhanced if embryo germination was done in non-selective medium. There are reports suggesting that antibiotics allow formation of transgenic embryos but interfere with embryo germination (Yao et al. 1995; Bretagnesagnard and Chupeau 1996). Hence, the use of subsequent germination medium with reduced levels or removal of hygromycin promises to enhance the regeneration frequency of transgenic plants. This approach will definitely require a fast and reliable technique to distinguish between transgenic and non-transgenic lines. One such approach would be the use of reporter genes like uidA and GFP along with the gene of interest. This will allow an initial screening of the transgenic and non-transgenic tissues at an early stage.
References
Abdullah H, Lizada MCC, Tan SC, Pantastico EB, Tongdee SC (1990) Storage of banana. In: Hassan A, Pantastico EB (eds) Fruit development postharvest physiology, handling and marketing in ASEAN. ASEAN Food Handling Bureau, Kuala Lumpur, pp 44–64
Asif MH, Nath P (2005) Expression of multiple forms of polygalacturonase gene during ripening in banana fruit. Plant Physiol Biochem 43:177–184. doi:10.1016/j.plaphy.2005.01.011
Assani A, Haicour R, Wenzel G, Foroughi-Wehr B, Bakry F, Cote FX, Ducreux G, Grapin AA (2002) Influence of donor material and genotype on protoplast regeneration of banana and plantain cultivars (Musa spp.). Plant Sci 162:355–362. doi:10.1016/S0168-9452(01)00562-3
Balint-Kurti P, Firoozabady E, Moy Y, Mercier J, Fong R, Wong L, Gutterson N (2001) Better bananas—the biotech way. INFOMUSA 10(1):VI (PROMUSA 2001). Available at http://bananas.bioversityinternational.org/files/files/pdf/publications/info10.1_en.pdf [Page number 58]
Becker DK, Dugdale B, Smith MK, Harding RM, Dale JL (2000) Genetic transformation of Cavendish banana (Musa spp. AAA group) cv. Grand Nain via microprojectile bombardment. Plant Cell Rep 19:229–234. doi:10.1007/s002990050004
Bradford MM (1976) A rapid and sensitive method for the quantification of microgram quantities of protein utilizing the principle of protein dye binding. Anal Biochem 72:248–254. doi:10.1016/0003-2697(76)90527-3
Bretagnesagnard B, Chupeau Y (1996) Selection of transgenic flax plants facilitated by spectinomycin. Transgenic Res 5:131–137. doi:10.1007/BF01969431
Broekaert WF, Cammue BPA, DeBolle M, Thevisen K, DeSamblanx G, Osborn RW (1996) Antimicrobial peptides from plants. Crit Rev Plant Sci 16:230–297. doi:10.1080/713608148
Chen Y, Lu L, Deng W, Yang X, McAvoy R, Zhao D, Pei Y, Luo K, Duan H, Smith W, Thammina C, Zheng X, Ellis D, Li Y (2006) In vitro regeneration and Agrobacterium-mediated genetic transformation of Euonymus alatus. Plant Cell Rep 25:1043–1051. doi:10.1007/s00299-006-0168-8
Clendennen SK, Kipp PB, May GD (1997) The role of ethylene in banana fruit ripening. In: Kanellis AK (ed) Biology and biotechnology of fruit ripening. Kluwer, Berlin, pp 141–148
Cote FX, Domergue R, Monmarson S, Schwendiman J, Teisson C, Escalant JV (1996) Embryogenic cell suspensions from the male flower of Musa AAA cv. Grand Nain. Physiol Plant 97:285–290. doi:10.1034/j.1399-3054.1996.970211.x
Dheda D, Dumortier F, Panis B, Vuylsteke D, Langhe E (1991) Plant regeneration in cell suspension cultures of the cooking banana cv. Bluggoe (Musa spp. ABB group). Fruits 46:125–135
Drury R, Hortensteiner S, Donnison J, Bird CR, Seymour GB (1999) Chlorophyll catabolism and gene expression in the peel of ripening banana fruits. Plant Physiol 107:32–38. doi:10.1034/j.1399-3054.1999.100105.x
Escalant JV, Teisson C, Cote F (1994) Amplified somatic embryogenesis from male flowers of triploid banana and plantain cultivars (Musa spp.). In Vitro Cell Dev Biol 30:181–186. doi:10.1007/BF02631441
Ganapathi TR, Higgs NS, Balint-Kurti PJ, Arntzen CJ, May GD, Van Eck JM (2001) Agrobacterium mediated transformation of embryogenic cell suspensions of banana cultivar Rasthali (AAB). Plant Cell Rep 20:157–162. doi:10.1007/s002990000287
Giovannoni J (2001) Molecular biology of fruit maturation and ripening. Annu Rev Plant Physiol Plant Mol Biol 52:725–749. doi:10.1146/annurev.arplant.52.1.725
Hood EE, Gelvin SB, Melchers LS, Hoekama A (1993) New Agrobacterium helper plasmid for gene transfer to plants. Transgenic Res 2:208–218. doi:10.1007/BF01977351
Jefferson RA (1987) Assaying chimeric plant genes: the GUS fusion system. Plant Mol Biol Rep 5:387–405. doi:10.1007/BF02667740
Khanna H, Becker D, Kleidon J, Dale J (2004) Centrifugation assisted Agrobacterium tumefaciens-mediated transformation (CAAT) of embryogenic cell suspensions of banana (Musa spp. Cavendish AAA and Lady finger AAB). Mol Breed 14:239–252. doi:10.1023/B:MOLB.0000047771.34186.e8
Marroquin CG, Paduscheck C, Escalant JV, Teisson C (1993) Somatic embryogenesis and plant regeneration through cell suspensions in Musa acuminate. In Vitro Cell Dev Biol 29P:43–46
May GD, Afza R, Mason HS, Wiecko A, Novak FJ, Arntzen CJ (1995) Generation of transgenic banana (Musa acuminata) plants via Agrobacterium-mediated transformation. Biotechnology 13:486–492. doi:10.1038/nbt0595-486
Medina-Suarez R, Manning K, Fletcher J, Aked J, Bird CR, Seymour GB (1997) Gene expression in the pulp of ripening bananas, two-dimensional sodium dodecyl-polyacrylamide gel electrophoresis of in vitro translation products and cDNA cloning of 25 different ripening-related mRNAs. Plant Physiol 115:453–461. doi:10.1104/pp.115.2.453
Murashige T, Skoog F (1962) A revised medium for rapid growth and bioassays with tobacco tissue cultures. Plant Physiol 15:473–497. doi:10.1111/j.1399-3054.1962.tb08052.x
Navarro C, Escobedo RM, Mayo A (1997) In vitro plant regeneration from embryogenic cultures of a diploid and a triploid, Cavendish banana. Plant Cell Tissue Organ Cult 51:17–25. doi:10.1023/A:1005965030075
Novak FJ, Afza R, Van Duren M, Perea-Dallos M, Conger BV, Xiaolang T (1989) Somatic embryogenesis and plant regeneration in suspension cultures of dessert (AA and AAA) and cooking (ABB) bananas (Musa spp.). Biotechnology 7:147–158. doi:10.1038/nbt0289-147
Opabode JT (2006) Agrobacterium-mediated transformation of plants: emerging factors that influence efficiency. Mol Biol Rep 1:12–20
Remy S, Francois I, Cammue BPA, Swennen R, Sagi L (1998) Co-transformation as a potential tool to create multiple and durable resistance in banana. Acta Hortic 461:361–365
Roux NS, Toloza A, Dolezel J, Panis B (2004) Usefulness of embryogenic cell suspension cultures for the induction and selection of mutants in Musa spp. In: Mohan Jain S, Swennen R (eds) Banana improvement: cellular, molecular biology and induced mutations. Science Publishers, USA, pp 33–43
Sagi L (2000) Genetic engineering of banana for disease resistance—future possibilities. In: Jones DR (ed) Diseases of banana, Abaca and Enset. CABI Publishers, UK, pp 465–515
Sagi L, Remy S, Panis B, Swennen R, Volckaert G (1994) Transient gene expression in electroporated banana (Musa spp. cv. Bluggoe, ABB group) protoplast isolated from regenerable embryogenic cell suspensions. Plant Cell Rep 13:262–266. doi:10.1007/BF00233316
Sagi L, Panis B, Remy S, Schoofs H, De Smet K, Swennen R, Cammue BPA (1995) Genetic transformation of banana and plantain (Musa spp.) via particle bombardment. Biotechnology 13:481–485. doi:10.1038/nbt0595-481
Sagi L, May GD, Remy S, Swennen R (1998) Recent development in biotechnological research on bananas (Musa spp.). Biotechnol Genet Eng Rev 15:313–327
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning, a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor
Schenk RU, Hildebrandt AC (1972) Medium and techniques for induction and growth of monocotyledonous plant cell cultures. Can J Bot 50:199–204. doi:10.1139/b72-026
Smith CJS, Watson CF, Morris PC, Bird CR, Seymour GB, Gray JE, Arnold C, Tucker GA, Schuch V, Harding S, Grierson D (1990) Inheritance and effect in ripening of antisense polygalacturonase genes in transgenic tomatoes. Plant Mol Biol 14:369–379. doi:10.1007/BF00028773
Sunil Kumar GB, Ganapathi TR, Revathi CJ, Srinivas L, Bapat VA (2005) Expression of hepatitis B surface antigen in transgenic banana plants. Planta 222:484–493. doi:10.1007/s00425-005-1556-y
Yao JL, Cohen D, Atkinson R, Richardson K, Morris B (1995) Regeneration of transgenic plants from the commercial apple cultivar ‘Royal Gala’. Plant Cell Rep 14:407–412. doi:10.1007/BF00234044
Acknowledgement
Antara Ghosh is thankful to Board of Research in Nuclear Sciences (BRNS), Department of Atomic Energy (DAE), Government of India for the financial assistance to carry out this work.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ghosh, A., Ganapathi, T.R., Nath, P. et al. Establishment of embryogenic cell suspension cultures and Agrobacterium-mediated transformation in an important Cavendish banana cv. Robusta (AAA). Plant Cell Tiss Organ Cult 97, 131–139 (2009). https://doi.org/10.1007/s11240-009-9507-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11240-009-9507-0