
Abstract
Marker gene elimination was investigated in hybrid aspen (Populus tremula L. × Populus tremuloides Michx.) using the FLP/FRT recombination system. The construct contained the FLP recombinase under control of a heat inducible promoter, the antibiotic resistance gene nptII driven by the CaMV 35S promoter, and a promoterless uidA gene. The construct was integrated into poplar via Agrobacterium-mediated transformation. The active FLP recombinase excised the nptII marker gene and combined the promoterless uidA gene with the CaMV 35S promoter to form an active uidA gene. For targeted transgene integration, two constructs were used. The first one carried FLP under control of the heat-inducible Gmhsp17.5-E promoter from soybean as well as an active nptII gene flanked by two FRT sites; the second contained the promoterless bar selection marker gene also flanked by two FRT sites. Following transformation and induction of FLP, the enzyme mediated a site-specific recombination at the FRT sites of both constructs. This recombination leads to an excision of the FLP and nptII gene from the first as well as an excision of the promoterless bar gene from the second construct. The promoterless bar gene reintegrated exactly at the former position of the FLP and nptII genes in the first construct to form an active bar gene. The FLP/FRT recombination system from yeast forms a promising basis for the production of antibiotic-free transgenic plants and a useful tool for directed integration of transgenes into plant genomes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Commercial use of genetically modified (GM) plants has been under debate since the first reports on the production of transgenic plants were published (e.g., Shaw et al. 1983; Caplan et al. 1983). Public acceptance of GM plants strongly depends on the plant species used and the trait engineered, but as we expect, acceptance can be increased due to the development of efficient risk assessment approaches. Biosafety research includes ecological/environmental as well as human and animal nutrition issues such as vertical and horizontal gene transfer, gene stability, and use of antibiotic/herbicide genes and effects of the novel traits on nontargeted organisms (Giridhar and Parimalan 2004; Hönicka and Fladung 2006; Craig et al. 2008).
To answer the important ecological and environmental questions, various GM plants have been tested in field studies in more than 30 countries throughout the world (Robischon 2006). Despite the sophisticated biosafety research that was carried out in the last 20 years, the use of herbicide or antibiotic selectable marker genes became a focus of the opponents of gene technology (Miki and McHugh 2004). Herbicide or antibiotic selectable marker genes, which are needed for the recognition of transgenic tissue, are unnecessary once transgenic plants have been established. Thus, various strategies have been suggested to remove marker genes from the genome of the transgenic plants including tobacco, rice, and poplar (Yoder and Goldsbrough 1994; Puchta 2000, 2003; Zubko et al. 2000; Kumar and Fladung 2001; de Vetten et al. 2003; Lutz et al. 2006; Zelasco et al. 2007; Nanto and Ebinuma 2008).
The most widely known general approach to eliminate genes is the use of site-specific recombination (Ow 2002). Site-specific recombination requires the action of a site-specific recombinase that can be supplied either by stable or transient transformation. Many reports have been published describing the successful removal of herbicide or antibiotic selectable marker genes in plants (Nanto and Ebinuma 2008; Lee et al. 2007), such as rice (Luo et al. 2008), tobacco (Chakraborti et al. 2008), citrus (Ballester et al. 2007), and poplar (Zelasco et al. 2007; Ebinuma et al. 2004).
Following the transfer of genes either through Agrobacterium-mediated transformation or particle bombardment, the integration of the foreign gene(s) is not predictable; thus, effects on (trans)gene stability and induction of undesired T-DNA integration events are likely to occur (Kumar and Fladung 2002b). A separate category of transgene expression or silencing concerns position effects, where genomic position of the integrated transgene affects its expression. Genomic position may have enhanced or repressive influences on transgene expression by flanking plant DNA and/or an unfavorable chromosomal location. This type of silencing presumably reflects the epigenetic state of neighboring host sequences or the relative tolerance of particular chromosomal regions to invasion by alien DNA (Matzke and Matzke 1998).
One strategy to address the issue of expression variability and gene silencing systematically is to precisely modify or target defined locations within the genome (reviewed in Vergunst and Hooykaas (1999) and Kumar and Fladung (2001)). Successful targeting of a single copy of a new gene to a defined position should allow production of transgenic plants in which the incoming gene can be expressed in a predictable pattern. Rather than selecting against position effect variation, these “recipient plants” would have been previously characterized for their individual pattern of position effect variation. The idea to use the reverse reaction of site-specific recombination systems to precisely insert transgenes into predefined positions of the tree genome has already been proposed (Tzfira and White 2005; Lyznik et al. 2003; Kumar and Fladung 2002a). To date, there are only a few reports describing the exchange of one gene with another one or the site-directed integration into a recombinase cutting site with a known location in the genome (Vergunst et al. 1998; Vergunst and Hooykaas 1998; Araki et al. 2002; Nanto et al. 2005; Louwerse et al. 2007).
In this study, we tested the site-specific recombination system FLP/FRT for the excision of a marker gene as well as the targeted integration of a transgene in a Populus-aspen hybrid. As Arabidopsis and rice, species of the genus Populus have become plant models due the availability of the complete genome sequence in P. trichocarpa (Tuskan et al. 2006) and the development of various genetic, genomic, and biochemical tools (Jansson and Douglas 2007). The expression of the FLP recombinase gene was controlled via the soybean heat inducible promoter Gmhsp17.5-E for both excision and integration. First, we confirmed earlier results and demonstrated that the FLP/FRT system can be used for the removal of an antibiotic marker gene. Second, similar to Louwerse et al. (2007), we used a two gene cassette system and replaced an nptII gene from one gene cassette with a promoterless bar gene located in an unlinked second cassette.
Following the excision of the FLP and nptII genes located between the two FRT-sites, the promoterless uidA came under control of the 35S promoter, which was validated by GUS staining. For integration, we replaced nptII gene from one gene cassette with a promoterless bar gene located in an unlinked second cassette to produce kanamycin-sensitive and BASTA-resistant plants.
Material and methods
Plasmids
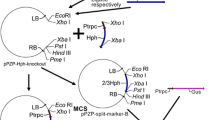
The excision plasmid pTex (Fig. 1a) contains the selection marker gene nptII and the FLP recombinase gene from yeast, which contains the intron Stls1 from potato at position 2768. FLP is driven by the heat shock promoter Gmhsp17.5-E from soybean. Two directly repeated FRT-sites flank, the 3′- and 5′-ends of the nptII marker and the FLP gene, with a 35S promoter and a promoterless uidA gene located outside, left and right of the FRT cassette. In the nonexcised situation, the 35S promoter controls nptII. Following heat-shock treatment, the Flp recombinase catalyzes site-specific recombination leading to excision of the fragment between the two FRT-sites. Thus, the promoterless uidA gene comes under the control of the 35S-promoter and becomes active (Fig. 1b).

Schematic representation of the binary vectors used for the excision experiments before (A) and after excision of the fragment between FRT-sites (A). A Molecular strategy for excision of the DNA located between two collateral orientated FRT-sites. The excision plasmid pTex (A) contains two functional gene items: the selection marker nptII and the FLP recombinase from yeast, which implies the intron Stls1 from potato at the position 2,768. The FLP is driven by the heat shock promoter Gmhsp17.5-E from soybean. Two collateral orientated FRT-sites are flanking at the 3′- and 5′-ends of the nptII selection marker and the FLP gene, with a 35S promoter and a promoterless uidA gene located outside left and right of the FRT cassette. In the nonexcised situation the 35S promoter controls the nptII gene. Following heat-shock treatment the FLP recombinase catalyses, the site-specific recombination leading to excision of the fragment between two FRT sites. Thus, the promoterless uidA gene gets under control of the 35S-promoter and becomes active (B). Arrows indicate gene-specific oligonucleotide primers. Relevant restriction enzyme cutting sites are introduced into the maps. Sequences surrounding the FRT sites are given above or below the constructs and were checked before or after excision by sequencing. (P-NOS nopaline synthase promoter, nos Nopaline synthase gene terminator, bar Phosphinothricin acetyltransferase gene, FLP FLP recombinase; Stls1 Stls1-Intron from potato, HSP GmHSP 17.5-E promoter, GUS β-glucuronidase gene, NptII Neomycin phosphotransferase gene, 35S-Pro 35S CaMV35S promoter, T35S CaMV35S terminator, OCS Octopine synthase gene terminator, LB and RB Left and right border sequences, FRT FRT sites)
For the investigation of targeted integration, two different constructs were used (Fig. 2a, b). The first construct pTin1 is similar to pTex (described above) but lacks the promoterless uidA gene at the 5′-end of the cassette (Fig. 2a). The second construct pTin2 contains a promoterless bar gene, flanked by two oppositely oriented FRT-sites (Fig. 2b). In addition, a hygromycin resistance gene (hpt) is located outside the FRT-sites as an additional plant selection marker. Following activation of Flp through heat-shock, both DNA fragments inside the FRT-sites are excised. In several cases, the promoterless bar gene from pTin2 integrates at the locus of the pTin1 T-DNA leading to a functional bar gene (Fig. 2c).

Schematic representation of the binary vectors pTin1 (A) and pTin2 (B) employed for site-directed cassette exchange experiments in poplar and the pTin1 construct after successful excision/integration (C). For the investigations on targeted integration, two constructs were used (A, B). The first construct pTin1 is similar to pTex as described in (A) but without the promoterless uidA gene at the 5′-end of the cassette. The second construct pTin2 contains a promoterless bar-gene, flanked by two oppositely oriented FRT sites. In addition, a hygromycin resistance gene (hpt) is located outside the FRT-sites as additional plant selection marker, following activating of the FLP through heat-shock both DNA fragments inside the FRT-sites excises. In few cases, the promoterless bar gene from pTin2 integrates in the locus of the pTin1 T-DNA leading to a functional bar gene (C). Arrows indicate gene-specific oligonucleotide primers. Relevant restriction enzyme cutting sites are introduced into the maps. Sequences surrounding the FRT-sites are given above or below the constructs and were checked before or after integration by sequencing. (P-NOS Nopaline synthase promoter, nos Nopaline synthase gene terminator, bar Phosphinothricin acetyltransferase gene, FLP FLP recombinase, Stls1 Stls1-Intron from potato, HSP GmHSP 17.5-E promoter, NptII Neomycin phosphotransferase gene, 35S-Pro, 35S CaMV35S promoter, T35S CaMV35S terminator, hpt Hygromycin phosphotransferase gene, OCS Octopine synthase gene terminator, LB and RB Left and right border sequences, FRT FRT sites)
All constructs were created and introduced into Agrobacterium strain GV3101 containing pMP90RK (Koncz et al. 1989) by DNA-Cloning Service (Hamburg, Germany).
Plant transformation
For Agrobacterium-mediated transformation experiments, a hybrid poplar line called Esch5 (Populus tremula L. × Populus tremuloides) was used. Plants were vegetatively propagated under sterile conditions in a growth chamber at 25°C, 24 h light (15–20 µE/m²/s).
The transformation of poplar leaf disks was performed using a leaf disk inoculation method (Fladung et al. 1997). Leaf disks infected with the excision construct pTex were transferred after cocultivation to WPM medium containing 1 ml/l Pluronic F-68, 10 µl/l Thidiazuron (601) supplemented with 50 mg/l kanamycin sulfate and 500 mg/l cefotaxime (601KC). Leaf disks infected with two Agrobacterium strains containing the two integration constructs pTin1 und pTin2 were cultivated on 601 medium with 50 mg/l kanamycin, 10 mg/l hygromycin, and 500 mg/l cefotaxime (601KHC). Shoots were regenerated and subcultivated on the respective media until molecular analysis.
Heat shock treatments
To activate the recombination system, transgenic poplar plants were incubated at 42°C for 3 h in growth chambers. Twenty four hours after the heat shock treatment, leaf disks were harvested and either stained with X-Gluc or plant material was stored at −70°C.
For selection of targeted transgene integration events, regenerative callus, leaves, and stems were cut into approximately 1 mm small pieces (microcalli) and transferred to fresh regeneration medium 601 supplemented with 10 mg/l hygromycin, 2 mg/l BASTA, and 500 mg/l cefotaxime (601HBC). The plant/calli material was cultivated up to 5 months at 25°C and continuous light in the growth chamber. Regenerated shoots were transferred to fresh 601HBC media and analyzed by PCR.
DNA extraction, PCR, and Southern analyses
Genomic DNA was isolated from poplar plants by the CTAB method of Dumolin et al. (1995).
The excision of the FLP/nptII fragment in the “excision construct” after heat shock treatment was verified by PCR using the primer pair TS29/35sF (5′-GAA ACC TCC TCG GAT TCC AT-3′) and TS30/GUSr (5′-TTT TTG ATT TCA CGG GTT GG-3′). Following successful excision, a fragment of 450 bp in size was obtained.
To validate the position of the bar and nptII genes in the “integration constructs” before and after the heat shock treatments, PCRs with different primer pairs were carried out. The primer pair 329/17 (5′-TCG GGA AAC CTC CTC GGA TT-3′/5′-AAG GCG ATA GAA GGC GAT GCG-3′) amplifies a 1.2-kb fragment spanning from the 35S-promoter and the npt-II gene, and the primer pair 329/459 (5′-TCG GGA AAC CTC CTC GGA TT-3′/5′-TGA AGT CCA GCT GCC AGA AAC-3′), a 0.8-kb fragment from the 35S-promoter, and the bar gene. Poplar lines with FLP/nptII removal and bar integration exactly at this position revealed a fragment of 800 bp with the primer pair 329/459 and no or weak PCR fragment with the primer pair 329/17.
For the monitoring of the PCR reaction, a genomic region of 250 bp was amplified using the primer pair 155/156 (5′-GAT CAT GCT TTA GGT GAT GAC-3′/5′-TGA AGC ATA CAC GAC AGA AGA-3′). PCR fragments for verification of excision and integration events were eluted from an agarose gel, and the DNA was sequenced (StarSeq, Mainz, Germany).
Integration of the “excision construct” in the genome of transgenic poplar plants and T-DNA copy number as well as the success of heat-shock treatments was monitored by Southern blot analyses using a DIG-labeled uidA specific DNA-probe. The primer pair used to amplify the uidA specific DNA-probe in a DIG-labeling reaction was GUS1 (5′-CAG GAA GTG ATG GAG CAT CAG-3′) and GUS2 (5′-TCG TGC ACC ATC AGC ACG TTA-3′; Fladung and Ahuja 1995). In Southern blot analyses of transgenic plants carrying the “integration constructs,” a DIG-labeled 35S-probe was used, generated with the primer pair 35S-f (5′-AGA TCA ACA TGG TGG AGC A-3) and 35S-r (5′-CTC CAA ATG AAA TGA ACT TCC-3′). For Southern blot analyses, 20–30 µg of genomic DNA was cleaved with restriction enzymes, separated on a 1% agarose gel, blotted on a nylon membrane, and hybridized with the DIG-labeled DNA probe (Fladung and Ahuja 1995).
Inverse PCR and TAIL-PCR
To locate the insertion sites of the transgenes in the plant genome, inverse PCR was used (Fladung 1999). Ten micrograms of genomic DNA was digested with HindIII and self-ligated with T4 DNA ligase (Fermentas, Germany). Primer pairs were used that were orientated away from each other and primed from the end of the T-DNA and upstream of the restriction sites.
TAIL-PCR was performed as described in Liu et al. (1995). All three arbitrary degenerate primers were tested in combination with three specific primers designed from the left and right borders of the constructs pTin1 and pTin2. These primers were: left border of both constructs: 35S-r-1: 5′–CGA GGA GGT TTC CCG ATA TTA–3′; 35S-r-2: 5′–CCT TTG TTG AAA AGT CTC AAT–3′; 35S-r-3: 5′–GAG ACT GTA TCT TTG ATA TTC –3′: For pTin1, the following primers were used: Ocs-f-1: 5′–CGG TTT CGG TTC ATT CTA ATG–3′; Ocs-f-2: 5′–CCC GTT ACT ATC GTA TTT TTA–3′; Ocs-f-3: 5′–ATT CTC CGT TCA ATT TAC TGA–3′. For pTin2 these primers were tested: Bar-f-1: 5′–TTG ACC GTG CTT GTC TCG ATG–3′; Bar-f-2: 5′–TCG GCC GGG CGT CGT TCT GGG–3′; Bar-f-3: 5′–AGA AAG TAT AGG AAC TTC GGA–3′.
Fragments obtained were eluted from agarose (Qiagen, Duesseldorf, Germany) and sequenced (StarSeq, Mainz, Germany). Sequences were blasted against the available (public) poplar genome of Populus trichocarpa v1.0 at http://genome.jgi-psf.org/Poptr1/Poptr1.home.html and http://blast.ncbi.nlm.nih.gov/Blast.cgi. Successful BLAST-results were used to position the T-DNAs on the physical map of P. trichocarpa. These positions were assigned to the Populus-aspen genome because of the high collinearity to the P. tremula/P.tremuloides genomes (Pakull et al. 2009).
GUS assay
Histochemical staining for GUS activity in transgenic plants was performed as described by Jefferson et al. (1987) to verify activation of the uidA gene after the heat treatment. Leaf segments were vacuum-infiltrated for 10 min in 1.5 mM x-Gluc in 100 mM sodium phosphate buffer, 10 mM EDTA, and 0.5% Triton X-100 (pH 7.0), then incubated at 37°C for 24 h. To remove chlorophyll and other pigments, leaf segments were soaked three times in 3:1 ethanol:acetic acid at 50°C for 20 min.
Results
Production of transgenic plants and molecular analysis
From Agrobacterium-mediated leaf transformation experiments, we were able to regenerate 23 independent transgenic poplar lines carrying the excision vector pTex (Fig. 1a). Transformation efficiency (based on number of explants infected with Agrobacterium in relation to independent transgenic lines obtained) was 2.3%. Integration of the construct was confirmed by PCR (not shown). Southern blot analyses using DIG-labeled uidA, FLP (both probes were tested only for lines TS131-#1 to -#16) and nptII probes confirmed the integration of the entire T-DNA cassette (SfiI digested genomic DNA) and revealed the presence of one to three copies of the nptII gene (SacI digested genomic DNA, Fig. 3). Some transgenic lines showed an additional DNA band of varying size in SfiI digested DNA indicating incomplete copies of the pTex construct. Ten transgenic lines identified as single copy lines as well as a few lines containing two and three gene copies, respectively, were tested in antibiotic marker excision experiments.

Southern blot analysis of transformed poplar lines with the excision plasmid pTex. A representative blot with SfiI and SacI restricted and nptII-probed DNA isolated from Agrobacterium strain used for transformation, negative control line Esch5, and seven pTex-transgenic lines TS131-#17 to -#23 is shown. With SfiI, the entire cassette of 6 kb is restricted. SacI has only one restriction site in the cassette that can be used for copy number determination. Genomic DNA was separated on Agarose gel, blotted, and hybridized with a DIG-labeled nptII probe. At positive control (Agrobacterium tumefaciens), Esch5 nontransformed control line
For targeted transgene integration (cassette exchange) double transformation experiments with pTin1 and pTin2 constructs were conducted resulting in integration of both T-DNAs. The first construct pTin1 contains the FLP gene under control of a heat-inducible promoter as well as the nptII gene to be exchanged. The second one pTin2 harbors a promoterless bar gene to replace the nptII gene. The double transformation experiments, using the integration constructs, pTin1 and pTin2 yielded 12 independent transgenic lines with a transformation efficiency of 1.6%. The integration of the T-DNA was confirmed by PCR and genomic DNA gel blot analyses (not shown). Eight single copy plants were used for the excision and integration experimental procedure.
Antibiotic marker excision experiments
The excision of the fragment between the FRT sites of the pTex construct following heat-shock treatment was demonstrated in single and multiple copy containing transgenic lines by GUS staining experiments, PCR, and Southern blot analyses. The leaves of pTex-transgenic poplar plants were subjected to GUS staining experiments before and after heat-shock (Fig. 4). Out of the 23 transgenic lines, 15 lines revealed the formation of blue color after heat-shock treatment of leaves. The intensity of blue color varied irrespective of pTex copy number, e.g., transgenic lines TS131-#4 and -#11 carrying one and three copies, respectively, revealed strongest GUS staining.

Transgenic and control poplar leaves after heat treatment at 42°C for 6 h and GUS-staining. a transgenic line TS131-#4 (single copy) without heat-shock, b and c transgenic lines TS131-#4 and -#8 after heat-shock
To verify the excision of the fragment between the FRT-sites in the heat-treated transgenic lines, isolated DNA was subjected to PCR (not shown) and Southern blot analyses. In Fig. 5a, a representative Southern blot is shown with single-copy containing transgenic lines. No signal indicating excision could be detected in PCR (not shown) or in Southern blot experiments in the non heat-shocked pTex transgenic lines. Following heat-shock, excision of the fragment between the FRT-sites reduces the size of the pTex-T-DNA from 7.0 to 3.2 kb in SfiI-restricted genomic DNA hybridized against uidA-probe (Fig. 5). However, weak signals of the original pTex construct could be detected in the Southern blots of the heat-shocked lines irrespective of the copy number. Sequencing of the PCR amplified fragment 35S-uidA confirmed excision of the fragment between the FRT sites (sequencing data from lines TS 131-#2 and TS 131-#19 are introduced in Fig. 1b) and revealed the presence of a remaining FRT site located between 35S promoter and the uidA gene. Heat-shocked pTex transgenic plant tissue or regenerative callus died on kanamycin-containing media (not shown).

Southern blot analysis of pTex transgenic poplar lines without and with heat-shock (HS) treatment. A representative blot with SfiI restricted and uidA-probed DNA isolated from Agrobacterium, negative control line Esch5, and the pTex-transgenic lines TS131-#1 (3 copies), -#4 (1 copy), -#5 (2 copies), -#7 (1 copy), -#8 (1 copy), -#15 (1 copy) is shown. Copy number was determined before with SacI restricted DNA blotted against nptII, uidA, and FLP probes. A new DNA fragment of 2.8 kb appears after heat-shock treatment. TS131 pTex transgenic poplar lines, Esch5 nontransformed control line, At Plasmid-DNA from Agrobacterium strain
Site-specific integration of transgenes
Correct exchange of one gene with another one was tested in eight independent transgenic poplar lines carrying both pTin constructs. To minimize the production of chimeras, the strategy of our experiment was to use regenerative calli, which were thoroughly crushed into microcalli for the heat-shock experiments (Fig. 6a). After heat shock, 16,422 (TS85-#2) to 51,392 (TS71-#3) microcalli were transferred to BASTA-containing regeneration media and cultivated for 5 months (Table 1). In seven of the eight double transgenic poplar lines, we detected varying numbers of regenerated (green) calli or plantlets on the BASTA-containing media (Fig. 6b). The number of green calli/plantlets varied between zero in TS79-#2 and 2,946 in TS79-#1 (Table 1). On control plates with 601 regeneration medium, supplemented with kanamycin, we never observed the formation of any green callus or plantlets.

Selection of pTin1 and pTin2 double transgenic poplar lines after heat shock treatment at 42°C for 6 h on 601HBC-media. a Microcalli without heat treatment. Plant cells are BASTA-sensitive and die on BASTA-containing media. b Microcalli after heat treatment, 6–8 weeks on BASTA-containing selection media. Few cell lines are BASTA-resistant and survive on BASTA-containing media. c Selected putative positive recombinants
To validate correct integration of the bar gene in pTin1, randomly three (TS71-#1) to 100 (TS79-#1, TS79-#4) regenerative calli/plantlets were selected and further subcultured on BASTA-containing media (Fig. 6c, Table 1). From these, DNA was isolated for PCR analyses (Fig. 7). The primer pair 329/17 amplifies a fragment of 1.2 kb only in nonheat shocked double transgenic plants and the control pTin1 plasmid (Fig. 7, upper gel). Lack of an amplification signal in heat-shocked double transgenic calli/plantlets indicates successful recombination and, simultaneously, presence of an 800-bp fragment using the primer pair 329/459 reveals integration of the bar gene in pTin1 (Fig. 7, lower gel). For all PCR experiments, a primer pair 155/156 amplifying a 250-bp genomic DNA fragment was added to monitor the PCR reaction.

PCR analyses of 17 randomly selected putative positive recombinants (8–24) of the heat-shock treated double transgenic poplar line TS79-#4 carrying pTin1 and pTin2. The primer pair 329/17 amplifies a PCR fragment of 1.1 kb only in case of no excision of the nptII-gene in pTin1. The genomic DNA of TS79-#4 without heat treatment and the positive control (At pbTin1) show such a band. Very weak bands are visible from the putative recombinants 20 and 23. In case of successful cassette exchange (excision of nptII from pTin1 and integration of bar gene at this position), a fragment of 800 bp in size is amplified with the primer pair 329/459. The genomic primer pair 155/156 is used as an endogenous control of the PCR reaction (380 bp). Esch5 nontransformed control line, At plasmid controls, HS heat shock treatment
All isolated green calli/plantlets were tested for excision of nptII and integration of bar gene. Using the strategy described above, 13–86% of the analyzed regenerants revealed the integration of the bar gene in pTin1 (Table 1). In some cases, the 800-bp amplification product was sequenced to verify the presence of the 35S-promoter, the FRT site, and the bar gene (not shown). Taken all together, we observed a targeted insertion frequency of 0% in TS79-#2 up to 6.4% in TS79-#1 (Table 1).
Sequencing of the 329/459 PCR-generated fragments of isolated green calli/plantlets of three independent transgenic lines (TS79-#1, -#4, and -#5) confirmed the formation of the newly arranged fragment comprising 35S-promoter, the FRT site, and the bar gene (see Fig. 2c).
Position of T-DNAs in the poplar genome
To identify the genomic position of the pTin1 and pTin2 T-DNAs, inverse-PCR analyses and TAIL-PCR experiments were carried out to sequence the left and right T-DNA flanking genomic regions (FSTs = flanking sequence tags). Table 2 summarizes the results of BLAST searches with the obtained flanking sequences and indicates the genomic positions of the T-DNAs in the P. trichocarpa map. These positions were assigned to the Populus-aspen genome because of the high collinearity of the P. trichocarpa genome to the one of P. tremula/P.tremuloides (Pakull et al. 2009).
Unfortunately, we could determine the insertion site of the plasmids only in four transgenic lines and out of these only in two lines, the location of both plasmids. For the transgenic line TS79-#2, it was not possible to exactly determine the linkage group (LGs) because very high BLAST similarity results were given for the two linkage groups LG IV and XI. Nevertheless, in TS79-#2, both possible integration sites for pTin2 are different from the genomic position of pTin1 located on LG I. TS79-#4 contains the two pTin plasmids also on two separate linkage groups, namely LG XIII and LG XVI.
Discussion
The development of a genetic system that allows the removal and also the site-specific integration of transgenes would be of great benefit for scientific purposes and for applied aspects of using transgenic plants. Here, we demonstrate for the first time for tree species that the FLP/FRT recombination system can be used for both marker gene excision and exchange of one gene for another one within the genome of Populus-aspen. The strategy of FLP-mediated site-specific recombination is based upon the activation of the FLP gene following heat shock treatments. This method employs Agrobacterium-mediated gene transfer for the integration of excision and integration constructs and kanamycin selection to identify transgenic events.
Excision
A gene construct for marker gene elimination, named pTex, contains two functional genes: the selectable marker nptII and the FLP recombinase under control of the heat inducible promoter HSP17.5-E from Glycine max. The nptII gene and the FLP recombinase are flanked by two FRT sites. Outside of the FRT-site, there is on one side a 35S promoter and on the other site, a promoterless uidA gene. Following heat activation, the FLP recombinase catalyzes the excision of the nptII- and the FLP-gene itself; thus, the promoterless uidA gene comes under control of the 35S-promoter and becomes active. Histochemical assay of GUS activity offers a simple, easy, and highly sensitive method for the detection of excision events in leaf pieces and whole plants. Furthermore, we were able to confirm excision events by Southern blots, by PCR analysis, and by sequencing the newly connected transgenes.
From 23 tested poplar lines, 15 (65%) revealed formation of blue color. We have never observed noninduced plants showing GUS-activity as reported by Cuellar and coworkers (2006). In transgenic potato, transformed with marker genes and a heat inducible cre recombinase gene flanked by two loxP sites; the authors were able to identify plants that show spontaneous excision of the test construct before the induction of the HSP promoter. These results underline the necessity of using a promoter that is inducible by only one signal.
All plants with GUS activity were subjected to PCR analyses to validate the presence of the newly connected 35S-uidA fragment. Sequencing the resulting PCR amplification products confirmed the excision of the nptII gene between the FRT sites and demonstrated the precise excision by formation of a remaining FRT site between the 35S promoter and the uidA gene. Irrespective of integrated pTex copy number, the excision in most of those lines seems not to be complete as indicated by Southern blot analyses. However, cultivation of heat-shock induced plant material on kanamycin-containing media revealed browning of the tissue within 4 weeks, similar to nontransgenic lines. It could be possible that excision of the nptII gene was not completed at the time of sample harvest for molecular analyses.
The band intensity on Southern blot seems to vary in strength, which could be further influenced by the integration site of the T-DNA within the genome or by nonoptimal expression of the wild type FLP recombinase gene in poplar. Codon distribution and genome GC content could play a role in successful heterologous gene expression in different organisms (Gustafsson et al. 2004) because rare codon gene expression can lead to incorporation of wrong amino acids, termination of translation or frame shift errors (Calderone et al. 1996). Changing the codon usage or the GC content increased heterologous gene expression in bacterial, animal, and plants (Wohlleben et al. 1988; Adang et al. 1993; Song and Niederweis 2007; Fedoryshyn et al. 2008). The GC content of the native yeast FLP recombinase (39%) is different from that of the poplar lines used in our experiments (45.3%). By increasing the GC-content of the FLP recombinase to 55%, we were able to enhance the expression of the FLP recombinase up to 22-fold in maize zygotic embryos (GC-content in maize: 55%, Becker, unpublished data).
Luo et al. (2007) demonstrated in transgenic tobacco containing fusions of the loxP and FRT sequences that the expression of either the FLP or the cre recombinase increased deletion efficiency. The authors suggested that fused loxP–FRT sequences might enhance the alignment of the recognition sequences, DNA binding or cleavage, or the formation of a Holliday junction or DNA recombinase complex.
Heterogeneous distribution of heat in our calli during the heat activation of the FLP recombinase might also influence the efficiency of excision events in our plants. Nonhomogenous distribution of heat produces a gradient of Gmhsp 17.5-E promoter activation in the calli, which is higher in the outer cell layers than in the central callus. Partial excision of the selectable marker genes was also observed by Cuellar and coworkers (2006) in potato and Thomsen and coworkers (2008) in Arabidopsis. These authors used a cre recombinase driven by a heat-inducible promoter, and they observed a partial excision of the marker gene nptII in 17% of regenerated shoots. In Arabidopsis, the frequency of partial excision was in the range of 51% (Thomsen et al. 2008), while in our Southern blot experiments, nearly 90% of all analyzed plants showed a noncomplete excision. However, our data did not explain why we were never able to regenerate calli or plantlets on kanamycin-containing media after heat-shock induction in excision experiments.
We determined the FRT recognition site after the excision by PCR amplification and sequencing (see Fig. 1b). The sequencing data demonstrate the fidelity and precision of the FLP site-specific recombinase because of the nature of the catalytic mechanism and the conservative character of this reaction. Indeed, it has been shown that cre-mediated recombination yields predominantly accurate recombination products (Dale and Ow 1991; Lakso et al. 1992).
Integration
Despite 25 years of experience in plant gene technology, targeted gene integration in higher plants is still beyond our control. Transgene integration by homologous integration is an inefficient way to direct the integration of a common gene to a specific region of the genome. However, the method of homologous integration of transgenes works very well in animal cells, fungi, and in the moss Physcomitrella patens (Schaefer and Zryd 1997; Mullins and Kang 2001). Therefore, research activities are intensified to establish new methods of targeted gene integration into the genome and to prevent variable gene expression. The development of site-directed integration methods, which allow the integration of a transgene into a well defined gene locus, is of great importance not only for fundamental research, but also for commercial crop improvement. It has been shown by several groups that targeting of a transgene to a similar genomic position in different experiments both in mammalian cells as well as in plants resulted in reproducible gene expression levels (Fukushige and Sauer 1992; Vergunst et al. 1998; Vergunst and Hooykaas 1998; Feng et al. 1999; Shrivastava and Ow 2004; Chawla et al. 2006; Nanto et al. 2005; Araki et al. 2002). Thus, from a large number of target sites for gene integration in plants, one can be selected following testing with reporter genes, which favors the expression of transgenes. Once such a genomic target site has been identified, in a second step, different relevant genes can be directed to this site.
To our knowledge, targeted integration using the FLP/FRT recombinase system has never been reported previously. There are already different successful reports using the cre/loxP recombinase system to induce a cre mediated cassette exchange between an exogenous supplied transgene, which is introduced by Agrobacterium-mediated transformation and contains one loxP site, against an already stable integrated transgene containing a second loxP site (Vergunst et al. 1998; Vergunst and Hooykaas 1998; Araki et al. 2002; Nanto et al. 2005; Louwerse et al. 2007).
In our strategy, we used Agrobacterium-mediated gene transfer to introduce two different T-DNAs into the poplar genome. The first T-DNA contains the FLP recombinase driven by an inducible promoter, and the second one harbors a promoterless herbicide resistance gene. Following recombinase induction, the herbicide resistance gene is expressed after specific integration downstream of the 35S promoter, formerly expressing the selectable marker gene nptII. In our experiments, the frequency of this cassette exchange varied between zero (line TS79-2) and 6.4% (line TS79-1). The highest frequency of 6.4% of cassette exchange is comparatively low in comparison to the 44% obtained in cassette exchange experiments in Arabidopsis using the cre/loxP recombinase system by Louwerse et al. (2007). Nevertheless, the estimated frequency of cassette exchange is only a preliminary result because, to estimate the correct frequency, it is necessary to start from single cells and not from microcalli.
It is also possible that the chromosomal location of the target FRT site was readily accessible to the FLP recombinase. The location of the target sites was observed to have an effect on recombination rates in other recombination systems (Dale and Ow 1990).
Our experiments demonstrate clearly that the FLP/FRT recombinase system has the potential to induce cassette exchange between two already integrated T-DNA molecules. The question now is: Is it also possible to direct exogenous supplied transgenes harboring one FRT site to a well-defined position into the genome where a second FRT site is still present?
Despite the fact that T-DNA may not be a perfect substrate for site-specific integration, it has been shown that T-DNA could be successfully targeted to a single genomic loxP site (Vergunst and Hooykaas 1998). In Arabidopsis, Vergunst and coworkers (1998) were able to target a chromosomally introduced lox site with a secondly applied T-DNA containing a promoterless nptII gene with a frequency of 1.2–2.3% when the cre recombinase is constitutively expressed. T-DNA can be used for gene targeting via homologous recombination (Lee et al. 1990; Offringa 1992). The cre/loxP mediated integration of the T-DNA into the genome requires conversion of the linear single-stranded form into a circular double-stranded form (Offringa et al. 1990; Tzfira et al. 2003; Marillonnet et al. 2004).
The cre/loxP system has also been used for site-specific integration or replacement of transfected DNA into a chromosomally positioned loxP site (Fukushige and Sauer 1992; Araki et al. 1997; Soukharev et al. 1999). The integration reaction is inefficient with wild-type loxP sites, due to the re-excision of the recombined product; therefore, mutant loxP sites have been developed to increase the efficiency of cre-mediated insertion or replacement (Louwerse et al. 2007).
In order to understand the effects of chromatin on T-DNA integration and transgene expression, it is instructive first to understand how T-DNA may choose target sites in the plant genome. Does T-DNA integration occur randomly or are there “hot” and “cold” spots where the T-DNA prefers to integrate? In situ analysis in Crepis capillaries and Petunia hybrid suggested that T-DNA integration occurs randomly among the chromosomes (Ambros et al. 1986; Wang et al. 1995). The analysis of thousands of Arabidopsis and rice tagged T-DNA mutant lines suggested that the integration did not occur in a random fashion (Feldmann 1991; Koncz et al. 1992; Krysan et al. 1999). In Arabidospsis, T-DNA integrates preferentially between genes, in 5′-gene regulatory regions, polyadenylation site regions, and A+T rich regions (Krysan et al. 2002; Szabados et al. 2002; Rosso et al. 2003; Schneeberger et al. 2005; Li et al. 2006). Also, nonrandom integration was observed in rice (Chen et al. 2003; Sallaud et al. 2004) and Populus (Fladung, unpublished data). All these analyses clearly show that T-DNA integration is common in gene-rich regions and is scant in gene-poor areas of the genome. T-DNA may preferentially target euchromatic chromosome regions and is mainly absent in heterochromatic regions.
Irrespective of the observed integration events, two double transgenic poplar lines were identified carrying the two pTin plasmids on two different linkage groups (Table 2). The line TS79-2 did not show any integration events (0%), while the line TS79-4 showed 1.4% of integration events, behind TS79-1 revealing the second-highest number of recombination events leading to cassette exchange. This could mean that location of the plasmids in the genome does not influence the success of cassette exchange.
Taken together, the application of site-specific recombination systems will lead to more sophisticated control of the genetic transformation process. Important consequences could be the production of environmentally safer transgenic plants with removed antibiotic- or herbicide-resistance genes as well as predictable transgene expression. We have simplified the recombination process by using regulated expression of the recombinase gene (Lyznik et al. 1989; Golic and Lindquist 1989; Logie and Stewart 1995). After successful transformation and selection, the activated recombinase could clean the transgenic genome from any unwanted foreign DNA sequences, including the recombinase gene itself. Furthermore, it is now possible to use the FLP/FRT recombinase system for targeted integration, too.
References
Adang MJ, Brody MS, Cardineau G, Eagan N, Roush RT (1993) The reconstruction and expression of a Bacillus thuringiensis cryIIIA gene in protoplasts and potato plants. Plant Mol Biol 21:1131–1145
Ambros PF, Matzke AJ, Matzke MA (1986) Localization of Agrobacterium rhizogenes T-DNA in plant chromosomes by in situ hybridization. EMBO J 5:2073–2077
Araki K, Araki M, Yamamura K (1997) Targeted integration of DNA using mutant lox sites in embryonic stem cells. Nucleic Acids Res 25:868–872
Araki K, Araki M, Yamamura K (2002) Site-directed integration of the cre gene mediated by Cre recombinase using a combination of mutant lox sites. Nucleic Acids Res 30:e103
Ballester A, Cervera M, Pena L (2007) Efficient production of transgenic citrus plants using isopentenyl transferase positive selection and removal of the marker gene by site-specific recombination. Plant Cell Rep 26:39–45
Calderone TL, Stevens RD, Oas TG (1996) High-level misincorporation of lysine for arginine at AGA codons in a fusion protein expressed in Escherichia coli. J Mol Biol 4(262):407–412
Caplan A, Herrera-Estrella L, Inzé D, Van Haute E, Van Montagu M, Schell J, Zambryski P (1983) Introduction of genetic material into plant cells. Science 222:815–821
Chakraborti D, Sarkar A, Mondal HA, Schuermann D, Hohn B, Sarmah BK, Das S (2008) Cre/lox system to develop selectable marker free transgenic tobacco plants conferring resistance against sap sucking homopteran insect. Plant Cell Rep 27:1623–1633
Chawla R, Ariza-Nieto M, Wilson AJ, Moore SK, Srivastava V (2006) Transgene expression produced by biolistic-mediated, site-specific gene integration is consistently inherited by the subsequent generations. Plant Biotechnol J 4:209–218
Chen S, Jin W, Wang M, Zhang F, Zhou J, Jia Q, Wu Y, Liu F, Wu P (2003) Distribution and characterization of over 1000 T-DNA tags in rice genome. Plant J 36:105–113
Craig W, Tepfer M, Degrassi G, Ripandelli D (2008) An overview of general features of risk assessments of genetically modified crops. Euphytica 164:853–880
Cuellar W, Gaudin A, Solórzano D, Casas A, Nopo L, Chudalayandi P, Medrano G, Kreuze J, Ghislain M (2006) Self-excision of the antibiotic resistance gene nptII using a heat inducible Cre-loxP system from transgenic potato. Plant Mol Biol 62:71–82
Dale EC, Ow DW (1990) Intra- and intermolecular site-specific recombination in plant cells mediated by the bacteriophage P1 recombinase. GENE 91:79–85
Dale EC, Ow DW (1991) Gene transfer with subsequent removal of the selection gene from the host genome. Proc Natl Acad Sci USA 88:10558–10562
Dumolin S, Demesure B, Petit RJ (1995) Inheritance of chloroplast and mitochondrial genomes in pedunculate oak investigated with an efficient PCR method. Theor Appl Genet 91:1253–1256
Ebinuma H, Sugita K, Matsunaga E, Endo S, Yamada-Watanabe K (2004) Asexual production of marker-free transgenic aspen using MAT vector systems. In: Kumar S, Fladung M (eds) Molecular genetics and breeding of forest trees. The Hawoth Press, New York, pp 309–338
Fedoryshyn M, Petzke L, Welle E, Bechthold A, Luzhetskyy A (2008) Marker removal from actinomycetes genome using Flp recombinase. Gene 419:43–47
Feldmann K (1991) T-DNA insertion mutagenesis in Arabidopsis: mutational spectrum. Plant J 1:71–82
FengYQ SJ, Alami R, Eisen A, Westerman KA, Leboulch P, Fiering S, Bouhassira EE (1999) Site-specific chromosomal integration in mammalian cells: highly efficient CRE recombinase-mediated cassette exchange. J Mol Biol 292:779–785
Fladung M (1999) Gene stability in transgenic aspen-Populus. I. Flanking DNA sequences and T-DNA structure. Mol Gen Genet 260:574–581
Fladung M, Ahuja MR (1995) ‘Sandwich’ method for non-radioactive hybridizations. Biotechniques 18:3–5
Fladung M, Kumar S, Ahuja R (1997) Genetic transformation of Populus genotypes with different chimaeric gene constructs: transformation efficiency and molecular analysis. Transgen Res 6:111–121
Fukushige S, Sauer B (1992) Genomic targeting with a positive-selection lox integration vector allows highly reproducible gene expression in mammalian cells. Proc Natl Acad Sci USA 89:7905–7909
Giridhar P, Parimalan R (2004) Genetically engineered crops: myths and canards about their biosafety and benefits—a review. Adv Plant Sci 17:353–360
Golic KG, Lindquist S (1989) The FLP recombinase of yeast catalyzes site-specific recombination in the Drosophila genome. Cell 59:499–509
Hönicka H, Fladung M (2006) Biosafety in Populus spp. and other forest trees: from non-native species to taxa derived from traditional breeding and genetic engineering. Trees 20:131–144
Gustafsson C, Govindarajan S, Minshull J (2004) Codon bias and heterologous protein expression. Trends Biotechnol 22:346–353
Jansson S, Douglas CJ (2007) Populus: a model system for plant biology. Ann Rev Plant Biol 58:435–458
Jefferson RA, Kavanagh TA, Bevan MW (1987) GUS-fusions: beta-glucuronidase as a sensitive and versatile gene fusion marker in higher plants. EMBO J 6:3901–3907
Koncz C, Martini N, Mayerhofer R, Koncz-Kalman Z, Korber H, Redei GP, Schell J (1989) High-frequency T-DNA-mediated gene tagging in plants. Proc Natl Acad Sci USA 86:8467–8471
Koncz C, Nemeth K, Redei GP, Schell J (1992) T-DNA insertional mutagenesis in Arabidopsis. Plant Mol Biol 20:963–976
Krysan PJ, Young JC, Sussman MR (1999) T-DNA as an insertional mutagen in Arabidopsis. Plant Cell 11:2283–2290
Krysan PJ, Young JC, Jester PJ, Monson S, Copenhaver G, Preuss D, Sussman MR (2002) Characterization of T-DNA insertion sites in Arabidopsis thaliana and the implications for saturation mutagenesis. OMICS 6:163–174
Kumar S, Fladung M (2001) Controlling transgene integration in plants. Trends Plant Sci 6:155–159
Kumar S, Fladung M (2002a) Gene targeting in plants. In: Jain SM, Brar DS, Ahloowalia BS (eds) Molecular techniques in crop improvement. Kluwer Academic Press, Dordrecht, pp 481–500
Kumar S, Fladung M (2002b) Transgene integration in aspen: structures of integration sites and mechanism of T-DNA integration. Plant J 31:543–551
Lakso M, Sauer B, Mosinger B Jr, Lee EJ, Manning RW, Yu SH, Mulder KL, Westphal H (1992) Targeted oncogene activation by site-specific recombination in transgenic mice. Proc Natl Acad Sci USA 89:6232–6236
Lee KY, Lund P, Lowe K, Dunsmuir P (1990) Homologous recombination in plant cells after Agrobacterium-mediated transformation. Plant Cell 2:415–425
Lee WK, Kim SH, Park YD (2007) Marker-free transgenic plants produced by autoexcised Cre-lox system. Horticult Environ Biotechnol 48:265–269
Li Y, Rosso MG, Ulker B, Weisshaar B (2006) Analysis of T-DNA insertion site distribution patterns in Arabidopsis thaliana reveals special features of genes without insertions. Genomics 87:645–652
Logie C, Stewart AF (1995) Ligand-regulated site-specific recombination. Proc Natl Acad Sci USA 92:5940–5944
Louwerse JD, van Lier MC, van der Steen DM, de Vlaam CM, Hooykaas PJ, Vergunst AC (2007) Stable recombinase-mediated cassette exchange in Arabidopsis using Agrobacterium tumefaciens. Plant Physiol 145:1282–93
Liu YG, Mitsukawa N, Oosumi T, Whittier RF (1995) Efficient isolation and mapping of Arabidopsis thaliana T-DNA insert junctions by thermal asymmetric interlaced PCR. Plant J 8:457–463
Luo K, Duan H, Zhao D, Zheng X, Deng W et al (2007) ‘GM-gene-deletor’: fused loxP-FRT recognition sequences dramatically improve the efficiency of FLP or CRE recombinase on transgene excision from pollen and seed of tobacco plants. Plant Biotechnol J 5:263–74
Luo KM, Sun M, Deng W, Xu S (2008) Excision of selectable marker gene from transgenic tobacco using the GM-gene-deletor system regulated by a heat-inducible promoter. Biotechnol Lett 30:1295–1302
Lutz KA, Svab Z, Maliga P (2006) Construction of marker-free transplastomic tobacco using the Cre-loxP site-specific recombination system. Nat Protocols 1:900–910
Lyznik LA, Ryan RD, Ritchie SW, Hodges TK (1989) Stable co-transformation of maize protoplasts with gusA and neo genes. Plant Mol Biol 13:151–161
Lyznik LA, Gordon-Kamm WJ, Tao Y (2003) Site-specific recombination for genetic engineering in plants. Plant Cell Rep 21:925–932
Marillonnet S, Giritch A, Gils M, Kandzia R, Klimyuk V, Gleba Y (2004) In planta engineering of viral RNA replicons: efficient assembly by recombination of DNA modules delivered by Agrobacterium. Proc Natl Acad Sci USA 101:6852–6857
Matzke AJM, Matzke MA (1998) Position effects and epigenetic silencing of plant transgenes. Curr Opin Plant Biol 1:142–148
Mullins ED, Kang S (2001) Transformation: a tool for studying fungal pathogens of plants. Cell Mol Life Sci 58:2043–52
Miki B, McHugh S (2004) Selectable marker genes in transgenic plants: applications, alternatives and biosafety. J Biotechnol 107:193–232
Nanto K, Ebinuma H (2008) Marker-free site-specific integration plants. Transgen Res 17:337–344
Nanto K, Yamada-Watanabe K, Ebinuma H (2005) Agrobacterium-mediated RMCE approach for gene replacement. Plant Biotechnol J 3:203–214
Offringa R (1992) Gene targeting in plants using Agrobacterium vector system. Ph.D. thesis, Leiden University, Netherlands
Offringa R, de Groot MJ, Haagsman HJ, Does MP, van den Elzen PJ, Hooykaas PJ (1990) Extrachromosomal homologous recombination and gene targeting in plant cells after Agrobacterium mediated transformation. EMBO J 9:3077–3084
Ow DW (2002) Recombinase-directed plant transformation for the post-genomic era. Plant Mol Biol 48:183–200
Pakull B, Groppe K, Meyer M, Markussen T, Fladung M (2009) Genetic linkage mapping in aspen (Populus tremula L. and P. tremuloides Michx.). Tree Gen Genom 5:505–515
Puchta H (2000) Removing selectable marker genes: taking the shortcut. Trend Plant Sci 5:273–274
Puchta H (2003) Marker-free transgenic plants. Plant Cell Tiss Org Cult 74:123–134
Robischon M (2006) Field trials with transgenic trees—state of the art and developments. In: Fladung M, Ewald D (eds) Tree transgenesis: recent developments. Springer, Berlin, pp 3–23
Rosso MG, Li Y, Strizhov N, Reiss B, Dekker K, Weisshaar B (2003) An Arabidopsis thaliana T-DNA mutagenized population (GABI-Kat) for flanking sequence tag-based reverse genetics. Plant Mol Biol 53:247–259
Schaefer DG, Zryd JP (1997) Efficient gene targeting in the moss Physcomitrella patens. Plant J 11:1195–206
Sallaud C, Gay C, Larmande P, Bès M, Piffanelli P, Piégu B, Droc G, Regad F, Bourgeois E, Meynard D, Périn C, Sabau X, Ghesquière A, Glaszmann JC, Delseny M, Guiderdoni E (2004) High throughput T-DNA insertion mutagenesis in rice: a first step towards in silico reverse genetics. Plant J 39:450–464
Schneeberger RG, Zhang K, Tatarinova T, Troukhan M, Kwok SF, Drais J, Klinger K, Orejudos F, Macy K, Bhakta A, Burns J, Subramanian G, Donson J, Flavell R, Feldmann KA (2005) Agrobacterium T-DNA integration in Arabidopsis is correlated with DNA sequence compositions that occur frequently in gene promoter regions. Funct Integr Genomics 5:240–253
Shaw CH, Leemans J, Shaw CH, van Montagu M, Schell J (1983) A general method for the transfer of cloned genes to plant cells. Gene 23:315–330
Song H, Niederweis M (2007) Functional expression of the Flp recombinase in Mycobacterium bovis BCG. Gene 399:112–119
Soukharev S, Miller JL, Sauer B (1999) Segmental genomic replacement in embryonic stem cells by double lox targeting. Nucleic Acids Res 27:e21
Srivastava V, Ow DW (2004) Marker-free site-specific gene integration in plants. Trends Biotechnol 22:627–629
Szabados L, Kovács I, Oberschall A, Abrahám E, Kerekes I, Zsigmond L, Nagy R, Alvarado M, Krasovskaja I, Gál M, Berente A, Rédei GP, Haim AB, Koncz C (2002) Distribution of 1000 sequenced T-DNA tags in the Arabidopsis genome. Plant J 32:233–242
Tzfira T, White C (2005) Towards targeted mutagenesis and gene replacement in plants. Trends Biotechnol 23:567–569
Tzfira T, Frankman LR, Vaidya M, Citovsky V (2003) Site-specific integration of Agrobacterium tumefaciens T-DNA via double-stranded intermediates. Plant Physiol 133:1011–1023
Thomsen JG, Yau YY, Blanvillain R, Chiniquy D, Thilmony R, Ow DW (2008) ParA resolvase catalyzes site-specific excision of DNA from the Arabidopsis genome. Transgen Res. doi:10.1007/s11248-008-9213-4
Tuskan GA, DiFazio S, Jansson S, Bohlmann J et al (2006) The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science 313:1596–1604
Vergunst AC, Hooykaas PJ (1998) Cre/lox-mediated site-specific integration of Agrobacterium T-DNA in Arabidopsis thaliana by transient expression of cre. Plant Mol Biol 38:393–406
Vergunst AC, Hooykaas PJ (1999) Recombination in the plant genome and its application in biotechnology. Crit Rev Plant Sci 18:1–31
Vergunst AC, Jansen LE, Hooykaas PJ (1998) Site-specific integration of Agrobacterium T-DNA in Arabidopsis thaliana mediated by Cre recombinase. Nucl Acids Res 26:2729–2734
de Vetten N, Wolters AM, Raemakers K, van der Meer I, ter Stege R, Heeres E, Heeres P, Visser R (2003) A transformation method for obtaining marker-free plants of a cross-pollinating and vegetatively propagated crop. Nat Biotechnol 21:439–442
Wang J, Lewis ME, Whallon JH, Sink KC (1995) Chromosomal mapping of T-DNA inserts in transgenic Petunia by in situ hybridization. Transgen Res 4:241–246
Wohlleben W, Arnold W, Broer I, Hillemann D, Strauch E, Puhler A (1988) Nucleotide sequence of the phosphinothricin N-acetyltransferase gene from Streptomyces viridochromogenes Tu494 and its expression in Nicotiana tabacum. Gene 70:25–37
Yoder JI, Goldsbrough AP (1994) Transformation systems for generating marker-free transgenic plants. Bio-Technol 12:263–267
Zelasco S, Ressegotti V, Confalonieri M, Carbonera D, Calligari P, Bonadei M, Bisoffi S, Yamada K, Balestrazzi A (2007) Evaluation of MAT-vector system in white poplar (Populus alba L.) and production of ipt marker-free transgenic plants by ‘single-step transformation’. Plant Cell Tiss Org Cult 91:61–72
Zubko E, Scutt C, Meyer P (2000) Intrachromosomal recombination between attP regions as a tool to remove selectable marker genes from tobacco transgenes. Nat Biotechnol 18:442–445
Acknowledgment
This research was funded by the Bundesministerium für Bildung und Forschung, Germany (Projektnummer PTJ-Bio/0313264T).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Communicated by R. Sederoff
Matthias Fladung and Tobias M. H. Schenk contributed equally to this work.
Rights and permissions
About this article
Cite this article
Fladung, M., Schenk, T.M.H., Polak, O. et al. Elimination of marker genes and targeted integration via FLP/FRT recombination system from yeast in hybrid aspen (Populus tremula L. × P. tremuloides Michx.). Tree Genetics & Genomes 6, 205–217 (2010). https://doi.org/10.1007/s11295-009-0241-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11295-009-0241-x