
Abstract
Removal of a selectable marker gene from genetically modified (GM) crops alleviates the risk of its release into the environment and hastens the public acceptance of GM crops. Here we report the production of marker-free transgenic rice by using a chemically regulated, Cre/loxP-mediated site-specific DNA recombination in a single transformation. Among 86 independent transgenic lines, ten were found to be marker-free in the T0 generation and an additional 17 lines segregated marker-free transgenic plants in the T1 generation. Molecular and genetic analyses indicated that the DNA recombination and excision in transgenic rice were precise and the marker-free recombinant T-DNA was stable and heritable.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Selectable marker genes are required to ensure the selection of transgenic plants during plant transformation. However, once transformation is accomplished, the presence of a marker gene in transgenic plants becomes undesirable, particularly as most of the selectable marker genes currently used confer resistance to antibiotics or herbicides. This raises several concerns regarding the field release of genetically modified (GM) crops. The presence of selectable antibiotic-resistant genes in the GM crops may lead to the horizontal transfer of these genes into pathogenic soil and gastrointestinal bacteria. Secondly, the genes conferring herbicide resistance can be transferred to closely related weed species through cross-pollination. In addition, new selectable markers are needed each time to pyramid the same crop variety with different desirable traits. This is, however, a limiting factor due to the scarce number of selectable marker genes that are suitable for each crop species. Therefore, the removal of selectable marker genes will not only lead to the elimination of potential environmental and health-related risks as well as technical barriers, but also increase the consumer acceptance of GM crops and their products.
Several strategies have been employed to remove selectable markers from transgenic plants (Ebinuma et al. 2001). These include the use of transposon-mediated repositioning of the gene of interest or the selectable marker gene (Goldsbrough et al. 1993; Cotsaftis et al. 2002), co-transformation and segregation of marker genes (McKnight et al. 1987; Komari et al. 1996; Daley et al. 1998), intrachromosomal homologous recombination between two homologous sequences (Zubko et al. 2000), and several site-specific DNA excision systems, including Cre/loxP from bacteriophage P1 (Dale and Ow 1991; Gleave et al. 1999; Hoa et al. 2002), Flp/frt from Saccharomyces cerevisiae (Lyznik et al. 1993; Lloyd and Davis 1994), R/RS from Zygosaccharomyces rouxii (Sugita et al. 2000; Endo et al. 2002) and Gin/gix from bacteriophage Mu (Maeser and Kahmann 1991).
In the Cre/loxP-mediated site-specific DNA excision system, Cre recombinase specifically recognizes and performs precise recombination and DNA excision between two directly repeated asymmetric 34-bp loxP recognition sites flanking the selectable marker gene (Dale and Ow 1991). Two main approaches have been used for introducing the Cre gene into transgenic plants. In one approach, Cre recombinase can be constitutively expressed from a transgenic locus that is introduced into the transgenic plants carrying the selectable marker gene by a further round of transformation or by out-crossing that enables the loss of the Cre gene in subsequent generations (Dale and Ow 1991; Gleave et al. 1999; Hoa et al. 2002). This approach, however, is laborious, expensive and time consuming. To overcome these shortcomings, an alternative approach was developed recently by including the Cre gene on the same DNA segment as the selectable marker gene flanked by two loxP sites (Zuo et al. 2001; Hoff et al. 2001). The expression of the Cre gene is activated or induced by a chemical inducer (Zuo et al. 2001) or heat shock (Hoff et al. 2001) only after the selectable marker gene is no longer required. The induced Cre recombinase “auto-excises” the DNA segment between the two loxP sites comprising the Cre gene itself and the selectable marker gene.
Rice (Oryza sativa) is one of the most important food crops in the world. It is therefore highly desirable to generate marker-free transgenic plants to hasten the public acceptance of GM rice as well as to protect the environment. Here, we report the successful excision of a selectable marker gene in transgenic rice plants using a chemically regulated, Cre/loxP-mediated DNA excision system (Zuo et al. 2001) and the conventional Agrobacterium-mediated transformation.
Materials and methods
Plasmid construction
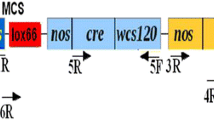
To construct a plasmid with a chemically regulated Cre/loxP system suitable for rice transformation, the G10-90 promoter in pX7-GFP (Guo et al. 2003) was replaced with a maize ubiquitin promoter from pSSZ32 (Kolesnik et al. 2004). To achieve this, pX7-GFP was digested with PmeI and MluI, which delete a 371-bp small PmeI-MluI fragment including 236 bp of the G10-90 promoter, the first loxP element and 79 bp of the XVE gene. The loxP element and the 79 bp of the XVE gene were then recovered in the intermediate construct pH-GFP by ligating the large PmeI-MluI fragment of pX7-GFP with a PmeI and MluI digested PCR fragment amplified from pX7-GFP with primers PmeI-F and MluI-R (Table 1). The PstI fragment (1992 bp) of the maize ubiquitin promoter from pSSZ32 (Kolesnik et al. 2004) was cloned into a pBluescript II KS vector [Stratagene (http://www.stratagene.com/homepage/)], released with EcoRV and SmaI digestion, and then inserted into pH-GFP at the PmeI site. The right orientation of the insertion of the maize ubiquitin promoter in pH-GFP was confirmed by DNA sequencing and the final construct was designated as pUH-GFP2 (Fig. 1). pUH-GFP2 was introduced into Agrobacterium tumefaciens AGL1 for rice transformation (Sambrook et al. 1989).

A schematic diagram of the T-DNA region of the vector pUH-GFP2 and Cre/loxP-mediated DNA recombination event (see Zuo et al. 2001 for details). Arrows inside squares indicate the direction of transcription. The cleavage sites for restriction enzymes EcoRV and XhoI are indicated. Region flanked by the two loxP sites in the upper diagram is an loxP fragment. pUbi Maize ubiquitin promoter, XVE chimeric transactivator containing the regulator domain of an estrogen receptor (Zuo et al 2000), hpt hygromycin phosphotransferase gene, cre-int bacteriophage P1 Cre recombinase gene with an intron, gfp green fluorescent protein gene, loxP specific recognition sites of Cre, OLexA-46 eight copies of LexA DNA binding site fused to the −46 CaMV 35 S minipromoter, P1–P4 primers used for PCR analysis
Rice transformation and induction by β-estradiol
Transformation of the Japonica rice cultivar Taipei 309 with A. tumefaciens AGL1(pUH-GFP2) was carried out following the procedures as described in the previous study (Yin and Wang 2000). Briefly, the co-cultivated rice calli were cultured on a selective medium (NB0 medium+250 mg/l cefotaxime+50 mg/l hygromycin+2 mg/l 2,4-D) at 26°C in the dark for 5–6 weeks. Hygromycin-resistant calli were sub-cultured on an inductive medium (NB0 medium+2 mg/l 2,4-D) containing 10 μM of β-estradiol for 2 weeks. The induced calli were then transferred to pre-regeneration medium (NB0 medium+1 mg/l 6-BA+2 mg/l NAA+5 mg/l ABA) and were allowed to grow for 3 more weeks. Later, white compact embryogenic calli were transferred to regeneration medium (NB0 medium+2 mg/l 6-BA+1 mg/l NAA+1 mg/l KT+1 mg/l IAA) and regenerated at 26°C with a 14-h light (2,000 lx) and a 10-h dark period. Regenerated plantlets were subsequently transplanted to the soil in pots and grown in a greenhouse.
Detection of GFP fluorescence
A fluorescent microscope (Leica MZ12 stereomicroscope with fluorescent module for GFP-plus using blue filters) was used to detect GFP fluorescence in induced calli and regenerated shoots. The selfed seeds (T1) were surface-sterilized, germinated on half-strength MS medium (Murashige and Skoog 1962) for 3 days in darkness and 2 days under light, and scored for GFP fluorescence using the fluorescent microscope.
Molecular techniques
The PCR primers used for screening DNA excision in transgenic plants are listed in Table 1 and their respective positions are shown in Fig. 1. The reaction mixture (20 μl) for PCR consisted of 10 ng of rice genomic DNA, 0.2 mM each of dNTPs, 0.2 μM of each primer, 2 μl of 10x PCR buffer and 0.5 unit of Taq polymerase from QIAGEN Taq PCR Core kit (QIAGEN, Germany). Thermal cycling was done at 94°C for 4 min followed by 35 cycles at 94°C for 45 s, 55°C for 45 s, and 72°C for 1 min. After the final cycle, the reaction mixture was maintained at 72°C for 5 min before completion. Reactions were performed using a PTC-100 programmable thermal controller (MJ Research., Mass.).
Southern analysis was carried out according to standard procedures (Sambrook et al. 1989). Approximately 2–5 μg of rice genomic DNA was digested using restriction enzymes EcoRV or XhoI, which have only one or two digestion sites within the T-DNA region of pUH-GFP2, respectively (Fig. 1). The digested DNA fragments were separated in a 0.8% agarose gel by electrophoresis and transferred onto a Hybond-N+ nylon membrane (Amersham Pharmacia, N.J.) by alkaline transfer. Hybridization was then performed sequentially with the same blot using probes to detect the hpt or gfp genes, or the maize ubiquitin promoter. The probes were amplified from pUH-GFP2 using primers as shown in Table 1. Labelling of the probes was done using Rediprime labelling kit from Amersham Pharmacia.
DNA fragments comprising the recombined loxP sites in marker-free transgenic plants were amplified using primers P1 and P4. The PCR products were sequenced using an ABI PRISM 377 DNA sequencer (Perkin-Elmer, Mass.). The DNA sequence was analysed using Sequencher 4.1 software (Gene Codes, Mich.).
Results
Production of transgenic rice and induction of DNA recombination
In the chemical-inducible Cre/loxP DNA recombination system, removal of the hygromycin-selectable marker gene, XVE as well as the cre-int cassettes (loxP fragment) flanked by two loxP sites was monitored by detecting GFP fluorescence in the transgenic calli or plants. Hygromycin-resistant transgenic calli of Taipei 309 that had been transformed with pUH-GFP2, were used for induction of loxP-fragment excision and GFP fluorescence. Initially the GFP fluorescence was very weak and appeared only as small spots in some portions of the calli. As time progressed, some of the induced calli appeared to have strong GFP fluorescence (Fig. 2b) as compared to no fluorescence observed from the untreated control (Fig. 2a). An initial induction test showed that the induction of GFP fluorescence by β-estradiol increased with greater concentrations of β-estradiol from 1 to 10 μM, and was saturated at 10 μM after 2 weeks of induction. We also found that there was no increase in GFP fluorescence even after a prolonged induction period by adding β-estradiol into the pre-regeneration and regeneration media.

β-estradiol-induced GFP fluorescence in pUH-GFP2-transformed calli, shoot and T1 plants of Taipei 309. a pUH-GFP2 transgenic calli subcultured on NB medium without β-estradiol. b pUH-GFP2-transformed calli subcultured on inductive medium with 10 μM β-estradiol for 14 days. c GFP fluorescent shoot regenerated from β-estradiol-induced transgenic calli. d Germinating seed of wild-type Taipei 309. e T1 individual with GFP fluorescence segregated from transgenic line 40
A total of 86 transgenic T0 plants were generated from the induction experiments. Visible GFP fluorescence was observed in 46 of the 86 transgenic T0 plants at the shoot stage during regeneration (Fig. 2c). However, visible GFP fluorescence was not detected in the remaining 40 T0 plants even though they were regenerated from the β-estradiol-induced calli that showed GFP fluorescence. Most of the T0 plants showed a normal morphological phenotype as compared to wild type Taipei 309.
PCR screening for loxP fragment excision in T0 plants
As the excision of the loxP fragment helps in juxtaposing the maize ubiquitin promoter and the downstream gfp gene, we carried out PCR analysis with primer pairs P1/P2, P3/P4 and P1/P4 to detect the recombination events in the 86 T0 plants. Partial PCR results in Fig. 3 showed that successful recombination between the two loxP sites yielded amplification of 468-bp P1–P4 fragments (Fig. 3; lanes 10 and 13), whereas presence of the loxP fragments in non-recombinant T-DNA led to amplification of 968-bp P1–P2 fragments (Fig. 3; lanes 2 and 5) and 689-bp P3–P4 fragments (Fig. 3; lanes 3 and 6). Based on the results of the PCR analysis, the 86 T0 plants could be assigned to one of the seven distinct categories (Table 2). Category A has ten T0 plants that showed amplification of only P1–P4 fragments. All these ten T0 plants also showed GFP fluorescence at the shoot stage and therefore they were putatively marker-free. Category B has 35 T0 plants that showed amplification of P1–P2, P3–P4 and P1–P4 fragments and 22 of the 35 T0 plants were observed to have GFP fluorescence at the shoot stage. These plants might contain more than one transgenic locus and at least one locus would have undergone successful excision of the loxP fragment. Categories C and D include three and 13 T0 plants, respectively, that showed amplification of P1–P4, and either P1–P2 or P3–P4 fragments. Moreover, two of the three T0 plants in category C and 12 of the 13 T0 plants in category D showed GFP fluorescence at the shoot stage. These GFP fluorescent T0 plants in categories C and D might have more than one transgenic locus and successful recombination could have occurred in at least one of the transgenic loci, while T-DNAs in additional loci may be truncated. Hence, it can be said that Cre/loxP-mediated DNA excision had occurred in 61 (or 69.3%; categories A, B, C and D) of the 86 transgenic lines, even though incomplete excision of the loxP fragments might occur in 51 lines (categories B, C and D). In addition, there were 25 T0 plants that showed amplification of either P1–P2 or P3–P4, or both fragments (categories E, F and G in Table 2). These plants might contain either intact or truncated T-DNA that has not undergone DNA recombination.

PCR analysis of genomic DNA prepared from non-recombinant T0 plant 27 (lanes 5–7), recombinant T0 plants 2 (lanes 8–10) and 3 (lanes 11–13). Control experiments using pUH-GFP2 as a template are shown as lanes 1–3. Primer pairs used for each PCR reaction are indicated on the top of each lane. The expected PCR products from different combinations of primer pairs are indicated on the right. No amplification was observed with the genomic DNA of wild-type Taipei 309 using any of the three primer pairs (P1/P2, P3/P4 or P1/P4) (result not shown). M DNA molecular marker of 1- kb ladders from Stratagene (USA)
Identification of marker-free individuals in the T0 generation
The ten putative marker-free T0 plants in category A were then subjected to Southern analysis for the excision of the loxP fragment. As expected, no hybridization band for the hpt gene was detected in these plants, indicating that the hpt genes had been removed together with the excised loxP fragments (Fig. 4a). Since there is only one EcoRV site in the loxP fragment as well as in the T-DNA region of pUH-GFP2, complete excision of the loxP fragment will remove the EcoRV sites from the T-DNA (Fig. 1). Therefore, probes for the gfp gene or the maize ubiquitin promoter will hybridize to the same fragment in a Southern blot filter with EcoRV digestion. Indeed, T0 plants 1, 2, 14, 31, 44, 50, 83 and 85 showed similar hybridization patterns with the probes for the gfp gene and the maize ubiquitin promoter (Fig. 4b, c). T0 plant 3 had two bands for the gfp gene but only one band for the maize ubiquitin promoter (Fig. 4b, c; lane 6). This extra gfp band might result from a truncated T-DNA that contained the gfp gene only. T0 plant 4 had one gfp band but showed no signal for the maize ubiquitin promoter (Fig. 4b, c; lane 7). This plant, however, should have a minimal length of the maize ubiquitin promoter upstream of the gfp gene as we could obtain P1–P4 fragment in a PCR analysis (data not shown). To further confirm the recombination events, we also detected the presence of a 1.4-kb recombined XhoI fragment (Fig. 1) in those putative marker-free plants. Southern blot analysis with the maize ubiquitin promoter probe showed that the 1.4-kb fragment was present in T0 plants 1, 2, 14, 31, 44, 50, 83, 85 but not in T0 plant 27 that contained two copies of non-recombinant T-DNA (Fig. 4d; T0 plants 3 and 4 were not shown due to insufficient DNA). Since there is a XhoI site in the middle of the pUbi probe (Fig. 1), two hybridized bands with at least one of 1.4 kb were detected for every copy of the maize ubiquitin promoter in the recombinant T-DNA when digested with XhoI. Southern analysis also showed that T0 plants 1, 83 and 85 contained an additional one to two copies of truncated T-DNA comprising the maize ubiquitin promoter only (Fig. 4d; lanes 4, 10 and 11)

Southern blot analysis of the putative marker-free transgenic T0 plants. Genomic DNAs from the ten putative marker-free transgenic T0 plants (1, 2, 3, 4, 14, 31, 44, 50, 83, 85) were digested with EcoRV, subjected to Southern blotting and probed sequentially with hpt (a), gfp (b) and maize ubiquitin promoter (c). Genomic DNA from the eight putative marker-free transgenic T0 plants (1, 2, 14, 31, 44, 50, 83, 85) was also digested with XhoI and subjected to Southern blot analysis with a probe for the maize ubiquitin promoter (d). Wild-type Taipei 309 (TP309) and non-recombinant T0 plant 27 were used as controls. Results of the PCR analysis with genomic DNA of each plant using primer pairs P1/P2, P3/P4 or P1/P4, and the GFP fluorescence of each plant are listed under c. M DNA molecular marker of λ DNA digested by HindIII, hpt hygromycin phosphotransferase gene, gfp green fluorescent protein gene, − negative PCR amplification, + positive PCR amplification, N negative GFP fluorescence, P positive GFP fluorescence
Genetic and molecular analyses with T1 progeny from the eight marker-free transgenic lines, which were marker-free in the T0 generation (T0 plants 2, 3, 14, 31, 44, 50, 83, 85), show that the marker-free recombinant T-DNA in these lines was heritable (Table 3). T0 plants 2, 31, 44 and 50 contained only one marker-free transgenic locus and the GFP fluorescence in the T1 progeny of the four lines was always co-segregated with the marker-free recombinant T-DNA. Figure 5 showed the co-segregation of GFP fluorescence, marker-free recombinant T-DNA and the amplification of PCR product with P1/P4 primer pair in the T1 progeny of transgenic line 44. T0 plant 14 had two copies of the marker-free recombinant T-DNA and Southern analysis showed that the GFP fluorescent T1 plants from this line inherited either one or two copies of the marker-free recombinant T-DNA (data not shown). As mentioned above, T0 plants 3, 83 and 85 contained both marker-free recombinant T-DNA and truncated T-DNA comprising the gfp gene or the maize ubiquitin promoter only. However, in the T1 generation, the GFP fluorescence was only co-segregated with the marker-free recombinant T-DNA (data not shown).

Co-segregation of marker-free recombinant T-DNA in transgenic T0 plant 44 [44(T 0 )] with GFP fluorescence and the amplification of PCR product with P1/P4 primer pair in the T1 generation (only the results of 28 T1 individuals from a single Southern blot filter are shown). Genomic DNAs from Taipei 309, 44(T 0 ) and its 28 T1 progeny were digested with EcoRV, subjected to Southern blotting and probed sequentially with hpt (a) and gfp (b) genes. Results of the PCR analysis with genomic DNA of each plant using primer pairs P1/P2, P3/P4 or P1/P4, and the GFP fluorescence of each plant are listed under b. For abbreviations, see Fig. 4
Segregation of marker-free progeny in the T1 generation
The 51 T0 plants in categories B, C and D in Table 2 were also subjected to Southern analysis. The transgenic lines from these 51 T0 plants showed the amplification of P1–P4 fragments in addition to P1–P2 and/or P3–P4 fragments. Some of the 51 lines had the potential to segregate marker-free transgenic plants with functional recombinant T-DNA in the subsequent generations, as at least 36 lines showed visible GFP fluorescence at the shoot stage during regeneration in the T0 generation. To confirm this assumption, we screened T1 plants of the 51 lines for GFP fluorescence at the seed germination stage. The 36 lines that showed visible GFP fluorescence in the T0 generation segregated GFP fluorescent T1 progeny. The remaining 15 lines did not show visible GFP fluorescence in either the T0 or T1 generation for unknown reasons. The T1 GFP fluorescent plants were further subjected to Southern analysis with probes for the gfp or hpt genes, or the maize ubiquitin promoter to screen for any marker-free transgenic plants. Marker-free transgenic plants were identified from the T1 progeny of 17 lines (Table 3). Figure 6 shows two examples of Southern blot hybridization with marker-free transgenic T1 plants segregated from those lines. In one example, T0 plant 40 had two copies of the hpt gene (Fig. 6a; lane 4), three copies of the gfp gene (Fig. 6b; lane 4) and one copy of the maize ubiquitin promoter (Fig. 6c; lane 4). In the T1 generation, six plants from line 40 showed visible GFP fluorescence (Table 3) and one of them, T1 plant 40-4, was confirmed to be marker-free in Southern analysis (Fig. 6a–d; lane 5). In another example, T0 plant 53 had one copy of the hpt gene (Fig. 6a; lane 6), four copies of the gfp gene (Fig. 6b; lane 6) and five copies of the maize ubiquitin promoter (Fig. 6c; lane 6). In the T1 generation, 26 of the 33 T1 plants from line 53 showed GFP fluorescence (Table 3) and two of them were marker-free. T1 plant 53-11 contained one copy of marker-free recombinant T-DNA while T1 plant 53-13 had two copies of marker-free recombinant T-DNA and one copy of truncated T-DNA that hybridized to the maize ubiquitin promoter probe only (Fig. 6; lanes 7, 8). In addition, the 1.4-kb recombined XhoI fragment was also detected in these marker-free T1 plants as well as in their corresponding T0 parents (Fig. 6d).

Southern blot analysis of the transgenic lines segregating marker-free transgenic plants in the T1 generation. Genomic DNAs from two transgenic T0 plants (40 and 53) and their T1 progeny (40-4, 53-11 and 53-13) were digested with EcoRV, subjected to Southern blotting and probed sequentially with the same blot using hpt (a), gfp (b) and maize ubiquitin promoter (c). The same set of DNA samples was also digested with XhoI and subjected to Southern blot analysis with a probe for the maize ubiquitin promoter (d). Genomic DNA from wild-type Taipei 309 and non-recombinant T0 plant 27 was used as a control. Results of the PCR analysis with genomic DNA of each plant using primer pairs P1/P2, P3/P4 or P1/P4, and the GFP fluorescence of each plant are listed under d. For abbreviations, see Figs. 1 and 4
DNA sequencing of the junction region at the recombined loxP site
The PCR products of P1/P4 fragments amplified from marker-free transgenic plants (T0 or T1) were sequenced and the partial sequence data is presented in Fig. 7. DNA sequence analysis clearly showed that the maize ubiquitin promoters were intact at the recombined regions and were contiguous with the loxP sites, followed by the gfp coding sequence. This was strictly in accordance with the theoretical recombination between the two loxP sites, leading to the removal of XVE, hpt and the cre-int cassette from T-DNA upon activation of XVE by β-estradiol.

Nucleotide sequence analysis of the junction regions at the recombined loxP sites in the marker-free transgenic plants. Sequences of the junction regions at the recombined loxP sites are shown below the two loxP sites of pUH-GFP2. Nucleotide sequences highlighted in bold small letters in pUH-GFP2 indicate the sequences that are retained after recombination. The loxP elements are indicated in bold capital letters. The starting codon of the gfp gene is presented in italicized capital letters
Discussion
Using β-estradiol-regulated, Cre/loxP-mediated site-specific DNA recombination, we have successfully removed the loxP fragments from the T-DNA in the transgenic cells and obtained marker-free transgenic plants in a single-step transformation. Among the 86 transgenic T0 plants, at least one copy of functional marker-free recombinant T-DNA should be present in the 46 plants as the visible GFP fluorescence was detected in the T0 and/or T1 generation. Furthermore, eight fertile T0 plants were found to be marker-free and an additional 17 plants segregated marker-free progeny in the T1 generation. In total, 29.1% of the transgenic T0 plants were marker-free or could segregate marker-free transgenic progeny. Our frequency of obtaining marker-free transgenic plants is comparable to that of a previous study (Hoa et al. 2002), in which the recombination frequency for selectable marker excision ranged from 26.0% (T0×T0) to 58.3% (T2×T2). In contrast to their study, the method used in our laboratory saves time and labour because it does not require two separate transformations followed by hybridization of the two categories of transgenic lines. Another highlight of this approach is that the expression of Cre recombinase in plant cells is tightly regulated by the XVE system and β-estradiol (Zuo et al. 2001). This reduces the risk of genome rearrangements at cryptic-target sites in either nuclear or organelle genomes due to the constitutive expression of Cre recombinase, which has been correlated with phenotypic aberrations in plants (Hajdukiewicz 2001; Coppoolse et al. 2003).
In a previous report (Zuo et al. 2001), DNA recombination in Arabidopsis was induced in all the transgenic plants with high efficiency in the germline cells (29–66%) of the transformed shoots (T0) or T1 seeds. In rice, we found that DNA recombination was more efficiently induced in the transformed cells (calli) rather than the germline cells of the transformed shoots (T0) or T1 seeds. We failed to identify marker-free plants in the T2 progeny by germinating transgenic T1 seeds that contained non-recombinant T-DNA on an inductive medium. One of the reasons could be that the inducer might not have accessed the germline cells of the T1 plants efficiently. In addition, in our induction experiments we observed that some induced calli were chimeric for GFP fluorescence after 2 weeks or longer induction, which indicated that there was a considerable number of non-recombinant cells cohered to the recombinant calli with GFP fluorescence probably due to the inducer inaccessibility or instability. This might lead to the regeneration of non-recombinant transgenic plants as there was no selection pressure in pre-regeneration or regeneration medium. In the present study there were at least 13 non-recombinant transgenic lines (category E in Table 2). Further more, due to the absence of selection pressure during pre-regeneration and regeneration of the induced calli, non-transgenic cells originally attached to the transgenic cells might also have proliferated, resulting in the regeneration of non-transgenic escapees. Indeed, in this study we found seven non-transgenic escapees which had no T-DNA insertion (data not shown). Nevertheless, the non-recombinant transgenic plants and non-transgenic escapees represented only a small portion of the total transgenic plants, which could be further reduced by modifying tissue culture procedures.
There were 19 additional T0 plants from categories B, C or D that segregated T1 progeny with GFP fluorescence. But none of the T1 progeny were marker-free as Southern analysis showed that they had multiple transgenic loci and the recombinant T-DNAs could not be separated from the non-recombinant T-DNAs in the T1 generation. One reason could be the small population size that was analysed in the T1 family due to the shortage or limited availability of T1 seeds. Another plausible reason was that, in some transgenic plants, the recombinant and non-recombinant T-DNAs were genetically linked to each other, which made it impractical to segregate marker-free transgenic plants in the subsequent generations. In both cases, incomplete DNA excision seemed to have occurred in these transgenic T0 plants with multiple transgenic loci. Compared to the high efficiency of DNA excision with multiple transgenic loci in Arabidopsis (Zuo et al. 2001), there was more incomplete DNA excision with multiple transgenic loci in the present study. In addition, a truncated T-DNA integration may also affect the efficiency of DNA recombination. DNA recombination would be impossible if the truncated T-DNA contained only one loxP site. Furthermore, truncated T-DNA with a single loxP site might cause chromosome rearrangement, such as translocation or deletion, upon induction for recombination when they are on different chromosomes in a transgenic plant (Qin et al. 1994). In the present study, in addition to the marker-free recombinant T-DNA, marker-free T0 transgenic plants 1, 3, 83 and 85 also contained one to two copies of truncated T-DNA comprising the gfp gene or the maize ubiquitin promoter only. Multiple transgenic loci and/or truncated T-DNA integration in rice seem to be correlated and unavoidable with the Agrobacterium-mediated transformation system (Yin and Wang 2000). Hence, we suggest generating and selecting marker-free transgenic plants with a single transgenic locus in the T0 generation. For instance, in the present study, T0 plants 2, 31, 44 and 50 were marker-free and contained a single transgenic locus. In addition, transgenic plants with a single transgenic locus are also advantageous in stabilizing transgene expression, since it has been found that multiple transgenic loci are more prone to transgene inactivation than single transgenic locus (Hobbs et al. 1993).
References
Coppoolse ER, Vroomen MJ, Roelofs D, Smit J, van Gennip F, Hersmus BJM, Nijkamp HJJ, van Kaaren MJJ (2003) Cre recombinase expression can result in phenotypic aberrations in plants. Plant Mol Biol 51:263–279
Cotsaftis O, Sallaud C, Breitler JC, Meynard D, Greco R, Pereira A, Guiderdoni E (2002) Transposon-mediated generation of T-DNA- and marker-free rice plants expressing a Bt endotoxin gene. Mol Breed 10:165–180
Dale EC, Ow DW (1991) Gene transfer with subsequent removal of the selection gene from the host genome. Proc Natl Acad Sci USA 88:10558–10562
Daley M, Knauf V, Summerfelt KR, Turner JC (1998) Co-transformation with one Agrobacterium tumefaciens strain containing two binary plasmids as a method for producing marker-free transgenic plants. Plant Cell Rep 17:489–496
Ebinuma H, Sugita K, Matsunaga E, Endo S, Yamada K, Komamine A (2001) Systems for the removal of a selection marker and their combination with a positive marker. Plant Cell Rep 20:383–392
Endo S, Sujita K, Sakai M, Tanaka H, Ebinuma H (2002) Single step transformation for generating marker-free transgenic rice using the ipt-type MAT vector system. Plant J 30:115–122
Gleave AP, Mitra DS, Mudge SR, Morris BAM (1999) Selectable marker-free transgenic plants without sexual crossing: transient expression of cre recombinase and use of a conditional lethal dominant gene. Plant Mol Biol 40:223–235
Goldsbrough AP, Lastrella CN, Yoder JI (1993) Transposition mediated re-positioning and subsequent elimination of marker genes from transgenic tomato. Biotechnology 11:1286–1292
Guo H-S, Fei J-F, Xie Q, Chua N-H (2003) A chemical-regulated inducible RNAi system in plants. Plant J 34:383–392
Hajdukiewicz PT (2001) Multiple pathways for Cre/lox-mediated recombination in plastids. Plant J 27:161–171
Hoa TTC, Bong BB, Huq E, Hodges TK (2002) Cre/lox site-specific recombination controls the excision of a transgene from the rice genome. Theor Appl Genet 104:518–525
Hobbs SLA, Warkentin TD, Delong CMO (1993) Transgene copy number can be positively or negatively associated with transgene expression. Plant Mol Biol 21:17–26
Hoff T, Schnorr KM, Mundy J (2001) A recombinase-mediated transcriptional induction system in transgenic plants. Plant Mol Biol 45:41–49
Kolesnik T, Szeverenyi I, Bachmann D, Kumar CS, Jiang S, Ramamoorthy R, Cai M, Ma ZG, Sundaresan V, Ramachandran S (2004) Establishing an efficient Ac/Ds tagging system in rice: large-scale analysis of Ds flanking sequences. Plant J 37:301–314
Komari T, Hiei Y, Saito Y, Murai N, Kumashiro T (1996) Vectors carrying two separate T-DNAs for co-transformation of higher plants mediated by Agrobacterium tumefaciens and segregation of transformants free from selection markers. Plant J 10:165–174
Lloyd AM, Davis RW (1994) Functional expression of the yeast FLP/FRT site-specific recombination system in Nicotiana tabacum. Mol Gen Genet 242:653–657
Lyznik LA, Mitchell JC, Hirayama L, Hodges TK (1993) Activity of yeast FLP recombinase in maize and rice protoplasts. Nucleic Acids Res 21:969–975
Maeser S, Kahmann R (1991) The Gin recombinase of phage Mu can catalyse site-specific recombination in plant protoplasts. Mol Gen Genet 230:170–176
McKnight T, Lillis M, Simpson R (1987) Segregation of genes transferred to one plant cell from two separate Agrobacterium strains. Plant Mol Biol 8:439–445
Murashige T, Skoog F (1962) A revised medium for rapid growth and bioassays with tobacco tissue cultures. Physiol Plant 15:473–497
Qin M, Bayley C, Stockton T, Ow DW (1994) Cre recombinase-mediated site-specific recombination between plant chromosomes. Proc Natl Acad Sci USA 91:1706–1710
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: a laboratory manual, 2nd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
Sugita K, Kasahara T, Matsunaga E, Ebinuma H (2000) A transformation vector for the production of marker-free transgenic plants containing a single copy transgene at high frequency. Plant J 22:461–469
Yin Z, Wang GL (2000) Evidence of mutiple complex patterns of T-DNA integration into the rice genome. Theor Appl Genet 100:461–470
Zubko E, Scutt C, Meyer P (2000) Intrachromosomal recombination between attP regions as a tool to remove selectable marker genes from tobacco transgenes. Nat Biotechnol 18:442–445
Zuo J, Niu QW, Chua NH (2000) An estrogen receptor-based transactivator XVE mediates highly inducible gene expression in transgenic plants. Plant J 24:265–273
Zuo J, Niu QW, Moller SG, Chua NH (2001) Chemical-regulated, site-specific DNA excision in transgenic plants. Nat Biotechnol 19:157–161
Acknowledgements
The authors would like to thank N. -H. Chua (Rockefeller University, USA) for providing the pX7-GFP vector, S. Ramachandran for providing the pSSZ32 vector, D. Eriksson, and J.-H. Lee for technical assistance, N. Naqvi, Y. Hong and M. Griffith for critically reading the manuscript. Our research was supported by the Temasek Life Sciences Laboratory, Singapore, and the Agri-Food and Veterinary Authority of Singapore.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by I.S. Chung
The first two authors contributed equally to the work
Rights and permissions
About this article
Cite this article
Sreekala, C., Wu, L., Gu, K. et al. Excision of a selectable marker in transgenic rice (Oryza sativa L.) using a chemically regulated Cre/loxP system. Plant Cell Rep 24, 86–94 (2005). https://doi.org/10.1007/s00299-004-0909-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00299-004-0909-5