
Abstract
The cubic MnCo2O4 was prepared by calcining MnCO3-2CoCO3⋅1.5H2O above 600 ∘C in air. The precursor and its calcined products were characterized by thermogravimetry and differential scanning calorimetry, Fourier transform infrared spectroscopy, X-ray powder diffraction, scanning electron microscopy, and vibrating sample magnetometer. The result showed that high-crystallized MnCo2O4 with cubic structure [space group Fd-3m(227)] was obtained when the precursor was calcined above 600 ∘C in air for 6 h. Magnetic characterization indicated that cubic MnCo2O4 behaved weak magnetic behavior at room temperature. The thermal process of the precursor in air experienced three steps, which are: first, the dehydration of 1.5 water molecules, then the decomposition of MnCO3-2CoCO3 into cubic MnO2 and cubic Co3O4, and at last the reaction of MnO2 with Co3O4 into cubic MnCo2O4. Based on the KAS equation, the values of the activation energies associated with the thermal process of MnCO3-2CoCO3⋅1.5H2O were determined.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Spinel mixed oxides with general formula AB2O4 can be considered strategic materials due to their electronic, magnetic, optical, catalytic properties, and electrochemical properties [1–10]. These materials contain either alkaline earth metal and 3d transition metals or only 3d transition metals. MnCo2O4 is very important mixed oxides with inverted spinel structure, which can be depicted in the chemical formula Co2+[Co2+Mn4+]O4 [11]. From X-ray absorption measurements, it has been confirmed that a number of valence states exist in the spinels containing Co and Mn and that the mean charge of Mn ion in these compounds is close to 4+ [12]. Compared with other spinel oxides, spinel MnCo2O4 has shown good catalytic activity for the oxygen reduction reaction in alkaline conditions [13]. Therefore, spinel MnCo2O4 is a promising candidate for the development of low-cost and practical electrically rechargeable metal–air batteries [14]. The quality of MnCo2O4 powders strongly depends on the synthesis method and conditions, which determine particle size, morphology, lattice parameters, stoichiometry, and average Mn valence of MnCo2O4 associated with catalytic and electrochemical performances [15, 16]. Preparations of high-quality samples with superfine particle size and/or doping have generally been considered to improve the catalytic and electrochemical performances of MnCo2O4.
To date, various methods of synthesizing spinel MnCo2O4 have been developed, including the Pechini method using a polymeric precursor [9], the refluxing method [10], the ceramic method [11], microwave synthesis [13], the hard template method [15], the sol-gel method [16], thermal decomposition of binary oxalates [12, 17], hydrothermal synthesis [18], the wet method [19], combustion reaction [20], and the polymeric precursors method [21]. The composition, morphology, and crystalline phases of MnCo2O4 associated with its properties are reportedly highly dependent on the starting materials, synthesis, and processing methods. For example, Liu and Yang [10] obtained cubic MnCo2O4 with nanospheres and stacked nanoplates morphology by a polyol-based precursor route using Mn(CH3COO)2⋅4H2O, Co(CH3COO)2⋅4H2O, and poly(vinyl-pyrrolidone) (PVP) as starting materials. The catalyst with spherical shape shows relatively higher activity than the plate for catalytic oxidation of CO to CO2. Yoon et al. [20] synthesized single phase MnCo2O4 by the combustion reaction using H2NCH2COOH, Mn(NO3)2⋅6H2O, and Co(NO3)2⋅6H2O as starting materials. Most researchers attempt to obtain single phase MnCo2O4 with high properties at the lowest possible cost. However, many MnCo2O4 synthesis methods are a complex process and have a high cost. Therefore, new synthesis methods for cubic MnCo2O4 are needed to be studied and innovated further. Besides, kinetics research of the thermal process of the MnCo2O4 precursor still has fewer reports in comparison with the synthesis of MnCo2O4.
This study aimed to prepare spinel MnCo2O4 by calcining MnCO3-2CoCO3⋅1.5H2O obtained via a solid-state reaction at low heating temperatures and to study the kinetics of thermal decomposition of the precursor. The kinetics of thermal decomposition of the precursor was studied using TG/DSC techniques. Non-isothermal kinetics of thermal decomposition of the precursor was interpreted by the Kissinger–Akahira–Sunose (KAS) equation [22–27]. The kinetic parameters (E a, A) and mechanisms of thermal decomposition of MnCO3-2CoCO3⋅1.5H2O were discussed for the first time.
2 Experimental
2.1 Reagent and Apparatus
All chemicals were of reagent-grade purity (purity >99.9 %). Thermogravimetry and differential scanning calorimetry (TG/DSC) measurements were taken using a Netzsch Sta 409 PC/PG thermogravimetric analyzer under a continuous flow of air (40 mL min−1) and sample mass was around 13 mg. X-ray powder diffraction (XRD) was performed using a Rigaku D/Max 2500V diffractometer equipped with a graphite monochromator and a Cu target. The radiation applied was Cu Kα (λ=0.15406 nm), operated at 40 kV and 50 mA. XRD scans were made from 5∘ to 70∘ in 2θ with a step size of 0.02∘. Fourier transform infrared (FT-IR) spectra of the precursor and its calcined products were recorded on a Nexus 470 FT-IR instrument. The morphologies of the synthesis products were observed using an S-3400 scanning electron microscope (SEM). The specific magnetization (M) of the calcined sample powders was carried out at room temperature using a vibrating sample magnetometer (Lake Shore 7410).
2.2 Preparation of MnCo2O4
The MnCo2O4 precursor was prepared by solid-state reaction at low heating temperatures [22, 23] using MnSO4⋅H2O, CoSO4⋅7H2O, and Na2CO3⋅10H2O as raw materials. In a typical synthesis, MnSO4⋅H2O (19.16 g), CoSO4⋅7H2O (63.73 g), Na2CO3⋅10H2O (71.38), and surfactant polyethylene glycol (PEG)-400 (4.0 mL, 50 vol.%) were placed in a mortar, and the mixture was thoroughly ground by hand with a rubbing mallet for 35 min. The grinding velocity was about 220 cycles/min, and the strength applied was moderate. The reactant mixture gradually became damp, and then a paste formed quickly. The reaction mixture was kept at room temperature for 1.5 h. The mixture was washed with deionized water to remove soluble inorganic salts until \(\mathrm{SO}_{4}^{2-}\) ion could not be visually detected with a 0.5 mol L−1 BaCl2 solution. The solid was then washed with a small amount of anhydrous ethanol and dried at 80 ∘C for 6 h. The resulting material was subsequently determined to be MnCO3-2CoCO3⋅1.5H2O. Cubic MnCo2O4 was obtained via calcining MnCO3-2CoCO3⋅1.5H2O above 600 ∘C in air for 6 h.
3 Method of Determining Kinetic Parameters and Mechanism Functions
3.1 Determination of Activation Energy by the KAS Equation
Activation energy of thermal decomposition of the solid compound can be obtained by the KAS equation (Eq. (1)).
where q is the heating rate (K min−1), T is the reaction temperature (K) in TG curve, E a is the activation energy (kJ mol−1) of thermal decomposition, R is the gas constant (8.314×10−3 kJ mol−1 K−1), A is the preexponential factor, and α is called the conversion degree.
The conversion degree (α) can be expressed as Eq. (2):
where m i, m f and m t are the initial, final, and current sample mass at the moment t, respectively. The g(α) is a function of α; it reveals the mechanism of reaction. The plot of ln(q/T 2) vs. 1/T corresponding to different values of α can be obtained by a linear regression of least-square method, respectively. Thus, reaction activation energy E a can be obtained from linear slope (−E a/R, Eq. (1)).
3.2 Determination of Most Probable Mechanism Functions
The following equation was used to estimate the most probable reaction mechanism of thermal decomposition of MnCO3-2CoCO3⋅1.5H2O, i.e., g(α) function [23, 28]:
where x=E a/(RT), \(h(x) = \frac{x^{4} + 18x^{3} + 86x^{2} + 96x}{x^{4} + 20x^{3} + 120x^{2} + 240x + 120}\), and β is the heating rate (K min−1). The conversions α corresponding to multiple rates at the same temperature are put into the left of Eq. (3), combined with 31 types of mechanism functions [29–31], the slope k and correlation coefficient r 2 are obtained from the plot of ln g(α) vs. lnβ. The probable mechanism function is the one for which the value of the slope k is near −1.00000 and correlation coefficient r 2 is better.
3.3 Calculation of Preexponential Factor A
The preexponential factor was estimated from Eq. (4) [32]:
where A is the preexponential factor (s−1), β is the heating rate (K min−1), g(α) is the most probable mechanism function determined by Eq. (3), E α is the activation energy (kJ mol−1) of thermal decomposition, R is the gas constant (8.314×10−3 kJ mol−1 K−1), and T max is the most rapid decomposition temperature (i.e., peak temperature in differential thermogravimetry (DTG ) curve, K).
4 Results and Discussion
4.1 TG/DTG/DSC Analysis of the Precursor
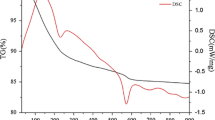
Figure 1 shows the TG/DTG/DSC curves of the precursor at four different heating rates in air from ambient temperature to 900 ∘C. The TG/DTG/DSC curves show that thermal decomposition of MnCO3-2CoCO3⋅1.5H2O below 900 ∘C occurs in three well-defined steps. For a heating rate of 10 K min−1, the first step starts at 20 ∘C, ends at 126 ∘C, and is characterized by a weak DTG peak at about 125 ∘C and a broad and weak endothermic DSC peak at 127 ∘C, which can be attributed to dehydration of one and a half water molecules from MnCO3-2CoCO3⋅1.5H2O (mass loss: observed, 7.15 %; theoretical, 7.11 %). The second decomposition step starts at 126 ∘C, ends at 306 ∘C, and is characterized by a strong DTG peak at 250 ∘C and a stronger DSC endothermic peak at 254 ∘C, attributed to reaction of MnCO3-2CoCO3 with 5/6O2 into MnO2, Co3O4, and of three CO2 molecules (mass loss: observed, 27.35 %; theoretical, 27.74 %). The third decomposition step starts at 306 ∘C, ends at 600 ∘C, and is characterized by a weak DTG peak at 366 ∘C, attributed to reaction of MnO2 with Co3O4 into MnCo2O4, and of 1/3O2 (mass loss: observed, 2.82 %; theoretical, 2.81 %). From Fig. 1a, the exothermic DSC peak at about 663 ∘C was ascribed to crystallization of cubic MnCo2O4.

TG/DTG/DSC curves of MnCO3-2CoCO3⋅1.5H2O at different heating rates in air
4.2 IR Spectroscopic Analysis of the Precursor and Its Calcined Samples
The FT-IR spectra of the precursor and its calcined sample are shown in Fig. 2. The precursor exhibited a strong and broad band at about 3431 cm−1 that can be assigned to symmetric and asymmetric stretching modes of water molecules [33–36]. The strong band at 1453 cm−1 was attributed to v 3 mode of carbonate, and the band at 828 cm−1 were assigned to v 2 mode of carbonate [37–39]. The band at 2340 cm−1 should be assigned to the asymmetric stretching of CO2 (g), which was attributed to the sample adsorbing CO2 from air [37, 40]. FT-IR spectra of the precursor and sample obtained at 150 ∘C are similar. Besides, FT-IR spectra of two samples obtained at 250 ∘C and 450 ∘C are similar. However, the FT-IR spectrum of the sample obtained at 600 ∘C has a little difference in comparison with those of the samples obtained at 250 ∘C and 450 ∘C. Such as, the bands at about 3431, 2027, 1638, and 1536 cm−1 became weak and/or disappeared, which implied that structure and composition of sample obtained at 600 ∘C has great difference in comparison with those of samples obtained at 250 ∘C and 450 ∘C. The spectrum of the calcined sample at 600 ∘C was in agreement with that of cubic MnCo2O4 from literature [16].

FT-IR spectra of MnCO3-2CoCO3⋅1.5H2O and its calcined samples
4.3 XRD Analysis of the Precursor and Its Calcined Samples
Figure 3 shows the XRD patterns of the precursor and its calcined samples. From Fig. 2a, the results showed that the precursor was a mixture containing rhombohedral CoCO3 [PDF card 11-0692, space group R-3c(167)] and amorphous MnCO3. The structure of the rhombohedral CoCO3 is still stable up to 150 ∘C. When the precursor was calcined at 250 ∘C and 450 ∘C for 6 h, the characteristic diffraction peaks of cubic MnO2 and Co3O4 appeared. However, when the precursor was calcined at 600 ∘C, strong intensity and smoothed baseline, and a wide and low diffraction pattern of the thermal decomposition product was observed. Except a weak diffraction peak of unknown compound, and three very weak diffraction peaks of MnO2 and/or Co3O4, which were located at 15.8, 37.0, 59.5, and 65.2∘ for 2θ, respectively, were observed; all other the diffraction peaks in the pattern of sample obtained at 600 ∘C were in agreement with those of cubic MnCo2O4, with space group Fd-3m(227) and the following cell parameters: a =b=c=0.8269 nm, α=β=γ=90∘, density =5.564 g cm−3, from PDF card 23-1237, which indicated that the sample obtained at 600 ∘C was cubic MnCo2O4.

XRD patterns of MnCO3-2CoCO3⋅1.5H2O and its calcined samples at different temperatures for 6 h
According to the Scherrer formula [22, 23]: D=Kλ/(βcosθ), where D is crystallite diameter, K=0.89 (the Scherrer constant), λ=0.15406 nm (wavelength of the X-ray used), β is the width of line at the half-maximum intensity, and θ is the corresponding angle. The resulting crystallite sizes of the products from the calcined precursor at 600, 700, and 850 ∘C in air for 6 h were 17.7, 27.8, and 47.9 nm, respectively. The crystallinity of cubic MnCo2O4 can be calculated via MDI Jade 5.0 software; the results showed that crystallinity of cubic MnCo2O4 obtained at 600, 700, and 850 ∘C were 78.15, 91.79, and 91.81 %, respectively.
4.4 SEM Analyses of the Calcined Samples
The morphologies of the calcined samples are shown in Fig. 4. From Fig. 4a, it can be seen that the calcined sample obtained at 600 ∘C was composed of approximately spherical grains; there is a soft agglomeration phenomenon among the particles of MnCo2O4, and the observed particles sizes are between 50 and 100 nm. With the increase of the calcination temperature, the crystallite in calcined sample was aggregated into larger grains further. Figure 4b and c show the SEM micrographs of samples obtained at 700 and 850 ∘C, respectively. The calcined sample at 700 ∘C still kept approximately spherical morphology, and particles sizes are mainly between 100 and 300 nm. However, the calcined sample obtained at 850 ∘C became polyhedral grains with a particle size of between 300 and 600 nm. The average crystallite sizes of the calcined samples determined by XRD were significantly smaller than the values determined by SEM. This can be attributed to that the values observed by the SEM technique gave the size of the secondary particles, which were composed of several or many crystallites by soft reunion, and the X-ray line broadening analysis disclosed only the size of single crystallite.

SEM micrographs of the products synthesized at different temperatures for 6 h: (a) 600 ∘C; (b) 700 ∘C; (c) 850 ∘C
4.5 Magnetic Properties of the Calcined Samples
The magnetic properties of the calcined samples obtained at different temperatures are shown in Fig. 5. It can be observed that three samples exhibited weak magnetic behaviors at room temperature, and the specific magnetization of powders increased with the increase of the applied field. The specific magnetizations of the powders obtained at 600, 700, and 850 ∘C were 1.16, 0.83, and 0.72 emu g−1 under an applied field of 19.5 kOe, respectively. That is, the larger the crystallinity of cubic MnCo2O4, the smaller is the specific magnetization. Compared with other methods [9], specific magnetization of cubic MnCo2O4 obtained at 700 ∘C in this paper has little difference with that from other synthesis methods. For instance, Borges et al. [9] obtained spinel MnCo2O4 with nearly spherical morphology by the Pechini method. The results showed that the sample obtained at 700 ∘C had a specific magnetization of about 1.00 emu g−1 under an applied field of 1 T and a measure temperature of 120 K.

The specific magnetization (M) as a function of field (H) of calcined samples at different temperatures in air for 6 h
4.6 Kinetics of Thermal Decomposition of the Precursor
In accordance with TG/DTG/DSC, IR, and XRD analyses of the precursor and its calcined products mentioned above, thermal decomposition of the precursor below 900 ∘C consists of three steps, which can be expressed as follows:
According to the non-isothermal method, the basic data of α and T collected from the TG curves of thermal decomposition of MnCO3-2CoCO3⋅1.5H2O at four heating rates (5, 10, 15, and 20 K min−1). According to Eq. (1), the isoconversional calculation procedure of the KAS equation was used. The corresponding KAS lines for different decomposition steps were obtained at different conversion degrees α and different heating rates β at first, and then reaction activation energy E a can be obtained from linear slope (−E a/R). The results are shown in Fig. 6. From Fig. 6, the average values of the activation energies associated with thermal decomposition of MnCO3-2CoCO3⋅1.5H2O were 88.24±13.61 for step 1, 96.36±7.12 for step 2, and 101.27±35.14 kJ mol−1 for step 3, respectively. The activation energy changes in steps 1 and 3 with α are higher than 10 %, and those in step 2 with α are lower than 10 %, so that we draw a conclusion that dehydration of 1.5 water molecules from MnCO3-2CoCO3⋅1.5H2O (step 1) and the reaction of MnO2 with Co3O4 into MnCo2O4 (step 3) could be multi-step reaction mechanisms; the reaction of MnCO3-2CoCO3 with 5/6O2 into MnO2 and Co3O4 (step 2) is simple reaction mechanisms [28, 41–43]. The activation energy of steps 2 and 3 is higher than that of step 1, which implies that the steps 2 and 3 of thermal decomposition of MnCO3-2CoCO3⋅1.5H2O may be interpreted as “slow” stages, while step 1 may be interpreted as a “fast” stage.

The dependence of E a on α at different thermal decomposition steps
Figure 7 shows the curves of α vs. t and dα/dt vs. t for step 2. From Fig. 7a, step 2 is sigmoidal model (sometimes also called autocatalytic) [32, 43], which represents the process of step 2 whose initial and final stages demonstrate the accelerating and decelerating behavior, respectively, so that the process rate reaches its maximum at some values of the extent of conversion. The result shows that rate for step 2 reaches its maximum at 24.54 min (Fig. 7b).

Curves of α vs. t and dα/dt vs. t at heating rate of 5 K min−1
We randomly choose several temperatures which corresponding conversions 0.10<α<0.90 at first, then conversions corresponding to an appropriate temperature for β=5, 10, 15, and 20 K min−1 were put into thirty-one types of mechanism functions [29–31]. The slope k, correlation coefficient r 2, and intercept B of linear regression of ln g(α) vs. ln β were obtained. The two mechanism functions of better correlation coefficient r 2 were determined to be probable mechanism functions at first, and then several temperatures were randomly chosen to calculate the slope k, correlation coefficient r 2, and intercept B of the two probable mechanism functions by the same method. Mechanism function in which the value of k is the closest to −1.00000 and the correlation coefficient r 2 is higher is chosen as mechanism function of thermal decomposition of MnCO3-2CoCO3⋅1.5H2O. The results showed that probable mechanism function integral form of thermal decomposition of MnCO3-2CoCO3⋅1.5H2O for step 2 was determined to be g(α)=[1−(1−α)1/3]2. Rate-determining mechanism for step 2 is three-dimensional diffusion.
The preexponential factor was obtained from Eq. (4), inserting the most probable mechanism function g(α), β, E a, R, and T max values. The results showed that the pre-exponential factor of thermal decomposition of MnCO3-2CoCO3⋅1.5H2O for step 2 was determined to be A=2.30×107 s−1.
In order to prove the validity of the kinetic mechanisms for step 2 of thermal decomposition of MnCO3-2CoCO3⋅1.5H2O, the comparisons were drawn between experimental data and results of the kinetic mechanism for every heating rate. The results are shown in Fig. 8. It can be found that the model-predicted plots were in agreement with the experimental plots, indicating that the mechanism function for step 2 of thermal decomposition of MnCO3-2CoCO3⋅1.5H2O is reliable.

Comparison of model results (dash line) with the experimental data (solid line) of the thermal decomposition of MnCO3-2CoCO3⋅1.5H2O for step 2 at different heating rates
5 Conclusions
We have successfully synthesized cubic MnCo2O4 via thermal decomposition of binary carbonates. XRD analysis suggested that high-crystallized MnCo2O4 with cubic structure was obtained when MnCO3-2CoCO3⋅1.5H2O was calcined above 600 ∘C in air for 6 h. The crystallite size of MnCo2O4 synthesized at 600 ∘C for 6 h was about 18 nm. Magnetic characterization indicated that cubic MnCo2O4 sample exhibited a weak magnetic behavior at room temperature. The thermal decomposition of the precursor in air experienced three steps, which are: first, the dehydration of 1.5 water molecules, then the decomposition of MnCO3-2CoCO3 into MnO2 and Co3O4, and at last the reaction of MnO2 and Co3O4 into cubic MnCo2O4. The values of the activation energies associated with thermal decomposition of MnCO3-2CoCO3⋅1.5H2O were determined to be 88.24±13.61, 96.36±7.12, and 101.27±35.14 kJ mol−1 for the first, second, and third thermal decomposition steps, respectively. Thermal decomposition of MnCO3-2CoCO3⋅1.5H2O for steps 1 and 3 could be multistep reaction mechanisms; reaction of MnO2 with Co3O4 into MnCo2O4 is simple reaction mechanisms (step 2).
References
Maqsood, A., Faraz, A.: J. Supercond. Nov. Magn. 25, 1025–1033 (2012)
Msomi, J.Z., Moyo, T., Abdallah, H.M.I.: J. Supercond. Nov. Magn. 25, 2643–2646 (2012)
Wu, X.H., Wu, W.W., Zhou, K.W., Cui, X.M., Liao, S.: J. Therm. Anal. Calorim. 110, 781–787 (2012)
Song, Q., Zhang, Z.J.: J. Am. Chem. Soc. 126, 6164–6168 (2004)
Pankhurst, Q.A., Pollard, R.J.: J. Phys. Condens. Matter 5, 8487–8508 (1993)
Abdallah, H.M.I., Moyo, T., Msomi, J.Z.: J. Supercond. Nov. Magn. 24, 669–673 (2011)
Gong, C., Bai, Y.J., Qi, Y.X., Lun, N., Feng, J.: Electrochim. Acta 90, 119–127 (2013)
Courtel, F.M., Abu-Lebdeh, Y., Davidson, I.J.: Electrochim. Acta 71, 123–127 (2012)
Borges, F.M.M., Melo, D.M.A., Câmara, M.S.A., Martinelli, A.E., Soares, J.M., de Araújo, J.H., Cabral, F.A.O.: J. Magn. Magn. Mater. 302, 273–277 (2006)
Liu, L., Yang, Y.Z.: Superlattices Microstruct. 54, 26–38 (2013)
Joy, P.A., Date, S.K.: J. Magn. Magn. Mater. 210, 31–34 (2000)
Bazuev, G.V., Korolyov, A.V.: J. Magn. Magn. Mater. 320, 2262–2268 (2008)
Nissinen, T., Valo, T., Gasik, M., Rantanen, J., Lampinen, M.: J. Power Sources 106, 109–115 (2002)
Koninck, M.D., Marsan, B.: Electrochim. Acta 53, 7012–7021 (2008)
Zhu, J.K., Gao, Q.M.: Microporous Mesoporous Mater. 124, 144–152 (2009)
Lavela, P., Tirado, J.L., Vidal-Abarca, C.: Electrochim. Acta 52, 7986–7995 (2007)
Gyrdasova, O.I., Bazuev, G.V., Grigorov, I.G., Koryakova, O.V.: Inorg. Mater. 42, 1126–1132 (2006)
Liu, H.W., Wang, J.: J. Electron. Mater. 41, 3107–3110 (2012)
Yamamoto, N., Higashi, S., Kawano, S., Achiwa, N.: J. Mater. Sci. Lett. 2, 525–526 (1983)
Yoon, M.Y., Lee, E.J., Song, R.H., Hwang, H.J.: Met. Mater. Int. 17, 1039–1043 (2011)
dos Santos, M.E., Ferreira, R.A., Lisboa-Filho, P.N., Peña, O.: J. Magn. Magn. Mater. 329, 53–58 (2013)
Wu, X.H., Zhou, K.W., Wu, W.W., Cui, X.M., Li, Y.N.: J. Therm. Anal. Calorim. 111, 9–16 (2013)
Wu, W.W., Li, Y.N., Zhou, K.W., Wu, X.H., Liao, S., Wang, Q.: J. Therm. Anal. Calorim. 110, 1143–1151 (2012)
Chrissafis, K.: J. Therm. Anal. Calorim. 95, 273–283 (2009)
Wu, X.H., Wu, W.W., Zhou, K.W., Qin, L.Q., Liao, S., Lin, Y.J.: J. Supercond. Nov. Magn. (2013). doi:10.1007/s10948-013-2297-y
Wu, W.W., Cai, J.C., Wu, X.H., Wang, K.T., Hu, Y.M., Wang, Q.: J. Supercond. Nov. Magn. (2013). doi:10.1007/s10948-013-2227-z
Boonchom, B., Vittayakorn, N.: J. Chem. Eng. Data 55, 3307–3311 (2010)
Zhou, K.W., Wu, W.W., Li, Y.N., Wu, X.H., Liao, S.: J. Therm. Anal. Calorim. 114, 205–212 (2013)
Vlaev, L., Nedelchev, N., Gyurova, K., Zagorcheva, M.: J. Anal. Appl. Pyrolysis 81, 253–262 (2008)
Liqing, L., Donghua, C.: J. Therm. Anal. Calorim. 78, 283–293 (2004)
Jiang, H.Y., Wang, J.G., Wu, S.Q., Wang, B.S., Wang, Z.Z.: Carbon 48, 352–358 (2010)
Vyazovkin, S., Burnham, A.K., Criado, J.M., Pérez-Maqueda, L.A., Popescu, C., Sbirrazzuoli, N.: Thermochim. Acta 520, 1–19 (2011)
Wu, W.W., Cai, J.C., Wu, X.H., Liao, S., Huang, A.G.: Powder Technol. 215–216, 200–205 (2012)
Li, Y.N., Wu, X.H., Wu, W.W., Wang, K.T., Liao, S.: J. Supercond. Nov. Magn. 26, 2153–2158 (2013)
Huang, J.W., Su, P., Wu, W.W., Li, Y.N., Wu, X.H., Liao, S.: J. Supercond. Nov. Magn. 25, 1971–1977 (2012)
Wu, W.W., Cai, J.C., Wu, X.H., Liao, S., Wang, K.T., Tao, L.: Adv. Powder Technol. 24, 154–159 (2013)
Zhou, K.W., Wu, X.H., Wu, W.W., Xie, J., Tang, S.Q., Liao, S.: Adv. Powder Technol. 24, 359–363 (2013)
Chandradass, J., Bae, D.S., Kim, K.H.: Adv. Powder Technol. 22, 370–374 (2011)
Wu, X.H., Wu, W.W., Cui, X.M., Liao, S.: J. Therm. Anal. Calorim. 107, 625–632 (2012)
Wu, X.H., Wu, W.W., Cui, X.M., Liao, S.: J. Therm. Anal. Calorim. 109, 163–169 (2011)
Budrugeac, P., Muşat, V., Segal, E.: J. Therm. Anal. Calorim. 88, 699–702 (2007)
Chaiyo, N., Muanghlua, R., Niemcharoen, S., Boonchom, B., Seeharaj, P., Vittayakorn, N.: J. Therm. Anal. Calorim. 107, 1023–1029 (2012)
Wu, W.W., Wang, K.T., Li, Y.N., Wu, X.H., Liao, S., Wang, Q.: J. Therm. Anal. Calorim. 112, 1391–1399 (2013)
Acknowledgements
This study was financially supported by the National Nature Science Foundation of China (Grant No. 21161002) and the Guangxi Nature Science Foundation of China (Grant No. 2011GXNSFA018036).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Wang, K., Wu, X., Wu, W. et al. Synthesis of Spinel MnCo2O4 by Thermal Decomposition of Carbonates and Kinetics of Thermal Decomposition of Precursor. J Supercond Nov Magn 27, 1249–1256 (2014). https://doi.org/10.1007/s10948-013-2430-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10948-013-2430-y