
Abstract
The thermolysis of the ill-known γ-MnC2O4·2H2O is investigated applying DTA, TG, DSC, FT-IR, SEM, BET and Rietveld XRD phase analyses for thorough characterization of the starting compound and its calcined products. The dehydration proceeds in one step with ∆H = 149 kJ mol−1 (reported for the first time). Primary decomposition product is Mn3O4 ([Mn2+Mn2 3+]O4) with high specific surface area. During its formation, the oxidation of Mn3+ species to Mn4+ starts, followed by oxidation of Mn2+–Mn3+, thus leading to formation of metastable Mn5O8 ([Mn2 2+Mn3 4+]O8) and Mn2O3. Before complete precursor decomposition under non-isothermal conditions, the reduction of Mn4+–Mn3+ occurs, forming an additional amount of Mn3O4. After isothermal annealing of γ-MnC2O4·2H2O in the range of 573–823 K, Mn3O4 is detected as a major product in all samples. Mn5O8 is identified after calcination at 573, 673, 723 K, and its amount decreases gradually in this order. Cubic Mn2O3 appears after heat treatment at 673 K; its content varies between 13 and 23 % in the range of 673–773 K and becomes close to that of Mn3O4 at 823 K. The application of the same experimental conditions as in the investigation of α-MnC2O4·2H2O and MnC2O4·3H2O allows an objective comparison of the nature and the peculiarities of the thermal decomposition of different crystal forms of manganese oxalate.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The thermal decomposition of 3d-transition metal oxalates has been subject of many investigations during the last decade due to their wide application as precursors for nanosized oxide materials [1–19]. A special attention is paid to the manganese oxides, which have a wide range of technological application with respect to their magnetic, electric and catalytic properties. Sparingly soluble manganese oxalate is an appropriate precursor for their preparation which explains the growing interest to the utilization and investigation of its thermolysis [1–11, 17–19]. Different in morphology, structure and oxidation state manganese oxides have been obtained by varying the preparation method, nature of solvents and/or using different surfactants at oxalate synthesis, changing the atmosphere, temperature and duration of heat treatment [1–9, 11, 17, 18]. Surprisingly, a little attention has been paid to the influence of the initial crystal structure of manganese(II) oxalate on the physicochemical characteristics of the oxides produced.
In the system containing Mn2+–C2O4 2−–H2O, three crystal forms of the oxalate could be obtained—monoclinic α-MnC2O4·2H2O (SG C2/c) [20], orthorhombic γ-MnC2O4·2H2O (SG P212121) [21] and orthorhombic MnC2O4·3H2O (SG Pcca) [22]. The difference in their structure is well described in Refs. [22, 23]. The procedures for preparation of a pure phase of the corresponding crystallohydrate from aqueous solutions are recommended in 1977 by Huizing [24]. The conclusion is made that the type of crystal form depends on the temperature and pH of the initial system, but at room temperature always the trihydrate is precipitated. However, we established that the nucleation mechanism is also a factor which determines the type of obtained crystallohydrate form [25], and we successfully synthesized pure phases of α-MnC2O4·2H2O and MnC2O4·3H2O from aqueous medium at pH = 3 and room temperature, by changing the concentration of initial solutions [18]. Up to our publication, however, the possibility of precipitation of crystal phase other than α-MnC2O4·2H2O was neglected for almost 30 years. Therefore, while the mechanism and products of thermal decomposition of the monoclinic dihydrate in air have been extensively studied by various methods [2, 3, 7, 11, 17, 26–35], the literature data for the other two forms are quite scarce. To the best of our knowledge, apart from our studies [18, 19], there are only a few papers about MnC2O4·3H2O [5, 9, 36, 37] and γ-MnC2O4·2H2O [11] and they are not always aimed on the investigation of their decomposition mechanism.
It is expected that the differences in the crystal structure of α-MnC2O4·2H2O, MnC2O4·3H2O and γ-MnC2O4·2H2O will affect the processes during decomposition, nature and physicochemical characteristics of the produced oxides. Our previous investigations by in situ magnetic measurements during thermolysis of α-MnC2O4·2H2O, MnC2O4·3H2O [18] and γ-MnC2O4·2H2O [19] proved that at the beginning of the process Mn(II) oxidation proceeds to a different extent. In Ref. [18], the non-isothermal and isothermal thermolysis of monoclinic α-MnC2O4·2H2O and orthorhombic MnC2O4·3H2O was compared and differences were discussed. The isothermal experiments were conducted at several temperatures in the range of 573–723 K. Later, Jia et al. [11] compare α-MnC2O4·2H2O and γ-MnC2O4·2H2O. The former is precipitated from aqueous solution and the latter from ethanolic solution. The authors establish that both kinetic parameters of thermal decomposition and the nature of the produced oxides (573 K, calcination for 5 h) depend on the used polymorphous form. The decomposition mechanism of γ-MnC2O4·2H2O, however, is not discussed in detail.
The scarce literature data for γ-MnC2O4·2H2O [11, 19] and absence of results, allowing objective comparison of the behavior of different manganese oxalate crystal forms under non-isothermal and isothermal conditions, provoked our interest. The aims of the present study are as follows: (1) to synthesize γ-MnC2O4·2H2O by deliberate modification of the recommended synthesis procedure [24]; (2) to investigate in detail the decomposition mechanism of this ill-known form of manganese oxalate; (3) to study the nature and the physicochemical characteristics of the oxides, produced at different temperatures; and (4) to compare the results with those for the other two forms—α-MnC2O4·2H2O and MnC2O4·3H2O.
To achieve the objectives of the work, a variety of experimental techniques were applied, as described in the experimental part. The thermal analysis and annealing of γ-MnC2O4·2H2O were conducted under the same conditions (equipments, starting sample mass, heating rates, temperatures and duration time) as in investigation of the other two crystal forms in order to ensure comparability and to check the influence of the initial crystal structure on both the decomposition mechanism of manganese(II) oxalate and the nature of the obtained oxides. Besides the purely scientific interest, the results will be useful from practical point of view because of unexplored possibility γ-MnC2O4·2H2O to be applied as precursor for manganese oxides production, as well as for deliberate choice of the initial crystallohydrate form depending on the intended objective.
Experimental
Synthesis of γ-MnC2O4·2H2O and oxide products
For preparation of the orthorhombic γ-MnC2O4·2H2O, water solutions of Mn(NO3)2 and (NH4)2C2O4 were poured simultaneously into the reaction vessel on stirring at room temperature. The mole ratio of Mn2+ to C2O4 2− was equal to 1.45 [24]. The acidity of starting solutions was adjusted in such a way that after mixing pH 4.0 ± 0.1 is achieved. After 15 min, the formed pinkish suspension was transferred to a flask and then was kept at 60 °C for 8 h. The warm suspension was filtered and washed several times with distilled water, and the product was dried in desiccator.
Although the initial reagents and mole ratio are the same as in the original synthesis procedure of Huizing [24], the temperature is lower by 20 °C, and the heating duration is much shorter than that recommended by him (at least 15 h). This is due to the difference in the degree of supersaturation created in the reaction mixture. The slow dropwise adding of Mn2+ solution to C2O4 2−, used in [24], creates an indefinite and gradually growing supersaturation during the oxalate nucleation and crystal growth. However, our experience [25] shows that the low degree of supersaturation leads to the formation of MnC2O4·3H2O, which in turn transforms via α-MnC2O4·2H2O into γ-MnC2O4·2H2O on heating or after prolonged aging in the supernatant liquor. Therefore, we consciously chose the simultaneously rapid mixing of the reagents, which ensures a high, well-defined supersaturation from the beginning of the crystallization and direct formation of a big portion of α-MnC2O4·2H2O even at room temperature.
Five samples from the prepared γ-MnC2O4·2H2O were annealed for 1 h in air at 573, 673, 723, 773 and 823 K. The products obtained were then tempered in a desiccator and weighed out. They are denoted in the text as G-573, G-673, G-723, G-773 and G-823, where the number corresponds to the annealing temperature.
Methods
Non-isothermal analysis of orthorhombic γ-MnC2O4·2H2O was performed on a Paulik–Erdey MOM OD-102 derivatograph, model Q 1500 D (Hungary). Differential thermal analysis (DTA), thermogravimetry (TG) and derivative thermogravimetry (DTG) were done in a static air atmosphere with sample mass of 0.200 g at a heating rate of 10 °C min−1 in the temperature range of 298–1273 K, using a standard corundum crucible. The reference sample was pure α-Al2O3. Differential scanning calorimetry (DSC) was carried out in a dynamic air atmosphere on PerkinElmer, model DSC 7 (USA) at a heating rate of 10 °C min−1 with background correction.
X-ray powder diffraction (XRD) patterns of the precursor and annealed samples were recorded in the angle interval 35°–115° (2θ), on a Philips PW 1050 diffractometer, equipped with Cu Kα tube and scintillation detector. Data for cell refinements and quantitative analysis were collected in θ–2θ, step-scan mode in the angle interval from 10 to 80o (2θ), at steps of 0.03o (2θ) and counting time of 3 s step−1. Quantitative analysis, unite cell refinement and estimation of crystallite size were carried out by BRASS—Bremen Rietveld Analysis and Structure Suite [38]. Le Bail whole pattern fitting [39] of γ-MnC2O4·2H2O was carried on in the framework of the same program and repeated in X’Pert HighScore Plus developed by PANalytical. Fourier transform infrared (FTIR) spectra of the oxide products were recorded on Shimadzu IR Prestige-21/FTIR-8000 spectrometer (Japan) in the range of 4000–400 cm−1 in Nujol mull. Background correction was made using a Nujol as reference. The morphological characterization of the initial γ-MnC2O4·2H2O and calcined samples was performed by scanning electron microscope (SEM) observation using a JEOL JSM-5510 (Japan). The determination of the specific surface area (S BET) was done by nitrogen adsorption using an apparatus “Surface area & micropore size analyzer” NOVA 1200e (Quantachrome Instruments, USA).
Results and discussion
Characterization of γ-MnC2O4·2H2O
The XRD pattern of the obtained γ-MnC2O4·2H2O is presented in Fig. 1. The strongest reflections of MnC2O4·3H2O (13.323°) and α-MnC2O4·2H2O (18.430°) are not observed. In addition, the performed Le Bail whole pattern fitting analysis shows a full coincidence with a reference sample (96426-ICSD) (Online Resource 1). Experimentally determined crystal lattice parameters are: a = 6.2561(4), b = 13.569(2) and c = 6.0957(8) Å. The high intensities and small FWHM of the diffraction peaks indicate the good crystallinity of the sample.

X-ray diffraction pattern of γ-MnC2O4·2H2O
This statement is confirmed by the low specific surface area (2.4 m2 g−1) and scanning electron images (Fig. 2). Tetragonal prisms are observed, in some cases with pyramidal termination. The shape is very different from both the smooth needle-like crystals of MnC2O4·3H2O and rose-like agglomerates of monoclinic α-MnC2O4·2H2O, prepared also from water solutions. The SEM observation suggests that the crystal growth of γ-MnC2O4·2H2O rods proceeds layer by layer. Such a mechanism could not be detected at previous synthesis procedure used by us, where γ-MnC2O4·2H2O was obtained via slow phase transformation of MnC2O4·3H2O at room temperature [19].

SEM micrographs of γ-MnC2O4·2H2O: a general view of the prismatic crystals; b pyramidal termination
Non-isothermal investigation of γ-MnC2O4·2H2O thermolysis
The DTA, TG and DTG profiles are displayed in Fig. 3. The regions from I to V, marked on the TG curve, correspond to the processes of dehydration (I), decomposition (II) and post-decomposition (III–V) phenomena.

DTA, TG and DTG profiles of γ-MnC2O4·2H2O at a heating rate of 10 °C min−1
In order to give a plausible interpretation of DTA, TG and DTG curves of γ-MnC2O4·2H2O thermal decomposition, it should take into account not only the expected changes in the mass and sign of the reaction enthalpy for the various redox reactions (some of them are given in [29, 40] and others can be calculated) but also: (1) the oxidation–reduction behavior of Mn3O4, Mn5O8, Mn2O3 as a function of the temperature and heating time, well described in Refs. [29, 40–44] and (2) the established dependence of the oxidation–reduction behavior of Mn3O4 (primary decomposition product) on the specific surface area, particle size of the samples and experimental conditions [41–43]; these factors influence significantly the transition temperatures, type of the intermediate oxides and final products.
Dehydration (region I) takes place in a range of 392–459 K, according to the DTG curve, which shows more clearly the changes occurring with the specimen. The water liberation proceeds in one step with peak maximum at 430 K. The dehydration of α-MnC2O4·2H2O proceeds in the same way (peak at 448 K), while for MnC2O4·3H2O three steps are observed (peaks at 393, 418 and 463 K) [18]. The experimental mass loss (ML) at the end of the steep slope of TG curve is 19 % and slowly increases to 19.4 % before to start the anhydrous oxalate decomposition. This is in a good agreement with the theoretical value (MLth) of 20 %.
The heat of dehydration was measured, and DSC curve is presented on the Fig. 4. For the studied orthorhombic γ-MnC2O4·2H2O, the determined enthalpy is 149 kJ mol−1, which is higher than that of monoclinic α-MnC2O4·2H2O (97 kJ mol−1) and orthorhombic MnC2O4·3H2O (59, 45 and 16 kJ mol−1). This result is in accordance with the deeper DTA dehydration peak of γ-MnC2O4·2H2O (Fig. 3) compared with that of α-MnC2O4·2H2O [18] and is evidence for the existence of stronger bonds in the structure of γ-MnC2O4·2H2O. The same conclusion was drawn by Mansilla et al. [23] based on the comparison of vibration frequencies in IR spectra of the three crystal forms of the manganese oxalate.

DSC curve of γ-MnC2O4·2H2O
According to the DTG curve (Fig. 3), the anhydrous oxalate is thermally stable within the range of 459–509 K. Decomposition starts with slowly decreasing mass in the beginning. Thereafter, an abrupt decrease in mass (ML = 56.6 %) is observed in region II on TG curve. The oxidative decomposition results in a broad asymmetric exo-peak with maximum at the beginning of the process (593 K). However, the corresponding DTG shows two well-defined peaks with maxima at 565 K (little shoulder at 573 K) and at 648 K, which implies that an additional reaction/s takes place during the decomposition, leading to a retardation of the mass loss. Based on the DTG curve, the region II can be divided into two subregions—IIa and IIb.
At the first stage IIa (509–613 K, DTG), the oxidative oxalate decomposition undoubtedly leads to the formation of Mn3O4 as a primary product [17, 26–28, 30]. However, Mn3O4 ([Mn2+Mn2 3+]O4) includes two types of Mnn+ ions: Mn2+ in the tetrahedral sites of the spinel structure and Mn3+ in the octahedral sites. It has been established [42, 43, 45] that (1) at low-temperature oxidation of Mn3O4 the oxidation of Mn3+ starts at lower temperatures than required for the oxidation of Mn2+ and (2) the Mn3O4 oxidation is facilitated by a specific surface area greater than 10 m2 g−1. As the measured specific surface area of our anhydrous oxalate is 16 m2 g−1 (that is 8 times higher than that of the dihydrate), it is logical to assume that the surface area of the primary formed Mn3O4 is high enough to facilitate the occurrence of oxidation–reduction phenomena. Therefore, immediately after receiving of Mn3O4 an exothermic oxidation of Mn3+ species to Mn4+ starts, followed by oxidation of Mn2+ species to Mn3+, thus leading to formation of metastable Mn5O8 ([Mn2 2+Mn3 4+]O8) and Mn2O3. This explains the observed retardation of the mass loss until 613 K (DTG curve), because the conversion of Mn3O4 to Mn5O8 and Mn2O3 is associated with mass gain.
Second stage IIb (613-656 K, DTG) can be attributed to the complete decomposition of γ-MnC2O4·2H2O, accompanied by an endothermic process of reduction of Mn4+ (from Mn5O8) to Mn3+. The latter process leads to the formation of an additional amount of Mn3O4 before the end of decomposition. In this temperature range and at non-isothermal conditions, transformation of Mn5O8 to cubic Mn2O3 could not be expected as predicted by the thermodynamic factors. According to Azzoni et al. [44], this reaction includes diffusion-limited structural readjustment and several hours at 823 K isothermal annealing are necessary to proceed. Consequently, the final product of γ-MnC2O4·2H2O decomposition (ML = 56.6 %) consists mainly of Mn2O3 (MLth = 55.89 %) and Mn3O4 (MLth = 57.38 %) which are in ratio almost 1:1. The expected theoretical values of mass loss for obtaining of the other manganese oxides are 51.40 % for MnO2, 55.04 % for Mn5O8 and 60.35 % for MnO.
The assumptions about the processes in regions IIa and IIb are supported by our previous in situ measurements of the magnetic properties during γ-MnC2O4·2H2O decomposition [19]. It has been established that at the beginning of anhydrous oxalate decomposition the effective magnetic moment of manganese (μ eff) is changed from 5.92 BM (typical for Mn2+) to 4.60 BM even at 1.3 % decrease in the mass, which is evidence for the existence of Mn3+ and Mn4+. After that, the increase in μ eff is observed due to the reduction of manganese to lower oxidation states.
Post-decomposition processes occur in regions III–V. After the end of decomposition, a sharp mass gain to overall mass loss of 55.8 % is observed in the range of 656–673 K (region III) and this process results in exo-peak with maximum at 673 K. It can be attributed to the oxidation of secondary formed Mn3O4 to additional quantity of Mn2O3. The observed mass gain of 0.8 % is lower than the half of theoretical one for this process (MLth = 3.4 %). It should be noted that at non-isothermal experiments it is difficult to determine which form of Mn2O3 is produced—γ-Mn2O3 or α-Mn2O3. Most probably, the gradual increase in the mass in region IV (additional 0.8 %) is connected with structural improvement and/or oxygenation of the formed Mn2O3, which ends at 813 K (overall ML = 55 %). The absence of further visible changes in the sample mass and temperature gradient up to the peak at 1173 K supports our assertion that Mn4+ species are reduced in the course of decomposition. If Mn5O8 is present in the sample, an endothermic effect at about 848 K should be observed on the DTA curve (accompanied by mass loss), which is typical for the transformation of Mn5O8 to cubic Mn2O3 [40, 41, 46].
Isothermal investigation of γ-MnC2O4·2H2O thermolysis
Characterization of the oxide products
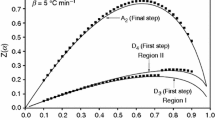
The XRD patterns of the manganese oxides, obtained from γ-MnC2O4·2H2O at different annealing temperatures, are depicted on the Fig. 5. All results are presented in frame of reference: 2θ versus normalized intensity. Main diffraction peaks of the phases are marked for comparison. As expected, increasing the annealing temperature leads to changes in the FWHM, intensity and position of the diffraction peaks as a result of the improved crystallinity and/or of the growing quantity of the corresponding phase. Due to the complex powder composition, Rietveld analysis was performed for estimating the phase quantities. An example is shown in Fig. 6, where the experimental and calculated patterns for sample G-673 are presented.

XRD patterns of the obtained oxides (the main reflections of the detected phases are presented; the arrow shows the position of main reflection of the precursor)

Rietveld analysis of XRD pattern of the sample G-673 (legend: solid lines calculated data, points experimental data; colored in the online version)
The results from the quantitative analyses of the XRD patterns are summarized in Table 1 together with experimentally determined mass loss of the samples, their specific surface area and crystallite size of the corresponding oxides. The crystal lattice parameters are given in Online Resource 2. The phases are specified as: tetragonal γ-Mn3O4—I 41/amd (141) (JCPDS 24-0734), monoclinic Mn5O8—C 1 2/m 1 (12) (JCPDS 39-1218) and cubic α-Mn2O3—I a-3 (206) (JCPDS 41-1442).
The data in Table 1 show a remarkable decrease in the specific surface area of the samples with increase in the temperature, which will influence the oxidation–reduction behavior of the primary decomposition product Mn3O4 [41–43] in addition to the thermally induced effects. An improvement in the crystallinity with increase in the temperature is established also for all of the detected phases, despite the variation in content of the corresponding phase. It should be noted that the experimentally determined mass losses are a little bit higher than the expected ones; however, their relative changes are in accordance with the detected phase compositions. The observed mass loss for sample G-573 (ML = 54.6 %) indicates that 1-h annealing at 573 K is not enough for complete decomposition of γ-MnC2O4·2H2O. However, its content could not be determined by Rietveld analysis due to the extremely low and diffuse diffraction peak of the oxalate and because of the low crystallinity of the sample. The absence of Mn2O3 in the sample G-573 shows that these conditions do not allow oxidation of Mn2+ (situated in the tetrahedral sites of the Mn3O4) to Mn3+. These experimental facts support the suppositions about the processes occurring at the first stage of the decomposition under non-isothermal thermolysis. For all other samples, the complete oxalate decomposition is achieved.
In order to confirm the nature of the products, obtained at different temperatures, the FT-IR spectra were recorded (Fig. 7). Differences are observed in the range of 700–400 cm−1, corresponding to the Mn–O vibrations. However, due to the multiphase composition of the samples and partial overlapping of the strongest vibration frequencies of the manganese oxides, it is difficult to distinguish the contribution of the different phases.

FT-IR spectra of the obtained oxides (colored in the online version)
The spectra of samples, annealed at 673, 723 and 773 K, are the simplest ones and contain two well-defined bands. The observed vibration frequencies are in very good agreement with the reported strongest bands for rod-like Mn3O4 crystals (483 and 605 cm−1 [47], 503 and 609 cm−1 [48], 517 and 603 cm−1 [49]). These bands are assigned to coupling between Mn–O stretching modes of tetrahedral and octahedral sites.
The sample G-573 contains Mn3O4 (major), Mn5O8 (minor) and traces of undecomposed oxalate. The data reported in the literature for the frequencies of Mn5O8 are at about 424 (w, sh), 509 (s, sh) and 602–607 (s, sh) cm−1 [40, 50], and those for anhydrous MnC2O4 are at 410 (s), 496 (m) and 514 (m) cm−1 [5]. In the FT-IR spectrum of this sample, a strong vibration at 606 cm−1 and wide spilled band at about 500 cm−1 (maximum at 489 cm−1 and shoulder at 506 cm−1) are observed. The broadened character of the band at 500 cm−1 is related to the superposition of Mn3O4 and Mn5O8 vibration frequencies. The impact of the MnC2O4 can be neglected due to its low content (<1 %) and medium band intensity. The existence of Mn5O8 is confirmed also by the value of the band at 606 cm−1. Its shift to 603 cm−1 as well as narrowing of the broad band around 500 cm−1 with increase in the annealing temperature could be explained by the decrease in the Mn5O8 content. The shoulder at 428 cm−1 in the spectrum of sample G-573 is assigned also to Mn5O8, and it overlaps the weak vibration of Mn3O4 at 414 cm−1.
In the FT-IR spectrum of sample G-823, new vibration frequencies arise, typical for cubic Mn2O3 [43, 51, 52] and related to the significant increase in its content in the obtained product. Absorption bands at 568 и 652 cm−1 are attributed to the Mn–O stretching vibration, and a shoulder at 520 cm−1 is assigned to Mn–O bending vibration of Mn2O3.
SEM images of the annealed samples are depicted on the Fig. 8. They clearly show the morphological changes after isothermal annealing. Generally, the produced oxides preserve, although slightly changed, the rod shape of the initial γ-Mn2C2O4·2H2O crystals, despite the high total mass loss (>55 %). However, cleavage of the crystals’ ends is observed, as well as bending of the ends of some crystals because the crystalline structure collapses abruptly under the rapid increase in the temperature prior to the isothermal annealing. On increasing the temperature, these processes are enhanced and even breakup of some of the primary crystals happens. In this way, the crystals approximately of the same length, but significantly thinner, are formed.

SEM micrographs of the oxides, obtained at 573, 673, 723, 773 and 823 K, are shown in rows a, b, c, d, e, respectively (left side topology, right side overview)
The changes in the surface topology of the initial smooth γ-MnC2O4·2H2O rods are more informative. After annealing at 573 K, the smoothness is preserved. The formation of deep parallel channels, observed at sample G-573, as well as the manner of ends’ cleavage confirms the assumption for layer by layer growth of the initial oxalate crystals. At 673 K, the surface seems to keep its smoothness, but higher magnification shows that the walls are covered or probably even consist of tiny spherical nanoparticles. Likely this is related to the complete oxalate decomposition and formation of Mn2O3 phase, which appears at this temperature.
After heating at 723 K, the surface roughness of thinner crystals increases and ganglia-like hills start to emerge uniformly throughout the crystal surface (most probably, this process occurs from the inner side of the breaking crystals, which are energetically richer and sensitive to small thermally induced fluctuations in the sample density and phase composition). The interface is very diffuse at 723 K but sharpens with the increase in temperature to 773 K and particularly to the “critical” temperature of 823 K, at which the amount of Mn2O3 becomes close to that of Mn3O4. All these facts allow us to describe the phenomena of hills appearance as a spinodal transformation of Mn3O4–Mn2O3, which is diffusion controlled process and is enhanced by the higher temperature. It facilitates the diffusion, resulting in a growth of the hills.
Mechanism of oxalate decomposition and formation of the oxides
The changes established in the phase composition and microstructure of the annealed samples reveal the processes taking place during γ-Mn2C2O4·2H2O decomposition and formation of the oxides. They are in accordance with the suppositions, made on the base of non-isothermal analysis, and can be summarized as follow:
-
1.
The primary and the major decomposition product is indeed Mn3O4.
-
2.
In the course of its formation, it begins to oxidize stepwise to Mn5O8 and cubic Mn2O3. For this reason, Mn3O4 content decreases continuously up to 723 K.
-
3.
The increase in the calcination temperature suppresses the oxidation of Mn3+ to Mn4+ and promotes the oxidation of Mn2+ to Mn3+, as it has been shown by Fritsch et al. [42] at low-temperature oxidation of Mn3O4. This explains the decrease in Mn5O8 content in the range of 573 ≤ T ≤ 723 K and disappearance of this phase at higher temperatures, as well as appearance of Mn2O3 after annealing at 673 K and increase in its content at 723 K.
-
4.
The oxidation of the formed Mn3O4 to cubic α-Mn2O3 is thermally activated process [40, 44] and a “critical” temperature is necessary for oxidation to occur substantially. It is known that this temperature depends on the crystallite size of Mn3O4 and annealing duration. In the case of γ-MnC2O4·2H2O decomposition, at 1-h annealing duration and heating rate applied in the present work, this critical temperature is above 773 K. Therefore, at 673, 723 and 773 K the amount of Mn2O3 remains relatively constant varying between 13 and 23 %.
-
5.
Upon heating at 773 K, the forming Mn3O4 oxidizes to cubic Mn2O3, without formation of Mn5O8 because at isothermal conditions the latter process is favored at low temperatures [42, 43]. Most probably, the mechanism of Mn3O4 oxidation at 723 K is the same, but the temperature is below the “critical” value. Therefore, the content of Mn3O4 is greatest for this sample.
Based on the results and their interpretation, a conclusion can be drawn that for the heating time applied, the nature of the oxide products depends on the inclination of Mn3O4 to be oxidized, which is influenced by the specific surface area and annealing temperatures applied.
Comparison of the thermal behavior of γ-MnC2O4·2H2O, α-MnC2O4·2H2O and MnC2O4·3H2O
A contribution of the present study, along with our previous reports [18, 19], is the investigation of the thermal behavior of the three oxalate forms under the identical conditions. This allows us to specify the peculiarities and the similarities in the thermolysis of the different forms. In addition, such a comparison allows clarifying the decomposition mechanism of the previous studied forms [18] and gives more precise explanation of some experimental results.
Under isothermal conditions, the behavior of γ-MnC2O4·2H2O is closer to that of the orthorhombic MnC2O4·3H2O than to the monoclinic α-MnC2O4·2H2O. After annealing of MnC2O4·3H2O at 573 K for 1 h, a single phase of Mn3O4 is detected. Mn2O3 phase appears after heat treatment at 673 K, and its amount increases at 723 K. If the precursor is α-MnC2O4·2H2O, two phases are detected at 573 K, being Mn3O4 (major) and Mn2O3 (traces). In our previous investigation [18], Mn5O8 was not detected by the XRD, probably due to the performed conventional XRD analysis. However, the presence of Mn4+ in all samples, produced in the studied range of 573–723 K, was established by the measurements of the magnetic properties of the obtained oxides. The results showed that Mn4+ content depends on the type of precursor used and decreases gradually with increase in annealing temperature from 573 to 723 K, as it was established and discussed in the present study.
The DTA curve of γ-MnC2O4·2H2O shows that processes in region II (decomposition) and region III (post-decomposition) result in two exothermic peaks with maxima at 593 and 673 K. This course of the curve is extremely similar to that of α-MnC2O4·2H2O (exo-peaks at 603 and 673 K) [18]. However, TG and DTG profiles of the two polymorphic forms are quite different. On TG curve of α-MnC2O4·2H2O, only one region is observed. In addition, DTG curve of α-MnC2O4·2H2O shows only one broad asymmetric peak in the course of decomposition, while on DTG of γ-MnC2O4·2H2O two clearly expressed peaks in region II are detected. Based on the comparison of the DTA, TG and DTG profiles, it can be concluded that the exo-peak at 673 K, observed on DTA curve of α-MnC2O4·2H2O, is a result of an additional oxidation process taking place before complete oxalate decomposition; for γ-MnC2O4·2H2O, it occurs in region III, after the end of decomposition. This is supported by the overall mass loss at the end of the decomposition, being 55.5 % for α-MnC2O4·2H2O and 56.6 % for γ-MnC2O4·2H2O. The experimental data reveal that the intermediate oxidation–reduction processes at monoclinic α-MnC2O4·2H2O proceed easier than at the orthorhombic γ-MnC2O4·2H2O. The same conclusion was drawn by us, based on the results from in situ magnetic measurements [18, 19].
The above-made juxtaposition shows that under non-isothermal conditions, the behavior of the orthorhombic γ-MnC2O4·2H2O is very similar to that of the monoclinic α-MnC2O4·2H2O, while under isothermal conditions it is closer to that of the orthorhombic MnC2O4·3H2O. This is due to different heating rates. For example, the temperature of 773 K is achieved for 47.5 min at non-isothermal experiments and for 3.5 min at isothermal. The isothermal annealing better accounts for the phase equilibria between the different manganese oxidation states; however, the applied heating time is not long enough to achieve the corresponding phases’ equilibria [43]. Hence, the extent of the occurring processes and type of the obtained products in both cases are determined by the kinetics, however, by the different kinetic factors. The elucidation of their nature could be an object of further studies.
Conclusions
Pure phase of pinkish γ-MnC2O4·2H2O is successfully obtained by modified synthesis procedure, and the thermolysis under non-isothermal and isothermal conditions was studied. The heat of γ-MnC2O4·2H2O dehydration (149 kJ mol−1) is reported for the first time. A decomposition mechanism is proposed and discussed in detail. A crucial role in the formation of final oxide products plays the inclination of the primary decomposition product Mn3O4 to be oxidized, depending on the temperature applied, heating rates and specific surface area. At low temperatures, the forming Mn3O4 starts to oxidize stepwise, thus leading to formation of metastable Mn5O8 and Mn2O3, along with the major product. The temperature increase suppresses the formation of Mn5O8 and under non-isothermal conditions the available Mn4+ ions in the system are reduced to Mn3+ before complete precursor decomposition.
Under isothermal experiments, Mn5O8 is formed until 723 K at heating rates applied in the present study. At higher temperature, Mn3O4 converts to cubic Mn2O3 without formation of Mn5O8. The “critical” temperature for the diffusion controlled transformation of Mn3O4 to Mn2O3 is above 773 K at experimental conditions used. This process is connected with substantial changes in the surface topology.
The comparison of non-isothermal and isothermal behaviors of studied γ-MnC2O4·2H2O and other two forms α-MnC2O4·2H2O and MnC2O4·3H2O is made. The peculiarities and similarities between them are pointed out.
References
Liu M, Zhang GJ, Shen ZR, Sun PC, Ding DT, Chen TH. Synthesis and characterization of hierarchically structured mesoporous MnO2 and Mn2O3. Solid State Sci. 2009;11:118–28.
Ahmad T, Ramanujachary KV, Lofland SE, Ganguli AK. Nanorods of manganese oxalate: a single source precursor to different manganese oxide nanoparticles (MnO, Mn2O3, Mn3O4). J Mater Chem. 2004;14:3406–10.
Thota S, Prasad B, Kumar J. Formation and magnetic behaviour of manganese oxide nanoparticles. Mater Sci Eng, B. 2010;167:153–60.
Davar F, Mohandes F, Salavati-Niasari M. Synthesis and characterization manganese oxide nanobundles from decomposition of manganese oxalate. Inorg Chim Acta. 2009;362:3663–8.
Gyrdasova OL, Krasil’nikov VN, Bazuev GV. Synthesis of micro- and nanosized manganese oxides from hydrated manganese oxalates and products of their chemical modification with ethylene glycol. Russ J Inorg Chem. 2009;54:1035–40.
Gnanam S, Rajendran V. Facile hydrothermal synthesis of alpha manganese sesquioxide (α-Mn2O3) nanodumb-bells: structural, magnetic, optical and photocatalytic properties. J Alloy Compd. 2013;550:463–70.
Yu CC, Zhang LX, Shi JL, Zhao JJ, Gao JH, Yan DS. A simple template-free strategy to synthesize nanoporous manganese and nickel oxides with narrow pore size distribution, and their electrochemical properties. Adv Funct Mater. 2008;18:1544–54.
Fang DL, Wu BC, Mao AQ, Yan Y, Zheng CH. Supercapacitive properties of ultra-fine MnO2 prepared by a solid-state coordination reaction. J Alloy Compd. 2010;507:526–30.
Frey K, Iablokov V, Sáfrán G, Osán J, Sajó I, Szukiewicz R, Chenakin S, Kruse N. Nanostructured MnOx as highly active catalyst for CO oxidation. J Catal. 2012;287:30–6.
Liu T, Shao G, Ji M, Ma Z. Composites of olive-like manganese oxalate on graphene sheets for supercapacitor electrodes. Ionics. 2014;20:145–9.
Jia ZG, Yue LH, Zheng YF, Xu ZD. Crystallization behavior and thermal decomposition characteristics of manganese(II) oxalate in organic-water binary solvent system. Chin J Inorg Chem. 2007;23:181–8.
Muraleedharan K, Kripa S. DSC kinetics of the thermal decomposition of copper (II) oxalate by isoconversional and maximum rate (peak) methods. J Thermal Anal Calorim. 2014;115:1969–78.
Hu C, Mi J, Shang S, Shangguan J. The study of thermal decomposition kinetics of zinc oxide formation from zinc oxalate dihydrate. J Thermal Anal Calorim. 2014;115:1119–25.
Wu X, Zhou K, Wu W, Cui X, Li Y. Magnetic properties of nanocrystalline CuFe2O4 and kinetics of thermal decomposition of precursor. J Therm Anal Calorim. 2013;111:9–16.
Zhou KW, Wu WW, Li YN, Wu XH, Liao S. Preparation of magnetic nanocrystalline Mn0.5Mg0.5Fe2O4 and kinetics of thermal decomposition of precursor. J Therm Anal Calorim. 2013;114:205–12.
Wu WW, Li YN, Zhou KW, Wu XH, Liao S, Wang Q. Nanocrystalline Zn0.5Ni0.5Fe2O4: preparation and kinetics of thermal process of precursor. J Therm Anal Calorim. 2012;110:1143–51.
Mohamed MA, Galwey AK, Halawy SA. A comparative study of the thermal reactivities of some transition metal oxalates in selected atmospheres. Thermochim Acta. 2005;429:57–72.
Donkova B, Mehandjiev D. Mechanism of decomposition of manganese(II) oxalate dihydrate and manganese(II) oxalate trihydrate. Thermochim Acta. 2004;421:141–9.
Donkova B, Mehandjiev D. In situ thermal magnetic investigation of γ-MnC2O4.2H2O decomposition. Compt Rend Acad Bul Sci. 2013;66:983–90.
Deyrieux R, Berro C, Peneloux A. Contribution à l´étude des oxalates de certains métaux bivalents. III. Structure cristalline des oxalates dihydratés de manganése, de cobalt, de nickel, et de zinc. Polymorphisme des oxalates dihydratés de cobalt et de nickel. Bull Soc Chim Fr. 1973;1:25–34 (in French).
Lethbridge ZAD, Congreve AF, Esslemont E, Slawin AMZ, Lightfoot P. Synthesis and structure of three manganese oxalates: MnC2O4.2H2O, [C4H8(NH2)2][Mn2(C2O4)3] and Mn2(C2O4)(OH)2. J Solid State Chem. 2013;172:212–8.
Wu WY, Song Y, Li YZ, You XZ. One-dimensional structure and long-range antiferromagnetic behaviour of manganese(II) oxalate trihydrate: {[Mn(μ-ox)(H2O)2].H2O}n. Inorg Chem Commun. 2005;8:732–6.
Mancilla N, Caliva V, D’Antonio MC, González-Baró AC, Baran EJ. Vibrational spectroscopic investigation of the hydrates of manganese(II) oxalate. J Raman Spectrosc. 2009;40:915–20.
Huizing A, van Hal HAM, Kwestroo W, Lahgereis C, van Loosdregt PC. Hydrates of manganese(II) oxalate. Mater Res Bull. 1977;12:605–11.
Donkova B. Investigation of the inclusion mechanism of 3d-elements in ZnC2O4.2H2O and its application for obtaining of oxide materials. Ph.D. Thesis. Sofia, Bulgaria, 2005, (in Bulg.).
Macklein ED. Influence of atmosphere on the thermal decomposition of some transition metal oxalates. J Inorg Nucl Chem. 1968;30:2689–95.
Dollimore D, Griffiths DL. Differential thermal analysis study of various oxalates in oxygen and nitrogen. J Therm Anal. 1970;2:229–50.
Dollimore D. The thermal decomposition of oxalates. Thermochim Acta. 1987;111:331–63.
Brown ME, Dollimore D, Galwey AK. The thermochemistry of the decomposition of manganese(II) oxalate dihydrate. Thermochim Acta. 1977;21:103–10.
Zaki MI, Nohman AK, Kappenstein C, Wahdan TM. Temperature-programmed characterization studies of thermochemical events occurring in the course of decomposition of MnII oxysalts. J Mater Chem. 1995;5:1081–8.
Nohman AK, Ismail HM, Hussein GA. Thermal and chemical events in the decomposition course of manganese compounds. J Anal Appl Pyrol. 1995;34:265–78.
Nikumbh AK, Athare AE, Pardeshi SK. Thermal and electrical properties of manganese(II) oxalate dehydrate and cadmium (II) oxalate monohydrate. Thermochim Acta. 1999;326:187–92.
Jacob MU, Perlamutter DD. Thermal decomposition of carbonates, carboxylates, oxalates, acetates, formates, and hydroxides. Thermochim Acta. 1981;49:207–18.
L’vov BV. Kinetics and mechanism of thermal decomposition of nickel, manganese, silver, mercury and lead oxalates. Thermochim Acta. 2000;364:99–109.
Boldyrev VV, Nev’yantsev IS, Mikhailov YI, Khairetdinov EF. K voprosu o myekhanizmye tyermichyeskogo razlozhyeniya oksalatov. Kinet Catal. 1970;11:367–73 (in Russ.).
Zaborenko KB, Kung SC, Melikhov LL, Portyanoi VA. Primenenie kompleksnogo ematzionno-tyermichyeskogo metoda dlya izucheniya razlozhyeniya oksalatov margantza i geleza. Radiokhimiya. 1964;6:749–55 (in Russ.).
Vannatta MW, Richards-Babb M, Sweeney RJ. Oxalate synthesis and pyrolysis: a colorful Introduction to stoichiometry. J Chem Educ. 2010;87:1225–9.
Birkenstock J, Fischer RX, Messner T. BRASS 2003: the Bremen Rietveld Analysis and Structure Suite. Ber DMG, Beih z Eur J Mineral. 2003;15:21–8.
Bail Le. Whole powder pattern decomposition methods and applications: a retrospection. Powder Diffr. 2005;20:316–26.
Zaki MI, Hasan MA, Pasupulety L, Kumari K. Thermochemistry of manganese oxides in reactive gas atmospheres: probing redox composition in the decomposition course MnO2 → MnO. Thermochim Acta. 1997;303:171–81.
Feitknecht W. Einfluss der teilchengrösse auf den mechanismus von festkörperreaktionen. Pure Appl Chem. 1964;9:423–40.
Fritsch S, Sarrias J, Rousset A, Kulkarni GU. Low-temperature oxidation of Mn3O4 hausmannite. Mater Res Bull. 1998;33:1185–94.
Gillot B, Gendouzi ME, Laarj M. Particle size effect on the oxidation-reduction behaviour of Mn3O4 hausmannite. Mater Chem Phys. 2001;70:54–60.
Azzoni CB, Mozzati MC, Galinetto P, Paleari A, Massarotti V, Capsoni D, Bini M. Thermal stability and structural transition of metastable Mn5O8: in situ micro-Raman study. Solid State Commun. 1999;112:375–8.
Gillot B. DTG curves of selective oxidation of submicrometer mixed valency spinels: data table for the oxidation temperature of transition metals and its relation to the cation-oxygen distance. J Solid State Chem. 1994;113:163–7.
Dhaouadi H, Ghodbane O, Hosni F, Touati F. Mn3O4 nanoparticles: synthesis, characterization and dielectric properties. ISRN Spectroscopy. 2012;article ID 706398:8.
Thapa JR, Chattopadhyay KK. Structural transformation from Mn3O4 nanorods to nanoparticles and band gap tuning via Zn doping. Mater Res Bull. 2012;47:813–9.
Wu Z, Yu K, Huang Y, Pan C, Xie Y. Facile solution-phase synthesis of gama-Mn3O4 hierarchical structures. Chem Cent J. 2007;1:8.
Li F, Wu J, Qin Q, Li Z, Huang X. Facile synthesis of γ-MnOOH micro/nanorods and their conversion to β-MnO2, Mn3O4. J Alloy Compd. 2010;492:339–46.
Ishii M, Nakahira M, Yamanaka T. Infrared absorption spectra and cation distributions in (Mn, Fe)3O4. Solid State Commun. 1972;11:209–12.
Busca G. The use of infrared spectroscopic methods in the field of heterogeneous catalysis by metal oxides. In: Jackson SD, Hargreaves JSJ, editors. Metal oxide catalysis. Weinheim: Wiley; 2009. p. 95–175.
Morales MR, Barbero BP, Cadús LE. Combustion of volatile organic compounds on manganese iron or nickel mixed oxide catalysts. Appl Catal B Env. 2007;74:1–10.
Acknowledgements
The authors are thankful to Mr. Nikola Dimitrov for the high quality of the SEM images, as well as to project UNION (DO-02-82/2008) for providing an apparatus PerkinElmer DSC 7.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Donkova, B., Avdeev, G. Synthesis and decomposition mechanism of γ-MnC2O4·2H2O rods under non-isothermal and isothermal conditions. J Therm Anal Calorim 121, 567–577 (2015). https://doi.org/10.1007/s10973-015-4590-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-015-4590-4