
Abstract
Cardioprotective strategies aim to salvage myocardium from ischemia/reperfusion injury and to reduce infarct size and its consequences. Different stimuli, acting at sites remote from the heart (remote conditioning), activate molecular self-defense mechanisms at the target organ heart as well as in other parenchymal organs. Remote conditioning of the heart has been established in many experimental studies and successfully translated to patients. Remote ischemic conditioning by short repetitive cycles of ischemia/reperfusion on an extremity reduces infarct size and improves the prognosis of patients with reperfused myocardial infarction. The present review focuses on three levels of remote conditioning and its resulting cardioprotection: I) at the stimulus level, electrical stimulation, chemical/pharmacological substances, mechanical trauma and cycles of ischemia/reperfusion act at sites remote from the heart, II) at the transfer level, neuronal and humoral mediators transfer the protective signal from the periphery to the heart, and III) at the target level, receptor activation and intracellular signal transduction ultimately affect protection of the myocardium and other organs, as established in different animal models and humans/patients. Remote conditioning is obviously a systemic response. Further mechanistic understanding is mandatory to translate the protection by remote conditioning more successfully to patients with cardiovascular disease.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Historical and conceptual background
In acute myocardial infarction [131], sustained coronary occlusion results most often from the rupture of an atherosclerotic plaque in an epicardial coronary artery and superimposed thrombosis (type I myocardial infarction) [157]. The only way to rescue the ischemic myocardium from impending infarction is timely reperfusion. However, reperfusion is a double-edged sword and does not only terminate ischemic injury, but also inflicts additional reperfusion injury [60, 75, 117, 168]. The final infarct size is therefore the aggregate of both ischemic and reperfusion injury. The final infarct size determines left ventricular function, remodeling [66] and the long-term outcome of patients surviving an acute myocardial infarction [102, 152].
Somewhat paradoxically, brief episodes of myocardial ischemia and reperfusion, which do not cause irreversible injury per se, can even protect from the consequences of prolonged myocardial ischemia and reperfusion by activation of molecular self-defense mechanisms [56]. Such conditioning can either precede (ischemic preconditioning) or follow (ischemic postconditioning) the infarct-causing index ischemia.
Interestingly, protection of the myocardium by conditioning can not only be induced locally by brief episodes of occlusion/ reperfusion in the target coronary artery but also from a distance (remote ischemic conditioning) [67]. In the original study in anesthetized dogs, infarct size in the left anterior descending coronary artery-perfused myocardium was reduced by preceding brief episodes of occlusion/reperfusion in the left circumflex coronary artery [123]. Initially, such protection by conditioning between two adjacent coronary vascular territories was considered as a laboratory curiosity. However, in subsequent studies such remote ischemic conditioning was not only confirmed, but also elicited from a larger distance and from different tissues and organs. Over the years, it became evident that remote conditioning is a systemic response which can be elicited from different tissues and organs, by a variety of stimuli (apart from ischemia/reperfusion) and protect different parenchymal organs from ischemia/reperfusion injury [67].
With respect to the time frame, remote conditioning is classified as pre- (before the target organ ischemia), per- (during the target organ ischemia) or post- (after the target organ ischemia) conditioning. When the time lag between the remote conditioning stimulus and the target ischemia is of longer duration than minutes, there is delayed remote ischemic conditioning [6]. Some studies suggest a dose-response relationship between stimulus strength and protection. In mice, number and duration of remote ischemia/reperfusion cycles in the hind limbs rather than ischemic tissue mass determined the magnitude of protection, as reflected by the reduction of infarct size: 4 to 6 cycles of 2–5 min ischemia / 2–5 min reperfusion on one or two hind limbs reduced infarct size, whereas with 6 cycles of 10 min ischemia and 5 min reperfusion, respectively, cardioprotection was not observed [78]. This finding is in line with the notion of “hyperconditioning”, i.e. an excessive conditioning protocol may cause additional damage to the target organ [164].
The signal transduction of remote conditioning is largely unclear in its details. Whereas the intramyocardial signal transduction of remote conditioning appears to be very similar to that of local conditioning, in particular the transfer signal from the remote tissue or organ to the heart or other target organs is still enigmatic [57]. Both neuronal and humoral mechanisms appear to be causally involved [56].
Remote ischemic conditioning is also operative in humans [61]. In the original proof-of-concept studies, transient ischemia/reperfusion by inflation and deflation of a blood pressure cuff on the arm protected from ischemia/reperfusion-induced endothelial dysfunction on the contralateral arm [86, 103]. In a further translational study, the myocardial injury by ischemic cardioplegic arrest during elective coronary artery bypass graft (CABG) surgery was taken as a surrogate of myocardial infarction, and the troponin release following CABG was reduced by remote ischemic preconditioning with 3 cycles of 5 min forearm ischemia and 5 min reperfusion [47]. Subsequently, infarct size (single photon emission computed tomography) was indeed reduced by remote ischemic perconditioning with brief cycles of forearm ischemia/reperfusion during the transport of patients with acute myocardial infarction in the ambulance to the hospital and before interventional reperfusion [9].
Signal transduction of remote conditioning
The remote stimulus acts, per definition, at a distance from the target organ. A variety of remote stimuli elicit target organ protection: a) electrical stimuli such as direct or percutaneous electrical nerve stimulation [110, 129] or electroacupuncture [37, 130, 159], b) chemical/pharmacological stimuli such as capsaicin [46, 79, 129] or autacoids (adenosine [99, 151], bradykinin [46, 136]), c) mechanical stimuli such as trauma i.e. by surgical skin incision [45, 46, 79], or d) cycles of ischemia/reperfusion of an extremity or organ [25, 68, 71, 126, 128, 142, 145, 151] which again comprise chemical and mechanical stimuli. Common to all these stimuli is an activation of peripheral sensory nerves. For the ischemia/reperfusion stimulus, it is not entirely clear to which extent the peripheral sensory nerve activation or additional mechanisms such hypoxia/acidosis or vascular shear stress contribute to remote conditioning [67, 88]. Currently it is impossible to compare the different stimuli and to estimate the most effective remote stimulus, since for none of the stimuli a stimulus strength-response relationship has been established. All such comparisons done so far [79, 130, 151] are therefore of limited value.
Neuronal and humoral components are involved in the signal transfer from the peripheral stimulus site to the target organ; they act in concert and interact on three different levels. Stimulus level: peripheral sensory nerves are directly activated and/or humoral factors are released which subsequently activate the peripheral sensory nerves [43]. Systemic level: the peripheral sensory afferent nerves travel to the spinal cord, project into autonomic centers of the central nervous system to result in activation of efferent vagal nerves [43]. Vagal nerve activation a) releases acetylcholine to activate muscarinic receptors in the target organs, and/or b) activates muscarinic receptors in non-target organs which release humoral factors which subsequently act on the target organs [7]. Target level: apart from and in addition to a) neuronal activation of receptors in the target organ, b) released humoral factors can activate the intrinsic nervous system of the target organ (e.g. intracardiac ganglia) [7, 121] or c) mediate a receptor-dependent or -independent response of the specific parenchymal cells of the target organ. Neither the quantitative amount nor the exact nature of neuronal and humoral mediators in remote ischemic conditioning are currently clear [43, 56, 57, 67].
The response to remote ischemic conditioning is obviously systemic because it occurs in all organs tested so far. Cell injury i.e. the consequence of ischemia/reperfusion is reduced by remote ischemic conditioning in the brain, the heart, the intestine, the kidneys, the liver, the lungs, the ovaries, the pancreas, the skeletal muscle, and the skin [15]. In different cell types, common intracellular signal transduction pathways are involved. However, a direct comparison of intracellular signaling between different cell types has not been performed so far, and signaling may to a certain extent be organ-specific. The intracellular signaling in cardiomyocytes is probably species-specific [56, 67]. Notwithstanding the specific signal transduction pathways, mitochondria are an important end-effector of protection [56, 64]. Ischemia/reperfusion injury causes, independently of the cell type and organ-specific susceptibility to this injury, cell death by different processes (necrosis, necroptosis, apoptosis and/or autophagy) [75, 85], and preserved mitochondrial function protects from such cell death.
In the following, we discuss the current knowledge about underlying mechanisms of remote conditioning, its stimuli, the signal transfer from the periphery to the target organ and the respective cellular response within the target organ. (Fig. 1) Most studies on cellular response/intracellular signal transduction by remote conditioning were performed in the myocardium. Therefore, we specifically focus on the target organ heart.

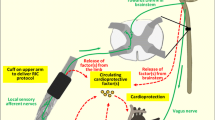
Remote conditioning - neuronal and humoral signal transfer from the source organ where the stimulus acts to the target organ heart
Remote stimulus
The different remote stimuli - electrical, chemical/pharmacological, mechanical and also cycles of ischemia/reperfusion - all act as noxious triggers which activate peripheral sensory nerves. A causal involvement of neuronal activation is evidenced by the response to stimuli which exclusively activate peripheral sensory nerves, e.g. electrical nerve stimulation or chemical/pharmacological nociceptor stimuli such as capsaicin [46, 79, 129]. The resulting cardioprotection is abolished by peripheral nerve transection or by local anesthesia [79, 129]. However, even in response to peripheral nerve stimulation humoral factor(s) are causally involved in signal transfer. Whether or not the humoral factor(s) are locally released as a direct response to the stimulus or are released from other neuronally activated organs is currently unclear [7, 57, 107, 120, 121]. So far, all studies investigating humoral signals in response to different stimuli used blood collected from the systemic circulation and its derivatives, such as plasma or plasma dialysate, respectively. Local release of humoral factor(s) was suggested in a study in mice where occlusion of the femoral vein draining the leg undergoing remote ischemic conditioning by 3 cycles of femoral artery occlusion and reperfusion abrogated infarct size reduction [100].
Electrical nerve stimulation (4 cycles of 5 min femoral nerve stimulation and 5 min rest) in rabbits induced the release of humoral factor(s) into the systemic circulation which reduced infarct size after transfer to an isolated acceptor rabbit heart [129]. Electroacupuncture at the forelimb reduced myocardial infarct size in rodents [37, 159] and induced the release of humoral factor(s) into the systemic circulation of rabbits, which then reduced infarct size after transfer to an isolated acceptor rabbit heart [130]. Cardioprotective humoral factor(s) were also released into the systemic circulation after cycles of transcutaneous electrical nerve stimulation of rabbit and human limbs [110]. Femoral nerve transection prevented the release of cardioprotective humoral factors after transcutaneous electrical nerve stimulation [129], but did not prevent the release of nitric oxide and subsequent infarct size reduction by remote ischemic conditioning [128], suggesting an additive/independent action of shear stress as stimulus. (Fig. 2)

Stimuli of remote conditioning: peripheral sensory nerve activation / vascular shear stress. DMSO dimethyl sulfoxide; NO nitric oxide, NOS nitric oxide synthase, PKC protein kinase C
Local chemical/pharmacological stimuli
Topical capsaicin selectively activates non-myelinated afferent C-fibers, and capsaicin when applied to the abdominal skin elicited infarct size reduction in rodents [46, 79]. Local anesthesia with lidocaine [79] or blockade of non-myelinated C-fiber activation by dimethyl sulfoxide (DMSO) abrogated the protection by application of capsaicin to the abdominal skin in rabbits [129]. C-fiber activation is nitric oxide-sensitive, and local application of a nitric oxide donor prior to nerve stimulation prevented the release of transferable cardioprotective humoral factor(s) [110]. Noxious mechanical stimuli also activate C-fiber nociceptors [33, 80]. Intra-arterial infusion of the autacoids adenosine [99] or bradykinin [136], in doses activating local sensory neurons without a systemic effect or direct activation of myocardial receptors, reduced myocardial infarct size in rodents. The infarct size reduction by adenosine or bradykinin can be elicited from different organs. Mesenteric artery infusion of adenosine [99] or bradykinin [136] in rats, femoral artery infusion of adenosine in rabbits [151] and in patients undergoing elective coronary angiography [24], but also application of bradykinin to the abdominal skin of rats [46] reduced infarct size [126]. In rats, in response to bradykinin application to the abdominal skin, two protein kinase C (PKC) isozymes (PKCε and PKCɣ) were involved in the local signal, and their inhibition abrogated the cardioprotection by bradykinin [46] (Fig. 2).
Trauma by transverse abdominal skin incision, i.e. partly a mechanical stimulus, reduced myocardial infarct size in rodents [46, 79] and dogs [45]. Such abdominal skin incision certainly activates sensory nerves. However, it is unclear to which extent other stimuli (interruption of blood vessels, edema and tissue compression) are also involved. This nociceptive, neuronal activation by skin incision involved also - as did topical bradykinin – a local activation of PKCε and PKCɣ [46]. Thus, there may be an interplay between bradykinin signaling and nerve activation. Indeed, protection in response to skin incision was prevented by blockade of bradykinin 2 receptors in mice and dogs [45, 79]. Since in these studies bradykinin 2 receptors were blocked systemically, it is unclear whether bradykinin is released and acts locally in response to the nociceptive stimulus or more downstream and systemically (Fig. 2).
Ischemia/reperfusion of an extremity or parenchymal organ reduces myocardial infarct size and induces the release of humoral factor(s) into the systemic circulation in rodents, pigs and humans, which elicit cardioprotection after transfer to an isolated acceptor heart [71, 142, 145]. Ischemia/reperfusion involves mechanical stimuli through vascular occlusion and tissue compression as well as chemical factors (adenosine and bradykinin) [88, 126]. Femoral nerve transection abrogated the infarct size reduction by 3 cycles of 5 min ischemia and 5 min reperfusion of one hind limb in rabbits [30]. Sensory nerve activation by remote ischemic conditioning with cycles of blood pressure cuff inflation/deflation of the arm was suggested by the lack of release of transferable humoral cardioprotective factors in humans with diabetic neuropathy as compared to diabetics without neuropathy and healthy controls [77]. However, it is not clear whether the stimulus or the transfer signal is absent in diabetic neuropathy.
Local adenosine receptor blockade by intra-arterial caffeine infusion abrogated the release of humoral factor(s) in response to remote ischemic conditioning in healthy volunteers by 3 cycles of 5 min forearm ischemia and 5 min reperfusion [24], indicating that adenosine mediates a local neuronal activation in response to ischemia/reperfusion.
The non-selective, acid-sensitive, cation channel transient receptor potential vanilloid 1 (TRPV1) is widely expressed on primary sensory nerves including sensory C-fibers [124]. Physical and/or chemical stimuli activate TRPV1 and subsequently trigger the release of neuropeptides such as calcitonin gene-related peptide (CGRP) and substance P from peripheral nerve terminals [112]. In rats, the infarct size reduction by remote ischemic conditioning with 3 cycles of 5 min ischemia and 5 min reperfusion on one hind limb was associated with a TRPV1-mediated CGRP and substance P release [38]. The TRPV1 antagonist capsazepine abolished the release of CGRP and substance P and the observed infarct size reduction [38]. However, capsazepine was applied systemically [38], and thus the participation of TRPV1 activation at other parts of the organism cannot be excluded. In rats, an intrathecal and intravenous administration of CGRP receptor antagonists abolished the infarct size reduction by intrathecal morphine administration [101], indicating that there is an interaction of CGRP- with opioid-mediated cardioprotection. Ischemia/reperfusion also induces tissue hypoxia, acidosis (during ischemia) and reactive hyperemia (during reperfusion), which trigger the release of local and humoral factor(s). Hypoxia of the perivascular parenchymal tissue may stimulate the expression of hypoxia-sensitive factors, which are subsequently released into the circulating blood [68]. The acidosis resulting from ischemia activates acid-sensitive sensory nociceptors of the vasculature [80]. The reactive hyperemia during reperfusion increases vascular shear stress and induces the release of humoral factor(s) from the endothelium such as nitric oxide [128]. A causal role for such nitric oxide release in response to vascular shear stress was evidenced by its prevention when reactive hyperemia was suppressed by controlled partial vascular compression with an ultrasonic probe in healthy volunteers undergoing remote ischemic conditioning by one cycle of 5 min blood pressure cuff inflation / 5 min deflation on the arm [128] (Fig. 2).
Signal transfer
Neuronal signal transfer
Neuronal signal transfer is certainly involved in remote conditioning of the heart [43], but details are not entirely clear.
Hexamethonium inhibits impulse transmission from preganglionic to postganglionic neurons of both the sympathetic and parasympathetic nerve systems. Hexamethonium abrogates cardioprotection by remote conditioning in some but not in all studies. Remote conditioning by surgical trauma in mice [79] and by one cycle of 15 min ischemia and 10 min reperfusion of the mesentery in rats [41] was abrogated by hexamethonium. Ganglionic blockade with trimetaphan abrogated also the protection of endothelium-dependent vasodilation by remote ischemic conditioning with 3 cycles of 5 min forearm ischemia and 5 min reperfusion in healthy volunteers [103]. However, the infarct size reduction by occlusion/reopening of the infrarenal aorta was not attenuated by hexamethonium in rats [163]. (Fig. 3)

Neuronal transfer of remote conditioning
In healthy humans with experimental ischemia/reperfusion in the forearm, remote ischemic conditioning of one leg by 2 cycles of 5 min ischemia and 5 min reperfusion attenuated the ischemia- induced sympathetic nerve activation in the muscle of the contralateral leg [94]. Optogenetic cell-specific silencing of neurons in the brainstem dorsal motor nucleus of the vagus nerve [107], but also bilateral cervical or subdiaphragmatic vagotomy abolished the infarct size reduction by remote ischemic conditioning with one cycle of 15 ischemia and 10 min reperfusion on both hind limbs in rats [7]. Conversely, electrical vagal nerve stimulation mimicked the effect of remote ischemic conditioning in rabbits [30] and pigs [160] and reduced infarct size. The infarct size reduction by electrical vagal nerve stimulation during myocardial ischemia and reperfusion was sensitive to nitric oxide and abolished by systemic inhibition of nitric oxide synthase [160] (Fig. 3).
The spinal cord also participates in neuronal signal transfer. In rats, blockade of spinal cord nerve transmission by intrathecal lidocaine abrogated the infarct size reduction by intrathecal application of morphine in 3 cycles of 5 min [105]. Also in rabbits, spinal cord section at T9-T10 level abolished the infarct size reduction by remote ischemic conditioning with 3 cycles of 5 min ischemia and 5 min reperfusion on one hind limb [30]. An intrathecal μ-opioid receptor antagonist abrogated the infarct size reduction by skin incision in rodents whereas antagonists of δ- or κ-opioid receptor subtypes did not [109, 165]. Such cardioprotection was associated with an adenosine release into spinal cord tissue of the T1-T5 region, and the μ-opioid receptor antagonist inhibited such adenosine release [109] (Fig. 3).
The involvement of supraspinal central nervous centers is contentious. Spinal cord integrity above C7 was not required for cardioprotection by skin incision in mice, but an intact spinal cord at T7 was required [79]. In rabbits, direct electrical stimulation of the spinal cord at C8-T2 before myocardial ischemia reduced infarct size [149]. However, the above studies and the involvement of brainstem vagal nuclei and vagal nerve activation support a mandatory role for supraspinal neuronal centers [7, 43, 57, 107].
Thus, given the current data it is unclear a) whether or not the brainstem must be involved, and b) which level(s) of the spinal cord are mandatory for signal transfer. Possibly, the involved neuronal level(s) depend on the remote stimulus and/or on the species under study.
Humoral signal transfer
In response to all remote stimuli, there is humoral signal transfer to the target organ heart. However, it is unclear whether the different stimuli operate via the same or different humoral factor(s) and how these factor(s) possibly interact. The core paradigm for the identification of humoral cardioprotective signals is the transfer of protection with blood, plasma or plasma dialysate from the donor organism which underwent a remote ischemic conditioning protocol to an isolated perfused acceptor heart that serves as a bioassay [28, 56, 71, 142]. In a more reductionist approach, coronary effluent after local ischemic conditioning of a donor heart (rat, rabbit) was used for transfer experiments to an isolated perfused acceptor (rat, rabbit) heart [29, 138]. Such transfer experiments have then been combined with analytical procedures and pharmacological inhibitor approaches. The analytical procedures comprised non-biased approaches such as gene microarrays and proteomic/metabolomic mass spectroscopy techniques. Other more hypothesis-driven studies have just focused on single humoral factors in isolation.
General properties of the humoral factor(s)
Cardioprotection in response to remote ischemic conditioning is transferable with whole blood from a donor to an acceptor rabbit [28], or with diluted plasma or plasma dialysate to an isolated perfused heart of the same species [142] or even across species [56, 145]. Neither the efficiency of cardioprotection by transfer of whole blood versus that of pure plasma nor that of plasma or plasma dialysate at different dilutions have been compared so far. Most studies used a plasma dialysate (< 12-14 kDa) with a final dilution of 1:10–1:50 for transfer experiments. Technical reasons (e.g. extensive foaming of undiluted plasma or adverse side effects by immune responses of the acceptor heart to plasma proteins of another species) are the background for the use of diluted plasma or plasma dialysate for such transfer experiments [71, 142]. However, as a side product of the successful humoral transfer with diluted dialysate, two properties of one or more humoral factor(s) are indirectly identified: a) its/their size is below <12-14 kDa (cut-off), and b) the original factor(s)` plasma concentration is effective in an up to 1:10–1:50 dilution. In plasma of pigs taken after vs. before remote ischemic conditioning by renal artery occlusion for 10 min and 20 min reperfusion no different expression of proteins and peptides >8-10 kDa was identified [95], supporting the notion that proteins and peptides <8 - 10 kDa are most relevant for the transfer of protection.
The protective, humoral factor(s) were further characterized as hydrophobic, thermolabile and lyophilizable. With a purified hydrophobic and with a lyophilized fraction of coronary effluent of rat and rabbit hearts undergoing local ischemic preconditioning the cardioprotection was transferable to an acceptor rat [138] or rabbit heart [29], respectively. Also, when rabbit plasma dialysate taken after remote ischemic conditioning by 4 cycles of 5 min ischemia and 5 min reperfusion on one hind limb was perfused through a hydrophobic column, the infarct size reduction in acceptor rabbit hearts was abrogated [142]. Infarct size reduction was abrogated when coronary effluent was heated at 70 °C [138]. These characteristics (hydrophobic, lyophilizable and thermolabile) suggest that some of the released humoral factor(s) are a peptide or protein.
After a single protocol of 3 cycles of 5 min forearm ischemia and 5 min reperfusion in healthy volunteers, plasma dialysate (<12-14 kDa) of blood taken at 5 min after and in the following 6 days following the protocol still reduced infarct size in isolated mouse hearts, and the cardioprotection disappeared no earlier than after 7 days [71]. Whether there is a circulating cardioprotective signal over one week following a single remote ischemic conditioning protocol or whether there is a rapid and continuous release of one or more factor(s) with a short half-life remains unclear from this study. A proteomic approach on plasma taken from healthy volunteers 15 min and 24 h after 3 cycles of 5 min forearm ischemia and 5 min reperfusion identified a time-dependent difference in up/down-regulated plasma proteins [54], indicating that different factors are appearing at a different time-scale, but this study neither identified these factors nor did it attribute a causal role to them in mediating cardioprotection.
Non- biased “omic” approaches
In response to remote ischemic conditioning in mice and humans by 4 cycles of 5 min ischemia and 5 min reperfusion on one hind limb or forearm, respectively, a microRNA (miRNA) array identified an association of increased systemically circulating miRNA144 with remote ischemic conditioning. The causal involvement of miRNA144 in cardioprotection was proven by induction of cardioprotection with exogenous miRNA144 and abrogation of infarct size reduction by remote ischemic conditioning with a specific antisense oligonucleotide [98]. An increase of plasma miRNA144, induced by remote ischemic conditioning with 4 cycles of 5 min forearm ischemia and 5 min reperfusion, was confirmed in patients with suspected ischemic coronary artery disease undergoing diagnostic cardiac positron emission tomography [122]. (Fig. 4)

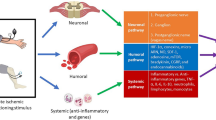
Humoral transfer of remote conditioning. CGRP calcitonin gene-related peptide; EPO erythropoietin; GLP-1 glucagon-like peptide-1; HIF-1α hypoxia-inducible factor-1α; IL-10 interleukin-10; miRNA microRNA; NO nitric oxide, NOS nitric oxide synthase, SDF-1α stromal cell-derived factor-1α
Several proteomic/metabolomic approaches did not identify a specific humoral protective factor or a “protective cocktail” of factor(s) after remote ischemia/reperfusion cycles in the limbs [70] or kidneys of rats [95], in adult healthy volunteers [52] and in children undergoing heart surgery for repair of Fallot’s tetralogy [55]. However, two different proteomic approaches on rat and human plasma after remote ischemic conditioning identified an increase of apolipoprotein A1 [54, 69]. In rats, this apolipoprotein A1 increase in response to one cycle of 10 min ischemia and 5 min reperfusion on one hind limb was associated with infarct size reduction [69]. Apolipoprotein A1 infusion before myocardial ischemia/reperfusion reduced infarct size [69] and shared intracellular signaling pathways with that of remote ischemic conditioning in rats [84]. Using a metabolomic approach in rat and patient plasma after remote ischemic conditioning by 4/3 cycles of 5 min hind limb/forearm ischemia and 5 min reperfusion, respectively, there was a decrease of ornithine and an increase of kynurenine and glycine in both species. In rats, the intraperitoneal infusion of exogenous kynurenine and glycine alone or in a combination 10 min before myocardial ischemia, respectively, reduced infarct size [20]. However, the doses of such exogenous amino acids were up to 10-fold higher than those determined in response to the remote ischemic conditioning stimulus, raising doubts about the causal involvement of kynurenine and glycine in the transfer of cardioprotection by remote ischemic conditioning (Fig. 4).
Hypothesis-driven approaches on individual humoral factors
Autacoids: Autacoids are endogenously released during local ischemic pre- and postconditioning, and exogenous application of autacoids induces cardioprotection. Adenosine as well as bradykinin receptor blockade abrogates the infarct size reduction by local conditioning [88]. Whether there is an endogenous release of autacoids into the systemic circulation in response to remote conditioning is not really clear.
Indeed, adenosine was increased in the coronary effluent of rabbits after local ischemic preconditioning, and this effluent reduced infarct size after transfer to another rabbit heart. Infarct size reduction was abrogated with an adenosine receptor blocker [97], suggesting the potential for adenosine to also contribute to the humoral transfer of remote conditioning. In rats in response to remote conditioning by skin incision, the infarct size reduction was associated with an adenosine release into spinal cord tissue [109], but circulating blood levels were not determined. In mice, in response to persistent brain ischemia through bilateral ligation of the internal carotid arteries there was an increase of arterial plasma, but not cerebral adenosine, and this increase in arterial adenosine was subsequently associated with infarct size reduction of isolated perfused mice hearts [137]. Persistent brain ischemia, however, is not really the same as remote ischemic conditioning which requires brief intermittent episodes of reversible ischemia and reperfusion.
In non-diabetic patients undergoing elective coronary angiography, intra-arterial infusion of exogenous adenosine was associated with the release of transferable cardioprotective factor(s). Transfer of plasma dialysate, taken after the arterial adenosine infusion, reduced infarct size in isolated perfused mouse hearts to an extent which could not be reproduced by the adenosine concentration in the dialysate [24]. Thus, adenosine in fact stimulated the release of cardioprotective factor(s), but adenosine itself was not the cardioprotective factor (Fig. 4).
Currently, there is no evidence that bradykinin is released into the systemic circulation in response to remote conditioning. Furthermore, the intravenous infusion of a bradykinin B(2) receptor antagonist did not abrogate the attenuation of ischemia/reperfusion-induced endothelial dysfunction in humans by remote ischemic conditioning by 3 cycles of 5 min forearm ischemia and 5 min reperfusion [119], suggesting that bradykinin is not a humoral transfer factor of cardioprotection.
Hormones: The glycoprotein hormone erythropoietin (EPO) (phylogenetically, EPO is related to the family of cytokines) is produced by the kidneys, the liver, the vasculature and the heart [36]. In mice, remote ischemic conditioning by 4 cycles of 5 min ischemia and 5 min reperfusion on one hind limb increased serum EPO and reduced infarct size. This increase in serum EPO was associated with increased EPO mRNA expression in the kidneys, and exogenous EPO antibodies before remote ischemic conditioning abolished the infarct size reduction [113]. Interestingly, there was an interaction between neuronal signal transduction and EPO release, since renal denervation abolished both the increase in serum EPO and the infarct size reduction by remote ischemic conditioning [113] (Fig. 4).
Recently, the incretin hormone glucagon-like peptide-1 (GLP-1) was reported as a potential humoral transfer factor. In rats, in response to remote ischemic conditioning by one cycle of ischemia/reperfusion of both hind limbs for 15 min, activation of the vagal nerves appeared to mediate a release of GLP-1 release from the gut. Subdiaphragmatic vagotomy abolished the infarct size reduction, and infarct size reduction was also achieved by a GLP-1 receptor agonist. Systemic muscarinic receptor M3 blockade abolished the infarct size reduction by the GLP-1 agonist [7]. Thus, GLP-1 appeared to be released from visceral organs which are innervated by the posterior gastric branch of the vagus nerve, and it involves the activation of muscarinic M3 receptors in the heart. However, the reported increase of circulating GLP-1 in response to remote ischemic conditioning was minimal and could not explain the observed protection [57]. Also, there was no distinction between the inactive full length GLP-1 and the active peptide of GLP-1 [72] (Fig. 4).
Cytokines/Chemokines: Local ischemic conditioning is associated with the inhibition of inflammatory responses [56]. In mice, 24 h after remote ischemic conditioning by 3 cycles of 5 min ischemia and 5 min reperfusion on one hind limb, the plasma concentration of the anti-inflammatory interleukin-10 (IL-10) was increased. The infarct size reduction by remote ischemic conditioning was abrogated, when the activation of IL-10 receptors was prevented by anti-IL-10 receptor antibodies or by a genetic knock out of IL-10 [13] (Fig. 4).
The chemokine stromal cell-derived factor-1α (SDF-1α) is abundant in ischemic tissue with a vascular and perivascular distribution after dermal ischemia in athymic nude mice [19]. In rats, SDF-1α was released into the plasma in response to remote ischemic conditioning by 3 cycles of 5 min ischemia and 5 min reperfusion on one hind limb. Inhibition of the SDF-1α chemokine 4 receptor attenuated the infarct size reduction by remote ischemic conditioning, but did not completely abrogate it [25] (Fig. 4).
Neuro-peptides: Opioid peptides are widely distributed in the central nervous system but they are also - eventually as a spillover from neuronal release- released into the blood in response to different remote stimuli [125]. A quantitative release of dynorphin, but not of met-enkephalin, into the plasma was reported in rats in response to remote ischemic conditioning by 3 cycles of 5 min ischemia and 5 min of reperfusion of one hind limb [172]. In healthy volunteers, however, remote ischemic conditioning by 4 cycles of 5 min forearm ischemia and 5 min reperfusion did not induce an increase of pro-opiomelanocortin derivates [8]. Non-specific opioid receptor blockade with naloxone abrogated the infarct size reduction by remote ischemic conditioning in mice and rats [51]. Also in patients undergoing primary percutaneous coronary intervention, the opioid receptor blocker naloxone abrogated the reduction of troponin release by remote ischemic conditioning with 3 cycles of 4 min forearm ischemia and 4 min reperfusion [132], emphasizing the relevance of opioids for cardioprotection by remote ischemic conditioning. (Fig. 4) However, whether the cardioprotection is mediated/transferred by circulating opioids or whether they are just a spillover of local neuronal release and the cardioprotection is actually mediated by local neuronal release in the myocardium is currently unclear.
In rats, the release of the neuropeptides CGRP and substance P into the plasma was triggered by the activation of TRPV1 in response to 3 cycles of 5 min ischemia and 5 min reperfusion of one hind limb. Both peptides were causally involved in the infarct size reduction by remote ischemic conditioning, and CGRP and substance P receptor antagonists abolished the infarct size reduction [38] (Fig. 4).
Amino acids: Several studies suggested that one or more amino acids may act as humoral transfer factors of protection. The enzyme hypoxia-inducible factor prolyl hydroxylase 2 or prolyl hydroxylase domain-containing protein 2 is encoded by the Egln1 gene and therefore termed Egln1. Egln1 is ubiquitously present in nearly all cell types and requires oxygen for its enzymatic activity [81]. In skeletal muscle, Egln1 converts α-ketoglutarate to succinate, and its activity is reduced with tissue hypoxia. Accordingly, the α-ketoglutarate concentration in the systemic circulation is increased with lack of Egln1 activity. In the liver, α-ketoglutarate is metabolized to kynurenic acid. In mice, the genetic deletion or pharmacological inhibition of Egln1, either systemically or in skeletal muscles, reduced infarct size. The humoral transfer of protection was determined in parabiosis experiments (n = 2) between donor mice with a skeletal muscle-specific Egln1 deletion and wild type recipient mice undergoing coronary occlusion/reperfusion. Infarct size was reduced in the recipient mice. Exogenous kynurenic acid also reduced infarct size [116]. In response to Egln1 deletion, the hypoxia-inducible factor-1α (HIF-1α) protein expression was increased [116], further relating these data to hypoxia-sensitive pathways. However, this particular study did not actually perform a single remote ischemic conditioning protocol and the conclusions of this study on mechanisms of remote ischemic conditioning are therefore largely speculative. As mentioned above, increased kynurenine was also determined in a metabolomic approach in rat and patient plasma after remote ischemic conditioning by 4/3 cycles of 5 min hind limb/forearm ischemia and 5 min reperfusion, respectively [20], and exogenous kynurenine and glycine also reduced infarct size in rats [20]. However, a causal relation between Egln1, kynurenic acid and remote ischemic conditioning remains to be established (Fig. 4).
Nitric oxide/nitrite: The role of nitric oxide as a humoral transfer signal of remote ischemic conditioning remains controversial. The oxidative metabolite of nitric oxide -nitrite- was increased in plasma in response to remote ischemic conditioning in rodents and humans. The nitric oxide release was prevented by genetic ablation of endothelial nitric oxide synthase but not by femoral nerve transection in mice [128]. Also in rats, systemic nitric oxide synthase inhibition abolished the infarct size reduction by 3 cycles of 5 min ischemia and 5 min reperfusion on one hind limb or one cycle of 15 min ischemia and 10 min reperfusion on both hind limbs, respectively [31, 139], whereas in rabbits, such systemic pre-treatment with a nitric oxide synthase inhibitor did not attenuate the protection by remote ischemic conditioning with 4 cycles of 5 min ischemia and 5 min reperfusion on one hind limb [151]. Two studies in humans used not nitrite but the sum of plasma nitrite and nitrate (NOx) to quantify the nitric oxide release in response to cycles of 5 min ischemia and 5 min reperfusion on one upper leg. Whereas in patients undergoing elective percutaneous coronary intervention NOx was released into coronary arterial blood after 3 cycles of 5 min forearm ischemia and 5 min reperfusion [3], there was no such release of NOx into the venous blood of healthy volunteers after 2 cycles of 5 min forearm ischemia and 5 min reperfusion [94]. Nitrite is more stable than the radical nitric oxide itself and enables therefore the transport of eNOS-derived nitric oxide with the circulating blood from the periphery to the heart [127] (Fig. 4).
Ribonucleic acid (RNA) degrading enzyme: With tissue injury, RNA is released into the extracellular space and it amplifies and propagates tissue injury. Circulating RNase1 degrades extracellular RNA and limits tissue damage [171]. Remote ischemic conditioning by 4 cycles of 5 min forearm ischemia and 5 min reperfusion increased endothelial RNase1 in the circulating blood in a smaller cohort of patients undergoing heart surgery [12]. However, troponin release was not reduced by the remote ischemic conditioning protocol in this particular patient cohort, and therefore a causal involvement of RNase1 in cardioprotection is still elusive (Fig. 4).
HIF-1α is potentially one common regulator of different humoral factors [68]. HIF-1α is a major regulator of hormone and cytokine signaling in response to hypoxia [106], and in HIF-1α heterozygous knockout mice the infarct size reduction by remote ischemic conditioning with 3 cycles of 5 min ischemia and 5 min reperfusion on one hind limb was abrogated [14]. EPO synthesis is initiated through HIF-1α [36]. However, currently it is unclear whether the EPO release in response to remote ischemic conditioning is also HIF-1α-dependent. The IL-10 release into the plasma in response to remote ischemic conditioning by 3 cycles of 5 min ischemia and 5 min reperfusion on one hind limb was abolished in mice with heterozygous deletion of HIF-1α [13]. Upregulation of HIF-1α expression in the hind limb after the remote ischemic conditioning protocol by 4 cycles of 5 min ischemia and 5 min reperfusion on one hind limb was confirmed in another study in mice, but this HIF-1α expression was not a prerequisite for infarct size reduction [83]. This study therefore does not establish a causal role for HIF-1α in the peripheral release of humoral factor(s) or its participation in humoral transfer. As mentioned above, hypoxia-induced Egln1 inhibition [116] may be one upstream regulator of HIF-1α.
Particulate carriers of humoral signals
Blood cells: Apart from and in addition to endothelial nitric oxide synthase, the nitric oxide synthase of red blood cells was also activated by remote ischemic conditioning with 4 cycles of 5 min forearm ischemia and 5 min reperfusion in healthy volunteers [44]. Thus, the nitric oxide synthase of red blood cells may therefore act as an additional, circulating source of circulating nitrite (Fig. 4).
In patients undergoing elective CABG surgery, remote ischemic conditioning by 3 cycles of 5 min forearm ischemia and 5 min reperfusion down-regulated the expression of bradykinin receptors 1 and 2 expression on leukocytes [135]. The expression of adhesion molecules on neutrophils was bradykinin receptor-dependent [140]. However, whether such differences in bradykinin receptor-dependent adhesion molecule expression result in decreased/increased leukocyte recruitment after myocardial infarction has not been examined so far.
Vesicles/particles: Extracellular vesicles/microparticles and exosomes are circulating extracellular lipid bilayer vesicles with a particle size of 50–1000 / 30–100 nm in diameter, respectively, and considered to be a carrier of information for intercellular communication. Extracellular vesicles/exosomes originate from different cell types and contain proteins, mRNAs, miRNAs, and DNAs [5, 74, 167]. Whether or not extracellular vesicles/exosomes and which particular type of them may contribute to myocardial protection in response to remote ischemic conditioning is currently not clear [26].
The total amount of extracellular vesicles was increased after local ischemic conditioning in the coronary effluent of isolated rat hearts, and administration of the coronary effluent reduced infarct size of isolated recipient hearts, whereas extracellular vesicles-depleted perfusate failed to reduce infarct size [42]. In contrast, after remote ischemic conditioning in mice and humans by 4 cycles of 5 min hind limb / forearm ischemia and 5 min reperfusion, respectively, the total amount of extracellular vesicles was not increased [98]. Extracellular vesicles originating from endothelial cells were increased in circulating blood after remote ischemic conditioning by one cycle of 10 min ischemia and 10 min reperfusion on one hind limb in rats and by 3 cycles of 5 min forearm ischemia and 5 min reperfusion in humans, respectively. However, these vesicles did not reduce infarct size when intravenously transferred to a donor rat [76] (Fig. 4).
Exosomes were increased in response to remote ischemic conditioning by 3 cycles of 5 min ischemia and 5 min reperfusion on one hind limb in rats or the forearm in humans, respectively. When such exosomes were transferred intravenously to an acceptor rat or ex-vivo to an isolated rat heart, the exosomes reduced infarct size, no matter whether they were taken before or after remote ischemic conditioning. In isolated cardiomyocytes, these exosomes activated toll-like receptor 4 and various kinases and induced an activation of cardioprotective heat shock protein27 [161].
Which specific vesicular content is responsible for or involved in the observed cardioprotection is not clear. The total DNA content [155] was unchanged, whereas the total mRNA content [154] was increased in extracellular vesicles in the coronary venous blood of pigs after local myocardial ischemic conditioning. In a more specific analysis, extracellular vesicles carried the cardioprotective miRNA144, but the miRNA144 content was unchanged after remote ischemic conditioning by 4 cycles of 5 min ischemia and 5 min reperfusion on one hind limb/forearm in mice and humans, respectively. The content of a miRNA144 precursor, however, was almost fourfold increased in these extracellular vesicles [98].
Differences of the particle type (extracellular vesicles/microparticles versus exosomes) and/or their cellular origin complicate the interpretation of the available data [5, 167].
Neuro-humoral interaction
There is a complex neuro-humoral interaction in remote ischemic conditioning’s signal transduction in the periphery, systemically and in the target organ. Activated afferent but also efferent nerves induce the release of humoral factor(s) and, vice versa, humoral factors activate afferent and efferent nerves. Within the myocardium, the intrinsic nervous system is activated by humoral factor(s) and probably also by locally released factors.
At the stimulus level, peripheral adenosine receptor activation after intra-arterial adenosine infusion in human volunteers was mandatory to induce the release of transferable cardioprotective factor(s) [24], suggesting that adenosine may activate peripheral sensory nerve fibers to ultimately cause the release of a humoral factor(s) and reflecting neuro-humoral interaction. (Fig. 5)

Neuro-humoral interaction in response to remote conditioning. CGRP calcitonin gene-related peptide; EPO erythropoietin; GLP-1 glucagon-like peptide-1; M3 muscarinic acetylcholine receptor M3
At the transfer level, in mice, the infarct size reduction by remote ischemic conditioning with 4 cycles of 5 min ischemia and 5 min reperfusion on one hind limb was markedly attenuated by femoral vein occlusion. Also, femoral nerve or sciatic nerve resection only partially abolished the infarct size reduction. Only combined femoral and sciatic nerve resection with femoral vein occlusion completely abolished the cardioprotective effect [100], indicating that there is a neuro-humoral interaction in the afferent signal transfer to the heart.
Efferent renal sympathetic nerve activation in response to remote ischemic conditioning by 4 cycles of 5 min ischemia and 5 min reperfusion on one hind limb was mandatory for the release of EPO into the systemic circulation in mice [113]. (Fig. 5) In rats, subdiaphragmatic vagotomy abolished the infarct size reduction by remote ischemic conditioning with one cycle of 15 ischemia and 10 min reperfusion on both hind limbs and, as mentioned above, GLP-1 was released from visceral organs innervated by the posterior gastric branch of the vagus nerve and was possibly the humoral factor to transfer protection to the heart [7]. Also, supporting the role of vagal gut innervation, in rats, infarct size reduction by remote ischemic conditioning with one cycle of 15 min ischemia and 10 min reperfusion on both hind limbs was not attenuated by section of hepatic, celiac or anterior gastric vagal branches, whereas bilateral cervical vagotomy, subdiaphragmatic vagotomy, gastric vagotomy or sectioning of the posterior gastric branch abolished the infarct size reduction [108]. In other studies in rats only cervical [31, 121] but not subdiaphragmatic vagotomy [31] abrogated the infarct size reduction by remote ischemic conditioning with 3/4 cycles of 5 min ischemia and 5 min reperfusion on one hind limb. However, these latter studies did not investigate a potential impact of vagal innervation on humoral factor release (Fig. 5).
Reflecting the participation of the central nervous system, remote ischemic conditioning by 3 cycles of 5 min ischemia and 5 min reperfusion of one hind limb in rats increased the CGRP and substance P mRNA and protein expression in the dorsal root ganglion, and both peptides were also increased in plasma and myocardial tissue and causally involved in infarct size reduction [38] (Fig. 5).
At the target organ level, humoral factors appear to reduce infarct size through recruitment of intracardiac ganglia. When isolated perfused rat hearts were treated with hexamethonium, the dialysate-mediated infarct size reduction was abrogated [121], and infarct size reduction was also abolished after transfer into an acceptor mouse heart which received an opioid antagonist [110], suggesting that humoral factors activate intracardiac ganglia. Also, systemic blockade of muscarinic receptor M3 abrogated the infarct size reduction by the GLP-1 analog exendin-4 which mimicked the cardioprotection by remote ischemic conditioning in rats [7], thus reflecting the action of GLP-1 on postganglionic cardiac cholinergic nerves. Indeed, intracardiac ganglia are activated directly by factors released from the myocardium which then, in turn, activate efferent post- ganglionic nerves [144]. Thus, even in isolated, centrally denervated hearts neuronal activation may be involved in cardioprotection [121] (Fig. 5).
Myocardial response
Incoming neuronal and humoral signals activate protective signaling cascades in the myocardium through sarcolemmal receptors or act receptor-independently [88]. The downstream intracellular signaling cascades then activate their intracellular target structure(s), notably the mitochondria.
Non- biased “omic” approaches on signal transduction
Up to know, no genomic/proteomic approach has identified an intracellular signal of particular relevance and with causal involvement in cardioprotection. Genomic profiling of myocardium taken 15 min after remote ischemic conditioning in mice identified an upregulation of genes against oxidative stress (Hadhsc, Prdx4, Fabp4) and of genes involved in cytoprotection (Hsp73). Pro-inflammatory genes (Egr-1, Dusp1, Dusp6) were downregulated [90]. Proteomic profiling for phosphorylated proteins found an increase of several phosphoproteins related to the sarcomeric Z-disk in myocardium of mice in response to remote ischemic conditioning by 4 cycles of 5 min ischemia and 5 min reperfusion on one hind limb [1]. Proteomic profiling of right atrial tissue of cardio-surgical patients, taken after remote ischemic conditioning by 4 cycles of 5 min forearm ischemia and 5 min reperfusion, identified an upregulation of thioredoxin and several cell stress-associated proteins [174], but this particular study did not report a cardioprotective effect. Neither a metabolomic nor a lipidomic approach nor a miRNA array have yet been performed in myocardial tissue after remote conditioning.
Hypothesis-driven approaches on signal transduction
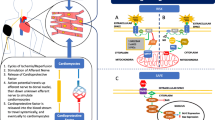
The signal transduction of remote ischemic conditioning in the myocardium is remarkably similar to that of local ischemic pre- and postconditioning. Autacoids and neurohormones (acetylcholine, opioids) activate G-protein coupled receptors. Reactive oxygen species such as nitric oxide initiate receptor-independent cardioprotective signaling. The activation of several proteins, notably protein kinases but also others, converges on the mitochondria and the cytoskeleton. The intracellular signal transduction is conceptually summarized in three major pathways: the endothelial nitric oxide synthase/protein kinase G (eNOS/PKG) pathway, the reperfusion injury salvage kinase (RISK) pathway and the survivor activating factor enhancement (SAFE) pathway [56, 88]. (Fig. 6)

Myocardial signal transduction - eNOS/PKG pathway in green, RISK pathway in yellow, and SAFE pathway in red. I,II,III, IV mitochondrial respiratory chain complexes I,II,III, IV; δ/κ δ- and κ- opioid receptors; A1 adenosine receptor A1; ALDH2 aldehyde dehydrogenase 2; Akt protein kinase B; B2 bradykinin receptor B2; CGRP calcitonin gene-related peptide; CX43 connexin43; CXCR4 chemokine 4 receptor; eNOS endothelial nitric oxide synthase; ERK 1/2 extracellular-regulated kinases 1/2; GLP-1 glucagon-like peptide-1 and its receptor; gp130 glycoprotein130; GSK3 ß glycogen synthase kinase 3ß; HIF-1α hypoxia-inducible factor-1α; IL-10 interleukin-10; KATP ATP-dependent potassium channel; M3 muscarinic receptor M3; miRNA microRNA; mPTP mitochondrial permeability transition pore; NO nitric oxide; OPA1 dynamin-like 120 kDa protein; PI3K phosphatidylinositol-4,5-bisphosphate 3-kinase; PKC protein kinase C; PKG protein kinase G; RISK reperfusion injury salvage kinases; ROS reactive oxygen species; SAFE survivor activating factor enhancement; SDF-1α stromal cell-derived factor-1α; STAT signal transducer and activator of transcription
Neurohormone receptors:In response to remote conditioning the following receptors are activated: In a cardiomyocyte cell line, the adenosine receptor A1 was activated with rabbit plasma dialysate after remote ischemic conditioning by by 4 cycles of 5 min ischemia and 5 min reperfusion on one hind limb [153]. In mice, the bradykinin receptor B2 was activated by traumatic remote conditioning (skin incision) and downstream of bradykinin receptor B2, myocardial PKC ɛ was activated whereas PKC δ was repressed [79]. The infarct size reduction by remote ischemic conditioning with one cycle of 15 min ischemia and 10 min reperfusion on both hind limbs was GLP-1 receptor- and muscarinic receptor M3 activation- dependent in rats [7]. The precise localization of the involved GLP-1 receptors, however, is currently unknown. The causal involvement of GLP-1 was proven by systemic GLP-1 receptor blockade, but the presence of GLP-1 receptors on cardiomyocytes is still contentious [115]. Muscarinic receptor activation may also trigger an increase in acetylcholine synthesis; a non-selective muscarinic receptor agonist increased the acetylcholine synthesis in cardiomyocytes in vitro [82]. Cardiomyocytes express choline acetyltransferase, and intramyocardial acetylcholine was increased by remote ischemic conditioning with 3 cycles of 3 min ischemia and 3 min reperfusion on one hind limb 24 h after myocardial infarction in mice [114]. However, in this study cardioprotection in response to remote ischemic conditioning was not examined.
Opioid receptors were activated in isolated rabbit cardiomyocytes with rabbit plasma dialysate taken after remote ischemic conditioning by 4 cycles of 5 min ischemia and 5 min reperfusion on one hind limb; whereas pre-treatment of cardiomyocytes with rabbit plasma dialysate after remote ischemic conditioning reduced the percentage of dead cardiomyocytes after simulated ischemia/reperfusion, unspecific blockade of opioid receptors with naloxone completely abolished this beneficial effect [142]. There is also an interaction of adenosine and opioid receptors. In a cardiomyocyte cell line, pharmacologically induced activation of δ- and κ-opioid receptors mimicked the protective effect on cellular viability induced by plasma dialysate after remote ischemic conditioning, and blockade of adenosine A1 receptors attenuated this cardioprotection [153].
EPO release is increased with remote ischemic conditioning [113]. EPO receptors are expressed in different tissues and cell types, including neurons and cardiomyocytes [133], and selective EPO receptor agonists, which did not stimulate erythropoiesis, reduced infarct size in rats [87]. However, the activation of EPO receptors in the cardioprotection by remote ischemic conditioning has not been directly proven so far.
Receptor(s) for CGRP and substance P as well as their downstream signaling which mediates infarct size reduction [124] have not been identified in the myocardium so far. Apart from the increase of plasmatic levels of both peptides, there was also an increase of both peptides in myocardial tissue in response to remote ischemic conditioning by 3 cycles of 5 min ischemia and 5 min reperfusion on both hind limbs in rats [38].
Cytokine/chemokine receptors:In rats, the chemokine receptor type 4 was activated by SDF-1α in response to remote ischemic conditioning by 3 cycles of 5 min ischemia and 5 min reperfusion on one hind limb [25]. In mice, anti-IL-10 receptor antibodies attenuated the late infarct size reduction by remote ischemic conditioning with 3 cycles of 5 min ischemia and 5 min reperfusion on one hind limb [13]; however, IL-10 receptors are ubiquitously expressed and their specific participation in the myocardium remains to be identified.
Cytosolic proteins:Different intracellular proteins are activated in response to remote conditioning. In mice, the eNOS/PKG was activated in response to remote ischemic conditioning by 3 cycles of 5 min ischemia and 5 min reperfusion on one hind limb; the key enzyme eNOS was phosphorylated 24 h after myocardial ischemia and reperfusion [13]. PKC was activated in rodents in response to abdominal surgical incision [46, 79]. The RISK pathway was activated regardless of what the specific remote stimulus was, in rodents [7, 11, 13, 46, 79, 145, 158, 166], and pigs [48] with the following key enzymes: protein kinase B/Akt [11, 13, 145, 158], phosphatidylinositol-4,5-bisphosphate 3-kinase [48], extracellular-regulated kinases 1/2 [145, 166], and glycogen synthase kinase 3ß [158]. One key enzyme of the SAFE pathway, the transducer and activator of transcription 3 was activated by remote ischemic conditioning in rats with one cycle of 15 min ischemia and 10 min reperfusion on both hind limbs [7] and in pigs with 4 cycles of 5 min ischemia and 5 min reperfusion on one hind limb [145]. When the protection was transferred via plasma from pigs to isolated rat hearts, both signaling pathways, RISK and SAFE were activated and causally involved in infarct size reduction [145]. Key enzymes of the RISK and the SAFE pathway were activated downstream of the muscarinic receptor M3 in the myocardium of rats in response to remote ischemic conditioning by one cycle of 15 min ischemia and 10 min reperfusion on both hind limbs [7]. In left ventricular myocardial biopsies of patients undergoing remote ischemic conditioning by 3 cycles of 5 min forearm ischemia and 5 min reperfusion before elective CABG surgery, none of the established cardioprotective proteins was activated, but only signal transducer and activator of transcription 5 was activated at early reperfusion [39, 65]. The intracellular signaling is obviously species-specific.
Apart from and in addition to the above mentioned cardioprotective signaling pathways, heat shock protein heme oxygenase-1 expression was increased in rats after repetitive remote ischemic conditioning by 4 cycles of 5 min ischemia and 5 min reperfusion on one hind limb on 1 to 3 consecutive days [173]. Associated with cardioprotection (reduced troponin T release) by remote ischemic conditioning by 4 cycles of 5 min forearm ischemia and 5 min reperfusion in patients undergoing heart surgery under cardiopulmonary bypass, HIF-1α protein expression was increased in right atrial tissue [2]. A causal involvement of HIF-1α in cardioprotection by remote conditioning, however, remains to be identified. As mentioned above, HIF-1α may be a regulator of different humoral mediators of protection.
In rats, remote ischemic conditioning by 4 cycles of 5 min ischemia and 5 min reperfusion on both hind limbs preserved connexin-43 phosphorylation and immunofluorescence staining, indicating a localization of phosphorylated connexin-43 at intercalated disks [10]. Exposure of cultured rabbit ventricular cardiomyocytes to plasma dialysate from rabbits with remote ischemic conditioning by 4 cycles of 5 min ischemia and 5 min reperfusion on one hind limb reduced hypoxia-induced cell-swelling. Experiments using inhibitors identified the activation of potassium- and chloride-channels which control volume and edema formation [27]. However, the role of chloride-channels and cell-volume control in cardioprotection remains somewhat contentious [62, 63].
In rats, miRNA1 was downregulated in response to remote ischemic conditioning by 3 cycles of 5 min ischemia and 5 min reperfusion on both hind limbs [32]. In mice, along with infarct size reduction by remote ischemic conditioning with 4 cycles of 5 min ischemia and 5 min reperfusion on one hind limb, miRNA144 was increased in myocardial tissue. A reduction of myocardial miRNA144 levels with a specific antisense oligonucleotide abrogated the observed infarct size reduction [98]. In association with remote ischemic conditioning by 3 cycles of 4 min forearm ischemia and 4 min reperfusion [73] or by 3 cycles of 5 min forearm ischemia and 5 min reperfusion [147], respectively, miRNA1 and miRNA195 were downregulated and miRNA388-3p was upregulated in right atrial myocardium of patients undergoing double valve replacement [73] or elective CABG surgery [147]. Up to now, all potential downstream pathways or interacting pathways of the miRNAs in conditioning rely on literature search and not on experimental data [4]. Further experiments are necessary to identify a potentially causal role and a relevance of miRNAs in remote conditioning.
Mitochondria as targets and potential effectors: Mitochondria have been identified as a major common intracellular target during local and remote ischemic conditioning [56, 64]. The preservation of mitochondrial integrity and function after ischemia/reperfusion is decisive for survival of cardiomyocytes and thus salvage of myocardium. Mitochondria are the major cellular source of ATP under aerobic conditions and therefore relevant for all cellular functions and cell survival [35, 56]. In ischemia/reperfusion, in contrast, mitochondrial permeability transition pore (mPTP) opening may initiate cell death by cytochrome C release and initiation of intracellular proteolysis.
In rats, myocardial infarct size reduction by remote ischemic conditioning with 4 cycles of 5 min ischemia and 5 min reperfusion on one hind limb was associated with preserved mitochondrial morphology [18], maintained mitochondrial membrane potential and increased mitochondrial manganese superoxide dismutase content [17]. Mitochondrial respiration was improved in rat hearts [35] and in right atrial appendages of patients undergoing elective CABG surgery [146, 147] in response to remote ischemic conditioning by 3 cycles of 5 min ischemia and 5 min reperfusion on one hind limb and by 4 cycles of 5 min forearm ischemia and 5 min reperfusion, respectively. Humoral transfer (pig to rat [40] or rabbit to rabbit [162]) of cardioprotection by remote ischemic conditioning with 4 cycles of 5 min ischemia and 5 min reperfusion on one hind limb improved mitochondrial function. Mitochondrial adenosine diphosphate-stimulated respiration was improved [40, 162], calcium retention capacity as an estimate of mPTP opening was increased [40], adenosine triphosphate (ATP) concentration was increased [40] and reactive oxygen species formation decreased [40]. The improved mitochondrial function in rat myocardium was associated with infarct size reduction [40]. Nitrosation and nitrosylation of mitochondrial membrane proteins are causally involved in cardioprotection [88]. In response to remote ischemic conditioning by 4 cycles of 5 min ischemia and 5 min reperfusion on one hind limb in mice, myocardial nitrite is increased [128]. Myocardial myoglobin reduces nitrite to bioactive nitric oxide, and nitric oxide subsequently inhibits mitochondrial complex I activity [53]. This nitrosation triggered a reduction of complex I activity and finally reduced myocardial reactive oxygen species formation in response to remote ischemic conditioning by 4 cycles of 5 min ischemia and 5 min reperfusion on one hind limb in mice [128]. In rats, remote ischemic conditioning by 4 cycles of 5 min ischemia and 5 min reperfusion on one hind limb increased mitochondrial dynamics and the expression of the mitochondrial fusion protein dynamin-like 120 kDa protein (OPA1); mitochondrial morphology was preserved [18]. Remote ischemic conditioning by 4 cycles of 5 min ischemia and 5 min reperfusion on one hind limb induced mitochondrial ATP-dependent K(+) channel opening in rat myocardium. The mitochondrial ATP-dependent K(+) channel blocker 5-hydroxydecanoic acid abolished the infarct size reduction by remote ischemic conditioning [93]. Aldehyde dehydrogenase 2 is located in the mitochondrial matrix and detoxifies aldehydes [21, 22]. Aldehyde dehydrogenase 2 was involved in the infarct size reduction by remote ischemic conditioning with 4 cycles of 5 min ischemia and 5 min reperfusion on one hind limb in rabbits and one forearm in humans, respectively. Inhibition of aldehyde dehydrogenase 2 by cyanamide abrogated the myocardial infarct size reduction in rabbits, and a single nucleotide polymorphism which inactivates aldehyde dehydrogenase 2 abrogated the attenuation of ischemia/reperfusion-induced endothelial dysfunction in humans [23] (Fig. 6).
Coronary vascular response
Ischemia/reperfusion-injury of the coronary microcirculation is characterized by increased vascular permeability and edema, endothelial dysfunction and impaired vasomotion, microembolization of atherothrombotic debris, and stasis with intravascular cell aggregates. Finally the vascular ischemia/reperfusion may result in its most severe form: destruction of the capillaries with hemorrhage [58]. Conversely, the coronary microcirculation is a target of cardioprotection, including that by remote conditioning. After ischemia/reperfusion, coronary flow and coronary flow reserve were improved in mice [71], pigs [141] and humans [89]. Red blood cell nitric oxide synthase activity was stimulated by remote ischemic conditioning with 4 cycles of 5 min forearm ischemia and 5 min reperfusion in healthy volunteers along with improved red blood cell deformability in vitro [44]. Leukocyte adhesion was reduced in healthy volunteers by remote ischemic conditioning with 3 cycles of 5 min forearm ischemia and 5 min reperfusion [143]. In healthy volunteers after ischemia/reperfusion injury (20 min ischemia with reperfusion) on one forearm [118], in patients undergoing coronary angiography [96] and in patients undergoing ablation for atrial fibrillation, remote ischemic conditioning with 3 cycles of 5 min forearm ischemia and 5 min reperfusion [150] attenuated the number of monocyte-platelet aggregates.
Translation of remote ischemic conditioning into clinical practice
Remote ischemic conditioning is also operative in humans. In the initial studies in healthy volunteers, endothelium-dependent vasodilation was taken as endpoint, and 3 preceding cycles of 5 min forearm ischemia and 5 min reperfusion attenuated the impairment of vasodilation following 20 min ischemia and reperfusion in the contralateral arm [86, 103, 104]. This protection of endothelium-dependent vasodilation was immediate, but a second window of protection was apparent 24 and 48 h later [103]. Protection was even seen when the remote ischemic conditioning procedure was performed during the sustained arm ischemia (perconditioning) [104], and activation of the autonomous nervous system and of ATP-dependent K(+) channels was causally involved [103, 104].
Subsequently, remote ischemic conditioning by 3 cycles of 5 min blood pressure cuff inflation and 5 min deflation was demonstrated to reduce myocardial injury, as reflected by troponin release, in patients undergoing CABG surgery under cardiopulmonary bypass and ischemic cardioplegic arrest [47]. Such protection by remote ischemic conditioning in patients undergoing cardiovascular surgery was confirmed in a number of, but not all studies [61, 134], and even an improved short-term [16] and long-term [156] clinical outcome became apparent from retrospective analyses. However, two recent large phase III trials on remote ischemic preconditioning in patients undergoing cardiovascular surgery in which clinical outcome was the primary endpoint were neutral, with respect to biomarker release and to clinical outcome [49, 111]. Unfortunately, all [111] or almost all [49] patients in these trials were operated under propofol anesthesia, although it was previously known that propofol specifically abrogates the protection by remote ischemic conditioning whereas volatile anesthesia does not [91, 92, 169]. Therefore, the actual potential of remote ischemic conditioning to confer protection in patients undergoing cardiovascular surgery remains unclear [59]. Interestingly, remote ischemic preconditioning protects against acute kidney injury in cardiosurgical patients when these are operated without the use of propofol [170].
Importantly, in patients who suffer from an acute myocardial infarction and undergo interventional or thrombolytic reperfusion, remote ischemic perconditioning reduces myocardial injury, as reflected by reduced biomarker release or reduced infarct size on imaging, in all studies published so far [61], and it even improves clinical outcome on retrospective analysis [148]. In acute myocardial infarction, the myocardial injury is obviously greater and protection more needed than in cardiovascular surgery, and patients with acute myocardial infarction have no propofol on board. A large phase III trial with clinical outcome as primary endpoint in patients with reperfused acute myocardial infarction is on its way [50].
While the available translational studies are encouraging, notably those in acute myocardial infarction, we need to develop a better understanding of the mechanisms and signal transduction of remote ischemic conditioning. Such better understanding will help us to avoid pitfalls in translational attempts such as the use of propofol and to deal with other confounding variables such as advanced age, and the multiple co-morbidities and co-medications in those patients who need the additional protection by remote ischemic conditioning the most [34].
References
Abdul-Ghani S, Heesom KJ, Angelini GD, Suleiman MS (2014) Cardiac phosphoproteomics during remote ischemic preconditioning: a role for the sarcomeric Z-disk proteins. Biomed Res Int 2014:767812. doi:10.1155/2014/767812
Albrecht M, Zitta K, Bein B, Wennemuth G, Broch O, Renner J, Schuett T, Lauer F, Maahs D, Hummitzsch L, Cremer J, Zacharowski K, Meybohm P (2013) Remote ischemic preconditioning regulates HIF-1alpha levels, apoptosis and inflammation in heart tissue of cardiosurgical patients: a pilot experimental study. Basic Res Cardiol 108:314. doi:10.1007/s00395-012-0314-0
Arroyo-Martinez EA, Meaney A, Gutierrez-Salmean G, Rivera-Capello JM, Gonzalez-Coronado V, Alcocer-Chauvet A, Castillo G, Najera N, Ceballos G, Meaney E (2016) Is local nitric oxide availability responsible for myocardial salvage after remote preconditioning? Arq Bras Cardiol 107:154–162. doi:10.5935/abc.20160100
Baars T, Skyschally A, Klein-Hitpass L, Cario E, Erbel R, Heusch G, Kleinbongard P (2014) microRNA expression and its potential role in cardioprotection by ischemic postconditioning in pigs. Pflügers Arch - Eur J Physiol 466:1953–1961. doi:10.1007/s00424-013-1429-3
Barile L, Moccetti T, Marban E, Vassalli G (2016) Roles of exosomes in cardioprotection. Eur Heart J. doi:10.1093/eurheartj/ehw304
Basalay M, Barsukevich V, Mastitskaya S, Mrochek A, Pernow J, Sjoquist PO, Ackland GL, Gourine AV, Gourine A (2012) Remote ischaemic pre- and delayed postconditioning - similar degree of cardioprotection but distinct mechanisms. Exp Physiol 97:908–917. doi:10.1113/expphysiol.2012.064923
Basalay M, Mastitskaya S, Mrochek A, Ackland GL, del Arroyo AG, Sanchez J, Sjoquist PO, Pernow J, Gourine AV, Gourine A (2016) Glucagon-like peptide-1 (GLP-1) mediates cardioprotection by remote ischaemic conditioning. Cardiovasc Res. doi:10.1093/cvr/cvw216
Birkelund T, Obad DS, Matejec R, Botker HE, Ravn HB (2015) Remote ischemic preconditioning does not increase circulating or effector organ concentrations of proopiomelanocortin derivates. Scand Cardiovasc J 49:257–263. doi:10.3109/14017431.2015.1046401
Bøtker HE, Kharbanda R, Schmidt MR, Bottcher M, Kaltoft AK, Terkelsen CJ, Munk K, Andersen NH, Hansen TM, Trautner S, Lassen JF, Christiansen EH, Krusell LR, Kristensen SD, Thuesen L, Nielsen SS, Rehling M, Sorensen HT, Redington AN, Nielsen TT (2010) Remote ischaemic conditioning before hospital admission, as a complement to angioplasty, and effect on myocardial salvage in patients with acute myocardial infarction: a randomised trial. Lancet 375:727–734. doi:10.1016/S0140-6736(09)62001-8
Brandenburger T, Huhn R, Galas A, Pannen BH, Keitel V, Barthel F, Bauer I, Heinen A (2014) Remote ischemic preconditioning preserves connexin 43 phosphorylation in the rat heart. J Transl Med 12:228. doi:10.1186/s12967-014-0228-8
Breivik L, Helgeland E, Aarnes EK, Mrdalj J, Jonassen AK (2011) Remote postconditioning by humoral factors in effluent from ischemic preconditioned rat hearts is mediated via PI3K/Akt-dependent cell-survival signaling at reperfusion. Basic Res Cardiol 106:135–145. doi:10.1007/s00395-010-0133-0
Cabrera-Fuentes HA, Niemann B, Grieshaber P, Wollbrueck M, Gehron J, Preissner KT, Boning A (2015) RNase1 as a potential mediator of remote ischaemic preconditioning for cardioprotection. Eur J Cardiothorac Surg 48:732–737 . doi:10.1093/ejcts/ezu519discussion 737
Cai ZP, Parajuli N, Zheng X, Becker L (2012) Remote ischemic preconditioning confers late protection against myocardial ischemia-reperfusion injury in mice by upregulating interleukin-10. Basic Res Cardiol 107:277. doi:10.1007/s00395-012-0277-1
Cai Z, Luo W, Zhan H, Semenza GL (2013) Hypoxia-inducible factor 1 is required for remote ischemic preconditioning of the heart. Proc Natl Acad Sci US A 110:17462–17467. doi:10.1073/pnas.1317158110
Candilio L, Malik A, Hausenloy DJ (2013) Protection of organs other than the heart by remote ischemic conditioning. J Cardiovasc Med (Hagerstown) 14:193–205. doi:10.2459/JCM.0b013e328359dd7b
Candilio L, Malik A, Ariti C, Barnard M, Di SC, Lawrence D, Hayward M, Yap J, Roberts N, Sheikh A, Kolvekar S, Hausenloy DJ, Yellon DM (2015) Effect of remote ischaemic preconditioning on clinical outcomes in patients undergoing cardiac bypass surgery: a randomised controlled clinical trial. Heart 10:185–192. doi:10.1136/heartjnl-2014-306178
Cao Y, Zhang SZ, Zhao SQ, Bruce IC (2011) The mitochondrial Ca(2+)-activated K(+) channel contributes to cardioprotection by limb remote ischemic preconditioning in rat. Life Sci 88:1026–1030. doi:10.1016/j.lfs.2011.03.011
Cellier L, Tamareille S, Kalakech H, Guillou S, Lenaers G, Prunier F, Mirebeau-Prunier D (2016) Remote ischemic conditioning influences mitochondrial dynamics. Shock 45:192–197. doi:10.1097/SHK.0000000000000500
Ceradini DJ, Kulkarni AR, Callaghan MJ, Tepper OM, Bastidas N, Kleinman ME, Capla JM, Galiano RD, Levine JP, Gurtner GC (2004) Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat Med 10:858–864. doi:10.1038/nm1075
Chao de la Barca JM, Bakhta O, Kalakech H, Simard G, Tamareille S, Catros V, Callebert J, Gadras C, Tessier L, Reynier P, Prunier F, Mirebeau-Prunier D (2016) Metabolic signature of remote ischemic preconditioning involving a cocktail of amino acids and biogenic amines. J Am Heart Assoc 5. doi:10.1161/JAHA.116.003891
Chen CH, Budas GR, Churchill EN, Disatnik MH, Hurley TD, Mochly-Rosen D (2008) Activation of aldehyde dehydrogenase-2 reduces ischemic damage to the heart. Science 321:1493–1495. doi:10.1126/science.1158554
Chen CH, Ferreira JC, Gross ER, Mochly-Rosen D (2014) Targeting aldehyde dehydrogenase 2: new therapeutic opportunities. Physiol Rev 94:1–34. doi:10.1152/physrev.00017.2013
Contractor H, Stottrup NB, Cunnington C, Manlhiot C, Diesch J, Ormerod JO, Jensen R, Bøtker HE, Redington A, Schmidt MR, Ashrafian H, Kharbanda RK (2013) Aldehyde dehydrogenase-2 inhibition blocks remote preconditioning in experimental and human models. Basic Res Cardiol 108:343. doi:10.1007/s00395-013-0343-3
Contractor H, Haarup Lie R, Cunnington C, Li J, Støttrup NB, Manlhiot C, Bøtker HE, Schmidt MR, Forfar JC, Ashrafian H, Redington A, Kharbanda RK (2016) Adenosine receptor activation in the “trigger” limb of remote pre-conditioning mediates human endothelial conditioning and release of circulating cardioprotective factor(s). J Am Coll Cardiol Basic Trans Science. doi:10.1016/j.jacbts.2016.06.002
Davidson SM, Selvaraj P, He D, Boi-Doku C, Yellon RL, Vicencio JM, Yellon DM (2013) Remote ischaemic preconditioning involves signalling through the SDF-1alpha/CXCR4 signalling axis. Basic Res Cardiol 108:377. doi:10.1007/s00395-013-0377-6
Davidson SM, Takov K, Yellon DM (2016) Exosomes and cardiovascular protection. Cardiovasc Drugs Ther. doi:10.1007/s10557-016-6698-6
Diaz RJ, Harvey K, Boloorchi A, Hossain T, Hinek A, Backx PH, Wilson GJ (2014) Enhanced cell volume regulation: a key mechanism in local and remote ischemic preconditioning. Am J Physiol Cell Physiol 306:C1191–C1199. doi:10.1152/ajpcell.00259.2013
Dickson EW, Reinhart CP, Renzi FP, Becker RC, Porcaro WA, Heard SO (1999) Ischemic preconditioning may be transferable via whole blood transfusion: preliminary evidence. J Thromb Thrombolysis 8:123–129
Dickson EW, Blehar DJ, Carraway RE, Heard SO, Steinberg G, Przyklenk K (2001) Naloxone blocks transferred preconditioning in isolated rabbit hearts. J Mol Cell Cardiol 33:1751–1756. doi:10.1006/jmcc.2001.1436
Donato M, Buchholz B, Rodriguez M, Perez V, Inserte J, Garcia-Dorado D, Gelpi RJ (2012) Role of the parasympathetic nervous system in cardioprotection by remote hindlimb ischemic preconditioning. Exp Physiol 98:425–434. doi:10.1113/expphysiol.2012.066217
Donato M, Goyeneche MA, Garces M, Marchini T, Perez V, Del Mauro J, Hocht C, Rodriguez M, Evelson P, Gelpi RJ (2016) Myocardial triggers involved in activation of remote ischaemic preconditioning. Exp Physiol 101:708–716. doi:10.1113/EP085535
Duan X, Ji B, Wang X, Liu J, Zheng Z, Long C, Tang Y, Hu S (2012) Expression of microRNA-1 and microRNA-21 in different protocols of ischemic conditioning in an isolated rat heart model. Cardiology 122:36–43. doi:10.1159/000338149
Dubin AE, Patapoutian A (2010) Nociceptors: the sensors of the pain pathway. J Clin Invest 120:3760–3772. doi:10.1172/JCI42843
Ferdinandy P, Hausenloy DJ, Heusch G, Baxter GF, Schulz R (2014) Interaction of risk factors, comorbidities and comedications with ischemia/reperfusion injury and cardioprotection by preconditioning, postconditioning, and remote conditioning. Pharmacol Rev 66:1142–1174. doi:10.1124/pr.113.008300
Ferko M, Kancirova I, Jasova M, Carnicka S, Murarikova M, Waczulikova I, Sumbalova Z, Kucharska J, Ulicna O, Ravingerova T, Ziegelhoffer A (2014) Remote ischemic preconditioning of the heart: protective responses in functional and biophysical properties of cardiac mitochondria. Physiol Res 63(Suppl 4):S469–S478
Foley RN (2008) Erythropoietin: physiology and molecular mechanisms. Heart Fail Rev 13:405–414. doi:10.1007/s10741-008-9083-0
Gao J, Fu W, Jin Z, Yu X (2007) Acupuncture pretreatment protects heart from injury in rats with myocardial ischemia and reperfusion via inhibition of the beta(1)-adrenoceptor signaling pathway. Life Sci 80:1484–1489. doi:10.1016/j.lfs.2007.01.019
Gao Y, Song J, Chen H, Cao C, Lee C (2015) TRPV1 activation is involved in the cardioprotection of remote limb ischemic postconditioning in ischemia-reperfusion injury rats. Biochem Biophys Res Commun 463:1034–1039. doi:10.1016/j.bbrc.2015.06.054
Gedik N, Thielmann M, Kottenberg E, Peters J, Jakob H, Heusch G, Kleinbongard P (2014) No evidence for activated autophagy in left ventricular myocardium at early reperfusion with protection by remote ischemic preconditioning in patients undergoing coronary artery bypass grafting. PLoS One 9:e96567. doi:10.1371/journal.pone.0096567
Gedik N, Maciel L, Schulte C, Skyschally A, Heusch G, Kleinbongard P (2016) Cardiomyocyte mitochondria as targets of humoral factors released by remote ischemic preconditioning. Arch Med Sci. doi:10.5114/aoms.2016.61789
Gho BCG, Schoemaker RG, van den Doel MA, Duncker DJ, Verdouw PD (1996) Myocardial protection by brief ischemia in noncardiac tissue. Circulation 94:2193–2200. doi:10.1161/01.CIR.94.9.2193
Giricz Z, Varga ZV, Baranyai T, Sipos P, Paloczi K, Kittel A, Buzas EI, Ferdinandy P (2014) Cardioprotection by remote ischemic preconditioning of the rat heart is mediated by extracellular vesicles. J Mol Cell Cardiol 68:75–78. doi:10.1016/j.yjmcc.2014.01.004
Gourine A, Gourine AV (2014) Neural mechanisms of cardioprotection. Physiology (Bethesda) 29:133–140. doi:10.1152/physiol.00037.2013
Grau M, Kollikowski A, Bloch W (2016) Remote ischemia preconditioning increases red blood cell deformability through red blood cell-nitric oxide synthase activation. Clin Hemorheol Microcirc 63:185–197. doi:10.3233/CH-152039
Gross GJ, Baker JE, Moore J, Falck JR, Nithipatikom K (2011) Abdominal surgical incision induces remote preconditioning of trauma (RPCT) via activation of bradykinin receptors (BK2R) and the cytochrome P450 epoxygenase pathway in canine hearts. Cardiovasc Drugs Ther 25:517–522. doi:10.1007/s10557-011-6321-9
Gross ER, Hsu AK, Urban TJ, Mochly-Rosen D, Gross GJ (2013) Nociceptive-induced myocardial remote conditioning is mediated by neuronal gamma protein kinase C. Basic Res Cardiol 108:381. doi:10.1007/s00395-013-0381-x
Hausenloy DJ, Mwamure PK, Venugopal V, Harris J, Barnard M, Grundy E, Ashley E, Vichare S, Di Salvo C, Kolvekar S, Hayward M, Keogh B, MacAllister RJ, Yellon DM (2007) Effect of remote ischaemic preconditioning on myocardial injury in patients undergoing coronary artery bypass graft surgery: a randomized controlled trial. Lancet 370:575–579. doi:10.1016/S0140-6736(07)61296-3
Hausenloy DJ, Iliodromitis EK, Andreadou I, Papalois A, Gritsopoulos G, Anastasiou-Nana M, Kremastinos DT, Yellon DM (2012) Investigating the signal transduction pathways underlying remote ischemic conditioning in the porcine heart. Cardiovasc Drugs Ther 26:87–93. doi:10.1007/s10557-011-6364-y
Hausenloy DJ, Candilio L, Evans R, Ariti C, Jenkins DP, Kolvekar S, Knight R, Kunst G, Laing C, Nicholas J, Pepper J, Robertson S, Xenou M, Clayton T, Yellon DM, Investigators ET (2015) Remote ischemic preconditioning and outcomes of cardiac surgery. N Engl J Med 373:1408–1417. doi:10.1056/NEJMoa1413534
Hausenloy DJ, Kharbanda R, Rahbek Schmidt M, Moller UK, Ravkilde J, Okkels Jensen L, Engstrom T, Garcia Ruiz JM, Radovanovic N, Christensen EF, Sorensen HT, Ramlall M, Bulluck H, Evans R, Nicholas J, Knight R, Clayton T, Yellon DM, Botker HE (2015) Effect of remote ischaemic conditioning on clinical outcomes in patients presenting with an ST-segment elevation myocardial infarction undergoing primary percutaneous coronary intervention. Eur Heart J 36:1846–1848
Headrick JP, See Hoe LE, Du Toit EF, Peart JN (2015) Opioid receptors and cardioprotection - 'opioidergic conditioning' of the heart. Br J Pharmacol 172:2026–2050. doi:10.1111/bph.13042
Helgeland E, Breivik LE, Vaudel M, Svendsen OS, Garberg H, Nordrehaug JE, Berven FS, Jonassen AK (2014) Exploring the human plasma proteome for humoral mediators of remote ischemic preconditioning - a word of caution. PLoS One 9:e109279. doi:10.1371/journal.pone.0109279
Hendgen-Cotta UB, Merx MW, Shiva S, Schmitz J, Becher S, Klare JP, Steinhoff HJ, Goedecke A, Schrader J, Gladwin MT, Kelm M, Rassaf T (2008) Nitrite reductase activity of myoglobin regulates respiration and cellular viability in myocardial ischemia-reperfusion injury. Proc Natl AcadSci USA 105:10256–10261. doi:10.1073/pnas.0801336105
Hepponstall M, Ignjatovic V, Binos S, Monagle P, Jones B, Cheung MH, d'Udekem Y, Konstantinov IE (2012) Remote ischemic preconditioning (RIPC) modifies plasma proteome in humans. PLoS One 7:e48284. doi:10.1371/journal.pone.0048284
Hepponstall M, Ignjatovic V, Binos S, Attard C, Karlaftis V, d'Udekem Y, Monagle P, Konstantinov IE (2015) Remote ischemic preconditioning (RIPC) modifies the plasma proteome in children undergoing repair of tetralogy of fallot: a randomized controlled trial. PLoS One 10:e0122778. doi:10.1371/journal.pone.0122778
Heusch G (2015) Molecular basis of cardioprotection: signal transduction in ischemic pre-, post- and remote conditioning. Circ Res 116:674–699. doi:10.1161/CIRCRESAHA.116.305348
Heusch G (2016) Remote ischemic conditioning – the enigmatic transfer of protection. Cardiovasc Res in press
Heusch G (2016) The coronary circulation as a target of cardioprotection. Circ Res 118:1643–1658. doi:10.1161/CIRCRESAHA.116.308640
Heusch G, Gersh BJ (2016) ERICCA and RIPHeart: two nails in the coffin for cardioprotection by remote ischemic conditioning? Probably not! Eur Heart J 37:200–202. doi:10.1093/eurheartj/ehv606
Heusch G, Gersh BJ (2016) The pathophysiology of acute myocardial infarction and strategies of protection beyond reperfusion: a continual challenge. Eur Heart J. doi:10.1093/eurheartj/ehw224
Heusch G, Rassaf T (2016) Time to give op on cardioprotection? A critical appraisal of clinical studies on ischemic pre-, post-, and remote conditioning. Circ Res 119:676–695. doi:10.1161/CIRCRESAHA.116.308736
Heusch G, Liu GS, Rose J, Cohen MV, Downey JM (2000) No confirmation for a causal role of volume-regulated chloride channels in ischemic preconditioning in rabbits. J Mol Cell Cardiol 32:2279–2285. doi:10.1006/jmcc.2000.1259
Heusch G, Cohen MV, Downey JM (2001) Ischemic preconditioning through opening of swelling-activated chloride channels ? Circ Res 89:e48
Heusch G, Boengler K, Schulz R (2008) Cardioprotection: nitric oxide, protein kinases, and mitochondria. Circulation 118:1915–1919. doi:10.1161/CIRCULATIONAHA.108.805242
Heusch G, Musiolik J, Kottenberg E, Peters J, Jakob H, Thielmann M (2012) STAT5 activation and cardioprotection by remote ischemic preconditioning in humans. Circ Res 110:111–115. doi:10.1161/CIRCRESAHA.111.259556
Heusch G, Libby P, Gersh B, Yellon D, Böhm M, Lopaschuk G, Opie L (2014) Cardiovascular remodeling in coronary artery disease and heart failure. Lancet 383:1933–1943. doi:10.1016/S0140-6736(14)60107-0
Heusch G, Botker HE, Przyklenk K, Redington A, Yellon DM (2015) Remote ischemic conditioning. J Am Coll Cardiol 65:177–195. doi:10.1016/j.jacc.2014.10.031
Heyman SN, Leibowitz D, Mor-Yosef Levi I, Liberman A, Eisenkraft A, Alcalai R, Khamaisi M, Rosenberger C (2016) Adaptive response to hypoxia and remote ischaemia pre-conditioning: a new hypoxia-inducible factors era in clinical medicine. Acta Physiol 216:395–406. doi:10.1111/apha.12613
Hibert P, Prunier-Mirebeau D, Beseme O, Chwastyniak M, Tamareille S, Lamon D, Furber A, Pinet F, Prunier F (2013) Apolipoprotein A-I is a potential mediator of remote ischemic preconditioning. PLoS One 8:e77211. doi:10.1371/journal.pone.0077211
Hibert P, Prunier-Mirebeau D, Beseme O, Chwastyniak M, Tamareille S, Pinet F, Prunier F (2014) Modifications in rat plasma proteome after remote ischemic preconditioning (RIPC) stimulus: identification by a SELDI-TOF-MS approach. PLoS One 9:e85669. doi:10.1371/journal.pone.0085669
Hildebrandt HA, Kreienkamp V, Gent S, Kahlert P, Heusch G, Kleinbongard P (2016) Kinetics and signal activation properties of circulating factor(s) from healthy volunteers undergoing remote ischemic pre-conditioning. J Am Coll Cardiol Basic Trans Science 1:3–13. doi:10.1016/j.jacbts.2016.01.007
Holst JJ (2007) The physiology of glucagon-like peptide 1. Physiol Rev 87:1409–1439. doi:10.1152/physrev.00034.2006
Hu Q, Luo W, Huang L, Huang R, Chen R (2016) Apoptosis-related microRNA changes in the right atrium induced by remote ischemic perconditioning during valve replacement surgery. Sci Rep 6:18959. doi:10.1038/srep18959
Iaconetti C, Sorrentino S, De Rosa S, Indolfi C (2016) Exosomal miRNAs in heart disease. Physiology (Bethesda) 31:16–24. doi:10.1152/physiol.00029.2015
Ibanez B, Heusch G, Ovize M, Van de Werf F (2015) Evolving therapies for myocardial ischemia/reperfusion injury. J Am Coll Cardiol 65:1454–1471. doi:10.1016/j.jacc.2015.02.032
Jeanneteau J, Hibert P, Martinez MC, Tual-Chalot S, Tamareille S, Furber A, Andriantsitohaina R, Prunier F (2012) Microparticle release in remote ischemic conditioning mechanism. Am J Physiol Heart Circ Physiol 303:H871–H877. doi:10.1152/ajpheart.00102.2012
Jensen RV, Stottrup NB, Kristiansen SB, Bøtker HE (2012) Release of a humoral circulating cardioprotective factor by remote ischemic preconditioning is dependent on preserved neural pathways in diabetic patients. Basic Res Cardiol 107:285. doi:10.1007/s00395-012-0285-1
Johnsen J, Pryds K, Salman R, Lofgren B, Kristiansen SB, Botker HE (2016) The remote ischemic preconditioning algorithm: effect of number of cycles, cycle duration and effector organ mass on efficacy of protection. Basic Res Cardiol 111:10. doi:10.1007/s00395-016-0529-6
Jones WK, Fan GC, Liao S, Zhang JM, Wang Y, Weintraub NL, Kranias EG, Schultz JE, Lorenz J, Ren X (2009) Peripheral nociception associated with surgical incision elicits remote nonischemic cardioprotection via neurogenic activation of protein kinase C signaling. Circulation 120:S1–S9. doi:10.1161/CIRCULATIONAHA.108.843938
Julius D, Basbaum AI (2001) Molecular mechanisms of nociception. Nature 413:203–210. doi:10.1038/35093019
Kaelin WG Jr, Ratcliffe PJ (2008) Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell 30:393–402. doi:10.1016/j.molcel.2008.04.009
Kakinuma Y, Akiyama T, Sato T (2009) Cholinoceptive and cholinergic properties of cardiomyocytes involving an amplification mechanism for vagal efferent effects in sparsely innervated ventricular myocardium. FEBS J 276:5111–5125. doi:10.1111/j.1742-4658.2009.07208.x
Kalakech H, Tamareille S, Pons S, Godin-Ribuot D, Carmeliet P, Furber A, Martin V, Berdeaux A, Ghaleh B, Prunier F (2013) Role of hypoxia inducible factor-1alpha in remote limb ischemic preconditioning. J Mol Cell Cardiol 65:98–104. doi:10.1016/j.yjmcc.2013.10.001
Kalakech H, Hibert P, Prunier-Mirebeau D, Tamareille S, Letournel F, Macchi L, Pinet F, Furber A, Prunier F (2014) RISK and SAFE signaling pathway involvement in apolipoprotein A-I-induced cardioprotection. PLoS One 9:e107950. doi:10.1371/journal.pone.0107950
Kalogeris T, Baines CP, Krenz M, Korthuis RJ (2012) Cell biology of ischemia/reperfusion injury. Int Rev Cell Mol Biol 298:229–317. doi:10.1016/B978-0-12-394309-5.00006-7
Kharbanda RK, Mortensen UM, White PA, Kristiansen SB, Schmidt MR, Hoschtitzky JA, Vogel M, Sorensen K, Redington AN, MacAllister AF (2002) Transient limb ischemia induces remote ischemic preconditioning in vivo. Circulation 106:2881–2883. doi:10.1161/01.CIR.0000043806.51912.9B
Kiss K, Csonka C, Paloczi J, Pipis J, Gorbe A, Kocsis GF, Murlasits Z, Sarkozy M, Szucs G, Holmes CP, Pan Y, Bhandari A, Csont T, Shamloo M, Woodburn KW, Ferdinandy P, Bencsik P (2016) Novel, selective EPO receptor ligands lacking erythropoietic activity reduce infarct size in acute myocardial infarction in rats. Pharmacol Res 113:62–70. doi:10.1016/j.phrs.2016.08.013
Kleinbongard P, Heusch G (2015) Extracellular signalling molecules in the ischaemic/reperfused heart - druggable and translatable for cardioprotection? Br J Pharmacol 172:2010–2025. doi:10.1111/bph.12902
Kono Y, Fukuda S, Hanatani A, Nakanishi K, Otsuka K, Taguchi H, Shimada K (2014) Remote ischemic conditioning improves coronary microcirculation in healthy subjects and patients with heart failure. Drug Des Devel Ther 8:1175–1181. doi:10.2147/DDDT.S68715
Konstantinov IE, Arab S, Li J, Coles JG, Boscarino C, Mori A, Cukerman E, Dawood F, Cheung MM, Shimizu M, Liu PP, Redington AN (2005) The remote ischemic preconditioning stimulus modifies gene expression in mouse myocardium. J Thorac Cardiovasc Surg 130:1326–1332. doi:10.1016/j.jtcvs.2005.03.050
Kottenberg E, Thielmann M, Bergmann L, Heine T, Jakob H, Heusch G, Peters J (2012) Protection by remote ischaemic preconditioning during coronary artery bypass grafting with isoflurane but not with propofol anesthesia - a clinical trial. Acta Anaesthesiol Scand 56:30–38. doi:10.1111/j.1399-6576.2011.02585.x
Kottenberg E, Musiolik J, Thielmann M, Jakob H, Peters J, Heusch G (2014) Interference of propofol with signal transducer and activator of transcription 5 activation and cardioprotection by remote ischemic preconditioning during coronary artery bypass grafting. J Thorac Cardiovasc Surg 147:376–382. doi:10.1016/j.jtcvs.2013.01.005
Kristiansen SB, Henning O, Kharbanda RK, Nielsen-Kudsk JE, Schmidt MR, Redington AN, Nielsen TT, Bøtker HE (2005) Remote preconditioning reduces ischemic injury in the explanted heart by a KATP channel-dependent mechanism. Am J Physiol Heart Circ Physiol 288:H1252–H1256. doi:10.1152/ajpheart.00207.2004
Lambert EA, Thomas CJ, Hemmes R, Eikelis N, Pathak A, Schlaich MP, Lambert GW (2016) Sympathetic nervous response to ischemia-reperfusion injury in humans is altered with remote ischemic preconditioning. Am J Physiol Heart Circ Physiol 311:H364–H370. doi:10.1152/ajpheart.00369.2016
Lang SC, Elsässer A, Scheler C, Vetter S, Tiefenbacher CP, Kübler W, Katus HA, Vogt AM (2006) Myocardial preconditioning and remote renal preconditioning. Identifying a protective factor using proteomic methods? Basic Res Cardiol 101:149–158. doi:10.1007/s00395-005-0565-0
Lanza GA, Stazi A, Villano A, Torrini F, Milo M, Laurito M, Flego D, Aurigemma C, Liuzzo G, Crea F (2016) Effect of remote ischemic preconditioning on platelet activation induced by coronary procedures. Am J Cardiol 117:359–365. doi:10.1016/j.amjcard.2015.10.056
Leung CH, Wang L, Nielsen JM, Tropak MB, Fu YY, Kato H, Callahan J, Redington AN, Caldarone CA (2014) Remote cardioprotection by transfer of coronary effluent from ischemic preconditioned rabbit heart preserves mitochondrial integrity and function via adenosine receptor activation. Cardiovasc Drugs Ther 28:7–17. doi:10.1007/s10557-013-6489-2
Li J, Rohailla S, Gelber N, Rutka J, Sabah N, Gladstone RA, Wei C, Hu P, Kharbanda RK, Redington AN (2014) MicroRNA-144 is a circulating effector of remote ischemic preconditioning. Basic Res Cardiol 109:423. doi:10.1007/s00395-014-0423-z
Liem DA, Verdouw PD, Ploeg H, Kazim S, Duncker D (2002) Sites of action of adenosine in interorgan preconditioning of the heart. Am J Physiol Heart Circ Physiol 283:H29–H37. doi:10.1152/ajpheart.01031.2001
Lim SY, Yellon DM, Hausenloy DJ (2010) The neural and humoral pathways in remote limb ischemic preconditioning. Basic Res Cardiol 105:651–655. doi:10.1007/s00395-010-0099-y
Ling Ling J, Wong GT, Yao L, Xia Z, Irwin MG (2010) Remote pharmacological post-conditioning by intrathecal morphine: cardiac protection from spinal opioid receptor activation. Acta Anaesthesiol Scand 54:1097–1104. doi:10.1111/j.1399-6576.2010.02295.x
Lonborg J, Vejlstrup N, Kelbaek H, Holmvang L, Jorgensen E, Helqvist S, Saunamaki K, Ahtarovski KA, Botker HE, Kim WY, Clemmensen P, Engstrom T (2013) Final infarct size measured by cardiovascular magnetic resonance in patients with ST elevation myocardial infarction predicts long-term clinical outcome: an observational study. Eur Heart J Cardiovasc Imaging 14:387–395. doi:10.1093/ehjci/jes271
Loukogeorgakis SP, Panagiotidou AT, Broadhead MW, Donald A, Deanfield JE, MacAllister RJ (2005) Remote ischemic preconditioning provides early and late protection against endothelial ischemia-reperfusion injury in humans. Role of the autonomic nervous system. J Am Coll Cardiol 46:450–456. doi:10.1016/j.jacc.2005.04.044
Loukogeorgakis SP, Williams R, Panagiotidou AT, Kolvekar SK, Donald A, Cole TJ, Yellon DM, Deanfield JE, MacAllister RJ (2007) Transient limb ischemia induces remote preconditioning and remote postconditioning in humans by a KATP channel dependent mechanism. Circulation 116:1386–1395. doi:10.1161/CIRCULATIONAHA.106.653782
Lu Y, Wong GT, Zhang Y, Hu J, Dong C (2014) Remote intrathecal morphine preconditioning is ineffective in the presence of neuraxial blockade with lidocaine. Kaohsiung J Med Sci 30:68–72. doi:10.1016/j.kjms.2013.10.007
Martin-Puig S, Tello D, Aragones J (2015) Novel perspectives on the PHD-HIF oxygen sensing pathway in cardioprotection mediated by IPC and RIPC. Front Physiol 6:137. doi:10.3389/fphys.2015.00137
Mastitskaya S, Marina N, Gourine A, Gilbey MP, Spyer KM, Teschemacher AG, Kasparov S, Trapp S, Ackland GL, Gourine AV (2012) Cardioprotection evoked by remote ischaemic preconditioning is critically dependent on the activity of vagal pre-ganglionic neurones. Cardiovasc Res 95:487–494. doi:10.1093/cvr/cvs212
Mastitskaya S, Basalay M, Hosford PS, Ramage AG, Gourine A, Gourine AV (2016) Identifying the source of a humoral factor of remote (pre)conditioning cardioprotection. PLoS One 11:e0150108. doi:10.1371/journal.pone.0150108
Mei B, Li W, Cheng X, Liu X, Gu E, Zhang Y (2016) Activating mu-opioid receptors in the spinal cord mediates the cardioprotective effect of remote preconditioning of trauma. Cardiol J. doi:10.5603/CJ.a2016.0062
Merlocco AC, Redington KL, Disenhouse T, Strantzas SC, Gladstone R, Wei C, Tropak MB, Manlhiot C, Li J, Redington AN (2014) Transcutaneous electrical nerve stimulation as a novel method of remote preconditioning: in vitro validation in an animal model and first human observations. Basic Res Cardiol 109:406. doi:10.1007/s00395-014-0406-0
Meybohm P, Bein B, Brosteanu O, Cremer J, Gruenewald M, Stoppe C, Coburn M, Schaelte G, Boning A, Niemann B, Roesner J, Kletzin F, Strouhal U, Reyher C, Laufenberg-Feldmann R, Ferner M, Brandes IF, Bauer M, Stehr SN, Kortgen A, Wittmann M, Baumgarten G, Meyer-Treschan T, Kienbaum P, Heringlake M, Schon J, Sander M, Treskatsch S, Smul T, Wolwender E, Schilling T, Fuernau G, Hasenclever D, Zacharowski K, Collaborators RIS (2015) A multicenter trial of remote ischemic preconditioning for heart surgery. N Engl J Med 373:1397–1407. doi:10.1056/NEJMoa1413579
Nilius B, Owsianik G, Voets T, Peters JA (2007) Transient receptor potential cation channels in disease. Physiol Rev 87:165–217. doi:10.1152/physrev.00021.2006
Oba T, Yasukawa H, Nagata T, Kyogoku S, Minami T, Nishihara M, Ohshima H, Mawatari K, Nohara S, Takahashi J, Sugi Y, Igata S, Iwamoto Y, Kai H, Matsuoka H, Takano M, Aoki H, Fukumoto Y, Imaizumi T (2015) Renal nerve-mediated erythropoietin release confers cardioprotection during remote ischemic preconditioning. Circ J 79:1557–1567. doi:10.1253/circj.CJ-14-1171
Oikawa S, Mano A, Takahashi R, Kakinuma Y (2015) Remote ischemic preconditioning with a specialized protocol activates the non-neuronal cardiac cholinergic system and increases ATP content in the heart. Int Immunopharmacol 29:181–184. doi:10.1016/j.intimp.2015.06.004
Okerson T, Chilton RJ (2012) The cardiovascular effects of GLP-1 receptor agonists. Cardiovasc Ther 30:e146–e155. doi:10.1111/j.1755-5922.2010.00256.x
Olenchock BA, Moslehi J, Baik AH, Davidson SM, Williams J, Gibson WJ, Pierce KA, Miller CM, Hanse EA, Kelekar A, Sullivan LB, Wagers AJ, Clish CB, Vander Heiden MG, Kaelin WG Jr (2016) EGLN1 inhibition and rerouting of alpha-ketoglutarate suffice for remote ischemic protection. Cell 164:884–895. doi:10.1016/j.cell.2016.02.006
Ovize M, Baxter GF, Di Lisa F, Ferdinandy P, Garcia-Dorado D, Hausenloy DJ, Heusch G, Vinten-Johansen J, Yellon DM, Schulz R (2010) Postconditioning and protection from reperfusion injury: where do we stand? Cardiovasc Res 87:406–423. doi:10.1093/cvr/cvq129
Pedersen CM, Cruden NL, Schmidt MR, Lau C, Bøtker HE, Kharbanda RK, Newby DE (2011) Remote ischemic preconditioning prevents systemic platelet activation associated with ischemia-reperfusion injury in humans. J Thromb Haemost 9:404–407. doi:10.1111/j.1538-7836.2010.04142.x
Pedersen CM, Schmidt MR, Barnes G, Bøtker HE, Kharbanda RK, Newby DE, Cruden NL (2011) Bradykinin does not mediate remote ischaemic preconditioning or ischaemia-reperfusion injury in vivo in man. Heart 97:1857–1861. doi:10.1136/heartjnl-2011-300323
Pickard JM, Botker HE, Crimi G, Davidson B, Davidson SM, Dutka D, Ferdinandy P, Ganske R, Garcia-Dorado D, Giricz Z, Gourine AV, Heusch G, Kharbanda R, Kleinbongard P, MacAllister R, McIntyre C, Meybohm P, Prunier F, Redington A, Robertson NJ, Suleiman MS, Vanezis A, Walsh S, Yellon DM, Hausenloy DJ (2015) Remote ischemic conditioning: from experimental observation to clinical application: report from the 8th biennial hatter cardiovascular institute workshop. Basic Res Cardiol 110:453. doi:10.1007/s00395-014-0453-6
Pickard JM, Davidson SM, Hausenloy DJ, Yellon DM (2016) Co-dependence of the neural and humoral pathways in the mechanism of remote ischemic conditioning. Basic Res Cardiol 111:50. doi:10.1007/s00395-016-0568-z
Pryds K, Nielsen RR, Hoff CM, Tolbod LP, Bouchelouche K, Li J, Schmidt MR, Redington AN, Frokiaer J, Botker HE (2016) Effect of remote ischemic conditioning on myocardial perfusion in patients with suspected ischemic coronary artery disease. J Nucl Cardiol. doi:10.1007/s12350-016-0709-7
Przyklenk K, Bauer B, Ovize M, Kloner RA, Whittaker P (1993) Regional ischemic "preconditioning" protects remote virgin myocardium from subsequent sustained coronary occlusion. Circulation 87:893–899. doi:10.1161/01.CIR.87.3.893
Randhawa PK, Jaggi AS (2015) TRPV1 and TRPV4 channels: potential therapeutic targets for ischemic conditioning-induced cardioprotection. Eur J Pharmacol 746:180–185
Randhawa PK, Jaggi AS (2016) Opioids in remote ischemic preconditioning-induced cardioprotection. J Cardiovasc Pharmacol Ther. doi:10.1177/1074248416660621
Randhawa PK, Jaggi AS (2016) Unraveling the role of adenosine in remote ischemic preconditioning-induced cardioprotection. Life Sci 155:140–146. doi:10.1016/j.lfs.2016.05.009
Rassaf T, Ferdinandy P, Schulz R (2014) Nitrite in organ protection. Br J Pharmacol 171:1–11. doi:10.1111/bph.12291
Rassaf T, Totzeck M, Hendgen-Cotta UB, Shiva S, Heusch G, Kelm M (2014) Circulating nitrite contributes to cardioprotection by remote ischemic preconditioning. Circ Res 114:1601–1610. doi:10.1161/CIRCRESAHA.114.303822
Redington KL, Disenhouse T, Strantzas SC, Gladstone R, Wei C, Tropak MB, Dai X, Manlhiot C, Li J, Redington AN (2012) Remote cardioprotection by direct peripheral nerve stimulation and topical capsaicin is mediated by circulating humoral factors. Basic Res Cardiol 107:241. doi:10.1007/s00395-011-0241-5
Redington KL, Disenhouse T, Li J, Wei C, Dai X, Gladstone R, Manlhiot C, Redington AN (2013) Electroacupuncture reduces myocardial infarct size and improves post-ischemic recovery by invoking release of humoral, dialyzable, cardioprotective factors. J Physiol Sci 63:219–223. doi:10.1007/s12576-013-0259-6
Reed GW, Rossi JE, Cannon CP (2016) Acute myocardial infarction. Lancet. doi:10.1016/S0140-6736(16)30677-8
Rentoukas I, Giannopoulos G, Kaoukis A, Kossyvakis C, Raisakis K, Driva M, Panagopoulou V, Tsarouchas K, Vavetsi S, Pyrgakis V, Deftereos S (2010) Cardioprotective role of remote ischemic periconditioning in primary percutaneous coronary intervention: enhancement by opioid action. J Am Coll Cardiol Cardiovasc Interv 3:49–55. doi:10.1016/j.jcin.2009.10.015
Sanchis-Gomar F, Garcia-Gimenez JL, Pareja-Galeano H, Romagnoli M, Perez-Quilis C, Lippi G (2014) Erythropoietin and the heart: physiological effects and the therapeutic perspective. Int J Cardiol 171:116–125. doi:10.1016/j.ijcard.2013.12.011
Sardar P, Chatterjee S, Kundu A, Samady H, Owan T, Giri J, Nairooz R, Selzman CH, Heusch G, Gersh BJ, Abbott JD, Mukherjee D, Fang JC (2016) Remote ischemic preconditioning in patients undergoing cardiovascular surgery: evidence from a meta-analysis of randomized controlled trials. Int J Cardiol 221:34–41. doi:10.1016/j.ijcard.2016.06.325
Saxena P, Aggarwal S, Misso NL, Passage J, Newman MA, Thompson PJ, d'Udekem Y, Praporski S, Konstantinov IE (2013) Remote ischaemic preconditioning down-regulates kinin receptor expression in neutrophils of patients undergoing heart surgery. Interact Cardiovasc Thorac Surg 17:653–658. doi:10.1093/icvts/ivt279
Schoemaker RG, van Heijningen CL (2000) Bradykinin mediates cardiac preconditioning at a distance. Am J Physiol Heart Circ Physiol 278:H1571–H1576
Schulte G, Sommerschild H, Yang J, Tokuno S, Goiny M, Lovdahl C, Johansson B, Fredholm BB, Valen G (2004) Adenosine a receptors are necessary for protection of the murine heart by remote, delayed adaptation to ischaemia. Acta Physiol Scand 182:133–143. doi:10.1111/j.1365-201X.2004.01350.x
Serejo FC, Rodrigues LF Jr, da Silva Tavares KC, de Carvalho AC, Nascimento JH (2007) Cardioprotective properties of humoral factors released from rat hearts subject to ischemic preconditioning. J Cardiovasc Pharmacol 49:214–220. doi:10.1097/FJC.0b013e3180325ad9
Shahid M, Tauseef M, Sharma KK, Fahim M (2008) Brief femoral artery ischaemia provides protection against myocardial ischaemia-reperfusion injury in rats: the possible mechanisms. Exp Physiol 93:954–968. doi:10.1113/expphysiol.2007.041442
Shigematsu S, Ishida S, Gute DC, Korthuis RJ (1999) Concentration-dependent effects of bradykinin on leukocyte recruitment and venular hemodynamics in rat mesentery. Am J Physiol Heart Circ Physiol 277:H152–H160
Shimizu M, Konstantinov IE, Kharbanda RK, Cheung MH, Redington AN (2007) Effects of intermittent lower limb ischaemia on coronary blood flow and coronary resistance in pigs. Acta Physiol (Oxf) 190:103–109. doi:10.1111/j.1748-1716.2007.01667.x
Shimizu M, Tropak M, Diaz RJ, Suto F, Surendra H, Kuzmin E, Li J, Gross G, Wilson GJ, Callahan J, Redington AN (2009) Transient limb ischaemia remotely preconditions through a humoral mechanism acting directly on the myocardium: evidence suggesting cross-species protection. Clin Sci (Lond) 117:191–200. doi:10.1042/CS20080523
Shimizu M, Saxena P, Konstantinov IE, Cherepanov V, Cheung MM, Wearden P, Zhangdong H, Schmidt M, Downey GP, Redington AN (2010) Remote ischemic preconditioning decreases adhesion and selectively modifies functional responses of human neutrophils. J Surg Res 158:155–161. doi:10.1016/j.jss.2008.08.010
Silvani A, Calandra-Buonaura G, Dampney RA, Cortelli P (2016) Brain-heart interactions: physiology and clinical implications. Philos Trans A Math Phys Eng Sci 374. doi:10.1098/rsta.2015.0181
Skyschally A, Gent S, Amanakis G, Schulte C, Kleinbongard P, Heusch G (2015) Across-species transfer of protection by remote ischemic preconditioning with species-specific myocardial signal transduction by reperfusion injury salvage kinase and survival activating factor enhancement pathways. Circ Res 117:279–288. doi:10.1161/CIRCRESAHA.117.306878
Slagsvold KH, Moreira JB, Rognmo O, Hoydal M, Bye A, Wisloff U, Wahba A (2014) Remote ischemic preconditioning preserves mitochondrial function and activates pro-survival protein kinase Akt in the left ventricle during cardiac surgery: a randomized trial. Int J Cardiol 177:409–417. doi:10.1016/j.ijcard.2014.09.206
Slagsvold KH, Rognmo O, Hoydal M, Wisloff U, Wahba A (2014) Remote ischemic preconditioning preserves mitochondrial function and influences myocardial microRNA expression in atrial myocardium during coronary bypass surgery. Circ Res 114:851–859. doi:10.1161/CIRCRESAHA.114.302751
Sloth AD, Schmidt MR, Munk K, Kharbanda RK, Redington AN, Schmidt M, Pedersen L, Sorensen HT, Bøtker HE (2014) Improved long-term clinical outcomes in patients with ST-elevation myocardial infarction undergoing remote ischaemic conditioning as an adjunct to primary percutaneous coronary intervention. Eur Heart J 35:168–175. doi:10.1093/eurheartj/eht369
Southerland EM, Milhorn DM, Foreman RD, Linderoth B, Dejongste MJ, Armour JA, Subramanian V, Singh M, Singh K, Ardell JL (2007) Preemptive, but not reactive, spinal cord stimulation mitigates transient ischemia-induced myocardial infarction via cardiac adrenergic neurons. Am J Physiol Heart Circ Physiol 292:H311–H317. doi:10.1152/ajpheart.00087.2006
Stazi A, Scalone G, Laurito M, Milo M, Pelargonio G, Narducci ML, Parrinello R, Figliozzi S, Bencardino G, Perna F, Lanza GA, Crea F (2014) Effect of remote ischemic preconditioning on platelet activation and reactivity induced by ablation for atrial fibrillation. Circulation 129:11–17. doi:10.1161/CIRCULATIONAHA.113.005336
Steensrud T, Li J, Dai X, Manlhiot C, Kharbanda RK, Tropak M, Redington A (2010) Pretreatment with the nitric oxide donor SNAP or nerve transection blocks humoral preconditioning by remote limb ischemia or intra-arterial adenosine. Am J Physiol Heart Circ Physiol 299:H1598–H1603. doi:10.1152/ajpheart.00396.2010
Stone GW, Selker HP, Thiele H, Patel MR, Udelson JE, Ohman EM, Maehara A, Eitel I, Granger CB, Jenkins PL, Nichols M, Ben-Yehuda O (2016) Relationship between infarct size and outcomes following primary PCI: patient-level analysis from 10 randomized trials. J Am Coll Cardiol 67:1674–1683. doi:10.1016/j.jacc.2016.01.069
Surendra H, Diaz RJ, Harvey K, Tropak M, Callahan J, Hinek A, Hossain T, Redington A, Wilson GJ (2013) Interaction of delta and kappa opioid receptors with adenosine A1 receptors mediates cardioprotection by remote ischemic preconditioning. J Mol Cell Cardiol 60:142–150. doi:10.1016/j.yjmcc.2013.04.010
Svennerholm K, Rodsand P, Hellmand U, Lundholm M, Waldenström A, Biber B, Ronquist G, Haney M (2015) Myocardial ischemic preconditioning in a porcine model leads to rapid changes in cardiac extracellular vesicle messenger RNA content. Int J Cardiol 8:62–67. doi:10.1016/j.ijcha.2015.05.006
Svennerholm K, Rodsand P, Hellman U, Waldenstrom A, Lundholm M, Ahren D, Biber B, Ronquist G, Haney M (2016) DNA content in extracellular vesicles isolated from porcine coronary venous blood directly after myocardial ischemic preconditioning. PLoS One 11:e0159105. doi:10.1371/journal.pone.0159105
Thielmann M, Kottenberg E, Kleinbongard P, Wendt D, Gedik N, Pasa S, Price V, Tsagakis K, Neuhäuser M, Peters J, Jakob H, Heusch G (2013) Cardioprotective and prognostic effects of remote ischaemic preconditioning in patients undergoing coronary artery bypass surgery: a single-Centre randomised, double-blind, controlled trial. Lancet 382:597–604. doi:10.1016/S0140-6736(13)61450-6
Thygesen K, Alpert JS, Jaffe AS, Simoons ML, Chaitman BR, White HD, Thygesen K, Alpert JS, White HD, Jaffe AS, Katus HA, Apple FS, Lindahl B, Morrow DA, Chaitman BR, Clemmensen PM, Johanson P, Hod H, Underwood R, Bax JJ, Bonow RO, Pinto F, Gibbons RJ, Fox KA, Atar D, Newby LK, Galvani M, Hamm CW, Uretsky BF, Gabriel SP, Wijns W, Bassand JP, Menasche P, Ravkilde J, Ohman EM, Antman EM, Wallentin LC, Armstrong PW, Simoons ML, Januzzi JL, Nieminen MS, Gheorghiade M, Filippatos G, Luepker RV, Fortmann SP, Rosamond WD, Levy D, Wood D, Smith SC, Hu D, Lopez-Sendon JL, Robertson RM, Weaver D, Tendera M, Bove AA, Parkhomenko AN, Vasilieva EJ, Mendis S, Bax JJ, Baumgartner H, Ceconi C, Dean V, Deaton C, Fagard R, Funck-Brentano C, Hasdai D, Hoes A, Kirchhof P, Knuuti J, Kolh P, McDonagh T, Moulin C, Popescu BA, Reiner Z, Sechtem U, Sirnes PA, Tendera M, Torbicki A, Vahanian A, Windecker S, Morais J, Aguiar C, Almahmeed W, Arnar DO, Barili F, Bloch KD, Bolger AF, Bøtker HE, Bozkurt B, Bugiardini R, Cannon C, de Lemos J, Eberli FR, Escobar E, Hlatky M, James S, Kern KB, Moliterno DJ, Mueller C, Neskovic AN, Pieske BM, Schulman SP, Storey RF, Taubert KA, Vranckx P, Wagner DR (2012) Third universal definition of myocardial infarction. Eur Heart J 33:2551–2567. doi:10.1093/eurheartj/ehs184
Tsai HJ, Huang SS, Tsou MT, Wang HT, Chiu JH (2015) Role of opioid receptors signaling in remote electrostimulation -induced protection against ischemia/reperfusion injury in rat hearts. PLoS One 10:e0138108. doi:10.1371/journal.pone.0138108
Tsou MT, Huang CH, Chiu JH (2004) Electroacupuncture on PC6 (Neiguan) attenuates ischemia/reperfusion injury in rat hearts. Am J Chin Med 32:951–965. doi:10.1142/S0192415X04002557
Uitterdijk A, Yetgin T, Te Lintel HM, Sneep S, Krabbendam-Peters I, van Beusekom HM, Fischer TM, Cornelussen RN, Manintveld OC, Merkus D, Duncker DJ (2015) Vagal nerve stimulation started just prior to reperfusion limits infarct size and no-reflow. Basic Res Cardiol 110:508. doi:10.1007/s00395-015-0508-3
Vicencio JM, Yellon DM, Sivaraman V, Das D, Boi-Doku C, Arjun S, Zheng Y, Riquelme JA, Kearney J, Sharma V, Multhoff G, Hall AR, Davidson SM (2015) Plasma exosomes protect the myocardium from ischemia-reperfusion injury. J Am Coll Cardiol 65:1525–1536. doi:10.1016/j.jacc.2015.02.026
Wang L, Oka N, Tropak M, Callahan J, Lee J, Wilson G, Redington A, Caldarone CA (2008) Remote ischemic preconditioning elaborates a transferable blood-borne effector that protects mitochondrial structure and function and preserves myocardial performance after neonatal cardioplegic arrest. J Thorac Cardiovasc Surg 136:335–342. doi:10.1016/j.jtcvs.2007.12.055
Weinbrenner C, Nelles M, Herzog N, Sarvary L, Strasser RH (2002) Remote preconditioning by infrarenal occlusion of the aorta protects the heart from infarction: a newly identified non-neuronal but PKC-dependent pathway. Cardiovasc Res 55:590–601. doi:10.1016/S0008-6363(02)00446-7
Whittaker P, Przyklenk K (2014) From ischemic conditioning to 'hyperconditioning': clinical phenomenon and basic science opportunity. Dose Response 12:650–663. doi:10.2203/dose-response.14-035
Wong GT, Lu Y, Mei B, Xia Z, Irwin MG (2012) Cardioprotection from remote preconditioning involves spinal opioid receptor activation. Life Sci 91:860–865. doi:10.1016/j.lfs.2012.08.037
Xin P, Zhu W, Li J, Ma S, Wang L, Liu M, Li J, Wei M, Redington AN (2010) Combined local ischemic postconditioning and remote perconditioning recapitulate cardioprotective effects of local ischemic preconditioning. Am J Physiol Heart Circ Physiol 298:H1819–H1831. doi:10.1152/ajpheart.01102.2009
Yellon DM, Davidson SM (2014) Exosomes: nanoparticles involved in cardioprotection? Circ Res 114:325–332. doi:10.1152/ajpheart.01102.2009
Yellon DM, Hausenloy DJ (2007) Myocardial reperfusion injury. N Engl J Med 357:1121–1135. doi:10.1056/NEJMra071667
Zangrillo A, Musu M, Greco T, Di Prima AL, Matteazzi A, Testa V, Nardelli P, Febres D, Monaco F, Calabro MG, Ma J, Finco G, Landoni G (2015) Additive effect on survival of anaesthetic cardiac protection and remote ischemic preconditioning in cardiac surgery: a Bayesian network meta-analysis of randomized trials. PLoS One 10:e0134264. doi:10.1371/journal.pone.0134264
Zarbock A, Schmidt C, Van Aken H, Wempe C, Martens S, Zahn PK, Wolf B, Goebel U, Schwer CI, Rosenberger P, Haeberle H, Gorlich D, Kellum JA, Meersch M, Renal RI (2015) Effect of remote ischemic preconditioning on kidney injury among high-risk patients undergoing cardiac surgery: a randomized clinical trial. JAMA 313:2133–2141. doi:10.1001/jama.2015.4189
Zernecke A, Preissner KT (2016) Extracellular ribonucleic acids (RNA) enter the stage in cardiovascular disease. Circ Res 118:469–479. doi:10.1161/CIRCRESAHA.115.307961
Zhang SZ, Wang NF, Xu J, Gao Q, Lin GH, Bruce IC, Xia Q (2006) Kappa-opioid receptors mediate cardioprotection by remote preconditioning. Anesthesiology 105:550–556
Zhou C, Li L, Li H, Gong J, Fang N (2014) Delayed remote preconditioning induces cardioprotection: role of heme oxygenase-1. J Surg Res. doi:10.1016/j.jss.2014.03.054
Zitta K, Meybohm P, Gruenewald M, Cremer J, Zacharowski KD, Scholz J, Steinfath M, Albrecht M (2015) Profiling of cell stress protein expression in cardiac tissue of cardiosurgical patients undergoing remote ischemic preconditioning: implications for thioredoxin in cardioprotection. J Transl Med 13:34. doi:10.1186/s12967-015-0403-6
Acknowledgments
PK and GH were supported by the German Research Foundation (GH: He 1320/18-1, 3; PK and GH: SFB 1116, B8).
Author information
Authors and Affiliations
Corresponding author
Additional information
An erratum to this article is available at http://dx.doi.org/10.1007/s00424-017-1936-8.
Rights and permissions
About this article

Cite this article
Kleinbongard, P., Skyschally, A. & Heusch, G. Cardioprotection by remote ischemic conditioning and its signal transduction. Pflugers Arch - Eur J Physiol 469, 159–181 (2017). https://doi.org/10.1007/s00424-016-1922-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-016-1922-6