
Abstract
Background
Coronary effluent from an isolated perfused heart undergoing ischemic preconditioning can be transferred to precondition another naïve isolated heart. We investigated the effects of this effluent on mitochondrial integrity and function following a global infarct model of ischemia/reperfusion and the role of adenosine in this model of remote preconditioning.
Methods and Results
Coronary effluent from isolated perfused rabbit hearts was collected prior to (control effluent) and during three cycles of 5-min ischemia and 10-min reperfusion (IPC effluent). Adenosine concentration was significantly increased in IPC effluent (2.6 ± 1.1 μM) versus control effluent (0.21 ± 0.06 μM, P < 0.01). Infarct size (% necrotic LV mass) after 30-min global ischemia and 90-min reperfusion was significantly reduced in hearts preconditioned with IPC effluent (IPCeff, 23 ± 7 %) and control effluent supplemented with 2.5 μM exogenous adenosine (Ceff + 2.5 μM ADO, 25 ± 10 %) when compared to control effluent perfused hearts (Ceff, 41 ± 8 %, P < 0.05). Compared to Ceff mitochondria, IPCeff mitochondria had preserved complex I/State3 and complex IV/State 3 respiration and outer membrane integrity, and reduced cytochrome c release. In contrast, Ceff + 2.5 μM ADO mitochondria had improved state 2 respiration and coupling to oxidative phosphorylation, reduced reactive oxygen species production and preserved outer membrane integrity. Administration of adenosine receptor blocker 8-(p-sulfophenyl)theophylline abolished the infarct limiting effect (46 ± 7 %) and the mitochondrial integrity and function preservation of IPC effluent.
Conclusion
Remote cardioprotection by IPC effluent preserves mitochondrial integrity and function in an adenosine receptor dependent mechanism, and although infarct size reduction can be mimicked by adenosine, IPC effluent contains additional factor(s) contributing to modulation of the mitochondrial response to ischemia/reperfusion injury.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Repetitive brief episodes of nonlethal ischemic stress applied to the myocardium, known as ischemic preconditioning (IPC), induces a potent cellular protective mechanism that renders the myocardium resistant against a subsequent potentially lethal ischemia/reperfusion (I/R) injury [1]. Remote ischemic preconditioning, in contrast, confers similar protection after repetitive brief periods of nonlethal ischemic stress applied to remote (non-cardiac) tissue [2–4]. The signaling pathways that mediate cardioprotection by IPC has been extensively characterized since its discovery (reviewed by Yang et al. [5]) but the extracellular factor(s) that mediates remote ischemic preconditioning is unknown.
We have previously shown that plasma-derived dialysate from remote ischemic preconditioned rabbits by brief episodes of hindlimb I/R elicited cardioprotection by a transferrable humoral factor(s) [6] and is associated with preservation of mitochondrial integrity and function after I/R injury [7]. Because such dialysate contains factors derived from multiple tissue beds, other investigators have used a single organ “transfer of cardioprotection” model in which an isolated crystalloid-perfused heart undergoes IPC and the coronary effluent (IPC effluent) is harvested and used to perfuse a second ‘naïve’ isolated heart with transfer of preconditioning [8, 9]. This model is useful because potential extraneous factors released from non-cardiac organs are excluded from the preparation. Furthermore, other investigators have demonstrated the presence of low molecular weight hydrophobic cardioprotective factors in the IPC effluent [9, 10] but the role of these factors on mitochondrial integrity and function following an ischemic insult is currently undefined.
Mitochondrial integrity and function is intrinsically related to energy production and recovery of myocardial performance after I/R injury. Complex I and complex III in the electron transport chain are major sources of reactive oxygen species (ROS’ production [11, 12] and ischemia-induced dysfunction at these two sites is associated with increased rate of ROS production [11]. I/R induced ROS production subsequently disrupts mitochondrial membrane integrity which is associated with the release of cytochrome c [13] and decreases the reducing capacity at cytochrome c oxidase [14]. Furthermore, cytochrome c is an early marker of pro-apoptotic stimuli that activates downstream mitochondrial apoptotic signaling cascades [15]. IPC has been demonstrated to protect against I/R injury through amelioration of ROS production [16], inhibition of mitochondrial permeability transition pore (MPTP) opening [17], and prevention of cytochrome c release [18].
The local endogenous release of bioactive substances (eg. adenosine, bradykinin, and opioids) from the myocardium following transient ischemia has been characterized as the major trigger of IPC [5]. Liu et al. [19] first demonstrated that blockade of the adenosine receptor could effectively abolish IPC and prior ischemic activation of A1 receptor by adenosine and its analogue mimicked IPC. Adenosine receptors are also critical mediators of remote ischemic preconditioning as systemic delivery of an adenosine receptor blocker abolishes the cardioprotection triggered by transient ischemia of non-cardiac tissue in the skeletal muscle [2] and kidney [20]. Given that adenosine is released into the coronary effluent after ischemic preconditioning [21], we hypothesized that adenosine mediates the infarct-limiting effect of IPC effluent.
The present study was therefore designed to determine if preconditioning with IPC effluent is associated with 1) improved cardiac hemodynamic recovery and diminished infarct size, 2) preservation of mitochondrial integrity and function, and 3) to examine if the adenosine receptor participates in this model of remote preconditioning.
Materials and Methods
All animals received humane care and treatment in accordance with the “Guide for the Care and Use of Laboratory Animals” (NIH publication No. 85–23, revised 1996) and all protocols were approved by the Animal Care and Use Committee of the Hospital for Sick Children in Toronto.
Effluent Donor Hearts Preparation
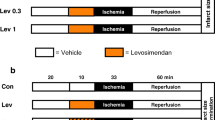
Donor hearts for coronary effluent production were obtained from male New Zealand White rabbits (Charles River, Saint-Constant, Quebec, Canada) weighing 3.0–3.5 kg. Rabbits were subjected to a single intramuscular injection of ketamine (15 mg/kg), acepromazine (0.29 mg/kg) and atropine (0.02 mg/kg), anaesthetized with pentobarbital (50 mg/kg), and heparinized. Hearts were quickly excised and retrogradely perfused on a Langendorff system by 95 % O2 and 5 % CO2 equilibrated Krebs-Henseleit Buffer (KHB) (in mM: 118 NaCl, 25 NaHCO3, 1.2 KH2O4, 4.7 KCl, 1.2 MgSO4, 1.8 CaCl2, and 11 glucose) at 37 °C. After a 30-min stabilization period, control effluent was collected (Fig. 1). Hearts then underwent ischemic preconditioning that consisted of 3 cycles of 5-min ischemia and 10-min reperfusion. IPC effluent was collected during each reperfusion interval. All effluents were harvested on ice and stored at 4 °C overnight before perfusion of recipient hearts. Effluents for nucleotide quantification were immediately stored at −80 °C.

Experimental design of effluent donor and recipient hearts on a Langendorff perfusion system. Control effluent was collected from effluent donor hearts following a 30 min stabilization period and IPC effluent was collected during the three cycles of 10 min reperfusion period following 5 min global ischemia. Effluent recipient hearts were pretreated with either control effluent, IPC effluent, control effluent supplemented with 2.5 μM exogenous adenosine, or IPC effluent with prior administration of adenosine receptor blocker 8-SPT. All effluent recipient hearts were then subjected to 30 min ischemia/90 min reperfusion for infarct assessment. For mitochondrial assessments, separate effluent recipient hearts were subjected to 30 min ischemia/30 min reperfusion. Normal perfused hearts without ischemia/reperfusion injury that were perfused with KHB for identical timeframe as effluent recipient hearts served as baseline controls for mitochondrial assessments. Ceff, Control effluent recipient hearts; IPCeff, IPC effluent recipient hearts; NH, non-ischemic perfused hearts; ADO, adenosine; KHB, Krebs-Henseleit Buffer; 8-SPT, 8-(p-sulfophenyl)theophylline
Quantitation of Effluent Nucleoside Levels
Control and IPC effluents (n = 10 in each group) were applied via an Autosampler (Leap Technologies HTS PAL) onto a Gemini-NX C18 column connected to an HPLC (Agilent) equipped with a binary pump (G1312A) and degasser (G132A). Mobile phases consisted of A: Milli-Q Water and B: Methanol. Adenosine and inosine were resolved using the following gradient 0 min: 5%B; 2 min.: 5%B; 7 min. 25 % B; 7.1. min. 5 % B; 9 min. 5 % B. Material from the column (flow rate 500 μL/min) was injected onto a Applied Biosystems (MDS Sciex, Toronto, ON, Canada) API4000 triple-quadruple mass spectrometer operating in positive MRM mode, with current set to 12, IS: 5500 and Temperature 700 °C (GS1: 40, GS2:40 and CAD: 8). Adenosine and inosine levels were monitored using parent to product ion transitions of 268.1–> 136.1 and 269.1–> 137.1 and with DF and CE set to 53, 27 and 35 and 19, respectively. Dwell time for each compound was set to 100 msec. For each batch of unknown samples, standard curves for adenosine and inosine (dissolved in KHB) spanning the range 2.5 nM to 500 nM were generated with an LOQ of 5 nM (average accuracy 100.1 %, range 92.8–103 %) for both compounds.
Effluent Recipient Heart Preparation
Prior to recipient heart perfusion, KHB, Control and IPC effluents were gassed with 95 % O2/5 % CO2 for 1 h at 37 °C. Perfusion buffers and effluents were analyzed for blood gases by Abbott i-STAT Portable Handheld device to ensure effluent conforms to the following parameters: pH 7.35 to 7.45, pO2 600 to 650 mmHG, and pCO2 35 to 40 mmHG. Upon excision, effluent recipient rabbit hearts were perfused on a flow-through Langendorff system and stabilized for 30-min with KHB perfusion. To determine the infarct size of effluent recipient hearts and whether the concentration of adenosine in the IPC effluent could precondition a naive isolated heart, hearts were perfused with either control effluent (Ceff), IPC effluent (IPCeff), or control effluent supplemented with 2.5 μM adenosine exogenous adenosine (Ceff + 2.5 μM ADO) to mimic adenosine concentration in IPC effluent (Fig. 1). Hearts were perfused by effluent for 30-min followed by 30-min global ischemia and 90-min reperfusion by KHB. To examine if adenosine receptor inhibition abolishes the infarct-limiting effect of IPC effluent, hearts were perfused with 100 μM 8-(p-sulfophenyl)theophylline [21] (IPCeff + 8-SPT) 5-min prior to and throughout IPC effluent perfusion for 30-min. 8-SPT was used as a non-specific adenosine receptor antagonist in this study as no specific adenosine receptor subtype has been characterized as the major trigger of remote ischemic preconditioning. Infarct sizes for all groups were assessed at the end of reperfusion.
LV Functional Assessment
Isovolumetric LV pressure changes were measured using a water-filled balloon placed in the LV cavity via the mitral valve, connected to a pressure transducer (MLT844; ADInstruments, Inc, Colorado Springs, CO). Balloon volume was adjusted to a left ventricular end-diastolic pressure (LVEDP) of 5 mmHg and kept constant throughout the entire experiment. Cardiac performance was assessed by LVEDP, LV developed pressure (LVDP), and peak positive and negative first derivatives LV pressure (±dP/dT). Heart rate and coronary flow rate were monitored with a small disk electrode probe (Harvard Apparatus, Holliston, MA) connected to an electrocardiographic amplifier (ML 136; ADInstruments, Colorado Springs, CO). Analog data was digitized and analyzed with Chart VI (ADInstruments, Colorado Springs, CO).
Infarct Size Assessment
At completion of reperfusion, the left ventricle from effluent recipient hearts was frozen and sliced into five 1 mm slices perpendicular to the apex-base axis with a rabbit heart chamber slicer (Zivic Instruments, Pittsburgh, PA). Slices were weighed and then stained in 1 % triphenyltetrazolium (TTC) (37 °C, 20 min) followed by 10 % neutral formalin fixation (room temperature, 2 h). Slices were scanned and digitized using a flatbed scanner. Color was standardized and the non-infarct (red) and infarct area representing necrosis (pale) were quantified by color pixel density (Adobe Photoshop CS4). Infarct size was expressed as a percentage of necrotic mass over total LV weight.
Mitochondrial Isolation
Because subsarcolemmal mitochondria (SSM) are more sensitive to ischemic damage when compared to interfibrillar mitochondria [14, 22], SSM was isolated for mitochondrial integrity and function evaluation. In separate effluent recipient hearts, SSM was isolated following 30-min ischemia and 30-min reperfusion to encapsulate the impact of the early phase of reperfusion on the mitochondria, which is critical in determining the severity of injury in the later phase (apoptosis versus necrosis). Mitochondria from time matched KHB perfused hearts (NH), which were not subjected to I/R injury, were used for baseline comparisons. SSM was isolated according to the protocol by Palmer et al. [23]. One gram of LV tissue was minced in buffer A (100 mmol/L KCl, 50 mmol/L 3-morpholino-propane-1-sulfonic acid (MOPS), 1.0 mmol/L MgSO4, 1.0 mmol/L EGTA, and 1 mmol/L adenosine triphosphate [pH 7.4] at 4 °C, and then homogenized in buffer A with 0.2 % bovine serum albumin with a loose Potter–Elvejhem homogenizer. The homogenate was centrifuged at 500 g, and the supernatant was combined with the supernatant from a second 500 g spin after the initial pellet was resuspended. The pooled supernatant was centrifuged at 3,000 g to sediment SSM, which was washed twice and resuspended in buffer containing 100 mmol/L KCl, 50 mmol/L MOPS, and 0.5 mmol/L EGTA). Since mitochondria are isolated from both viable and non-viable myocardium, the purity and quality of the mitochondria were assessed by the recovery rate per 1 g of LV tissue and citrate synthase activity per mg of mitochondria.
Mitochondrial Oxygen Consumption Measurement
Mitochondrial oxygen consumption was measured by a Clark-type electrode (Instech Laboratories, Plymouth, PA) in buffer containing (in mM): 100 KCl, 50 MOPS, 1.0 EGTA, 5.0 KH2PO4 and 5 % defatted BSA at 30 °C. Respiratory Complex I (State 2 and 3), complex II and complex IV (State 3) were measured as previously described by Ricci et al. [24]. Complex I, II and IV respiratory control ratio were expressed as State 3 normalized to State 2 oxygen consumption rates. State 4 respiration was not measured as a consequence of our protocol with the addition of ADP and respiratory inhibitors.
Mitochondrial Outer Membrane Permeability
Integrity of the mitochondrial outer membrane was assessed by monitoring mitochondrial oxygen consumption after administration of exogenous cytochrome c (10 μM) into the respiratory chamber during measurement of complex IV mitochondrial respiration [25]. The subsequent increase in complex IV activity reflects the permeabilization of outer mitochondrial membrane [26].
Cytochrome C Content
Mitochondrial cytochrome c content was determined by the difference of oxidized and reduced spectra measured by Agilent 8453 UV-visible Spectrophotometer (Agilent Technologies, Waldbronn, Germany) in a dual cuvette method as described by William [27].
Mitochondrial ROS Production
Reactive oxygen species (ROS) production was monitored in triplicates and standardized against a negative control from reduced dichlorofluorescein (H2DCF) oxidation (exCitation/emission, 490/525 nm) by a microplate spectroflurometer (Spectra MAX Gemini EM, Molecular Devices, Sunnyvale, CA) after incubating 50 mg of mitochondria energized by malate/glutamate (both 2.5 mM) in mitochondrial respiratory buffer with 10 μM CM-H2DCF diacetate (Molecular Probes, Eugene, OR) at room temperature for 30 min.
Statistics
Data were expressed as mean ± S.D. Planned two group comparisons (effluent adenosine and inosine concentrations) were performed by t-test. Infarct data and mitochondrial function analysis comparison between groups were performed by one-way ANOVA followed by Tukey’s post hoc test. Hemodynamic data were compared among groups by 2-factor ANOVA with replication and Tukey’s post hoc test was applied to identify differences between groups when significance was achieved. The correlation between infarct size and adenosine/inosine concentration in the effluent was determined by Pearson’s correlation coefficient. P < 0.05 was considered statistically significant.
Results
Adenosine and Inosine Levels in the Coronary Effluent of Donor Hearts
Quantification of nucleotide levels in the coronary effluent (Table 1) from normal perfused hearts and hearts undergoing three cycles of ischemic preconditioning showed that preconditioning increased adenosine concentration in the IPC effluent by more than 10-fold (P < 0.01 vs Control effluent), and inosine concentration by approximately 7-fold (P < 0.01 vs Control effluent).
Hemodynamics
Baseline hemodynamics obtained during the stabilization period were not significantly different between all groups (Table 2). Compared to control effluent recipient hearts, perfusion by IPC effluent and 2.5 μM exogenous adenosine increased the coronary flow prior to ischemia. However, this increase was transient and did not persist after reperfusion. ± dP/dT was also increased during effluent perfusion for IPC effluent and 2.5 μM exogenous adenosine treated hearts compared to control effluent and IPCeff + 8-SPT perfused hearts. After 30-min global ischemia, left ventricular performance and contractility were significantly better preserved in IPC effluent and 2.5 μM exogenous adenosine recipient hearts compared to control effluent recipient hearts throughout the course of reperfusion. Administration of adenosine receptor blocker 8-SPT prior to perfusion by IPC effluent abolished the protection against left ventricular dysfunction that was observed in IPC effluent recipient hearts.
Myocardial Infarction
Consistent with previous reports [8, 9], infarct size in IPC effluent recipient hearts (23 ± 7 %) was significantly reduced compared to control effluent recipient hearts (41 ± 8 %, P < 0.01) after 30 min ischemia and 90 min reperfusion (Fig. 2). Antecedent perfusion with control effluent supplemented with 2.5 μM adenosine resulted in infarct size reduction similar to IPC effluent (25 ± 10 %, P < 0.01 vs. Ceff). Adenosine receptor inhibitor treatment by 8-SPT prior to preconditioning by IPC effluent completely abolished the infarct limiting effect of IPC effluent (IPCeff + 8-SPT: 46 ± 7 %). Infarct size was not significantly different between IPCeff + 8-SPT and Ceff hearts. The correlation between infarct size and adenosine concentration was found non-significant by Pearson’s Correlation Coefficient statistical test (P = 0.205 for control effluent and P = 0.143 for IPC effluent). The correlation between infarct size and inosine concentration was also non-significant by Pearson’s Correlation Coefficient statistical test (P = 0.225 for control effluent and P = 0.276 for IPC effluent).

Infarct size assessed by TTC staining expressed as % of necrotic mass over total LV weight after 30 min ischemia and 90 min reperfusion in effluent recipient hearts. IPC effluent and 2.5 μM adenosine preconditioned hearts had significantly reduced infarct size compared to control effluent recipient hearts following ischemia/reperfusion injury. Administration of adenosine receptor blocker 8-SPT abolished the cardioprotection by IPC effluent. n = 5–6 per group. * P < 0.01 vs C
Mitochondrial Respiration Assessment
The relative mitochondrial recovery rate per 1 g of LV tissue (mg protein/g wet weight) and citrate synthase activity per mg of mitochondria were used as indicators of isolation purity and were not significantly different between all groups (Table 3). These values were consistent with a previous study from Lesnefsky et al. [14].
Ischemia and reperfusion in control effluent recipient hearts resulted in a significant deficit in mitochondrial complex I/State 3 respiration (P < 0.01 vs. NH) and complex IV/State 3 respiration (P < 0.05 vs. NH) (Fig. 3a). However, pretreatment with IPC effluent attenuated the deficits in complex I (P < 0.01 vs. IPCeff) and complex IV respiration (P < 0.05 vs. IPCeff). Administration of 8-SPT with IPC effluent abolished the improvements in complex I (P < 0.01 vs IPCeff) and complex IV (P < 0.05 vs IPCeff) respiration that was observed in IPC effluent recipient hearts. Perfusion with 2.5 μM adenosine prior to I/R failed to preserve complex I (P < 0.01 vs NH) and complex IV respirations (P < 0.01 vs NH). Complex II/State 3 respiration was not different among all groups.

Isolated subsarcolemmal mitochondria respirometry assessment after 30 min ischemia and 30 min reperfusion in effluent recipient hearts. n = 5–7 per group. a Mitochondrial oxygen consumption of Complex I, II, and IV in State 3 respiration. Complex I and complex IV respiration were better preserved in IPC effluent preconditioned hearts after ischemia/reperfusion injury compared to control effluent recipient hearts. * P < 0.01 vs NH. # P < 0.05 vs NH. ^ P < 0.01 vs IPCeff. ɸ P < 0.05 vs IPCeff. b Mitochondrial oxygen consumption in State 2 respiration. 2.5 μM adenosine preconditioned hearts had improved state 2 respiration compared to control effluent recipient hearts. * P < 0.01 vs NH and Ceff. # P < 0.05 vs IPCeff. c Respiratory control ratio as an indicator of the coupling between respiration and oxidative phosphorylation is represented by State 3/State2. Respiratory control ratio of complex I was preserved in IPC effluent preconditioned hearts compared to control effluent recipient hearts. In contrast, complex I, II, and IV respiratory control ratio was better preserved in 2.5 μM adenosine preconditioned hearts compared to control effluent recipient hearts. * P < 0.01 vs NH. ^ P < 0.05 vs IPCeff. ɸ P < 0.05 vs Ceff, IPCeff + 8-SPT . # P < 0.01 vs NH, C, IPCeff, IPCeff + 8-SPT. d Proportional increase in Complex IV activity after addition of exogenous cytochrome c was significantly lower in IPC effluent and 2.5 μM adenosine preconditioned hearts compared to control effluent recipient hearts indicating integrity of the outer mitochondrial membrane was preserved following ischemia/reperfusion. * P < 0.01 vs NH. # P < 0.05 vs Ceff
Mitochondrial State 2 respiration however was significantly improved in 2.5 μM adenosine recipient hearts (P < 0.01 vs Ceff and NH, P < 0.05 vs IPCeff, Fig. 3b), suggesting inner membrane permeability and proton leakage were decreased following I/R. No significant increase in mitochondrial State 2 respiratory activity was observed in all groups, indicating that I/R did not impair the integrity of the mitochondrial inner membrane.
The coupling between respiration and oxidative phosphorylation as represented by the respiratory control ratio (State 3/State 2) in control effluent and IPCeff + 8-SPT recipient hearts were significantly lower for complex I after I/R injury (P < 0.05 vs. NH, Fig. 3c). Perfusion with IPC effluent (P < 0.05 vs. Ceff) and 2.5 μM adenosine (P < 0.01 vs. Ceff) significantly attenuated this deficit. Respiratory control ratios for complex II and complex IV were also improved in mitochondria of 2.5 μM adenosine recipient hearts due to improved State 2 respiration but not improved State 3 respiration (P < 0.01 vs. all groups) albeit with a high degree of standard deviation in the complex IV RCR. Respirometry rates in the current study were consistent with our previous reports [7, 28].
Mitochondrial Membrane Integrity and Cytochrome c Retention
Administration of exogenous cytochrome c reversed the deficit in the complex IV/State 3 respiration of mitochondria from control effluent and IPCeff + 8-SPT recipient hearts (P < 0.01 vs. NH, Fig. 3d) suggesting that outer mitochondrial membrane permeabilization contributes to the decline in complex IV activity. IPC effluent and 2.5 μM ADO effluent recipient hearts maintained the integrity of the mitochondrial outer membrane as exogenous cytochrome c failed to elicit additional complex IV activity. Correlating with the notion of increased permeabilization of the outer mitochondrial membrane in control effluent recipient hearts was the significantly diminished total mitochondrial cytochrome c content when compared to non ischemic hearts (P < 0.01 vs. NH, Fig. 4). Conversely, total mitochondrial cytochrome c content in IPC effluent recipients hearts was well preserved (P < 0.01 vs. Ceff) but was not preserved in 2.5 μM adenosine recipient hearts (P < 0.01 vs NH). Adenosine receptor blockade with IPC effluent recipient hearts abolished the preservation of mitochondrial outer membrane integrity and total cytochrome content that were observed in IPC effluent recipient hearts.

Total cytochrome c content in the mitochondria following ischemia/reperfusion. Cytochrome c was preserved in IPC effluent preconditioned hearts but not in 2.5 μM adenosine preconditioned hearts compared to control effluent recipient hearts. * P < 0.01 vs NH. ^ P < 0.01 vs IPCeff. n = 5–7 per group
Mitochondrial ROS Production
I/R injury resulted in a significant increase in mitochondrial ROS production measured by H2DCF in control effluent recipient hearts (P < 0.05 vs. NH) (Fig. 5). Preconditioning with 2.5 μM adenosine significantly attenuated the increase in ROS production (P < 0.05 vs. Ceff), supporting a role for reduction of oxidative stress in adenosine mediated cardioprotection. Mitochondrial ROS production however was not significantly reduced in IPC effluent recipient hearts (P = 0.11 vs. Ceff) after I/R injury.

Mitochondrial ROS production measured by H2DCF was significantly attenuated in 2.5 μM adenosine preconditioned hearts but not in IPC effluent preconditioned hearts when compared to control effluent recipient hearts. # P < 0.05 vs NH. * P < 0.05 vs Ceff. n = 5–7 per group
Discussion
Based on the remote preconditioning model described by Dickson et al. [8] which utilized coronary effluent from an ex vivo preconditioned heart to precondition a second naive heart, we evaluated the hypothesis that endogenously released adenosine could be responsible for effluent-mediated transfer of cardioprotection between isolated perfused hearts. The novel findings in the present study include: (1) the concentration of adenosine in the IPC effluent is 10-fold greater than control effluent and supplementation of this concentration of adenosine in control effluent mimics IPC effluent associated infarct size reduction and left ventricle functional recovery. This protection by IPC effluent can be abolished with adenosine receptor inhibition indicating the participation of adenosine receptors in this mode of preconditioning. (2) In contrast to adenosine supplementation, IPC effluent induces a unique profile of alterations in mitochondrial function in the reperfusion period compared to adenosine preconditioned hearts, which suggests that adenosine alone is not entirely responsible for the preservation of mitochondrial function. IPC effluent likely contains other factor(s) in addition to adenosine that confer mitochondrial resistance to I/R injury.
Dickson et al. [29] provided evidence supporting the hypothesis that the protective mechanism in a model of coronary effluent-mediated transfer of cardioprotection requires an intact opioid receptor system as administration of opioid receptor antagonist naloxone prior to IPC effluent preconditioning abolished cardioprotection. However, pretreatment with exogenous Met- and Leu-enkephalin which was found up-regulated in the IPC effluent failed to elicit a protective response. Since adenosine is rapidly released from the hydrolysis of ATP during hypoxia in isolated hearts [21, 30], we hypothesized that endogenously released adenosine in the IPC effluent is responsible for the transfer of preconditioning. In the present study, adenosine content was measured in the entire collection of IPC effluent aggregated during the three 10-min reperfusion periods after three cycles of 5-min ischemia. With this protocol, adenosine concentration was increased 10-fold in comparison to control effluent and this concentration was cardioprotective. Our observation combined with Dickson’s report [29] suggest the possibility of adenosine and opioid receptor crosstalk that has been implicated in IPC [31] and remote ischemic preconditioning [32].
Ischemia induced membrane permeability and release of cytochrome c resulting in reduction of mitochondrial respiration at cytochrome c oxidase contributes to the decline in myocardial performance following reperfusion injury [14]. We demonstrate that cardioprotection by IPC effluent is associated with mitochondrial integrity and function preservation as evidenced by maintenance of mitochondrial membrane integrity, prevention of cytochrome c release, and preservation of complex I and complex IV respiration. Although the reduction in infarct size by IPC effluent could be reproduced by 2.5 μM adenosine, mitochondrial function in complex I and complex IV state 3 respirations and cytochrome c content in adenosine preconditioned hearts were not well preserved compared to IPC effluent preconditioned hearts. However, cardioprotection by adenosine was associated with lowered ROS production and improved RCR due to a significantly lower State 2 respiration but not improved State 3 respiration. The mechanism by which adenosine improves coupling however is unclear. Our results suggest that other factors present in the IPC effluent preserve mitochondrial integrity and function that is independent of adenosine.
It is well established that opening of the MPTP during reperfusion has a critical impact on determining the fate of the myocardium [33]. An important core component of the MPTP is the adenine nucleotide translocase (ANT), w hich is also responsible for the majority of the basal proton leak across the inner membrane [34]. Preconditioning with adenosine has been demonstrated to induce translocation of protein kinase C epsilon to the mitochondria [35] and protein kinase c epsilon interacts with ANT to inhibit opening of the MPTP [36]. It is not known whether this interaction results in reduced proton leak across the inner membrane to induce a hypercoupled state but Scorrano et al. [37] provided evidence that the inner membrane potential facilities the state of the MPTP and that uncoupling enhances the propensity towards opening. Our observation that state 2 respiration is lower in adenosine preconditioned hearts suggests that the lower proton leakage and improved coupling may protect the mitochondria by decreasing the propensity towards opening of the MPTP. Since adenosine preconditioned hearts have decreased cytochrome c content but preserved outer membrane permeability, this suggests that transient opening of the MPTP during reperfusion results in the release of cytochrome c but early closure may have protected the myocardium by preventing necrotic cell death [33]. Furthermore, although mild uncoupling is recognized as an innate protective mechanism through reduced ROS production and hypercoupling may predispose the mitochondria to enhanced ROS production, adenosine preconditioned hearts have lower ROS production suggesting that adenosine may have up regulated the anti-oxidative activity of manganese superoxide dismutase, which have been suggested as a protective mechanism in delayed adenosine preconditioning [38]. Reduced ROS production with adenosine may also contribute to inhibition of MPTP opening as demonstrated in a study by Clarke et al. [39] in which reduction in oxidative stress may inhibit MPTP opening from ischemic preconditioned hearts at 3 min post reperfusion. We are unable to detect differences in exogenous calcium induced propensity to MPTP opening between groups (data not shown) and it is possible that we have missed the critical time point at which inhibition of the MPTP opening occurs due to our protocol which includes mitochondrial isolation at 30 min post reperfusion. Other studies have noted that the MPTP reseals as early as 25 min after reperfusion in the isolated rat heart [40] and 30 min after cardioplegic arrest in the isolated rabbit heart [28].
Preservation of mitochondrial outer membrane integrity, cytochrome c, and state 3 respiration in IPC effluent preconditioned hearts may largely be contributed to activation of the mitochondrial KATP channel as previous studies from our group have demonstrated the importance of mitochondrial KATP channels in mediating remote ischemic preconditioning as specific inhibition by 5-hydroxydecanoic acid abolished cardioprotection [41]. Opening of the mitochondrial KATP channel may prevent reperfusion injury by reducing accumulation of Ca2+ in the mitochondria during ischemia through membrane depolarization [42, 43], and by maintaining ATP synthesis through preserved oxidative phosphorylation [44]. We have observed a trend towards a reduction in ROS production (P = 0.11) in IPC effluent perfused hearts, which may be indicative of mild uncoupling from opening of the mitochondrial KATP channel resulting in reduction of ROS production. Taken together, these observations suggest that IPC effluent may precondition the myocardium in a different pathway than adenosine alone and induce cardioprotection through a different mechanism in the mitochondria, which involves preservation of cytochrome c and respiration.
Other investigators have pursued the identification of these cardioprotective factor(s) in the IPC effluent using similar isolated rat heart perfusion models. A study by Serejo et al. [9] has shown that IPC effluent contains low molecular weight hydrophobic cardioprotective factors higher than 3.5 kDa while Breivik et al. [10] have demonstrated that cardioprotection was lost when hearts were perfused with a hydrophobic IPC effluent fraction containing proteins higher than 30 kDa. Characterization by liquid chromatography-tandem mass spectrometry (LC-MS/MS) of the coronary effluent following five cycles of 5 min ischemia and 11 min reperfusion in the isolated rat heart by Koomen et al. [45] identified 185 unique proteins (66 % intracellular proteins), with functions related to metabolism, structure/contractility, and oxidative stress response. We have performed a preliminary peptidomic/metabolomic analysis of the coronary effluent using LC-MS analysis of control and IPC effluent (n = 2) concentrated by isocratic elution from C18 reverse phase column and analyzed on an Orbitrap LTQ MS instrument. In this preliminary survey (m/z scan from 310 to 1,300), we were able to identify 50–60 peptides of which 8–10 were increased by more than 50 % relative to the levels found in control effluent (Supplementary Figure 1). Additionally, we detected 406 uncharacterized ions corresponding to either metabolites or peptides, 25 % of which were increased more than 1.5 fold in the IPC effluent. The identities of these factor(s) associated with remote cardioprotection remain known and will require further studies to characterize the physiological significance of the changes in metabolite and proteins that are found in the IPC effluent.
Inosine, a metabolic breakdown product arising from adenosine deamination and a potent activator of the adenosine A1 [46] and A3 receptor [47] was also significantly elevated in IPC effluent. It is not clear whether the concentration of inosine found in IPC effluent (5.6 μM) would produce a similar level of protection when used in combination with adenosine to perfuse a naive heart although Naydenova et al. [46] demonstrated that inosine at a concentration of 5 μM was sufficient to precondition HL-1 cardiomyocytes through adenosine A1 receptor activation. However, Peart et al. [48] demonstrated that pretreatment with inosine (20 μM) plus hypoxanthine (10 μM) prior to I/R in an isolated mouse heart model failed to alter pre-ischemic contractile function and had no effect on post-ischemic functional recovery (contractile and coronary flow).
Adenosine however remains a critical mediator in remote ischemic preconditioning as systemic delivery of 8-SPT abolishes the cardioprotection triggered by brief ischemia of non-cardiac tissue in the skeletal muscle [2] and kidney [20]. Furthermore, intra-arterial (but not intravenous) injection of adenosine into a limb leads to cardioprotection and the plasma dialysate taken from adenosine pretreated rabbit is able to protect a naïve heart against I/R injury [49]. These observations suggest the possibility that endogenous adenosine release from ischemic preconditioning in the heart or the limb may induce the secondary release of cardioprotective humoral factor(s).
In summary, transient ischemia liberates endogenous protective substances into the coronary effluent of isolated hearts. This effluent provides potent cardioprotection in a naïve recipient heart, an effect which can be abolished with an adenosine receptor antagonist. Addition of exogenous adenosine to control effluent recapitulated the reduction in infarct size and left ventricle recovery but preserved mitochondrial function with a different profile than IPC effluent. Our study reinforces the notion that multiple innate cardioprotective factors are released after ischemic preconditioning and the identity of these factors remain to be identified.
References
Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation. 1986;74(5):1124–36.
Oxman T, Arad M, Klein R, Avazov N, Rabinowitz B. Limb ischemia preconditions the heart against reperfusion tachyarrhythmia. Am J Physiol. 1997;273(4 Pt 2):H1707–12.
Pell TJ, Baxter GF, Yellon DM, Drew GM. Renal ischemia preconditions myocardium: role of adenosine receptors and ATP-sensitive potassium channels. Am J Physiol. 1998;275(5 Pt 2):H1542–7.
Konstantinov IE, Li J, Cheung MM, Shimizu M, Stokoe J, Kharbanda RK, et al. Remote ischemic preconditioning of the recipient reduces myocardial ischemia-reperfusion injury of the denervated donor heart via a Katp channel-dependent mechanism. Transplantation. 2005;79(12):1691–5.
Yang X, Cohen MV, Downey JM. Mechanism of cardioprotection by early ischemic preconditioning. Cardiovasc Drugs Ther. 2010;24(3):225–34.
Shimizu M, Tropak M, Diaz RJ, Suto F, Surendra H, Kuzmin E, et al. Transient limb ischaemia remotely preconditions through a humoral mechanism acting directly on the myocardium: evidence suggesting cross-species protection. Clin Sci (Lond). 2009;117(5):191–200.
Wang L, Oka N, Tropak M, Callahan J, Lee J, Wilson G, et al. Remote ischemic preconditioning elaborates a transferable blood-borne effector that protects mitochondrial structure and function and preserves myocardial performance after neonatal cardioplegic arrest. J Thorac Cardiovasc Surg. 2008;136(2):335–42.
Dickson EW, Lorbar M, Porcaro WA, Fenton RA, Reinhardt CP, Gysembergh A, et al. Rabbit heart can be “preconditioned” via transfer of coronary effluent. Am J Physiol. 1999;277(6 Pt 2):H2451–7.
Serejo FC, Rodrigues Jr LF, da Silva Tavares KC, de Carvalho AC, Nascimento JH. Cardioprotective properties of humoral factors released from rat hearts subject to ischemic preconditioning. J Cardiovasc Pharmacol. 2007;49(4):214–20.
Breivik L, Helgeland E, Aarnes EK, Mrdalj J, Jonassen AK. Remote postconditioning by humoral factors in effluent from ischemic preconditioned rat hearts is mediated via PI3K/Akt-dependent cell-survival signaling at reperfusion. Basic Res Cardiol. 2010;106(1):135–45.
Chen Q, Moghaddas S, Hoppel CL, Lesnefsky EJ. Ischemic defects in the electron transport chain increase the production of reactive oxygen species from isolated rat heart mitochondria. Am J Physiol Cell Physiol. 2008;294(2):C460–6.
Chen Q, Vazquez EJ, Moghaddas S, Hoppel CL, Lesnefsky EJ. Production of reactive oxygen species by mitochondria: central role of complex III. J Biol Chem. 2003;278(38):36027–31.
Borutaite V, Jekabsone A, Morkuniene R, Brown GC. Inhibition of mitochondrial permeability transition prevents mitochondrial dysfunction, cytochrome c release and apoptosis induced by heart ischemia. J Mol Cell Cardiol. 2003;35(4):357–66.
Lesnefsky EJ, Tandler B, Ye J, Slabe TJ, Turkaly J, Hoppel CL. Myocardial ischemia decreases oxidative phosphorylation through cytochrome oxidase in subsarcolemmal mitochondria. Am J Physiol. 1997;273(3 Pt 2):H1544–54.
Jiang X, Wang X. Cytochrome C-mediated apoptosis. Annu Rev Biochem. 2004;73:87–106.
Crestanello JA, Lingle DM, Kamelgard J, Millili J, Whitman GJ. Ischemic preconditioning decreases oxidative stress during reperfusion: a chemiluminescence study. J Surg Res. 1996;65(1):53–8.
Javadov SA, Clarke S, Das M, Griffiths EJ, Lim KH, Halestrap AP. Ischaemic preconditioning inhibits opening of mitochondrial permeability transition pores in the reperfused rat heart. J Physiol. 2003;549(Pt 2):513–24.
Lundberg KC, Szweda LI. Preconditioning prevents loss in mitochondrial function and release of cytochrome c during prolonged cardiac ischemia/reperfusion. Arch Biochem Biophys. 2006;453(1):130–4.
Liu GS, Thornton J, Van Winkle DM, Stanley AW, Olsson RA, Downey JM. Protection against infarction afforded by preconditioning is mediated by A1 adenosine receptors in rabbit heart. Circulation. 1991;84(1):350–6.
Takaoka A, Nakae I, Mitsunami K, Yabe T, Morikawa S, Inubushi T, et al. Renal ischemia/reperfusion remotely improves myocardial energy metabolism during myocardial ischemia via adenosine receptors in rabbits: effects of “remote preconditioning”. J Am Coll Cardiol. 1999;33(2):556–64.
Goto M, Cohen MV, van Wylen DG, Downey JM. Attenuated purine production during subsequent ischemia in preconditioned rabbit myocardium is unrelated to the mechanism of protection. J Mol Cell Cardiol. 1996;28(3):447–54.
Chen Q, Camara AK, Stowe DF, Hoppel CL, Lesnefsky EJ. Modulation of electron transport protects cardiac mitochondria and decreases myocardial injury during ischemia and reperfusion. Am J Physiol Cell Physiol. 2007;292(1):C137–47.
Palmer JW, Tandler B, Hoppel CL. Biochemical properties of subsarcolemmal and interfibrillar mitochondria isolated from rat cardiac muscle. J Biol Chem. 1977;252(23):8731–9.
Ricci JE, Gottlieb RA, Green DR. Caspase-mediated loss of mitochondrial function and generation of reactive oxygen species during apoptosis. J Cell Biol. 2003;160(1):65–75.
Lee AC, Zizi M, Colombini M. Beta-NADH decreases the permeability of the mitochondrial outer membrane to ADP by a factor of 6. J Biol Chem. 1994;269(49):30974–80.
Mootha VK, Wei MC, Buttle KF, Scorrano L, Panoutsakopoulou V, Mannella CA, et al. A reversible component of mitochondrial respiratory dysfunction in apoptosis can be rescued by exogenous cytochrome c. EMBO J. 2001;20(4):661–71.
William JN. A method for the simultaneous quantitative estimation of cytochrome a, b, c1 and and c in mitochondrial. Arch Biochem Biophys. 1964;107:537–43.
Leung CH, Wang L, Fu YY, Yuen W, Caldarone CA. Transient mitochondrial permeability transition pore opening after neonatal cardioplegic arrest. J Thorac Cardiovasc Surg. 2011;141(4):975–82.
Dickson EW, Blehar DJ, Carraway RE, Heard SO, Steinberg G, Przyklenk K. Naloxone blocks transferred preconditioning in isolated rabbit hearts. J Mol Cell Cardiol. 2001;33(9):1751–6.
Schrader J, Haddy FJ, Gerlach E. Release of adenosine, inosine and hypoxanthine from the isolated guinea pig heart during hypoxia, flow-autoregulation and reactive hyperemia. Pflugers Arch. 1977;369(1):1–6.
Peart JN, Gross GJ. Adenosine and opioid receptor-mediated cardioprotection in the rat: evidence for cross-talk between receptors. Am J Physiol Heart Circ Physiol. 2003;285(1):H81–9.
Surendra H, Diaz RJ, Harvey K, Tropak M, Callahan J, Hinek A, et al. Interaction of delta and kappa opioid receptors with adenosine A1 receptors mediates cardioprotection by remote ischemic preconditioning. J Mol Cell Cardiol. 2013;60:142–50.
Halestrap AP, Clarke SJ, Javadov SA. Mitochondrial permeability transition pore opening during myocardial reperfusion–a target for cardioprotection. Cardiovasc Res. 2004;61(3):372–85.
Brand MD, Pakay JL, Ocloo A, Kokoszka J, Wallace DC, Brookes PS, et al. The basal proton conductance of mitochondria depends on adenine nucleotide translocase content. Biochem J. 2005;392(Pt 2):353–62.
Yang Z, Sun W, Hu K. Molecular mechanism underlying adenosine receptor-mediated mitochondrial targeting of protein kinase C. Biochim Biophys Acta. 2012;1823(4):950–8.
Baines CP, Song CX, Zheng YT, Wang GW, Zhang J, Wang OL, et al. Protein kinase Cepsilon interacts with and inhibits the permeability transition pore in cardiac mitochondria. Circ Res. 2003;92(8):873–80.
Scorrano L, Petronilli V, Bernardi P. On the voltage dependence of the mitochondrial permeability transition pore. A critical appraisal. J Biol Chem. 1997;272(19):12295–9.
Dana A, Jonassen AK, Yamashita N, Yellon DM. Adenosine A(1) receptor activation induces delayed preconditioning in rats mediated by manganese superoxide dismutase. Circulation. 2000;101(24):2841–8.
Clarke SJ, Khaliulin I, Das M, Parker JE, Heesom KJ, Halestrap AP. Inhibition of mitochondrial permeability transition pore opening by ischemic preconditioning is probably mediated by reduction of oxidative stress rather than mitochondrial protein phosphorylation. Circ Res. 2008;102(9):1082–90.
Kerr PM, Suleiman MS, Halestrap AP. Reversal of permeability transition during recovery of hearts from ischemia and its enhancement by pyruvate. Am J Physiol. 1999;276(2 Pt 2):H496–502.
Kristiansen SB, Henning O, Kharbanda RK, Nielsen-Kudsk JE, Schmidt MR, Redington AN, et al. Remote preconditioning reduces ischemic injury in the explanted heart by a KATP channel-dependent mechanism. Am J Physiol Heart Circ Physiol. 2005;288(3):H1252–6.
Wang L, Cherednichenko G, Hernandez L, Halow J, Camacho SA, Figueredo V, et al. Preconditioning limits mitochondrial Ca(2+) during ischemia in rat hearts: role of K(ATP) channels. Am J Physiol Heart Circ Physiol. 2001;280(5):H2321–8.
Murata M, Akao M, O’Rourke B, Marban E. Mitochondrial ATP-sensitive potassium channels attenuate matrix Ca(2+) overload during simulated ischemia and reperfusion: possible mechanism of cardioprotection. Circ Res. 2001;89(10):891–8.
Fryer RM, Eells JT, Hsu AK, Henry MM, Gross GJ. Ischemic preconditioning in rats: role of mitochondrial K(ATP) channel in preservation of mitochondrial function. Am J Physiol Heart Circ Physiol. 2000;278(1):H305–12.
Koomen JM, Wilson CR, Guthrie P, Androlewicz MJ, Kobayashi R, Taegtmeyer H. Proteome analysis of isolated perfused organ effluent as a novel model for protein biomarker discovery. J Proteome Res. 2006;5(1):177–82.
Naydenova Z, Rose JB, Coe IR. Inosine and equilibrative nucleoside transporter 2 contribute to hypoxic preconditioning in the murine cardiomyocyte HL-1 cell line. Am J Physiol Heart Circ Physiol. 2008;294(6):H2687–92.
Jin X, Shepherd RK, Duling BR, Linden J. Inosine binds to A3 adenosine receptors and stimulates mast cell degranulation. J Clin Invest. 1997;100(11):2849–57.
Peart J, Matherne GP, Cerniway RJ, Headrick JP. Cardioprotection with adenosine metabolism inhibitors in ischemic-reperfused mouse heart. Cardiovasc Res. 2001;52(1):120–9.
Steensrud T, Li J, Dai X, Manlhiot C, Kharbanda RK, Tropak M, et al. Pretreatment with the nitric oxide donor SNAP or nerve transection blocks humoral preconditioning by remote limb ischemia or intra-arterial adenosine. Am J Physiol Heart Circ Physiol. 2010;299(5):H1598–603.
Grants
This study was supported by The Leducq Foundation (grant number 06/CVD) and the Heart and Stroke Foundation of Ontario.
Conflicts of Interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Supplementary Figure 1
Ions detected in the effluent by reverse phase liquid chromatography mass spectrometry. Intensity of select ions in IPC effluent (n = 2) relative to Control effluent (n = 2) is shown on the y-axis. Mass to charge (m/z) ratio for detected ions are shown on the x-axis. This ratio is a reflection of the molecular weight of the individual ions detected by the mass spectrometer (LTQ Orbitrap). (JPEG 12 kb)
Rights and permissions
About this article
Cite this article
Leung, C.H., Wang, L., Nielsen, J.M. et al. Remote Cardioprotection by Transfer of Coronary Effluent from Ischemic Preconditioned Rabbit Heart Preserves Mitochondrial Integrity and Function via Adenosine Receptor Activation. Cardiovasc Drugs Ther 28, 7–17 (2014). https://doi.org/10.1007/s10557-013-6489-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10557-013-6489-2