
Abstract
Key message
Marker-free transgenic plants can be generated with high efficiency by using the Cre/ lox P self-excision system controlled by the pollen- and embryo-specific Arabidopsis DLL promoter.
Abstract
In this work, we aimed to study the feasibility of using the pollen- and embryo-specific DLL promoter of the At4g16160 gene from Arabidopsis thaliana in a Cre/loxP self-excision strategy. A Cre/loxP self-excision cassette controlled by the DLL promoter was introduced into the tobacco genome via Agrobacterium-mediated transformation. No evidence for premature activation of the Cre/loxP system was observed in primary transformants. The efficiency of nptII removal during pollen and embryo development was investigated in transgenic T1 progenies derived from eight self- and four cross-pollinated T0 lines, respectively. Segregation and rooting assays were performed to select recombined T1 plants. Molecular analyses of these plants confirmed the excision event in all analysed T0 lines and marker-free transgenic T1 plants were obtained with efficiency of up to 96.2 %. The Arabidopsis DLL promoter appears to be a strong candidate to drive Cre-mediated recombination not only in tobacco as a model plant, but also in other plant species.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Selectable marker genes enable selection of the small number of cells that are able to include foreign DNA. The presence of such genes in the genome of transgenic plants, especially those encoding for resistance to antibiotics or herbicides, has caused considerable public concern about their potential adverse impact on human health and environment (Costa-Font et al. 2008; Nicolia et al. 2014).
Several approaches to eliminate selectable markers have been described including co-transformation, transposon-based transgene excision, homologous recombination or site-specific recombination (reviewed by Scutt et al. 2002; Gidoni et al. 2008; Woo et al. 2011 or Tuteja et al. 2012). Over the recent years studies have focused on the site-specific Cre/loxP recombination system. The first commercial marker-free transgenic maize (LY038) has been developed (Ow 2007).
The Cre/loxP recombination system consists of the gene for cre recombinase and of two loxP sites. Cre recombinase mediates a recombination event ending with excising the DNA sequence placed between two directly oriented loxP sites (Gilbertson 2003). Individual approaches exploiting the Cre/loxP system differ in duration of cre expression, which can be constitutive, transient or temporal. The constitutively expressed cre gene is delivered to targeted loxP sites by cross-pollination (Odell et al. 1990; Hoa et al. 2002; Chakraborti et al. 2008) or re-transformation (Dale and Ow 1991; Russel et al. 1992). To reduce undesired long-term effect of the CRE protein (Coppoolse et al. 2003), approaches based on transient and temporal cre expression were developed. Transient cre expression has been achieved by application of purified Cre protein (Will et al. 2002) and virus- or Agrobacterium-mediated cre expression (Kopertekh and Schiemann 2005; Kopertekh et al. 2012).
In the temporal expression approach, the cre recombinase and the selectable marker genes are placed between two loxP sites as a part of the same T-DNA. Upon activation, the Cre recombinase removes the marker gene as well as its own sequence. The excision event can be controlled by using inducible promoters activated by heat shock (Zhang et al. 2003; Liu et al. 2005; Wang et al. 2005; Cuellar et al. 2006; Roy et al. 2008), chemically (Zuo et al. 2001; Sreekala et al. 2005; Zhang et al. 2006, 2009; Petri et al. 2012; García-Almodóvar et al. 2014), by salicylic acid (Ma et al. 2008, 2009); or by using promoters of genes with a high degree of specific expression such as microspore NTM19 (Mlynarova et al. 2006), pollen/seed-specific PAB5 (Luo et al. 2007), embryo-specific APP1 (Li et al. 2007), germline-specific SDS and AP1 (Verweire et al. 2007), flower-specific OsMADS45 (Bai et al. 2008), seed-specific CRUC (Moravcikova et al. 2008; Boszorádová et al. 2014), seed-specific napin (Kopertekh et al. 2009) or embryo-globulin REG-2 (Chong-Pérez et al. 2013).
However, efficiency of excision varies greatly. For example, when cre expression was regulated by the embryo-specific CRUC promoter, an excision efficiency of 10.2 % was achieved (Moravcikova et al. 2008). Unexpected complications with premature excision of the marker gene were observed suggesting that a more reliable promoter is needed. TAIR database searches identified the promoter DLL of Arabidopsis At4g16160 gene as highly tissue-specific and robust towards premature activation. The activity of DLL in Arabidopsis is specifically limited to developing pollen grains (Honys and Twell 2004), young ovules and developing embryos (Drea et al. 2006). Experiments performed on transgenic tobacco using the gus gene-reporter system (Jopcik et al. 2014) showed activity of DLL in pollen grains and germinating tubes as well from the middle torpedo stage of the developing embryo. No GUS activity was detected in leaves and stems. In addition, among the five pollen- and/or embryo-specific promoters tested, only the DLL promoter showed activity consistent with the predicted pattern (Jopcik et al. 2014).
Therefore, in this work, we studied the feasibility of using the promoter DLL from Arabidopsis to drive Cre-mediated excision of the selectable nptII gene in tobacco. The efficiency of nptII removal in pollen and embryo was investigated in transgenic T1 progenies of selected self- and cross-pollinated T0 plants.
Materials and methods
Vector construction
The DLL promoter sequence was previously isolated as a 1644 bp PCR fragment of the At4g16160 gene from Arabidopsis thaliana (Jopcik et al. 2013).
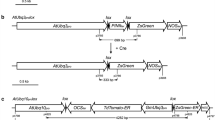
The pZP6 construct (Fig. 1a) was prepared by cloning dCaMV35S/gus/nosT/loxP/DLL/cre INT /nosT and nosP/nptII/nosT/loxP fragments into the low copy number binary vector pUN (Vaculkova et al. 2007) as described by Polóniová et al. (2012).

The plant binary vector pZP6 used in the transformation experiments. a T-DNA configuration consisting of the β-glucuronidase (gus) gene driven by the double dCaMV 35S promoter (d35S); the intron-containing cre recombinase gene (cre INT) under control of DLL promoter and the neomycin phosphotransferase (nptII) gene driven by the nos promoter. All three genes were terminated by the nosT. Black arrows indicate the presence and orientation of the loxP sites. The restriction site used for Southern blot analyses is indicated as well as predicted fragment size. The positions of primers used for PCR analyses are indicated as P1/P2, P3/P4, P5/P6, P9/P8 and P7/P8. b The T-DNA configuration generated after excision of the floxed DNA. The positions of primers used for PCR analyses are indicated as P9/P8
The plasmid pZP6 was evaluated for stability and recombination events in Escherichia coli DH5αF′ according to the protocol by Mlynarova and Nap (2003).
The binary vector pZP6 was introduced into Agrobacterium tumefaciens strain AGL0 and its stability was verified by restriction analyses after re-transformation into E. coli.
Plant material and transformation
Tobacco (Nicotiana tabacum cv. Petit Havana SR1) was transformed using the leaf disc transformation protocol described by Mlynarova et al. (1994). The transformed tobacco tissues were selected on medium with 50 mg l−1 kanamycin. Regenerated shoots were rooted in the presence of 50 mg l−1 kanamycin. Selected transgenic plants were transferred to the soil, cultivated in greenhouse conditions and used to conduct self- and cross-pollination with the wild type.
β-Glucuronidase assays
Histochemical GUS assays were conducted as described by Jefferson et al. (1987). Leaf explants/seedlings were incubated in 1 mmol l−1 5-bromo-4-chloro-3-indolyl glucuronide (X-gluc, Duchefa, Netherlands), 50 mmol l−1 phosphate buffer (pH 7) at 37 °C in the dark overnight. To improve colour contrast, the tissues were washed in 70 % (v/v) ethanol.
Fluorimetric GUS assays were performed as described by Mlynarova et al. (1994). GUS activity was expressed in picomoles of methylumbelliferone released per min per μg of soluble protein. The concentration of proteins was determined according to Bradford (1976).
Segregation assays
Two segregation assays were used to test for kanamycin resistance in progenies of the transgenic plants. In the germination assay, surface-sterilised seeds (about 100 seeds per plant) germinated on MS medium (Murashige and Skoog 1962) containing 1 % (w/v) sucrose, 0.8 % (w/v) agar and 50 mg l−1 kanamycin. Three weeks later, the seedlings were evaluated for kanamycin resistance. Green seedlings were considered to be kanamycin-resistant (KmR) and yellow or pale seedlings as kanamycin-sensitive (KmS).
A rooting assay was carried out according to Moravcikova et al. (2008). Surface-sterilised tobacco seeds (about 100 seeds per plant) were germinated on MS medium (Murashige and Skoog 1962) containing 1 % (w/v) sucrose and 0.8 % (w/v) agar under non-selective conditions. After 2 weeks the seedlings were transferred into 100-ml glass culture vessels onto fresh MS medium and grown for 6 weeks. Next, roots were removed from plants and each plant was multiplied vegetatively into two clones. The first clone was allowed to re-root in the presence of 50 mg l−1 kanamycin. The plants that formed roots in 3 weeks were evaluated as kanamycin-resistant (R+), whereas non-rooted plantlets were considered kanamycin-sensitive (R−). The second clone was rooted without the presence of antibiotics. These plants were used for DNA analyses.
PCR analyses
Genomic DNA from the leaf tissues of tobacco plants was isolated using the protocol of Chen et al. (1992). The primers used in PCR analyses, the size of expected amplicons and other relevant descriptions are listed in Table 1. The respective positions of the primers are given in Fig. 1. The PCR reaction mixture of 25 µl contained 100–200 ng of DNA template, 20 pmol of each primer, 0.2 mmol l−1 dNTPs, 1× PCR buffer, 2.5 mmol l−1 MgCl2 and 1 U FIREPol Taq DNA polymerase (Solid BioDyne, Estonia). Step one of the PCR was performed at 95 °C for 4 min, followed by 35 cycles of 95 °C for 45 s, 62 or 64 °C (see Table 1) for 45 s and 72 °C for 90 s. The last step was performed at 72 °C for 10 min.
Some of the PCR amplicons were isolated using the QIAEX® II Gel Extraction Kit (Qiagen, Germany), cloned into the pGEMT-easy vector and commercially sequenced. Alignment of the obtained sequences was performed using the CLUSTALW2 program (Thompson et al. 1994).
Southern blot analyses
Genomic DNA (10 μg) was digested with restriction enzyme KpnI, separated on a 1 % (w/v) agarose gel and blotted onto a positively charged nylon membrane (Roche, Switzerland). An NPT-specific probe was prepared using PCR with the primer set P3/P4 and non-radioactively labelled using the DIG Probe Synthesis Kit (Roche, Switzerland). Hybridisation was performed in DIG easy hyb hybridisation solution (Roche, Switzerland) at 42 °C according to the manufacturer’s instructions. Hybridisation signals were visualised by DIG Nucleic Acid Detection Kit (Roche, Switzerland).
Results
The pollen- and embryo-specific Arabidopsis DLL promoter was used to drive the cre recombinase. The T-DNA of the corresponding plant transformation vector pZP6 is given in Fig. 1a. Figure 1b shows the final outcome of Cre-mediated excision in transgenic tobacco plants.
Generation and proof of T0 plants
The pZP6 T-DNA was introduced into the tobacco genome via Agrobacterium tumefaciens. Putative transgenic shoots were initially screened for kanamycin resistance and for GUS activity. Transformation efficiency (56.6 %) was normal; therefore, we assumed that no premature activity of the DLL promoter occurred during regeneration of transformed cells.
A set of histochemically GUS-positive T0 plants were subjected to PCR analyses with primers P1/P2, P3/P4, P5/P6 and P7/P8. PCR products corresponding to the predicted sizes of 744 bp (P1–P2), 552 bp (P3–P4), 589 bp (P5–P6) and 781 bp (P7–P8) were detected in all analysed T0 plants. Examples of the PCR analyses with the primer sets P5/P6 and P7/P8 are given in Fig. 2a and b respectively.

Photographs of ethidium bromide-stained 1 % agarose gels with the PCR products obtained on transgenic T0 plants. a PCR results with primers P5/P6 that amplified a 589 bp gus-loxP-DLL fragment. b PCR results with primers P7/P8 that amplified a 781 bp nptII-loxP-RB end fragment. c PCR results with primers P9/P8 that amplified a 716 bp gus-DLL fragment of the non-excised T-DNA. Lane M contains 100 bp DNA ladder (Fermentas) as a size marker, lanes 2–19 represent PCR products of individual transgenic T0 plants, NT non-transformed plant, ZP6 plasmid pZP6 used for plant transformation
Further, PCR analyses were used to detect possible ectopic activation of DLL and thus premature excision of the nptII gene. Unfortunately, there were limited alternatives for designing the proper primer set (Fig. 3a). By using primers P9/P8, two PCR products could be generated, the first of 716 bp (P9–P8) derived from non-recombined T-DNA templates and the second of 653 bp (P9–P8) from the recombined T-DNA templates. In all analysed T0 plants, only a 716 bp P9–P8 PCR product was detected (Fig. 2c) pointing to lack of premature excision of the nptII gene in the T0 plants. The P9–P8 amplicon (line T0-2) was isolated and sequenced. Pairwise alignments showed the identity of the amplicon with the corresponding non-recombined sequence of the pZP6 T-DNA (Fig. 3b).

The position of primers P9/P8 in the non- and recombined pZP6 T-DNA. a Using primers P9/P8 a 716-bp derived from the non-recombined T-DNA and a 653 bp from the recombined T-DNA PCR products could be generated. b Multiple alignment of the P9–P8 fragment from the transgenic tobacco line T0-2 and the P9-P8 of T1-2/32 derived from the self-pollinated line T0-2 with the corresponding sequence of the plasmid pZP6. Alignment was generated using the CLUSTALW2 program. Nucleotides which are conserved in all aligned sequences are marked by asterisk. The letters in the box show the sequence of primers P9 and P8. Dashes show the loxP sequences
The T0 plants were screened by Southern blot hybridisation using the NPT probe after digestion of DNA with KpnI to determine the number of the right border fragments. Based on the restriction map of pZP6 (Fig. 1a), the probe was expected to hybridise with fragments larger than a 2.2-kb border fragment. The number of detected fragments corresponded to the number of integrated transgenes. The number of nptII gene copies varied from 1 to 5 (Fig. 4).

Southern blot analyses on transgenic T0 tobacco plants. KpnI-digested DNA was non-radioactively probed with the NPT-specific probe. The bands (>2.2 kb) correspond to the number of independent transgene copies. Lanes 2–19 represent individual transgenic T0 plants. NT non-transformed plant
Eight of the analysed T 0 plants with single- or two-copy integrations were identified, transferred to in vivo conditions and subjected to self- and cross-pollinations.
Efficiency of nptII gene excision from transgenic pollen and embryo
The T 1 progenies of eight selected self-pollinated T 0 plants were investigated for the presence of the nptII gene by germination and rooting assays. The scheme of the predicted genetic segregation upon self-pollination of a single-copy T 0 plant is given in Fig. 5a.

Predicted genetic segregation of a single-copy transgenic T0 plant upon a self-pollination and upon b crossing of a single-copy transgenic T0 plant as a pollen donor to the wild-type (WT) plant. The scheme of Mlynarova et al. (2006) was adopted and modified. G β-Glucuronidase gene, C cre recombinase gene, N neomycin phosphotransferase gene
The number of seedlings sensitive to kanamycin (KmS) ranged from 93.5 (T0-2) to 100 % (T0-15). These seedlings were also screened histochemically for GUS activity. The number of (GUS+, KmS) seedlings ranged from 91.5 (T0-2) to 99.0 % (T0-9). The exception was line T0-15 that produced progeny of GUS-positive seedlings that were all sensitive to kanamycin (Table 2; Table S1—Online resource 1).
In rooting assays, 8-week-old plants derived from seeds germinated under non-selective conditions had roots removed and were allowed to re-root in the presence of kanamycin. The proportion of T 1 plants that did not form any roots (R−) varied from 51.4 (T0-6) to 89.0 % (T0-9). None of the analysed T0 lines produced completely (R−) T1 plants. The number of (GUS+, R−) T 1 plants ranged from 17.0 % (T0-6) to 86.4 % (T0-9) (Table 2; Table S2—Online resource 1).
To study the excision of the nptII gene at molecular level, genomic DNAs from all phenotypically marker-free (GUS+, R−) T1 plants were isolated and assayed by PCR. The analyses were carried out using the primer sets P1/P2, P9/P8 and P7/P8. PCR amplifications of a 744 bp P1–P2 fragment and a 781 bp P7–P8 fragment confirmed the presence of the gus gene and nptII-loxP-RB end sequences, respectively. An example of PCR is given in Fig. 6. As is shown in Fig. 3a, using the primer set P9/P8 could generate a 716 bp P9–P8 amplicon derived from the non-recombined and/or 653 bp P9–P8 amplicon generated from the recombined T-DNA.

Photographs of ethidium bromide-stained 1 % agarose gels with the PCR products obtained on tobacco T1 plants. a PCR results with primers P1/P2 that amplified an internal 742-bp fragment of the gus gene. b PCR results with primers P9/P8 that amplified a 653-bp fragment derived from the recombined T-DNA and/or a 716-bp P9–P8 fragment derived from the non-recombined T-DNA. c PCR results with primers P7/P8 that amplified a 781 pb nptII-loxP-RB end fragment. Lane M contains 1 kb DNA ladder (Fermentas) as a size marker, lanes 3–21 represent PCR products of individual transgenic T1 plants. The plants were sorted into three groups A, B and C. NT non-transformed tobacco plant, ZP6 plasmid pZP6 used for plant transformation
The post-excision (653 bp) P9–P8 fragment was observed in 99.0 % (292 of 295) of T1 plants (Table 3). Upon sequencing (T1-2/32), the P9–P8 sequence was proven to be identical with the expected sequence (Fig. 3b). Based on the PCR results, the 295 analysed T1 plants were divided into three groups (Table 3). Group A contains 241 plants that amplified both the P1–P2 and post-excision P9–P8 fragments, thus these plants carried the gus gene but not the nptII gene. These plants were considered marker-free. The number of marker-free T1 plants varied between individual lines from 3.8 % (T0-6) to 81.5 % (T0-9). Group B comprises 51 plants that showed amplification of P1–P2, P7–P8 and both P9–P8 fragments. These T1 plants were regarded as chimeric for the nptII gene. Group C includes three plants that showed amplification of P1–P2 and P9–P8 fragments (non-recombined T-DNA) and the P7–P8 fragment. The absence of a 653-bp P9–P8 fragment indicated no transgene excision because of a failure of the Cre/loxP system.
Taken together, Cre-mediated excision occurred in 292 out of 295 (groups A and B) analysed T1 plants derived from eight self-pollinated T0 lines. Of these, 241 plants were marker-free (group A) and 51 plants as were chimeric (group B).
Efficiency of nptII gene excision from transgenic pollen
To investigate the excision of the nptII gene directly from transgenic pollen, pollen grains of selected T0 lines (T0-2, T0-8, T0-9, and T0-19) were used to pollinate wild-type tobacco plants. The T1 plants were first investigated for the presence/absence of the nptII gene by germination and rooting assays. The scheme of the predicted genetic segregation on cross-pollination of a single-copy T0 plant is given in Fig. 5b.
In germination assays, the number of seedlings evaluated as kanamycin-sensitive was greater than 89 %. Two lines WTxT0-9 and WTxT0-19 produced progeny in which all histochemically GUS-positive seedlings were also kanamycin-sensitive (Table 4; Table S3—Online Resource 2). At the same time, the number of T1 plants that did not form any roots (R−) ranged from 78.0 (WTxT0-2) to 98.9 % (WTxT0-9). None of the analysed T0 lines produced completely (R−) T1 plants. The number of (GUS+, R−) T1 plants ranged from 48.4 (WTxT0-19) to 98.1 % (WTxT0-9) (Table 4; Table S4—Online Resource 2). These plants were considered putatively marker-free and further analysed.
In total, 137 (GUS+, R−) T1 plants were subjected to PCR analyses with the primer sets P1/P2, P9/P8 and P7/P8. The post-excision P9–P8 fragment was detected in 99.3 % (136 of 137) analysed T1 plants. As described above, the T1 plants were divided into three groups. Data are summarised in Table 5. A total of 129 plants were sorted into group A and were considered marker-free. The number of marker-free plants varied between individual lines from 40.0 (WTxT0-19) to 96.2 % (WTxT0-9). Group B consisted of seven plants in which the nptII gene was only partially removed. These plants were chimeric for the nptII gene. Group C contained one plant in which the nptII gene was not excised because of a failure of the Cre/loxP system.
Taken together, Cre-mediated excision in pollen occurred in 136 of 137 T1 plants analysed. There were 129 plants that were marker-free (group A) and seven plants were chimeric (group B).
Detailed analyses of the self- and cross-pollinated T0-2 line
For a comprehensive view of excision events in transgenic progeny, both (GUS+, R−) (Tables 3, 5) and (GUS+, R+) T1 plants from the self- and cross-pollinated line T0-2 were analysed by PCR with the primer sets P1/P2, P9/P8, P7/P8. In all analysed (GUS+, R+) T1 plants the P7–P8 fragment and both P9–P8 fragments were detected pointing to chimerism (Table 6). Excision occurred in all histochemically GUS-positive progenies of the self-pollinated T0-2 line and in 98.0 % of plants derived from WTxT0-2 line. However, only 46.4 and 49.0 % of self- and cross-pollinated plants were marker-free, respectively.
Discussion
The aim of this study was to evaluate the feasibility of the tissue-specific Arabidopsis DLL promoter in the Cre/loxP self-excision strategy. Corresponding gene characteristics (At4g16160, Arabidopsis eFP Browser database) with respect to cell types and expression level suggest its activation during pollen and embryo development but not in somatic cells. Indeed, recent work by Jopcik et al. (2014) showed no evidence for activation of DLL either in developing calli or in vegetative organs. Activity was detected strictly in pollen grains and tubes and in embryos at middle torpedo stage. Our work has confirmed that the DLL promoter is a strong candidate for use in the Cre/loxP self-excision strategy. Analyses of T0 plants did not detect any premature excision of the nptII gene (Fig. 2c). This result is very encouraging since ectopic activation of another candidate, the CRUC promoter, has previously been shown to hamper its usability due to the unexpected promoter–enhancer interaction (Boszorádová et al. 2014).
The ability of DLL to drive excision directly in pollen and embryos was further investigated in T1 plants derived from eight self- and four cross-pollinated T0 lines. In both types of pollination experiments, excision of the nptII gene was expected to result in sensitivity to kanamycin. However, in germination assays, there were kanamycin-sensitive as well as kanamycin-resistant seedlings detected in progenies of most self- and cross-pollinated T0 lines (Tables 2, 4). Similarly, the results of rooting assays confirmed imperfect marker gene excision (Tables 2, 4). Nevertheless, the number of phenotypically marker-free T1 plants was lower (Table 2). We assume that it could coincide with higher sensitivity of germinated seedlings to kanamycin. Probably, among the kanamycin-sensitive seedlings were also chimeric for the nptII gene with lower survival potential. In contrast, plants at later developmental stages, especially those chimeric for transgene, may withstand a certain level of selection agent, yet are unable to root. At the same time, the results of the rooting assays were consistent with the data obtained by PCR (Table 6).
In our previous self-excision experiments (Moravcikova et al. 2008) with the CRUC promoter active in developing embryos, excision efficiency was 10.2 % and an incomplete excision of the marker gene was found in most of the T1 plants. In this study, the application of the promoter DLL resulted in up to 96.2 % marker-free plants (Tables 3, 5). Detailed analyses of the GUS-positive T1 plants (self-pollinated T0-2 line) showed that excision occurred in all plants, but chimeric plants were still detected (Table 6).
Since strong activity of DLL in a uninucleate microspore is assumed, only marker-free T1 plants (cross-pollination) are theoretically expected (Fig. 5b). However, plants with incomplete excision of the nptII gene were also observed. We hypothesise that the activity of DLL in transgenic tobacco was shifted from the uninucleate microspore to a later stage of pollen development. It could coincide with a slightly different activity pattern of DLL in pollen of transgenic tobacco and Arabidopsis as an original organism. A delay in the transgene expression controlled by tissue-specific promoters has been reported (Odell et al. 1994; Jopcik et al. 2014).
The relatively high number of marker-free plants is likely to be a result of the cre expression in pollen rather than in both pollen and embryo. For example, the self-pollinated line T0-8 generated marker-free T1 plants with efficiency of 65.6 % (Table 3). When the line T0-8 was used as a pollen donor to a wild type, 89.4 % excision efficiency was achieved (Table 5). We assume this phenomenon is a result of non-uniform expression of the cre gene in an embryo. An embryo represents a more complex multicellular structure than bicellular pollen. To gain marker-free plants, the cre gene has to be sufficiently expressed in every embryogenic cell. A relationship between the cre expression level and recombination efficiency has been found in other studies (Marjanac et al. 2008).
In summary, marker-free plants were obtained from all transgenic tobacco lines with an excision efficiency up to 96.2 %, depending on the type of pollination experiment (Tables 3, 5). At the same time, differences in excision efficiency among individual transgenic lines likely resulted from the position effect of the transgene insertion in the tobacco genome (Matzke and Matzke 1998).
Most previously reported self-excision recombination systems have relied mainly on the activity of embryo-specific promoters (Li et al. 2007; Moravcikova et al. 2008; Kopertekh et al. 2009; Chong-Perez et al. 2013). To date, only few pollen- and embryo-specific promoters have been investigated (Luo et al. 2007). This study adds the DLL promoter to the list of candidates for efficient generation of marker-free plants.
Our study has proved the feasibility of the pollen- and embryo-specific DLL promoter to generate marker-free T1 plants. Our results using tobacco as a model plant are promising for potential use in other sexually propagated plant species. Marker-free transgenic plants generated in this way can contribute to biosafety and greater acceptability of GM plants to the public.
Author contribution statement
JL, IM and JM designed the entire experiments. The experiments were carried out by ZP, MJ, JL and JM. JM and IM wrote and edited the manuscript. All authors have read and approved the final manuscript.
References
Bai X, Wang Q, Chu C (2008) Excision of a selective marker in transgenic rice using a novel Cre/loxP system controlled by a floral specific promoter. Transgenic Res 17:1035–1043
Boszorádová E, Libantová J, Matušíková I, Moravčíková J (2014) Application of Arabidopsis tissue-specific CRUC promoter in the Cre/loxP self-excision strategy for generation of marker-free oilseed rape: potential advantages and drawbacks. Acta Physiol Plant 36:1399–1409
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Chakraborti D, Sarkar A, Mondal HA, Schuermann D, Hohn B, Sarmah BK, Das S (2008) Cre/lox system to develop selectable marker-free transgenic tobacco plants conferring resistance against sap sucking homopteran insect. Plant Cell Rep 27:1623–1633
Chen J, Greenblatt IM, Dellaporta SL (1992) Molecular analysis of Ac transposition and DNA-replication. Genetics 130:665–676
Chong-Perez B, Reyes M, Rojas L, Ocana B, Ramos A, Kosky RG, Angenon G (2013) Excision of a selectable marker gene in transgenic banana using a Cre/lox system controlled by an embryo specific promoter. Plant Mol Biol 83:143–152
Coppoolse ER, de Vroomen MJ, Roelofs D, Smit J, van Gennip F, Hersmus BJM, Nijkamp HJJ, van Haaren MJJ (2003) Cre recombinase expression can result in phenotypic aberrations in plants. Plant Mol Biol 51:263–279
Costa-Font M, Gil JM, Traill WB (2008) Consumer acceptance, valuation of and attitudes towards genetically modified food: review and implications for food policy. Food Policy 33:99–111
Cuellar W, Gaudin A, Solorzano D, Casas A, Nopo L, Chudalayandi P, Medrano G, Kreuze J, Ghislain M (2006) Self-excision of the antibiotic resistance gene nptII using a heat inducible Cre–loxP system from transgenic potato. Plant Mol Biol 62:71–82
Dale EC, Ow DW (1991) Gene-transfer with subsequent removal of the selection gene from the host genome. P Natl Acad Sci USA 88:10558–10562
Drea SC, Lao NT, Wolfe KH, Kavanagh TA (2006) Gene duplication, exon gain and neofunctionalization of OEP16-related genes in land plants. Plant J 46:723–735
García-Almodóvar R, Petri C, Padilla I, Burgos L (2014) Combination of site-specific recombination and a conditional selective marker gene allows for the production of marker-free tobacco plants. Plant Cell Tiss Organ Cult 116:205–215
Gidoni D, Srivastava V, Carmi N (2008) Site-specific excisional recombination strategies for elimination of undesirable transgenes from crop plants. In Vitro Cell Dev Biol Plant 44:457–467
Gilbertson L (2003) Cre–lox recombination: cre-active tools for plant biotechnology. Trends Biotechnol 21:550–555
Hoa TTC, Bong BB, Huq E, Hodges TK (2002) Cre/lox site-specific recombination controls the excision of a transgene from the rice genome. Theor Appl Genet 104:518–525
Honys D, Twell D (2004) Transcriptome analysis of haploid male gametophyte development in Arabidopsis. Genome Biol 5:R85
Jefferson RA, Kavanagh TA, Bevan MW (1987) Gus fusions—beta-glucuronidase as a sensitive and versatile gene fusion marker in higher-plants. EMBO J 6:3901–3907
Jopcik M, Bauer M, Moravcikova J, Boszoradova E, Matusikova I, Libantova J (2013) Plant tissue-specific promoters can drive gene expression in Escherichia coli. Plant Cell Tiss Organ Cult 113:387–396
Jopcik M, Moravcikova J, Matusikova I, Libantova J (2014) Spacer length-dependent protection of specific activity of pollen and/or embryo promoters from influence of CaMV 35S promoter/enhancer in transgenic plants. Plant Cell Tiss Organ Cult 118:507–518
Kopertekh L, Schiemann J (2005) Agroinfiltration as a tool for transient expression of cre recombinase in vivo. Transgenic Res 14:793–798
Kopertekh L, Broer I, Schiemann J (2009) Developmentally regulated site-specific marker gene excision in transgenic B. napus plants. Plant Cell Rep 28:1075–1083
Kopertekh L, Saint Paul V, Krebs E, Schiemann J (2012) Utilization of PVX-Cre expression vector in potato. Transgenic Res 21:645–654
Li Z, Xing A, Moon BP, Burgoyne SA, Guida AD, Liang H, Lee C, Caster CS, Barton JE, Klein TM, Falco SC (2007) A Cre/loxP-mediated self-activating gene excision system to produce marker gene free transgenic soybean plants. Plant Mol Biol 65:329–341
Liu HK, Yang C, Wei ZM (2005) Heat shock-regulated site-specific excision of extraneous DNA in transgenic plants. Plant Sci 168:997–1003
Luo K, Duan H, Zhao D, Zheng X, Deng W, Chen Y, Stewart CN Jr, McAvoy R, Jiang X, Wu Y, He A, Pei Y, Li Y (2007) GM-gene-deletor: fused loxP-FRT recognition sequences dramatically improve the efficiency of FLP or CRE recombinase on transgene excision from pollen and seed of tobacco plants. Plant Biotech J 5:263–274
Ma BG, Duan XY, Ma CX, Niu JX, Zhang HP, Pan LZ (2008) Salicylic-acid-induced self-excision of the marker gene nptII from transgenic tomato using the Cre–loxP system. Plant Mol Biol Rep 26:199–212
Ma BG, Duan XY, Niu JX, Ma C, Hao QN, Zhang LX, Zhang HP (2009) Expression of stilbene synthase gene in transgenic tomato using salicylic acid-inducible Cre/loxP recombination system with self-excision of selectable marker. Biotechnol Lett 31:163–169
Marjanac G, De Paepe A, Peck I, Jacobs A, De Buck S, Depicker A (2008) Evaluation of CRE-mediated excision approaches in Arabidopsis thaliana. Transgenic Res 17:239–250
Matzke AJM, Matzke MA (1998) Position effects and epigenetic silencing of plant transgenes. Curr Opin Plant Biol 1:142–148
Mlynarova L, Nap JP (2003) A self-excising Cre recombinase allows efficient recombination of multiple ectopic heterospecific lox sites in transgenic tobacco. Transgenic Res 12:45–57
Mlynarova L, Loonen A, Heldens J, Jansen RC, Keizer P, Stiekema WJ, Nap JP (1994) Reduced position effect in mature transgenic plants conferred by the chicken lysozyme matrix-associated region. Plant Cell 6:417–426
Mlynarova L, Conner AJ, Nap JP (2006) Directed microspore-specific recombination of transgenic alleles to prevent pollen-mediated transmission of transgenes. Plant Biotech J 4:445–452
Moravcikova J, Vaculkova E, Bauer M, Libantova J (2008) Feasibility of the seed specific cruciferin C promoter in the self excision Cre/loxP strategy focused on generation of marker-free transgenic plants. Theor Appl Genet 117:1325–1334
Murashige I, Skoog F (1962) A revised medium for rapid growth and bioassays with tobacco tissue cultures. Plant Physiol 15:473–497
Nicolia A, Manzo A, Veronesi F, Rosellini D (2014) An overview of the last 10 years of genetically engineered crop safety research. Crit Rev in Biotechnol 34:77–88
Odell J, Caimi P, Sauer B, Russell S (1990) Site-directed recombination in the genome of transgenic tobacco. Mol Gen Genet 223:369–378
Odell JT, Hoopes JL, Vermerris W (1994) Seed-specific gene activation mediated by the Cre/lox site-specific recombination system. Plant Physiol 106:447–458
Ow DW (2007) GM maize from site-specific recombination technology, what next? Curr Opin in Biotech 18:115–120
Petri C, Lopez-Noguera S, Wang H, Garcia-Almodovar C, Alburquerque N, Burgos L (2012) A chemical-inducible Cre–LoxP system allows for elimination of selection marker genes in transgenic apricot. Plant Cell Tissue Organ Cult 110:337–346
Polóniová Z, Jopčík M, Matušíková I, Libantová J, Moravčíková J (2012) Preparation of plant transformation vector containing “self-excision” Cre/loxP system. J Microbiol Biotech Food Sci 1:563–572
Roy SD, Saxena M, Bhomkar PS, Pooggin M, Hohn T, Bhalla-Sarin N (2008) Generation of marker-free salt tolerant transgenic plants of Arabidopsis thaliana using the gly I gene and cre gene under inducible promoters. Plant Cell Tiss Org 95:1–11
Russell SH, Hoopes JL, Odell JT (1992) Directed excision of a transgene from the plant genome. Mol Gen Genet 234:49–59
Scutt CP, Zubko E, Meyer P (2002) Techniques for the removal of marker genes from transgenic plants. Biochimie 84:1119–1126
Sreekala C, Wu L, Gu K, Wang D, Tian D, Yin Z (2005) Excision of a selectable marker in transgenic rice (Oryza sativa L.) using a chemically regulated Cre/loxP system. Plant Cell Rep 24:86–94
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL-W—improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680
Tuteja N, Verma S, Sahoo RK, Raveendar S, Reddy INBL (2012) Recent advances in development of marker-free transgenic plants: regulation and biosafety concern. Journal Biosci 37:167–197
Vaculkova E, Moravcikova J, Matusikova I, Bauer M, Libantova J (2007) A modified low copy number binary vector pUN for Agrobacterium-mediated plant transformation. Biol Plantarum 51:538–540
Verweire D, Verleyen K, De Buck S, Claeys M, Angenon G (2007) Marker-free Transgenic plants through genetically programmed auto-excision. Plant Physiol 145:1220–1231
Wang Y, Chen BJ, Hu YL, Li JF, Lin ZP (2005) Inducible excision of selectable marker gene from transgenic plants by the Cre/lox site-specific recombination system. Transgen Res 14:605–614
Will E, Klump H, Heffner N, Schwieger M, Schiedlmeier B, Ostertag W, Baum C, Stocking C (2002) Unmodified Cre recombinase crosses the membrane. Nucl Acid Res 30(12):e59
Woo H-J, Suh S-C, Cho Y-G (2011) Strategies for developing marker-free transgenic plants. Biotechnol Bioprocess Eng 16:1053–1064
Zhang W, Subbarao S, Addae P, Shen A, Armstrong C, Peschke V, Gilbertson L (2003) Cre/lox-mediated marker gene excision in transgenic maize (Zea mays L.) plants. Theor Appl Genet 107:1157–1168
Zhang YY, Li HX, Bo OY, Lu YG, Ye ZB (2006) Chemical-induced autoexcision of selectable markers in elite tomato plants transformed with a gene conferring resistance to lepidopteran insects. Biotechnol Lett 28:1247–1253
Zhang Y, Liu H, Li B, Zhang JT, Li YZ, Zhang HX (2009) Generation of selectable marker-free transgenic tomato resistant to drought, cold and oxidative stress using the Cre/loxP DNA excision system. Transgen Res 18:607–619
Zuo JR, Niu QW, Moller SG, Chua NH (2001) Chemical-regulated, site-specific DNA excision in transgenic plants. Nat Biotechnol 19:157–161
Acknowledgments
Authors thank Anna Fábelová for in vitro plant care. Funding was supported by Scientific Grant Agency of the Ministry of Education of Slovak Republic and Slovak Academy of Sciences VEGA 2-0090-14.
Conflict of interest
The authors declare they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by Zeng-Yu Wang.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Polóniová, Z., Jopčík, M., Matušíková, I. et al. The pollen- and embryo-specific Arabidopsis DLL promoter bears good potential for application in marker-free Cre/loxP self-excision strategy. Plant Cell Rep 34, 469–481 (2015). https://doi.org/10.1007/s00299-014-1726-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00299-014-1726-0