
Abstract
In the present paper, attempts were made to explore the possibility of employing ultraviolet (UV) irradiation in citrus asymmetric fusion for transfer of limited amount of favorable traits from a desirable cultivar to a target one. Exposure of Satsuma mandarin (Citrus unshiu Marc.) embryogenic protoplasts to UV at an intensity of 300 μW cm−2 led to reduced viability, especially under long irradiation duration. The protoplasts could not grow during culture when they were irradiated for over 30 s. Terminal deoxynucleotidyl transferase biotin-dUTP nick end labeling (TUNEL) assay revealed extensive DNA fragmentation in the UV-irradiated protoplasts compared with those without UV treatment. Electrofusion between UV-irradiated protoplasts of Satsuma mandarin (donor) with those of Jincheng (C. sinensis Osbeck, recipient), a local cultivar of superior quality, gave rise to regeneration of several lines of shoots, which failed to root despite enormous endeavors. Ploidy analysis via flow cytometry and chromosome counting showed that four selected shoots were either diploid, triploid or tetraploid. Random amplified polymorphism DNA (RAPD) and amplified fragment length polymorphism (AFLP) confirmed the shoots, irrespective of their ploidy level, as putative somatic hybrids. Cleaved amplified polymorphism sequences (CAPS) demonstrated that the shoots predominantly got their cytoplasmic components, in terms of chloroplast (cp) and mitochondrion DNA, from Jincheng, along with possible recombination of cpDNA in some shoot lines. The current data indicated that UV-based asymmetric fusion could also be employed in citrus somatic hybridization with the intention of creating novel germplasms, which may provide an alternative approach for cultivar improvement.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Jincheng (Citrus sinensis Osbeck) is one of the local citrus cultivars of superior quality, which, however, is not satisfactorily tolerant to cold, a harsh environment stress limiting plant growth and productivity. Therefore, improvement of its stress tolerance has been an important issue for extending producing acreage. Satsuma mandarin (C. unshiu Marc.) is the most cold-tolerant cultivar of commercial importance in Citrus since it can survive −11.11°C. Besides, it is more resistant than sweet orange to canker, gummosis, psorosis and melanose. Therefore, transfer of traits concerning biotic and abiotic stress tolerance from Satsuma mandarin to Jincheng is anticipated. Nevertheless, it is hard to produce sexual hybrids between them due to polyembryony in both species, which necessitates the development of alternative methods. Somatic hybridization via protoplast fusion paves the way for circumventing the barrier of polyembryony and recovering novel germplasms (Grosser and Gmitter 2005). In that tolerant genes are predominantly carried in few chromosomes, fusion of two intact cells may give rise to a somatic hybrid inheriting both desirable and undesirable traits from the fusion parents, leading to unexpected performance of the somatic hybrids (Grosser et al. 2000). In order to minimize the incorporation of unwanted traits into somatic hybrid, asymmetric fusion, also called gamma-fusion or donor–recipient fusion, has been proposed (Samoylov and Sink 1996; Oberwalder et al. 1997; Forsberg et al. 1998b; Liu et al. 2005). Since the first work on asymmetric fusion between Nicotiana tabacum and N. sylvestris was reported (Zelcer et al. 1978), this fusion pattern has been realized in a wide range of plant species, leading to production of a variety of asymmetric hybrids (Dudits et al. 1987; Spangenberg et al. 1994; Gerdemann-Knörck et al. 1995; Liu et al. 2005; Davey et al. 2005). It proves to be an effective way for transferring limited amount of genetic materials from donor to recipient, as has been shown by successful transfer of genes coding for resistance to black leg and club root from Brassica nigra into B. napus (Gerdemann-Knörck et al. 1995), methotrexate and 5-methyltryptophan resistance from carrot to tobacco (Dudits et al. 1987) and chromosomes with genes resistant to diseases from wild potato to cultivated one (Xu et al. 1993), among others. In asymmetric fusion, the donor protoplasts were commonly exposed to ionizing irradiation provided by either X-rays or γ-rays before fusion to cause chromosome loss (Dudits et al. 1987; Liu and Deng 2002). Regeneration of many asymmetric hybrids demonstrated that the two rays are applicable and powerful for inducing loss of donor nuclear genomes. However, they require complicated and expensive equipment for emission of the rays, which is not easily and readily accessible in many laboratories. In this regard, exploitation of other simple means that could be the substitute of X-rays and γ-rays is important and as well necessary for asymmetric fusion.
Ultraviolet (UV) has been regarded as an alternative for directed or selective transfer of donor chromosomes. Atanassov et al. (1991) for the first time carried out asymmetric fusion with donor protoplasts exposed to UV, which resulted in regeneration of highly asymmetric hybrids. Since then, UV irradiation has been increasingly used in asymmetric fusion (Vlahova et al. 1997; Forsberg et al. 1998a, b; Yue et al. 2001; Xia et al. 2003; Xiang et al. 2003; Wang et al. 2003; Cheng et al. 2004, 2006), from which some valuable germplasms have been recovered. For instance, Wang et al. (2003) developed novel oilseed rape via asymmetric fusion between Brassica napus and UV-irradiated Crambe abyssinica, which contained significantly greater amounts of erucic acid than the recipient, B. napus. Xia et al. (2003) obtained asymmetric hybrids between UV-irradiated Agropyron elongatum and wheat, exhibiting evidently higher salt tolerance than wheat. All of these, taken together, seem to suggest that UV irradiation could be used as a replacement of ionizing irradiation to favor chromosome elimination and to accelerate production of novel materials.
As for Citrus, so far more than 300 somatic hybrids have been regenerated from a large spectrum of combinations (Grosser et al. 2000; Takami et al. 2004; Wu et al. 2005; Grosser and Gmitter 2005; Olivares-Fuster et al. 2005; Liu et al. 2005; Guo et al. 2006). It is noted that most of the somatic hybrids came from symmetric fusion, whereas very few were derived from asymmetric fusion based on X-rays (Vardi et al. 1987, 1989; Liu and Deng 2000, 2002). Whether UV irradiation can be used for citrus asymmetric fusion to get somatic hybrids remains unclear. Therefore, in the present paper, an attempt was first made to identify DNA fragmentation of citrus cells exposed to UV irradiation and then to electrofuse protoplasts between UV-irradiated Satsuma mandarin and Jincheng with the intention of establishing a strategy for transfer of limited genetic component aimed at cultivar improvement in the future.
Materials and methods
Plant materials
Embryogenic calluses of Guoqing No.1 (donor, G1), one type of Satsuma mandarin, and Jincheng (recipient, JC) were maintained on solid MT (Murashige and Tucker 1969) basal medium containing 40 g l−1 sucrose and 7 g l−1 agar (pH 5.8). Their suspension cultures were established by culturing the callus in liquid medium containing MT supplemented with 0.5 g l−1 malt extract (ME, Sigma), 1.5 g l−1 glutamine and 30 g l−1 sucrose (pH 5.8). After subculture for four cycles at 12-day intervals, the suspension cells were used for protoplast isolation.
Isolation and purification of protoplasts
Protoplasts of both the donor and the recipient were isolated from 6-day-old suspension cultures and then purified following a method reported previously (Liu and Deng 1999; Zhang et al. 2006). About 1 g of callus was incubated overnight on a shaker (30 rpm) at 28°C in 3 ml isolation buffer containing 1.5 ml enzyme solution (1% cellulase Onozuka R-10, 1.5% macerozyme, 12.8% mannitol, 0.12% 2, (N-morpholino) ethane sulfonic acid, 0.26% CaCl2, pH 5.6) and 1.5 ml of equilibrium medium (MT + 0.7 M mannitol + 1.5 g l−1 ME). After enzyme incubation, the mixture was passed through two layers of stainless steel sieves and then centrifuged with 26% sucrose/13% mannitol gradient for 6 min at 88 g. The purified protoplasts were washed twice with electrofusion solution (ES) composed of 0.7 M mannitol and 0.25 mM CaCl2.
Treatment of donor protoplasts
Protoplasts of G1 were placed in 15 mm × 60 mm Petri dishes to form a thin layer and placed under a UV lamp with wavelength of 254 nm and irradiation intensity of 300 μW cm−2 for 15, 30, 60, 120 and 240 s, respectively. After irradiation, the protoplasts were washed with ES and then adjusted to a density of 5 × 105 ml−1 before fusion.
Analysis of protoplast viability
Viability of protoplast was determined via a dual staining method using both fluorescein diacetate (FDA) and Hoechst 33258 (H33258) based on previous reports (Zeng et al. 2006; Meadows and Potrykus 1981). FDA is a commonly used fluorescence dye for checking viability, while H33258 can be used to detect nuclei of both live and dead cells. Five microliters of FDA solution (1 mg of FDA in 1 ml acetone) were added to 20 μl of suspended protoplasts, in which H33258 was also added to a final concentration of 0.5 mg l−1. The protoplasts were incubated for 5 min in the dark, followed by observation with UV excitation under a fluorescence microscope (BX61, Olympus, Japan). Ten observation fields were counted in each treatment. Protoplast viability was expressed as NFDA × 100/NH33258, where NFDA and NH33258 were the numbers of cells stained with FDA and H33258, respectively.
Detection of nuclear DNA fragmentation
DNA fragmentation was detected by terminal deoxynucleotidyl transferase biotin-dUTP nick end labeling (TUNEL) using the TUNEL Apoptosis Detection Kit (Nanjing Keygen Biotech. Co., Ltd., China). For this purpose, protoplasts of G1 subjected to UV irradiation for 240 s and those without UV treatment (considered as control) were sampled 12 h after culture, compared with staining pattern of the protoplasts collected at the onset of culture. The protoplasts were fixed in 4% paraformaldehyde (v/v) for 25 min, followed by wash with 1 × phosphate buffered saline solution (PBS) for three times. All observations were performed under a light fluorescence microscope (BX61, Olympus, Japan) and photographs were taken using an Olympus DP 70 CCD camera.
Electrofusion, culture and regeneration of fusion products
The donor and recipient protoplasts were mixed at 1:1 ratio and fused in a somatic hybridizer (SSH-2, Shimadzu, Japan) with fusion parameters modified from Liu and Deng (2002). In brief, the protoplasts were aligned with an alternate current strength of 95 V cm−1 at a frequency of 1 MHz for 60 s, followed by a direct current (1250 V cm−1) for 40 μs. Totally five pulses at 0.5 s intervals were used, after which the fusion products were kept still for about 20 min so that the fusants could be spherical. Thereafter, the fusion products were collected by centrifugation for 5 min at 70 g, and the resulting pellet was resuspended in protoplast culture medium (PCM, MT added with 0.15 M sucrose, 0.45 M mannitol and 80 g l−1 adenine, pH 5.8) to a final density of 106 ml−1. The protoplasts were cultured using the solid embedding culture according to Liu and Deng (1999), and kept in dark at growth chamber (28°C). Mini-calluses of 1–2 mm in size were picked carefully and cultured on solid EME500 (MT + 50 g l−1 sucrose + 500 mg l−1 ME + 7 g l−1 agar, pH 5.8). Embryoids developed from protoplast-derived callus were cultured on solid EME1500 (MT + 50 g l−1 sucrose + 1500 mg l−1 ME + 7 g l−1 agar, pH 5.8) for further growth. The cotyledonous embryoids were transferred to shoot-induction medium (SIM, MT + 0.5 mg l−1 N6-benzyl aminopurine + 0.5 mg l−1 kinetin + 0.1 mg l−1 α-naphthaleneacetic acid + 30 g l−1 sucrose + 7 g l−1 agar, pH 5.8) to induce shoots.
Ploidy analysis via flow cytometry and chromosome counting
Ploidy of the regenerated shoots was determined by flow cytometry (FCM) and chromosome counting according to our previous methods (Liu and Deng 2002; Liu et al. 2002; Xu et al. 2004, 2005, 2006). For FCM, young leaves were chopped in 0.5 ml of nuclear extraction buffer (solution A of High Resolution Kit, Partec, Germany) and incubated for 3 min, which was then filtered through a nylon sieve with mesh diameter of 30 μm (CellTrics™, Partec, Germany), followed by addition of 2 ml of 4′, 6-diamidino-2-phenylindole (DAPI, solution B of the kit). Two minutes later, the samples were measured with a flow cytometer (PA-I, Partec, Germany). Relative fluorescence intensity of diploid G1 (the donor herein) was used as a standard, which was compared with those of the samples to calculate their ploidy.
For chromosome counting, a haematoxylin staining method was employed (Liu and Deng 1999). Young shoot tips were collected and pretreated with 1,4-dichlorobenzene for 3 h, fixed in Carnoy (one part glacial acetic acid: three parts ethanol) for 16–20 h, then softened for 15 min with 5 M HCl, followed by 1-h treatment with 4% animonium sulfate. The samples were fully rinsed and then incubated in 0.5% hematoxylin for 3 h before chromosome examination. Chromosomes of at least ten cells with good metaphases were recorded under a compound microscope (Olympus, BH-2).
Isolation of total DNA and molecular analysis of the regenerated shoots
Total DNA was extracted from the regenerated shoots and calluses of fusion parents using CTAB based on Liu et al. (2002) and dissolved in 200–400 μl TE buffer (10 mM Tris–HCl and 0.1 mM EDTA). DNA quality was confirmed by either electrophoresis or spectrophotometry (data not shown) before use for molecular analysis through RAPD (random amplification of polymorphism DNA), AFLP (amplified fragment length polymorphism) and CAPS (cleaved amplification polymorphisms).
Random amplified polymorphism DNA (RAPD)
For RAPD analysis, a PTC-200 thermal cycler (MJ Research, USA) was used with four decamer random primers (OPA-07, OPV-07, OPS-13 and OPA-04) in the same cocktail as reported before (Liu and Deng 1999). Amplification was performed with the following conditions: 1 cycle of 94°C, 3 min, 38 cycles of 94°C, 45 s, 36°C, 45 s and 72°C, 1 min and 1 cycle of 72°C for 10 min. Electrophoretic separation of the amplified DNA fragment was performed in 1.6% agarose gel containing 0.5 μg ml−1 ethidium bromide. The gels were visualized and photographed with UV light.
Amplified fragment length polymorphism (AFLP)
AFLP was carried out according to Pang et al. (2006) with minor modifications. Total DNA (250 ng) of each sample was digested with EcoRI (3 U) and MseI (3 U) at 37°C for 3 h, followed by incubation at 65°C for 20 min before adapters for EcoRI and MseI were added. Pre-amplification using a +1 primer (EcoRI + A, MseI + C) was then carried out and the pre-amplified products were diluted 50 times and used as template for selective amplification with three primer combinations (MC04/E04, MC05/E04, MC06/E04; E and M representing EcoRI and MseI, respectively). The resultant PCR products were separated by electrophoresis in 6% (w/v) denatured polyacrylamide gel, which was silver-stained according to the manual of Silver Staining Kit (Promega, USA).
Cleaved amplified polymorphism sequence (CAPS)
CAPS analysis with five chloroplast and three mitochondrial universal primers was done as depicted before (Xu et al. 2004, 2006). Reaction mix consisted of 100 ng of genomic DNA, 1 × reaction buffer, 2 mM MgCl2, 0.2 mM dNTP, 1.0 U Taq polymerase and 0.1 μM forward primer and 0.1 μM reverse primer. PCR amplifications were conducted in the PTC-200 thermal cycler with the programmes same as Xu et al. (2006). The PCR products were digested with restriction enzymes (HinfI, HindIII, TasI, TaqI, RasI) for 3–4 h, followed by fractionation in 2.0% agarose gel containing 0.5 μg ml−1 ethidium bromide at 2.5 v cm−1 for 2–3 h and visualized under UV transillumination.
Results
Effect of UV irradiation on protoplast viability and culture of donor protoplasts
With UV excitation under a fluorescence microscope FDA-positive cells gave green fluorescence, and H33258-positive cells were blue (Fig. 1a). When they were added to cell population simultaneously the live cells would be stained with both FDA and H33258, whereas the dead ones were only stained with H33258. Therefore, under UV activation, the viable cells were yellowish due to overlapping of blue and green color, while the dead cells were blue (Fig. 1b). Calculation based on the dual staining method showed that the donor protoplasts had a transient viability of 90.6% without UV irradiation (Fig. 2). However, when they were exposed to UV for different duration, their transient viability was reduced with the exception of irradiation for 15 s. Viability of the protoplasts irradiated for 240 s was significantly decreased (79.4%) compared with the other treatments. Within 6–9 days after culture, the control protoplasts and those irradiated for 15 s recovered the first division and finally developed into callus (data not shown), whereas those irradiated for 30 s and longer duration did not divide, but burst (Fig. 1c), indicating that high dosage of UV irradiation has caused great injury to the cells, in agreement with the work on Arabidopsis protoplasts by Danon and Gallois (1998). Since irradiation for 30 s slightly reduced the viability and in the meantime arrested cell division, it was selected to treat the donor protoplasts in this research.

Viability analysis via dual staining with FDA and Hoechst 33258 and culture of UV-irradiated Guoqing No. 1 (Citrus unshiu Marc.) protoplasts. a FDA-positive protoplasts exhibited green fluorescence (arrow) under UV excitation. b Observation of the protoplasts stained with FDA and Hoechst 33258, in which dead and live cells are shown in blue (solid arrow) and yellow (open arrow), respectively, under UV excitation. c Protoplasts exposed to UV for 240 s at an intensity of 300 μW cm−2 burst 4 days after culture. Bars = 50 μm

Effects of UV irradiation for different duration at an intensity of 300 μW cm−2 on viability (%) of Guoqing No. 1 (Citrus unshiu Marc.) protoplasts. Data were collected from ten independent observation fields, expressed as mean ± SE shown by bars
DNA fragmentation and cell morphology of UV-irradiated protoplasts
TUNEL reaction offers the possibility of visualizing in situ DNA fragmentation and allows the monitoring of cells undergoing DNA damage at a given time (Danon and Gallois 1998). In TUNEL assay, cells with DNA damage fragmentation are brown, and the color intensity could indirectly reflect degree of cell injury. Herein, we also used this methodology to identify if UV caused DNA fragmentation in the donor cells. In order to ensure the validity of TUNEL analysis, we used a positive control composed of protoplasts treated with DNase I, an endonuclease that nonspecifically cleaves DNA to release di-, tri- and oligonucleotide products with 5′-phosphorylated and 3′-hydroxylated ends. It demonstrated that protoplasts in the positive control exhibited high level of DNA fragmentation, in which nearly all of the cells were brown (Fig. 3a), confirming that the TUNEL assay here was reliable and informative. Protoplasts without UV treatment, which could be regarded as negative control, did not show obvious staining at the onset of culture (Fig. 3b). Likewise, the protoplasts without UV irradiation were not stained 12 h after culture in TUNEL analysis (Fig. 3c), whereas those exposed to UV exhibited strong TUNEL-positive signals at the same time point (Fig. 3d), implying that UV has caused extensive DNA fragmentation (DNA damage) in the donor cells.

Analysis of DNA fragmentation in Guoqing No. 1 (Citrus unshiu Marc.) protoplasts treated with or without UV via TUNEL assay. a TUNEL assay of protoplasts treated with DNase I (positive control). b, c TUNEL assay of protoplasts without UV treatment at the onset (negative control) of and 12 h after culture. d TUNEL assay of protoplasts exposed to UV (240 s, 300 μW cm−2) that were collected 12 h after culture. e, f UV-treated cells with crescent-shaped nucleus (e, shown by solid arrow) or nucleus migrating to cell periphery (f, shown by open arrow). bars = 50 μm (black) or 10 μm (white)
It has been shown that TUNEL-positive cells contained three types of nuclei, normal-looking round nuclei, elongated, crescent-shaped nuclei and fragmented nuclei migrated to the cell periphery (Danon and Gallois 1998). When the above-mentioned dual staining method was used to observe the UV-treated protoplasts, cells with nuclei of the last two types mentioned above were also observed (Fig. 3e, f), which meant that such cell morphology might be a common case in UV-irradiated cells.
Asymmetric fusion, culture and regeneration
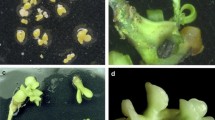
Protoplasts of G1 exposed to UV for 30 s were electrofused with those of JC. Mini-calluses were regenerated from the fusion products within 30–40 days after solid embedding culture. They were picked with care and regenerated into single cell-derived lines on EME500 (Fig. 4a), which subsequently developed into embryoids 50–60 days after culture on EME1500 (Fig. 4b). When the cotyledonous embryoids were transferred to EIM, shoots could be recovered in every cell line. Surprisingly, the regenerated shoots showed abnormal morphology compared with the fusion parents (Fig. 4c), which could not develop into normal ones despite enormous efforts.

Regeneration of callus (a), embryoids (b) and shoots (c) from asymmetric fusion between UV-irradiated Guoqing No. 1 (Citrus unshiu Marc.) protoplasts and Jincheng (C. sinensis Osbeck) protoplasts. bars = 1 cm
Ploidy and molecular analysis of the regenerated shoots
Four shoot lines randomly selected from the regenerated ones, which were designated as GJ1, GJ2, GJ3 and GJ4 (GJ is the abbreviation for G1 and JC), were subjected to ploidy analysis and hybrid nature identification.
Fluorescence intensity of the diploid control was set to 50, which was compared with those of the four shoot lines to determine their ploidy. FCM showed that fluorescence intensity of GJ1, GJ2, GJ3 and GJ4 were 82.11, 46.72, 55.75 and 97.82, respectively, which indicated that GJ1 and GJ4 were triploid and tetraploid, respectively, while both GJ2 and GJ3 were diploid (Fig. 5a–d). Results of chromosome counting via haematoxylin staining supported the analysis of FCM (Fig. 5e–g), implying that UV-based asymmetric fusion has led to regeneration of shoots with different ploidy.

Ploidy analyses of the regenerated shoots via flow cytometry (a–d) and chromosome counting (e–g). a FCM histogram of diploid control, Guoqing No. 1 (Citrus unshiu Marc.). FCM histograms of the regenerated shoots, GJ1 (b), GJ2 (c) and GJ4 (d), respectively. e–g Metaphase cells with 18 (from GJ2), 27 (from GJ1) and 36 (from GJ4) chromosomes, respectively. bars = 5 μm
RAPD was first employed to investigate whether the four shoots were of hybrid origin. Two out of the four random primers, OPA-04 and OPA-07, revealed polymorphism between the fusion parents. In the amplification products of these two polymorphic primers, all of the shoots shared the same banding patterns as that of the recipient, JC, and no polymorphic bands present in the donor were observed (Fig. 6a, b).

Banding patterns of the regenerated shoots (GJ1, GJ2, GJ3 and GJ4), coupled with their fusion parents, Guoqing No. 1 (G1) and Jincheng (JC), in RAPD (a–b) and AFLP (c) analysis. RAPD amplification by random primers OPA-04 (a) and OPA-07 (b). c AFLP analysis by primer pair MC06/E04. Open and filled arrows indicate bands specific to the donor (G1) and the recipient (JC), respectively
Since RAPD analysis with the polymorphic primers could not clearly confirm the hybridity of the regenerated shoots, another highly polymorphic marker, AFLP, was adopted. Only one pair of the primers (MC06/E04) distinguished the donor from the recipient, which was therefore used for investigating hybrid nature of the shoots. As shown in Fig. 6c, bands specific to G1 were observed in the regenerated shoots, which indicated that genetic material of the donor was also present in the shoots. AFLP, in concert with RAPD analysis, showed that the shoots were truly somatic hybrids derived from asymmetric fusion between UV-irradiated G1 and JC.
In order to analyze cytoplasmic constitution of the hybrid shoots, CAPS analysis with universal primers for chloroplast and mitochondrial genomes was carried out. As described before (Liu et al. 2002; Xu et al. 2004, 2006), no difference was found between the fusion parents in the amplified products of both types of primers. When the amplified products were digested with restriction enzymes, two polymorphic chloroplast markers (trnH-trnK/HinfI and trnH-trnK/TasI) and one polymorphic mitochondrial marker (18S-5S rRNA/TasI) were detected. As far as the polymorphic chloroplast markers were concerned, the hybrid shoots showed banding profiles similar to that of the recipient, JC (Fig. 7a, b). It is noted that novel bands absent in the fusion parents were detected in GJ2 and GJ3 produced by trnH-trnK/TasI, indicating possible recombination of cpDNA in these two lines, as has been reported before (reviewed in Liu et al. 2005). As for the mitochondrial polymorphic marker the hybrid shoots shared the same banding profiles as that of JC (Fig. 7c). CAPS analysis showed that all of the hybrids inherited their cytoplasmic components predominantly from the recipient.

CAPS analysis of chloroplast (a–b) and mitochondrial DNA (c) composition of the regenerated shoots (GJ1, GJ2, GJ3 and GJ4), coupled with their fusion parents, Guoqing No. 1 (G1) and Jincheng (JC). Chloroplast banding patterns produced by polymorphic primer/enzyme combinations, trnH-trnK/HinfI (a) and trnH-trnK/TasI (b). The arrows show novel bands present in the hybrids. c Mitochondrial banding profile produced by polymorphic primer/enzyme combination, 18S-5S rRNA/TasI
Discussion
A key issue on employing UV in asymmetric fusion is whether it can truly induce extensive fragmentation of donor chromosomes and promote subsequent elimination. In the present paper, effects of UV irradiation on protoplast transient viability was studied based on a dual staining method. It showed that protoplasts exposed to UV irradiation for short duration (15 s) at an intensity of 300 μW cm−2 did not alter their viability obviously and could grow normally, while UV irradiation for over 30 s led to slight (30–120 s) or remarkable (240 s) decrease in protoplast viability and completely arrested the mitotic division, implying a dose-dependent inhibitory effect of UV on cell growth (Xiang et al. 2003). TUNEL assay revealed occurrence of extensive DNA damage in the UV-irradiated protoplasts, suggesting that UV could undoubtedly induce chromosome fragmentation, in line with the illustration in Arabidopsis thaliana by Danon and Gallois (1998). Although we did not compare parallel effects on DNA breakage between UV and other radiation rays in the same experiment, our result validated an earlier work by Hall et al. (1992) who concluded that UV could lead to pronounced DNA fragmentation at a biological dosage same as γ-rays. Herein, it is noticed that fusion between UV-irradiated G1 and JC, both of which were diploids, gave rise to several lines of hybrid shoots, in which two were diploids and one was triploid in addition to a tetraploid one. Regeneration of diploid and triploid hybrid shoots seems tempting to indicate that extensive chromosome loss has taken place following fusion of two diploid parents. Despite the fact that we could not definitely identify the parental origin of chromosomes in the hybrids, the current data showed that highly asymmetric somatic hybrids (cybrids, GJ2 and GJ3) have been successfully regenerated. Production of highly asymmetric somatic hybrids via UV-dependent fusion has been previously reported in an array of plant species (Forsberg et al. 1998a; Xia et al. 2003). All of these, taken together, suggested that UV was a possible means for inducing fragmentation of donor genomes and could be used for limited transfer of donor DNA. However, it is of interest to note that one out of four analyzed shoots (GJ4) was a tetraploid, implying that there might be no chromosome elimination in this line, although the donor was irradiated with UV, which might be ascribed to the following reasons. Firstly, UV irradiation did not cause DNA fragmentation in all of the protoplasts due to its weak penetration capacity and/or different sensitivity to UV irradiation of the protoplasts, leading to some escapes in the population, which, when fused with the recipient, would produce tetraploid cells. As has been described elsewhere (Liu et al. 2005; Oberwalder et al. 1997, 1998; Xiang et al. 2003), irradiation-derived chromosome elimination in asymmetric fusion is not merely correlated with irradiation, but can be influenced by several other internal or external factors, such as genotypes, phylogenetic relatedness between the fusion parents, physiological status of the explants used for protoplast isolation. Secondly, DNA repair after UV irradiation was responsible for restoration of cells without chromosome loss. It is known that plant cells have evolved certain strategies, such as nucleotide excision repair and photoreactivation, to repair DNA after UV irradiation in order to maintain normal function (Ishibashi et al. 2006). If the repair happened soon after UV irradiation and before the fusion with recipient protoplasts, no chromosome elimination would be available in the hybrid cells, which developed into a cell without chromosome elimination.
Although we obtained several lines of shoots from asymmetric fusion, they were unexpectedly abnormal in morphology and were recalcitrant to rooting. Aberrant growth in somatic hybridization may be caused by several factors, such as somatic incompatibility at the chromosomal level or physiological inconsistency between the fusion parents (Harms 1983). Since the fusion parents we used herein were phylogenetically related (Herrero et al. 1996), somatic incompatibility might not be the main reason for abnormal growth of the hybrids. Alternatively, such unexpected phenomenon may be largely due to inhibitory effect of UV irradiation prior to fusion and culture, which has also been described before. For example, Forsberg et al. (1998a) reported that low shoot regeneration frequency was observed when they performed fusion between UV-irradiated Arabidopsis thaliana and Brassica napus. In another report, Xiang et al. (2003) showed that only albino plants could be obtained when UV-irradiated Avena sativa were fused with wheat (Triticum aestivum). In the current study, donor protoplasts were exposed to UV for 30 s at 300 μW cm−2, the lowest division-arresting intensity, before they were electrofused with the recipient protoplasts. Although treatment at this time point did not significantly reduce the transient viability of the donor protoplasts (88.3% vs. 90.6%), it is surmised that the irradiation might have caused overdamage to the donor protoplasts. As a result, genomic and physiological complementation between the donor and the recipient was insufficient to recover normal growth and development of the fusants (Liu and Deng 2002), leading to aberrant growth of the hybrids, as has been previously delineated in different combinations (Samoylov and Sink 1996; Oberwalder et al. 1997; Forsberg et al. 1998a; Liu and Deng 2002). Therefore, exploitation of appropriate irradiation duration at the fixed intensity will be conducive to regeneration of normal hybrid plants in the future.
In the present work, the donor protoplasts could not develop into plants when they were cultured alone due to mitotic arrest by UV irradiation. On the other hand, no special treatment was posed on the recipient protoplasts, which exhibited good potential of embryogenesis and could regenerate into plantlets under the applied culture condition. In this regard, theoretically, pure recipient colonies were present in the regenerants of fusion products. However, all of the four shoot lines derived from the fusion event we randomly selected for detailed analysis were confirmed as hybrids. The reasons underlying this phenomenon remained to be determined, which may be related to regeneration advantage of hybrid cells. It has been shown that the somatic hybrids were more vigorous and had higher capacity for embryogenesis (Guo and Grosser 2005), implying that the somatic hybrids grew and developed at a faster speed than the unfused and homofused recipient cells. As a consequence, the first batch of regenerants may be of hybrid origin, which has been recently confirmed using green fluorescence protein (GFP) as a visual marker (Guo and Grosser 2005). Such regeneration advantage, to some degree, was a timesaving and effective measure that could facilitate early selection and enrichment of somatic hybrids, as has been reported elsewhere (Polgár et al. 1993; Oberwalder et al. 1998; Cai et al. 2006).
In conclusion, the current data demonstrated that UV could truly cause extensive DNA damage and led to chromosome elimination after asymmetric fusion. To the best of our knowledge, this is the first report on utilizing UV irradiation in citrus somatic hybridization. Although several issues await for future resolution, this strategy may be an alternative for citrus asymmetric fusion with the intention of transferring partial genomes from candidate sources in an attempt to expedite cultivar improvement via cell fusion in the long run.
Abbreviations
- AFLP :
-
Amplified fragment length polymorphism
- CAPS :
-
Cleaved amplified polymorphism sequence
- DAPI :
-
4′, 6-Diamidino-2-phenylindole
- EIM :
-
Embryoid-induction medium
- FCM :
-
Flow cytometry
- FDA :
-
Fluorescein diacetate
- H33258 :
-
Hoechst 33258
- MT :
-
Murashige and Tucker
- RAPD :
-
Random amplified polymorphism DNA
- RFLP :
-
Restriction fragment length polymorphism
- rpm:
-
Revolutions per minute
- SSR :
-
Simple sequence repeat
- TUNEL :
-
Terminal deoxynucleotidyl transferase biotin-dUTP nick end labeling
- UV :
-
Ultraviolet
References
Atanassov A, Dimantov D, Atanassov I, Dragoeva A, Vassileva Z, Vitanov V, Jankulova M, Djilianov D (1991) Transfer of resistance to tomato spotted wilt virus (TSWV) from wild Nicotiana species to N. tabacum via somatic hybridization. Physiol Plant A23, poster session 8, No. 130
Cai XD, Liu X, Guo WW (2006) GFP expression as an indicator of somatic hybrids between transgenic Satsuma mandarin and calamondin at embryoid stage. Plant Cell Tiss Organ Cult 87:245–253
Cheng AX, Xia GM, Zhi DY, Chen HM (2004) Intermediate fertile Triticum aestivum (+) Agropyron elongatum somatic hybrids are generated by low doses of UV irradiation. Cell Res 14:86–91
Cheng A, Cui H, Xia GM (2006) Construction of a primary RH panel of Italian ryegrass genome via UV-induced protoplast fusion. Plant Biol 8:1–7
Danon A, Gallois P (1998) UV-C radiation induces apoptotic-like changes in Arabidopsis thaliana. FEBS Lett 437:131–136
Davey MR, Anthony P, Power JB, Lowe KC (2005) Plant protoplasts: status and biotechnological perspectives. Biotechnol Adv 23:131–171
Dudits D, Maroy E, Praznovszky T, Olah Z, Gyorgyey J, Cella R (1987) Transfer of resistance traits from carrot into tobacco by asymmetric somatic hybridization: regeneration of fertile plants. Proc Natl Acad Sci USA 84:8434–8438
Forsberg J, Dixelius C, Lagercrantz U, Glimelius K (1998a) UV dose-dependent DNA elimination in asymmetric somatic hybrids between Brassica napus and Arabidopsis thaliana. Plant Sci 131:65–76
Forsberg J, Lagercrantz U, Glimelius K (1998b) Comparison of UV light, X-ray and restriction enzyme treatment as tools in production of asymmetric somatic hybrids between Brassica napus and Arabidopsis thaliana. Theor Appl Genet 96:1178–1185
Gerdemann-Knörck M, Nielen S, Tzscheetzsch C, Iglisch J, Schieder O (1995) Transfer of disease resistance within the genus Brassica through asymmetric somatic hybridization. Euphytica 85:247–253
Grosser JW, Gmitter FG (2005) Applications of somatic hybridization and cybridization in crop improvement, with citrus as a model. In Vitro Cell Dev Biol Plant 41:220–225
Grosser JW, Ollitrault P, Olivares-Fuster O (2000) Somatic hybridization in Citrus: an effective tool to facilitate variety improvement. In Vitro Cell Dev Biol Plant 36:434–449
Guo WW, Grosser JW (2005) Somatic hybrid vigor in Citrus: Direct evidence from protoplast fusion of an embryogenic callus line with a transgenic mesophyll parent expressing the GFP gene. Plant Sci 168:1541–1545
Guo WW, Cheng YJ, Chen CL, Deng XX (2006) Molecular analysis revealed autotetrploid, diploid and tetraploid cybrid plants regenerated from an interspecific somatic fusion in Citrus. Sci Hortic 108:162–166
Hall RD, Rouwendal GJA, Krens FA (1992) Asymmetric somatic cell hybridization in plants I. The early effects of (sub)lethal doses of UV and gamma radiation on the cell physiology and DNA integrity of cultured sugarbeet (Beta vulgaris L) protoplasts Mol Gen Genet 234:306–314
Harms CT (1983) Somatic incompatibility in the development of higher plant somatic hybrids. Q Rev Biol 58:325–353
Herrero H, Asins MJ, Pina JA, Carbonell EA, Navarro L (1996) Genetic diversity in the orange subfamily Aurantioideae. II. Genetic relationships among genera and species. Theor Appl Genet 93:1327–1334
Ishibashi T, Kimura S, Furukawa T, Skaguchi K (2006) DNA repair mechanisms in UV-B tolerant plants. JARQ 40:107–113
Liu JH, Deng XX (1999) Regeneration of hybrid calluses via donor-recipient fusion between Microcitrus papuana and Citrus sinensis. Plant Cell Tiss Organ Cult 59:81–87
Liu JH, Deng XX (2000) Regeneration and characterization of plants derived from protoplast asymmetric fusion in Citrus. Acta Bot Sin 42:1144–1149
Liu JH, Deng XX (2002) Regeneration and analysis of citrus interspecific mixoploid hybrid plants from asymmetric somatic hybridization. Euphytica 125:13–20
Liu JH, Pang XM, Cheng YJ, Meng HJ, Deng XX (2002) Molecular characterization of the nuclear and cytoplasmic genomes of intergeneric diploid plants from cell fusion between Microcitrus papuana and rough lemon. Plant Cell Rep 21:327–332
Liu JH, Xu XY, Deng XX (2005) Intergeneric somatic hybridization and its application to crop genetic improvement. Plant Cell Tiss Organ Cult 82:19–44
Meadows MG, Potrykus I (1981) Hoechst 33258 as a vital stain for plant cell protoplasts. Plant Cell Rep 1:77–79
Murashige T, Tucker D (1969) Growth factors requirement of citrus tissue cultures. Proc Int Citrus Symp 3:1155–1161
Oberwalder B, Ruoß B, Schilde-Rentschler L, Hemleben V, Ninnemann H (1997) Asymmetric fusion between wild and cultivated species of potato (Solanum spp.) -detection of asymmetric hybrids and genome elimination. Theor Appl Genet 94:1104–1112
Oberwalder B, Schilde-Rentschler L, Ruoß B, Wittemann S, Ninnemann H (1998) Asymmetric protoplast fusions between wild species and breeding lines in potato-effect of recipients and genome stability. Theor Appl Genet 97:1347–1354
Olivares-Fuster O, Duran-Vila N, Navarro L (2005) Electrochemical protoplast fusion in citrus. Plant Cell Rep 24:112–119
Pang XM, Wen XP, Hu CG, Deng XX (2006) Genetic diversity of Poncirus accessions as revealed by amplified fragment length polymorphism (AFLP). J Hortic Sci Biotechnol 81:269–275
Polgár Z, Preiszner J, Dudits D, Fehér A (1993) Vigorous growth of fusion products allows highly efficient selection of interspecific potato somatic hybrids: molecular proofs. Plant Cell Rep 12:399–402
Samoylov YM, Sink K (1996) The role of irradiation dose and DNA content of somatic hybrid calli in producing asymmetric plants between an interspecific tomato hybrid and eggplant. Theor Appl Genet 92:850–857
Spangenberg G, Vallés MP, Wang ZY, Montavon P, Nagel J, Potrykus I (1994) Asymmetric somatic hybridization between tall fescue (Festuca arundinacea Schreb.) and irradiated Italian ryegrass (Lolium multiflorum Lam.) protoplasts. Theor Appl Genet 88:509–519
Takami K, Matsumara A, Yahata M, Imayama T, Kunitake H, Komatsu H (2004) Production of intergeneric somatic hybrids between round kumquat (Fortunella japonica Swingle) and Morita navel orange (Citrus sinensis Osbeck). Plant Cell Rep 23:39–45
Vardi A, Breiman A, Galun E (1987) Citrus cybrids: production by donor-recipient protoplast-fusion and verification by mitochondrial-DNA restriction profiles. Theor Appl Genet 75:51–58
Vardi A, Arzee-Gonen P, Frydman-Shani A, Bleichman S, Galun E (1989) Protoplast-fusion mediated transfer of organelle from Microcitrus into Citrus and regeneration of novel alloplasmic trees. Theor Appl Genet 78:741–747
Vlahova M, Hinnisdaels S, Frulleux F, Claeys M, Atanassov A, Jacobs M (1997) UV irradiation as a tool for obtaining asymmetric somatic hybrids between Nicotiana plumbaginifolia and Lycopersicon esculentum. Theor Appl Genet 94:184–191
Wu JH, Ferguson AR, Mooney PA (2005) Allotetraploid hybrids produced by protoplast fusion for seedless triploid Citrus breeding. Euphytica 141:229–235
Wang YQ, Sonntag K, Rudloff E (2003) Development of rapeseed with high erucic acid content by asymmetric somatic hybridization between Brassica napus and Crambe abyssinica. Theor Appl Genet 106:1147–1155
Xia GM, Xiang FN, Zhou AF, Wang H, He SX, Chen HM (2003) Asymmetric somatic hybridization between wheat (Triticum aestivum L.) and Agropyron elongatum (Host) Nevski. Theor Appl Genet 107:299–305
Xiang FN, Xia GM, Chen HM (2003) Effect of UV dosage on somatic hybridization between common wheat (Triticum aestivum L.) and Avena sativa L. Plant Sci 164:697–707
Xu YS, Murto M, Dunckley R, Jone MGK, Pehu E (1993) Production of asymmetric hybrids between Solanum tuberosum and irradiated S. brevidens. Theor Appl Genet 85:729–734
Xu XY, Liu JH, Deng XX (2004) Production and characterization of intergeneric diploid cybrids derived from symmetric fusion between Microcitrus papuana Swingle and sour orange (Citrus aurantium). Euphytica 136:115–123
Xu XY, Liu JH, Deng XX (2005) FCM, SSR and CAPS analysis of intergeneric somatic hybrid plants between Changshou kumquat and Dancy tangerine. Bot Bull Acad Sci 46:93–98
Xu XY, Liu JH, Deng XX (2006) Isolation of cytoplasts from Satsuma mandarin (Citrus unshiu Marc.) and production of alloplasmic hybrid calluses via cytoplast-protoplast fusion. Plant Cell Rep 25:533–539
Yue W, Xia GM, Zhi DY, Chen HM (2001) Transfer of salt tolerance from Aeleuropus littorulis sinensis to wheat (Triticum aestivum L.) via asymmetric somatic hybridization. Plant Sci 161:259–266
Zelcer A, Aviv D, Galun E (1978) Interspecific transfer of cytoplasmic male sterility by fusion between protoplasts of normal Nicotiana sylvestris and X-irradiated protoplasts of male sterile N. tabacum. Z Pflanzenphysiol 90:397–407
Zeng SH, Chen CW, Hong L, Liu JH, Deng XX (2006) In vitro induction, regeneration and analysis of autotetraploids derived from protoplasts and callus treated with colchicine in Citrus. Plant Cell Tiss Organ Cult 87:85–93
Zhang QH, Liu JH, Deng XX (2006) Isolation of microprotoplasts from a partially synchronized suspension culture of Citrus unshiu. J Plant Physiol 163:1185–1192
Acknowledgments
This research was partially supported by Natural Science Foundation of China, IRT 0548, the Ministry of Education, China, and Hubei Provincial Natural Science Foundation. The authors are grateful to Dr. JH Wu (HortResearch, New Zealand) for his critical reading of the paper.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by A. Atanassov.
Rights and permissions
About this article
Cite this article
Xu, XY., Hu, ZY., Li, JF. et al. Asymmetric somatic hybridization between UV-irradiated Citrus unshiu and C. sinensis: regeneration and characterization of hybrid shoots. Plant Cell Rep 26, 1263–1273 (2007). https://doi.org/10.1007/s00299-007-0350-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00299-007-0350-7