
Abstract
Key message
A novel high-density consensus wheat genetic map was obtained based on three related RIL populations, and the important chromosomal regions affecting yield and related traits were specified.
Abstract
A prerequisite for mapping quantitative trait locus (QTL) is to build a genetic linkage map. In this study, three recombinant inbred line populations (represented by WL, WY, and WJ) sharing one common parental line were used for map construction and subsequently for QTL detection of yield-related traits. PCR-based and diversity arrays technology markers were screened in the three populations. The integrated genetic map contains 1,127 marker loci, which span 2,976.75 cM for the whole genome, 985.93 cM for the A genome, 922.16 cM for the B genome, and 1,068.65 cM for the D genome. Phenotypic values were evaluated in four environments for populations WY and WJ, but three environments for population WL. Individual and combined phenotypic values across environments were used for QTL detection. A total of 165 putative additive QTL were identified, 22 of which showed significant additive-by-environment interaction effects. A total of 65 QTL (51.5 %) were stable across environments, and 23 of these (35.4 %) were common stable QTL that were identified in at least two populations. Notably, QTkw-5B.1, QTkw-6A.2, and QTkw-7B.1 were common major stable QTL in at least two populations, exhibiting 11.28–16.06, 5.64–18.69, and 6.76–21.16 % of the phenotypic variance, respectively. Genetic relationships between kernel dimensions and kernel weight and between yield components and yield were evaluated. Moreover, QTL or regions that commonly interact across genetic backgrounds were discussed by comparing the results of the present study with those of previous similar studies. The present study provides useful information for marker-assisted selection in breeding wheat varieties with high yield.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Common wheat (Triticum aestivum L.) is one of the most important staple crops worldwide, and an understanding of its genetics is critical for improving yield stability. Collectively, phenotyping, genotyping with molecular markers, and molecular map construction constitute the essential steps in identifying quantitative trait locus (QTL) (Lörz and Wenzel 2004; Heidari et al. 2011). Hence, detailed molecular genetic maps can facilitate QTL analysis. The first wheat SSR map with 279 loci was reported by Röder et al. (1998). Almost simultaneously, Stephenson et al. (1998) mapped 53 novel SSR loci (with the prefix PSP) to two genetic maps. Soon afterwards, various types of polymerase chain reaction (PCR)-based molecular markers were widely used to construct a wheat genetic map (Nachit et al. 2001; Sourdille et al. 2003; Yu et al. 2004; Suenaga et al. 2005; Liu et al. 2005; Torada et al. 2006; Li et al. 2007b). Diversity arrays technology (DArT), a novel means of molecular marker development, combines a complexity reduction method with hybridization-based polymorphism detection using high-throughput, solid-state platforms. This technique can generate hundreds of high-quality genomic dominant markers with high efficiency (http://www.diversityarrays.com/). Thus far, several wheat genetic maps with DArT markers have been reported (Akbari et al. 2006; Mantovani et al. 2008; Peleg et al. 2008; Francki et al. 2009; Wang et al. 2011b; Huang et al. 2012). Integrating the genetic maps obtained from different related mapping populations not only results in a relatively high-density consensus genetic map but also facilitates the comparison of common QTL across different mapping populations.
One of the main objectives in wheat breeding is to obtain high yield, which is quantitatively inherited and significantly influenced by the environment. Grain yield in cereals can be dissected into several components, which are also under QTL control but have higher heritabilities than grain yield itself (Bezant et al. 1997; Yano and Sasaki 1997; Kato et al. 2000; Hai et al. 2008). Furthermore, individual traits correlated with wheat grain yield are often controlled by analogous genomic regions (Quarrie et al. 2006; Huang et al. 2004, 2006; Li et al. 2007b; Hai et al. 2008; Cuthbert et al. 2008). Productive spikes per unit area, kernels per spike, and kernel weight are the so-called three yield components that together determine the yield of wheat. Among the three yield components, kernel weight shows the highest heritability, varying from 59 to 80 % (Xiao and He 2003), indicating that selection for kernel weight in early breeding generations is highly effective. Kernel weight is greatly influenced by kernel dimensions (KD) such as kernel length and kernel width. Therefore, it is of utmost interest to obtain more information about the underlying genetic control of KD traits (Breseghello and Sorrells 2006; Sun et al. 2009; Cui et al. 2011a, b).
With the rapid development of molecular marker technology in wheat, increasing numbers of QTL studies have been conducted to dissect the genetic basis of yield and its components. All 21 wheat chromosomes have been shown to harbor factors affecting yield (Supplementary Table S1). For example, Campbell et al. (1999), Ammiraju et al. (2001), Dholakia et al. (2003) and Tsilo et al. (2010) detected QTL for kernel size on chromosomes 1A, 1D, 2A, 2B, 2D, 3B, 5A, 5B, 6B, 7A, and/or 7B using recombinant inbred line (RIL) populations. Börner et al. (2002) documented two stable QTL for grain number on chromosomes 4A and 7D and one stable QTL for kernel weight on chromosome 5A. Based on advanced backcross QTL (AB-QTL) analysis, Huang et al. (2003, 2004) and Narasimhamoorthy et al. (2006) reported QTL for grain yield on chromosomes 1A, 1B, 2A, 2B, 2D, 3B, 3D, 4D, 5A, 5B, 6B, 6D, and/or 7D; for kernel weight on 1B, 1D, 2A, 2D, 3A, 3B, 3D, 4B, 4D, 5B, 6A, 7A, 7B, and/or 7D; for kernels per spike on 1D, 2A, 3D, 6A, 7A, and/or 7D; and for tiller number on 1B, 2A, 2D, 3B, 4D, 5D, 6D, and/or 7A. Using doubled haploid (DH) mapping populations, Huang et al. (2006), McCartney et al. (2005) and Heidari et al. (2011) detected QTL for grain yield on chromosomes 2A, 2B, 3D, 4A, 4D, 5A, 6A, 6D, 7A, and/or 7B and for grain weight on 2A, 3D, 2B, 2D, 3B, 4A, 4B, 4D, 5A, and/or 6D. Kirigwi et al. (2007) documented a QTL for grain yield in the proximal region of chromosome 4AL under reduced moisture, whereas Snape et al. (2007) reported a stable QTL for yield on chromosome 6A under both irrigated and non-irrigated conditions. McIntyre et al. (2010) reported four putative QTL for yield under irrigated and rainfed conditions, which all colocalized with QTL for yield components. Ramya et al. (2010) reported 10 QTL for grain weight and 15 QTL for kernel dimensions, and pleiotropic QTL were found on chromosomes 2B, 2D, 4B, and 5B. Using F 2:3 mapping populations consisting of 237 lines, Wang et al. (2011a) detected ten QTL for kernel number on chromosomes 1A, 2D, 3B, 4A, 4B, 5A, 5B, 7A, and 7B and seven QTL for kernel weight on chromosomes 1A, 4B, 5A, 5B, 6A, 6B, and 7B, of which three chromosomal regions on 5A, 6A, and 4B harbor QTL clusters involving all six yield-related traits. Using two different mapping populations, Kumar et al. (2007) detected 173 and 521 QTL for yield and yield-contributing traits distributed across 19 wheat chromosomes except 5B and 7D; however, only one QTL was common between the two mapping populations. Using genome-wide association mapping analysis based on linkage disequilibrium, Wang et al. (2012) and Zhang et al. (2012) verified numerous previously reported QTL. Common QTL across different mapping populations were rarely defined or explored because only a single mapping population was used. The incidence of common QTL between different genetic backgrounds implies both the reliability and importance of the corresponding genomic region, thus providing theoretical guidance in molecular breeding.
In the present study, three related RIL populations were used in map construction and QTL mapping analysis for yield-related traits. The aims of this study were to (1) develop a consensus wheat molecular genetic map with PCR-based and DArT markers, (2) identify the genetic factors affecting yield-related traits, and (3) provide useful information for marker-assisted selection (MAS) in wheat breeding for increased yield.
Materials and methods
Plant materials, field arrangement and trait evaluation
Three related RIL populations were used in the present study, which were created by crossing the common parental line Weimai 8 with Jimai 20 (WJ), Yannong 19 (WY), and with Luohan 2 (WL), respectively. The WJ and WY populations were reported in our previous studies (Cui et al. 2011a, b), whereas this is the first report describing the WL population. In total, 175 of the 485 WJ-derived RILs, 172 of the 229 WY-derived RILs, and 179 of the 302 WL-derived RILs were used in the present study, and these RILs were randomly subsampled from their initial populations. Relevant information about the WJ and WY populations and their parents can be found in previous reports (Cui et al. 2011a, b). Moreover, Weimai 8 is characterized by its high level of resistance to powdery mildew and moderate resistance to yellow rust and leaf rust, and Jimai 20 is characterized by its high level of resistance to powdery mildew and moderate resistance to yellow rust and sheath blight. Luohan 2, a common wheat variety that is highly drought tolerant, was produced in 2001 by the Crop Research Institute, Luoyang Municipal Academy of Agricultural Sciences, Henan, China. Luohan 2 is characterized by its high level of resistance to leaf rust and moderate resistant to yellow rust and bacterial blight. Among the four parental lines, Weimai 8 has the largest kernels (Cui et al. 2011a).
The WJ and WY populations together with their parents were evaluated in four environments in Shandong Province, China: Tai’an in 2008–2009 (E1), and Tai’an in 2009–2010 (E2), Zao’zhuang in 2009–2010 (E3) and Ji’ning in 2009–2010 (E4). The four environments have been detailed in our previous reports (Cui et al. 2011a). The WL population and its parents were evaluated only in E1, E2, and E4.
In each plot, five representative plants were selected from the middle row as samples for measuring spike number per plant (SNPP), kernel number per spike (KNPS), and kernel weight per plant (KWPP). Seeds were thoroughly cleaned, and all non-wheat materials and broken kernels were removed before measuring kernel weight. KWPP was determined in grams by weighing all kernels of each representative plant. Thousand-kernel weight (TKW) was determined in grams by weighing two samples containing 1,000 kernels from each plot. Two samples of 20 kernels from each plot were lined up length-wise along a ruler with a precision of 0.1 mm, to measure kernel length (KL), and then the kernels were arranged breadth-wise to measure kernel width (KW). All lengths are reported in centimeters. KNPS, TKW, KL, and KW were evaluated in E1, E2, E3, and E4 in both the WY and WJ populations; in the WL population, these traits were evaluated in E1, E2, and E4. SNPP and KWPP were evaluated in E1, E2, and E3 in both the WY and WJ populations; in the WL population, these traits were evaluated in E1 and E2.
Analysis of molecular and biochemical markers
Polymorphic PCR-based and biochemical markers, including G-SSR, EST-SSR, ISSR, STS, SRAP, RAPD, and high molecular weight glutenin subunits (HMW-GS) at Glu-a1, Glu-b1, and Glu-d1, were detected using the method previously reported by Cui et al. (2012). All of the RILs were genotyped with PCR-based and biochemical markers. Seedling leaves were used to prepare the DNA for DArT analysis using the recommended DNA extraction method (http://www.diversityarrays.com/cgi-bin/order/login.pl). RILs of the subsets that were randomly subsampled from their initial populations and their parents were assayed using the wheat DArT array ‘Wheat PstI (TaqI) 2.3 D’ (http://www.triticarte.com.au/). The scores of all markers were converted into genotype codes according to the scores of the parents (‘A’ for the common parental line Weimai 8, ‘B’ for the remaining three parental lines, and ‘–’ for the missing data and heterozygous genotype amplified by PCR-based markers in the map construction and integration using MAPMAKER 3.0 and JoinMap 4.0; ‘2’ for the common parental line Weimai 8, ‘0’ for the remaining three parental lines, ‘1’ for heterozygous genotypes amplified by PCR-based markers, and ‘−1’ for the missing data in the QTL analysis by IciMapping 3.2).
Map construction and integration
Individual maps for each population were constructed using MAPMAKER 3.0 (Lander et al. 1987). First, 21 groups were defined using the ‘MAKE CHROMOSOME’ command. The ‘ANCHOR’ command was then used to locate marker loci on chromosomes based on identified Chinese Spring nullisomic–tetrasomic lines and public genetic maps in GrainGenes 2.0 and http://www.cerealsdb.uk.net/search_reads.htm. The remaining loci were assigned to chromosomes using the ‘ASSIGN’ command at a log-of-odds (LOD) score of 3.0 with distances less than 50 cM. Linkage analysis was performed using JoinMap 4.0 (Biometris, Wageningen, the Netherlands; http://www.kyazma.nl/) with a minimum LOD score of 3.0 and a recombination frequency of 0.45. Groups that had the same linkage with at least two common loci were loaded into the same project via JoinMap 4.0. Based on the common markers between individual maps, the ‘COMBINE GROUPS FOR MAP INTEGRATION’ function from the ‘JOIN’ menu was used for map integration. Three separate steps were performed for map integration. First, markers that were common between at least two maps were defined as anchor markers and used to link corresponding linkage groups on individual maps. Subsequently, the consensus order of the anchor markers was calculated from their relative positions on each individual map. Lastly, the integrated order of all loci was determined, beginning with the most common marker and adding the closest markers one at a time. The centimorgan (cM) map unit was calculated using the Kosambi mapping function (Kosambi 1944). The integrative map was drawn using MapChart 2.2 (http://www.biometris.nl/uk/Software/MapChart/).
Data analysis and QTL mapping
Basic statistical analysis of the phenotypic data in the three RIL populations was performed using the SPSS13.0 software (SPSS, Chicago, IL, USA; http://en.wikipedia.org/wiki/SPSS). Heritability (H 2) was calculated using the GLM procedure of SAS according to Knapp et al. (1985). The formula for estimating H 2 is H 2 = 1 − M 2/M 1, where M 1 and M 2 indicate the variance of genotype and genotype × environment, respectively. Due to the number of replications (r = 1), it was impossible to estimate genotype × environment interaction variance in the present study. The mean variance for residual error is represented as M 2 (Huang et al. 2006).
Using IciMapping 3.2 (http://www.isbreeding.net/), QTL scanning was performed by inclusive composite interval mapping (ICIM) through stepwise regression by considering all marker information simultaneously (Li et al. 2007a). In both the WJ and WY populations, the phenotypic values of the RILs in E1, E2, E3, and E4 were used for individual environment QTL mapping; in the WL population, the phenotypic values of the RILs in E1, E2, and E4 were used. Missing phenotypes were deleted using the ‘Deletion’ command, whereas the missing genotypic data were speculated according to its closest linkage marker scores. The walking speed chosen for all QTL was 1.0 cM, and the probability in stepwise regression (p value inclusion threshold) was 0.001. The threshold LOD scores were calculated using 1,000 permutations with a type 1 error of 0.05. Moreover, combined (C) QTL analysis across environments was conducted with the aim of identifying QTL with additive-by-environment (A by E) interaction effects, using 2.5 as the threshold LOD score (the default value) (Li et al. 2007a).
Nomenclature of QTL
All QTL were named as follows: italic uppercase ‘Q’ denotes ‘QTL’; letters following it before the dash are the abbreviation of the corresponding trait; next, after the dash is the wheat chromosome on which the corresponding QTL is distributed; if more than one QTL for a certain trait dispersed along a certain chromosome, a serial number, viz. 1, 2, 3, etc., is used after the chromosome name to describe their order, from the short arm to the long arm.
A QTL that was significant in just one family was defined as rare QTL, whereas those significant in at least two families were designated as common QTL (Li et al. 2011). We defined a stable QTL as one that was significant in at least two different environments of E1, E2, E3, and E4. A QTL with significant A by E interaction effect is defined as that where the LOD value for A by E is ≥2.5.
Results
The novel integrative genetic map
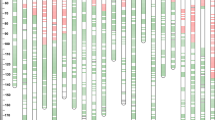
A total of 883 (504 DArT loci), 855 (477 DArT loci), and 786 (430 DArT loci) polymorphic loci were used for linkage analysis in the WL, WY, and WJ populations, respectively. Of these loci, 871, 834, and 751 from the WL, WY, and WJ populations, respectively, were assigned to 21 wheat chromosomes. Due to the 1BL/1RS translocation of the common parent Weimai 8, 69 1RS- or 1BS-specific loci showed co-segregation in the RILs; these were excluded from the linkage analysis and map construction. Overall, 1,127 loci distributed on all 21 wheat chromosomes were mapped to the integrative genetic map (Fig. 1). Of these loci, 576 were DArT markers, most of which were clustered in wheat genome. The 548 PCR-based polymorphic loci included 279 G-SSR loci, 158 EST-SSR, 62 STS, 22 ISSR, 15 SRAP, and 12 RAPD. Three biochemical markers, Glu-a1, Glu-b1, and Glu-d1, were accurately mapped to their corresponding chromosomes. In total, 496 of the 1,127 loci were common to at least two of the three individual maps (Fig. 1). The integrative genetic map covered 2,976.8 cM, including 986.0 cM (33.12 %) of the A genome, 922.2 cM (30.98 %) of the B genome, and 1,068.7 cM (35.90 %) of the D genome. The average distance between adjacent loci was 2.6 cM. Most markers were mapped to the A genome (40.56 %) and the B genome (40.11 %), each with an average of 65 markers per chromosome. The remaining markers (19.33 %) were mapped to the D genome, with an average of 31 markers per chromosome. The chromosome size ranged from 59.2 cM for chromosome 1BL to 217.3 cM for chromosome 4A, with an average of 141.8 cM per chromosome. The number of loci per chromosome ranged from 95 on chromosome 2B to 14 on chromosome 4D, with a mean of 54 loci per chromosome. Chromosome 2B had the highest average marker density with 1 marker per 1.3 cM, whereas chromosome 4D had the lowest average marker density, with an average of 12.4 cM between adjacent loci. Although good coverage of the genome was obtained, gaps of over 30 cM between the adjacent loci remained on chromosomes 2D, 3D, 4A, 4B, 4D, and 5D (Fig. 1).




The novel integrative genetic map and location of QTL for yield and its related traits based on three related RIL populations of between Weimai 8 and Luohan 2 (WL), Weimai 8 and Yannong 19 (WY), and Weimai 8 and Jimai 20 (WJ). The approximate centromere position is indicated by bright green color on the corresponding chromosomal segment. The short arms are at the top. The positions of the marker loci and the QTL are listed to the left of the corresponding chromosomes. The names of the marker loci are listed to the right of the corresponding chromosomes. Different colors indicate that a locus is owned by only a certain population or shared by at least two RIL populations. For more details, see marker loci symbols at the bottom of figure. DArT markers are prefixed with ‘wPt’, and the remaining markers are PCR-based markers. The intervals of QTL were LOD >2.0 with LOD peak values more than 2.5. Different colors of the QTL symbols indicate that the QTL is detected in the WL, WY, or WJ, and different stripes of QTL symbol indicate a QTL for TKW, KL, KW, KNPS, SNPP, or KWPP. For more details, see QTL symbols at the bottom of figure (color figure online)
Phenotypic performance of the six yield and yield-related traits
The final six yield and yield-related traits for the three RIL populations and the parents in all environments are shown in Supplementary Table S2. Weimai 8 was characterized by more KNPS, larger kernels, higher TKW, higher KWPP, and less SNPP compared with Jimai 20, Yannong 19, and Luohan 2. In addition, both RIL lines and parents in E3 had the lowest phenotypic values for TKW, KL, KW, SNPP, and KWPP among all of the trials in both WJ and WY populations. This result might be due to the outbreak of gibberellic disease in Zao’zhuang in 2009–2010, which appeared in the watery stage and milk ripe stage. The phenotypic variation in all the six traits among the RIL lines was obvious in all three populations and segregated continuously. Strong transgressive segregation was observed for all six traits in all environments, indicating that alleles with positive effects are distributed among the parents. The estimated broad-sense heritability of the six traits ranged from 12.22 to 71.32 %. TKW, KL, and KNPS had higher broad-sense heritabilities, ranging from 45.94 to 71.32 %; however, KW, SNPP, and especially KWPP showed lower broad-sense heritabilities, ranging from 12.22 to 36.56 %.
The phenotypic correlations between kernel weight and KD and between yield and its three components are listed in Table 1. Both KL and KW were significantly positively correlated with TKW in all environments; similarly, KNPS, TKW, and SNPP were significantly positively correlated with KWPP. Higher positive correlation coefficients were observed between TKW and KW in all environments, indicating a strong stable genetic association between TKW and KW. SNPP and KWPP exhibited strong stable genetic associations in all environments.
QTL mapping for the six yield and yield-related traits
A total of 39, 25, 18, 28, 15, and 8 putative additive QTL were detected in individual environment QTL mapping analysis for TKW, KL, KW, KNPS, SNPP, and KWPP, respectively. These QTL were distributed across all 21 wheat chromosomes and individually exhibited 3.10–44.45 % of the phenotypic variance (Supplementary Table S3; Fig. 1). A total of 31 of the 39 putative additive QTL for TKW, 24 of 25 for KL, 15 of 18 for KW, 27 of 28 for KNPS, 12 of 15 for SNPP, and 6 of 8 for KWPP were confirmed in combined QTL analysis across environments (Supplementary Tables S3 and S3; Fig. 1). Combined QTL analysis across environments detected 11 additional QTL for TKW, 9 for KL, 10 for KW, and 1 each for SNPP and KWPP (Supplementary Table S4; Fig. 1).
Stable QTL across environments
A total of 20 stable QTL were identified for TKW, all of which were significant in combined QTL analysis across environments (Table 2; Supplementary Table S4; Fig. 1). Eleven of these QTL were verified in at least two populations; however, most of them showed stability across environments in only a single RIL population. QTkw-2B.3, QTkw-3A.1, QTkw-3A.3, QTkw-3B.2, QTkw-3D.2, QTkw-5B.1, QTkw-6A.2, QTkw-6D, and QTkw-7B.1 were major stable QTL that accounted for >10 % of the phenotypic variation. It is important to note that QTkw-5B.1, QTkw-6A.2, and QTkw-7B.1 were common major stable QTL that were verified in at least two RIL populations.
A total of 13 stable QTL were detected for KL, and all were significant in combined QTL analysis across environments (Table 2; Supplementary Table S4; Fig. 1). Of these, eight QTL were common QTL that were verified in more than one population. QKl-1BL.1, QKl-2D.1, QKl-2D.2, and QKl-6B.3 showed stability across environments in at least two RIL populations. QKl-2B.1, QKl-2D.1, QKl-2D.2, QKl-3A.1, QKl-6B.2, and QKl-7A.1 were major stable QTL that individually explained >10 % of the phenotypic variation.
Seven QTL for KW were stable QTL, and all were reproducibly identified in combined QTL analysis across environments (Table 2; Supplementary Table S4; Fig. 1). Of these, QKw-2B.3 and QKw-6B.2 were major stable QTL, individually exhibiting 10.12–13.11 and 11.22–24.94 % of the phenotypic variation, respectively.
In total, 17 stable QTL for KNPS were identified, and all were significant in combined QTL analysis across environments (Table 2; Supplementary Table S4; Fig. 1). Of these, QKnps-2A.2, QKnps-2D.2, QKnps-4A, and QKnps-5A were common QTL. QKnps-1BL, QKnps-2A.2, QKnps-3A.1, QKnps-4A, QKnps-4B.1, QKnps-5A, QKnps-6A, and QKnps-7B.1 were major QTL.
QTL mapping detected five stable QTL for SNPP, and all were significant in combined QTL analysis across environments (Table 2; Supplementary Table S4; Fig. 1). Only two QTL (QSnpp-2B.2 and QSnpp-4A.1) individually accounted for >10 % of the phenotypic variation as major QTL.
Only three stable QTL were detected for KWPP, and all were significant in combined QTL analysis across environments (Table 2; Supplementary Table S4; Fig. 1). All three (QKwpp-4A.1, QKwpp-4A.2, and QKwpp-5D) were major QTL.
QTL with significant additive-by-environment interaction effects
Combined QTL analysis across environments indicated that 2 (4.0 %) of the 50 QTL for TKW, 11 (32.4 %) of 34 for KL, 1 (3.6 %) of 28 for KW, 4 (14.3 %) of 28 for KNPS, 1 (6.3 %) of 16 for SNPP, and 3 (33.3 %) of 9 for KWPP showed significant A by E interaction effects (Table 3). Of these, QTkw-4A.1, QKl-2A.1, QKl-2D.1, QKl-2D.2, QKl-5A.3, QKl-6B.2, QKl-7B.1, and QKwpp-4A.1 were significant in more than one environment. Notably, QKl-7B.1, a stable QTL with significant A by E interaction effects, was significant in all four environments in the WJ population, with additive effect values that were positive in E3 but negative in E1, E2, and E4 (Table 3). Fourteen of the above 22 QTL were major QTL, individually accounting for 8.02–44.45 % of the phenotypic variation.
Common QTL consistent with favorable alleles from Weimai 8 across populations
Eight of the 18 common QTL for TKW, 4 of 12 for KL, and 1 of 6 for KNPS showed positive additive effects consistently across populations, with positive additive effects from Weimai 8 of 0.17–2.36 g, 0.001–0.024 cm, and 0.55–3.22, respectively (Table 4). Common QTL for KW, SNPP, and KWPP could not be consistently detected with favorable alleles from Weimai 8 across the populations. Interestingly, favorable alleles of QTkw-1BL.1, QTkw-3B.2, and QTkw-7B.1 originated from Weimai 8 in all the three RIL populations.
Discussion
The novel integrative genetic map
Both wheat molecular breeding and genomics research require accurate and detailed genetic maps. By comparing the positions of common markers across different mapping populations, map integration of different genetic maps not only produces a relatively high-density consensus genetic map but also enhances the accuracy of linear relationships among markers, which is of great value for future fine mapping or even map-based cloning of major QTL. Moreover, a consensus integrative genetic map obtained from related mapping populations facilitates the definition of common QTL. In the present study, three related RIL populations were used for map construction and integration. The novel consensus integrative wheat genetic map covers 74 % of the wheat genome, assuming that the final size of wheat genome is approximately 4,000 cM as predicted by Sourdille et al. (2003). Both PCR-based and DArT molecular markers were mapped to the novel wheat genetic map. Consistent with previous reports, DArT markers were highly clustered, which might be due to the presence of redundant clones in the genomic representation (Akbari et al. 2006; Semagn et al. 2006; Mantovani et al. 2008; Peleg et al. 2008; Francki et al. 2009; Wang et al. 2011b).
Although the new genetic map covers all 21 wheat chromosomes fairly evenly across the A, B, and D genomes, the D genome contains the fewest loci (19.33 %). This finding agrees with other hexaploid wheat maps, in which fewer markers were found in the D genome, especially on chromosome 4D (Röder et al. 1998; Sourdille et al. 2003; Liu et al. 2005; Suenaga et al. 2005; Torada et al. 2006). A targeted isolation from a diploid ancestor might effectively saturate the molecular markers on the D genome (Röder et al. 1998). Although our map contains 1,127 loci, some regions have inadequate coverage and large gaps. The addition of more markers, particularly for D genome chromosomes, will provide more complete genome coverage, thus ensuring powerful QTL detection (Sourdille et al. 2003).
Common QTL and stable QTL
The existence of a common QTL across different mapping populations implies that the QTL will have a consistent stable effect in many genetic backgrounds if it is used in wheat breeding as donor. In contrast, a rare QTL with identifiable genetic effects that segregates in just one family may have a stable effect only in limited genetic backgrounds (Li et al. 2011). In the present study, 36.0 % of the 50 QTL for TKW, 35.9 % of the 34 for KL, 7.1 % of the 28 for KW, and 21.4 % of the 28 for KNPS were common to at least two of the three RIL populations (Supplementary Tables S3 and S4; Fig. 1). Common QTL that are consistent with favorable alleles from the common parent across related mapping populations will be more effective in MAS in wheat breeding programs compared with rare QTL. Therefore, the common QTL listed in Table 4 might be of great value in wheat breeding programs designed to improve yield.
The expression of a stable QTL is less affected by the environment. In general, a major QTL that is consistent across environments is of great value for MAS in breeding varieties adapted to various ecological environments. Therefore, the nine major stable QTL for TKW, six for KL, two each for KW and SNPP, eight for KNPS, and three for KWPP, all of which are detailed in the “Results” section and listed in Table 2, should be of great value in the genetic improvement of wheat yield.
Common major stable QTL that are consistent with favorable alleles from the common parent consistently across populations are of greater value for MAS, with higher efficiencies in breeding varieties adapted to various ecological environments. As a major QTL, QTkw-7B.1 accounted for >10 % of the phenotypic variance and exhibited stability across environments in all three RIL populations. In addition, Weimai 8 alleles of this QTL were consistently associated with increased TKW in all three RIL populations (Tables 2, 4). Thus, this QTL should be precisely mapped and cloned from Weimai 8. QTkw-6A.2, QKl-2D.1, and QKnps-2A.2, the three major QTL for TKW, KL, and KNPS, respectively, exhibited stability across environments in the two different RIL populations. Moreover, the common parent Weimai 8 contributed favorable alleles of all three QTL consistently across populations (Tables 2, 4). These three QTL are also worthy of precise mapping or even map-based cloning.
Genetic relationships between kernel dimensions and kernel weight and between yield components and yield
Kernel weight is greatly influenced by KL and KW. To determine the genetic relationships between TKW and KD at the individual QTL level, multivariate conditional QTL analysis for kernel weight with respect to KD has been conducted using WY and WJ populations (Cui et al. 2011a). The results indicated that KW had a stronger influence on TKW than KL at the QTL level, which is consistent with the phenotypic correlation analysis in the present study based on WL, WY, and WJ populations (Table 1). In addition, 31 (62.0 %) of the 50 QTL for TKW colocalized with that for KL and/or KW, which partially confirms the previous conditional QTL mapping analysis (Supplementary Table S5; Fig. 1). Moreover, 73.2 % of the pleiotropic QTL for pairwise traits of TKW and KD showed positive or negative additive effects simultaneously in the corresponding population, confirming the significant positive correlation between TKW and KD (Table 1; Supplementary Table S5).
TKW, KNPS, and SNPP were all significantly positively correlated with KWPP. The present study showed that SNPP and KWPP exhibited the strongest stable positive genetic associations in all environments (Table 1), indicating that SNPP should be enhanced in wheat breeding programs designed to increase yield in accordance with practical wheat breeding. However, SNPP showed a lower level of broad-sense heritability, ranging from 19.28 to 23.75 % in the three RIL populations, indicating the lower efficiency of selection in earlier generation for genetic improvement of SNPP in wheat breeding programs. Six (75.0 %) of the eight QTL for KWPP colocalized with QTL for TKW, KNPS, and/or SNPP, and all but one had simultaneous positive or negative additive effects on pairwise traits of KWPP and its components in the corresponding population (Supplementary Table S6; Fig. 1), consistent with their significant positive correlations between these traits.
Comparison of the present study with previous research
The WJ and WY populations, which include 485 and 229 lines, respectively, have been utilized in traditional (Cui et al. 2012; Li et al. 2012) and conditional (Cui et al. 2011a, b, 2013) QTL analysis in our previous studies. Only biochemical and PCR-based markers were included in the previous molecular genetic maps, with an average density of one marker per 8.5 and 8.4 cM, respectively, in the WJ and WY populations. Map integration was hampered by the limited common loci among genetic maps. As mentioned above, a consensus integrative map can be used to define a common or rare QTL across mapping populations. The novel integrative map revealed that 36.0 % of the 50 QTL for TKW, 35.9 % of the 34 for KL, 7.1 % of the 28 for KW, and 21.4 % of the 28 for KNPS, respectively, were common QTL. The larger number of common markers and the consensus integrative map might account for the larger number of common QTL detected for yield-related traits in the present study compared with that of our previous QTL analysis (Cui et al. 2011a, 2012, 2013).
In individual environment QTL mapping analysis, 20 (51.3 %) of the 39 QTL for TKW, 13 (52 %) of the 25 for KL, 7 (38.9 %) of the 18 for KW, 17 (60.7 %) of the 28 for KNPS, 5 (33.3 %) of the 15 for SNPP, and 3 (37.5) of the 8 for KWPP were repeatedly detected across environments. Moreover, all of the stable QTL were significant in combined QTL analysis across environments. Despite these findings, the repeatability of the QTL analysis across environments decreased in the present study compared with that of our previous QTL analysis using a larger population size (Cui et al. 2011a, 2012, 2013). It should be noted that TKW, KL, KW and KNPS were only evaluated in three environments in the WL population, whereas SNPP and KWPP were only measured in three environments in both the WY and WJ populations and only in two environments in the WL population, which might have caused the decreased repeatability mentioned above. In addition, the reduced population size might also account for this lower repeatability to some extent.
QTL analysis for yield and its related traits has been performed in many previous studies. To infer whether there were common interacting QTL or regions across genetic backgrounds, the results of our study were compared with those of previous similar studies on wheat. In total, 21 of the 24 important chromosomal regions associated with major stable QTL detected herein were coincident with those of previous studies; however, some of them had effects on different yield components (Supplementary Table S7). For example, both QKnps-1BL and QTkw-1BL.1 were mapped to the Glu-b1–wPt-0944 interval. Huang et al. (2003), Quarrie et al. (2005), Verma et al. (2005), Cuthbert et al. (2008), Wang et al. (2009, 2012), Golabadi et al. (2010), Zheng et al. (2010) and Cui et al. (2011a) also detected QTL for yield and/or its related traits in this chromosomal region. QSnpp-2B.2, QKw-2B.3, QKnps-2B.3, and QTkw-2B.3 were mapped to chromosomal regions similar to those associated with yield and/or its related traits reported by Varshney et al. (2000), Groos et al. (2003), Verma et al. (2005), Huang et al. (2006), Hai et al. (2008), Golabadi et al. (2010), Ramya et al. (2010), Heidari et al. (2011), Tang et al. (2011) and Cui et al. (2011a). The remaining common chromosomal regions associated with yield and/or its related traits across genetic backgrounds are presented in Supplementary Table S7. The coincidence of QTL across different mapping populations not only implies the reliability of the QTL reported herein but also highlights the importance of these chromosomal regions in wheat breeding programs designed to increase yield. The markers flanking common QTL across different mapping populations should be further verified via marker–trait association mapping using natural mapping populations, with the aim of uncovering the positive alleles and in turn providing effective markers for MAS in wheat breeding.
Markers that are more closely linked with some of the common QTL that interact across genetic backgrounds have been reported herein. For example, QKnps-1BL was previously mapped to an interval of over 10 cM by Verma et al. (2005) and Zheng et al. (2010); in the present study, it was mapped between 10 and 17.8 cM on chromosome 1BL, with Xswes18 and Glu-b1 at the LOD score peak. QSnpp-2B.2 was mapped to between 49 and 53 cM with Xwmc154 and Xgwm388 as flanking markers; Tang et al. (2011) mapped this QTL to Xgwm429–Xbarc373, with a confidence interval of >20 cM. Based on composite interval mapping (CIM) analysis using 30 molecular markers, Golabadi et al. (2010) reported that the closest marker to this QTL was Xbarc45, which was approximately 10 cM distal from the confidence interval reported herein. As pleiotropic/coincident QTL of QSnpp-2B.2, QTkw-2B.3, QKw-2B.3, and QKnps-2B.3 were also mapped to this chromosomal region with a support interval of <5 cM. QKnps-4A was mapped to confidence intervals of ≤5 cM in both the WY and WJ populations; it was previously mapped to Xwmc468–Xbarc170, with a support interval of approximately 15 cM (Tang et al. 2011). Based on genome-wide association mapping of 262 members of the Chinese wheat mini core collection, Zhang et al. (2012) found that Xcfe273 had a strong effect on KNPS; in the present study, QKnps-6A was mapped to the Xcfe273.1–wPt-731054 interval (a confidence interval of 5 cM). Xissr808 was present at the peak of the LOD profile and therefore was regarded as the closest marker to QKnps-6A. Aside from the high QTL mapping resolution of ICIM, the high-density molecular genetic map also contributed the short confidence intervals of the QTL reported herein (Li et al. 2007a). Markers that are closely linked to QTL increase the efficiency of MAS in wheat breeding programs designed to improve yield.
To our knowledge, the present study is the first report of ten novel major stable yield-related QTL (Supplementary Table S7). Of these, QKl-2B.1, QTkw-3A.1, QKnps-3A.1, QKwpp-4A.1, QKnps-5A, and QKwpp-5D colocalized or were linked with previously reported QTL for other yield-related traits. The chromosomal regions containing QTkw-3D.2, QSnpp-4A.1, QKwpp-4A.2, and QKl-6B.2 were first reported to harbor factors affecting the yield-related traits described herein. Interestingly, QSnpp-4A.1 and QKl-6B.2 were mapped to confidence intervals of ≤5 cM and are therefore of great value for MAS.
Moreover, six chromosomal regions related to major stable QTL harbor well-known genes (Supplementary Table S7; http://wheat.pw.usda.gov/GG2/index.shtml). For example, Rf3 and Yr24 are approximately 6.0 and 5.0 cM distal from Glu-b1, respectively, which is one flanking marker of QKnps-1BL. Near the major stable QTL cluster of QSnpp-2B.2, QKw-2B.3, and QTkw-2B.3, Yr27, YrCN19, and Ne2 are located approximately 1.0, 4.0, and 5.0 cM, respectively, away from Xbarc13. Rht-B1 is approximately 3.0 cM away from Xgwm495, which is a flanking marker of QKnps-4B.1. The frost tolerance gene Fr-A2 is approximately 6.0 cM from Xbarc40, which is one flanking marker of QKnps-5A. Xgwm544.1 and Xgwm335, two flanking markers of QTkw-5B.1, are approximately 9.0 and 1.0 cM, respectively, from the hybrid necrosis gene Nei; moreover, Stb1, a potentially durable gene for resistance to septoria tritici blotch in wheat, is 1.0 cM distal from Xgwm335. The chromosomal region containing QTkw-7B.1, a common major stable QTL, contains the genes Rht13, Sr25, Lr19, and Psy-B1. Strong selection in breeding programs and pyramiding breeding might account for the colocalization of QTL for yield with known genes. Further study on the relationships between yield and resistance is warranted because a large number of resistance genes colocalize with QTL for yield.
In conclusion, a novel wheat genetic map with both PCR-based and DArT molecular markers was constructed based on three related RIL populations. A total of 165 putative additive QTL for six yield-related traits were reported, 22 of which showed significant A by E interaction effects, individually exhibiting 3.10–44.45 % of the phenotypic variance. Of these, 65 QTL (51.5 %) were stable QTL that showed significance in no less than two different environments, and 23 of 65 (35.4 %) were common stable QTL that were identified in at least two RIL populations. Ten novel major stable yield-related QTL were first reported in the present study. The genetic relationships between KD and kernel weight and between yield components and yield were evaluated. In addition, QTL or regions that commonly interact across genetic backgrounds were extensively discussed by comparing the results of the present study with those of previous similar studies on wheat. This study provides useful information for genetic improvement of wheat yield.
Abbreviations
- TKW:
-
Thousand-kernel weight
- KL :
-
Kernel length
- KW:
-
Kernel width
- KNPS:
-
Kernel number per spike
- SNPP:
-
Spike number per plant
- KWPP:
-
Kernel weight per plant
- WL:
-
Recombinant inbred line population derived from the cross between Weimai 8 and Luohan 2
- WY:
-
Recombinant inbred line population derived from the cross between Weimai 8 and Yannong 19
- WJ:
-
Recombinant inbred line population derived from the cross between Weimai 8 and Jimai 20
References
Akbari M, Wenzl P, Caig V, Carling J, Xia L, Yang S, Uszynski G, Mohler V, Lehmensiek A, Kuchel H, Hayden MJ, Howes N, Sharp P, Vaughan P, Rathmell B, Huttner E, Kilian A (2006) Diversity arrays technology (DArT) for high-throughput profiling of the hexaploid wheat genome. Theor Appl Genet 113:1409–1420
Ammiraju JSS, Dholakia BB, Santra DK, Singh H, Lagu MD, Tamhankar SA, Dhaliwal HS, Rao VS, Gupta VS, Ranjekar PK (2001) Identification of inter simple sequence repeat (ISSR) markers associated with seed size in wheat. Theor Appl Genet 102:726–732
Bezant J, Laurie D, Pratchett N, Chojecki J, Kearsey M (1997) Mapping QTLs controlling yield and yield components in a spring barley (Hordeum vulgare L.) cross using marker regression. Mol Breed 3:29–38
Börner A, Schumann E, Fürste A, Cöter H, Leithold B, Röder MS, Weber WE (2002) Mapping of quantitative trait locus determining agronomic important characters in hexaploid wheat (Triticum aestivum L.). Theor Appl Genet 105:921–936
Breseghello F, Sorrells ME (2006) Association mapping of kernel size and milling quality in wheat cultivars (Triticum aestivum L.). Genetics 172:1165–1177
Campbell KG, Bergmem CJ, Gualberto DG, Anderson JA, Giroux MJ, Hareland G, Fulcher RG, Sorrells ME, Finney PL (1999) Quantitative trait loci associated with kernel traits in a soft × hard wheat cross. Crop Sci 39:1184–1195
Cui F, Ding AM, Li J, Zhao CH, Li XF, Feng DS, Wang XQ, Wang L, Gao JR, Wang HG (2011a) Wheat kernel dimensions: how do they contribute to kernel weight at an individual QTL? J Genet 90:409–425
Cui F, Li J, Ding AM, Zhao CH, Wang L, Wang XQ, Li SS, Bao YG, Li XF, Feng DS, Kong LR, Wang HG (2011b) Conditional QTL mapping for plant height with respect to the length of the spike and internode in two mapping populations of wheat. Theor Appl Genet 122:1517–1536
Cui F, Ding AM, Li J, Zhao CH, Wang L, Wang XQ, Qi XL, Li XF, Li GY, Gao JR, Wang HG (2012) QTL detection of seven spike-related traits and their genetic correlations in wheat using two related RIL populations. Euphytica 186:177–192
Cui F, Zhao CH, Li J, Ding AM, Li XF, Bao YG, Li JM, Ji J, Wang HG (2013) Kernel weight per spike: what contributes to it at the individual QTL level? Mol Breed 31:265–278
Cuthbert JL, Somers DJ, Brũlé-Babel AL, Brown PD, Crow GH (2008) Molecular mapping of quantitative trait loci for yield and yield components in spring wheat (Triticum aestivum L.). Theor Appl Genet 117:595–608
Dholakia BB, Ammiraju JSS, Singh H, Lagu MD, Rörder MS, Rao VS, Dhaliwal HSD, Ranjekar PKR, Gupta VS (2003) Molecular marker analysis of kernel size and shape in bread wheat. Plant Breed 122:392–395
Francki MG, Walker E, Crawford AC, Broughton S, Ohm HW, Barclay I, Wilson RE, McLean R (2009) Comparison of genetic and cytogenetic maps of hexaploid wheat (Triticum aestivum L.) using SSR and DArT markers. Mol Genet Genomics 281:181–191
Golabadi M, Arzani A, Mirmohammadi Maibody SAM, Sayed Tabatabaei BE, Mohammadi SA (2010) Identification of microsatellite markers linked with yield components under drought stress at terminal growth stages in durum wheat. Euphytica 177:207–221
Groos C, Robert N, Bervas E, Charmet G (2003) Genetic analysis of grain protein-content, grain yield and thousand-kernel weight in bread wheat. Theor Appl Genet 106:1032–1040
Hai L, Guo HJ, Wagner C, Xiao SH (2008) Genomic regions for yield and yield parameters in Chinese winter wheat (Triticum aestivum L.) genotypes tested under varying environments correspond to QTL in widely different wheat materials. Plant Sci 175:226–232
Heidari B, Sayed-Tabatabaei EB, Saeidi G, Kearsey M, Suenaga K (2011) Mapping QTL for grain yield, yield components, and spike features in a doubled haploid population of bread wheat. Genome 54:517–527
Huang XQ, Cöster H, Ganal MW, Röder MS (2003) Advanced backcross QTL analysis for the identification of quantitative trait loci alleles from wild relatives of wheat (Triticum aestivum L.). Theor Appl Genet 106:1379–1389
Huang XQ, Kempf H, Ganal MW, Röder MS (2004) Advanced backcross QTL analysis in progenies derived from a cross between a German elite winter wheat variety and a synthetic wheat (Triticum aestivum L.). Theor Appl Genet 109:933–943
Huang XQ, Cloutier S, Lycar L, Radovanovic N, Humphreys DG, Noll JS, Somers DJ, Brown PD (2006) Molecular detection of QTL for agronomic and quality traits in a doubled haploid population derived from two Canadian wheats (Triticum aestivum L.). Theor Appl Genet 113:753–766
Huang BE, Cavanagh C, Rampling L, Kilian A, Andrew Geroge (2012) iDArTs: increasing the value of genomic resources at no cost. Mol Breed 30:927–938
Kato K, Miura H, Sawada S (2000) Mapping QTLs controlling grain yield and its components on chromosome 5A of wheat. Theor Appl Genet 101:1114–1121
Kirigwi FM, Ginkel MV, Brown-Guedira G, Gill BS, Paulsen GM, Fritz AK (2007) Markers associated with a QTL for grain yield in wheat under drought. Mol Breed 20:401–413
Knapp SJ, Stroup WW, Ross WM (1985) Exact convidence intervals for heritability on a progeny mean basis. Crop Sci 25:192–194
Kosambi DD (1944) The estimation of map distances from recombination values. Ann Eugen 12:172–175
Kumar N, Kulwal PL, Balyan HS, Gupta PK (2007) QTL mapping for yield and yield contributing traits in two mapping populations of bread wheat. Mol Breed 19:163–177
Lander ES, Green P, Abrahamson J, Barlow A, Daly MJ, Lincoln SE, Newberg L (1987) MAPMAKER: an interactive computer package for constructing primary genetic linkage maps of experimental and natural populations. Genomics 1:174–181
Li HH, Ye GY, Wang JK (2007a) A modified algorithm for the improvement of composite interval mapping. Genetics 175:361–374
Li SS, Jia JZ, Wei XY, Zhang XC, Li LZ, Chen HM, Fan YD, Sun HY, Zhao XH, Lei TD, Xu YF, Jiang FS, Wang HG, Li LH (2007b) A intervarietal genetic map and QTL analysis for yield traits in wheat. Mol Breed 20:167–178
Li HH, Bradbury P, Ersoz E, Buckler ES, Wang JK (2011) Joint QTL linkage mapping for multiple-cross mating design sharing one common parent. PLoS One 6:e17573
Li J, Cui F, Ding AM, Zhao CH, Wang XQ, Wang L, Bao YG, Qi XL, Li XF, Gao JR, Feng DS, Wang HG (2012) QTL detection of seven quality traits in wheat using two related recombinant inbred line populations. Euphytica 183:207–226
Liu ZH, Anderson JA, Hu J, Friesen TL, Rasmussen JB, Faris JD (2005) A wheat intervarietal genetic linkage map based on microsatellite and target region amplified polymorphism markers and its utility for detecting quantitative trait loci. Theor Appl Genet 111:782–794
Lörz H, Wenzel G (eds) (2004) Molecular marker systems in plant breeding and crop improvement. Springer, Berlin, Heidelberg
Mantovani P, Maccaferri M, Sanguineti MC, Tuberosa R, Catizone I, Wenzl P, Thomson B, Carling J, Huttner E, Ambrogio ED, Kilian A (2008) An integrated DArT-SSR linkage map of durum wheat. Mol Breed 22:629–648
McCartney CA, Somers DJ, Humphreys DG, Lukow O, Ames N, Noll J, Cloutier S, McCallum BD (2005) Mapping quantitative trait loci controlling agronomic traits in the spring wheat cross RL4452 × ‘AC Domain’. Genome 48:870–883
McIntyre CL, Mathews KL, Rattey A, Chapman SC, Drenth J, Ghaderi M, Reynolds M, Shorter R (2010) Molecular detection of genomic regions associated with grain yield and yield-related components in an elite bread wheat cross evaluated under irrigated and rainfed conditions. Theor Appl Genet 120:527–541
Nachit MM, Elouafi I, Pagnotta MA, Ei SA, Iacono E, Labhilili M, Asbati A, Azrak M, Hazzam H, Benscher D, Khairallah M, Ribaut J-M, Tanzarella OA, Porceddu E, Sorrells ME (2001) Molecular linkage map for an intraspecific recombinant inbred population of durum wheat (Triticum turgidum L. var. durum). Theor Appl Genet 102:177–186
Narasimhamoorthy B, Gill BS, Fritz AK, Nelson JC, Brown-Guedira GL (2006) Advanced backcross QTL analysis of a hard winter wheat × synthetic wheat population. Theor Appl Genet 112:787–796
Peleg Z, Saranga Y, Suprunova T, Ronin YW, Röder MS, Kilian A, Korol AB, Fahima T (2008) High-density genetic map of durum wheat × wild emmer wheat based on SSR and DArT markers. Theor Appl Genet 117:103–115
Quarrie SA, Steed A, Calestani C, Semikhodskii A, Lebreton C, Chinoy C, Steele N, Pljevljakusiæ D, Waterman E, Weyen J, Schondelmaier J, Habash DZ, Farmer P, Saker L, Clarkson DT, Abugalieva A, Yessimbekova M, Turuspekov Y, Abugalieva S, Tuberosa R, Sanguineti M-C, Hollington PA, Aragués R, Royo A, Dodig D (2005) A high-density genetic map of hexaploid wheat (Triticum aestivum L.) from the cross Chinese Spring × SQ1 and its use to compare QTLs for grain yield across a range of environments. Theor Appl Genet 110:865–880
Quarrie SA, Quarrie SP, Radosevic R, Rancic D, Kaminska A, Barnes JD, Leverington M, Ceoloni C, Dodig D (2006) Dissecting a wheat QTL for yield present in a range of environments: from the QTL to candidate genes. J Exp Bot 57:2627–2637
Ramya P, Chaubal A, Kulkarni K, Gupta L, Kadoo N, Dhaliwal HS, Chhuneja P, Lagu M, Gupta V (2010) QTL mapping of 1000-kernel weight, kernel length, and kernel width in bread wheat (Triticum aestivum L.). J Appl Genet 51:421–429
Röder MS, Korzun V, Wendehake K, Tixier MH, Leroy P, Ganal MW (1998) A microsatellite map of wheat. Genetics 149:2007–2023
Semagn K, Bjørnstad Å, Skinnes H, Marøy AG, Tarkegne Y, William M (2006) Distribution of DArT, AFLP, and SSR markers in a genetic linkage map of a doubled-haploid hexaploid wheat population. Genome 49:545–555
Snape JW, Foulkes MJ, Simmonds J, Leverington M, Fish LJ, Wang YK, Ciavarrella M (2007) Dissecting gene × environmental effects on wheat yields via QTL and physiological analysis. Euphytica 154:401–408
Sourdille P, Cadalen T, Guyomarc’h H, Snape JW, Perretant MR, Charmet G, Boeuf C, Bernard S, Bernard M (2003) An update of the Courtot × Chinese Spring intervarietal molecular marker linkage map for the QTL detection of agronomic traits in wheat. Theor Appl Genet 106:530–538
Stephenson P, Bryan G, Kirby J, Collins A, Devos K, Busso C, Gale M (1998) Fifty new microsatellite loci for the wheat genetic map. Theor Appl Genet 97:946–949
Suenaga K, Khairallah M, William HM, Hoisington DA (2005) A new intervarietal linkage map and its application for quantitative trait locus analysis of ‘‘gigas’’ features in bread wheat. Genome 48:65–75
Sun XY, Wu K, Zhao Y, Kong FM, Han GZ, Jiang HM, Huang XJ, Li RJ, Wang HG, Li SS (2009) QTL analysis of kernel shape and weight using recombinant inbred lines in wheat. Euphytica 165:615–624
Tang YL, Li J, Wu YQ, Wei HT, Li CS, Yang WY, Chen F (2011) Identification of QTL for yield-related traits in the recombinant inbred line population derived from the cross between a synthetic hexaploid wheat- derived variety Chuanmai 42 and a Chinese elite variety Chuannong 16. Agric Sci China 10:1665–1680
Torada A, Koike M, Mochida K, Ogihara Y (2006) SSR-based linkage map with new markers using an intraspecific population of common wheat. Theor Appl Genet 112:1042–1051
Tsilo TJ, Hareland GA, Simsek S, Chao S, Anderson JA (2010) Genome mapping of kernel characteristics in hard red spring wheat breeding lines. Theor Appl Genet 121:717–730
Varshney RK, Prasad M, Roy JK, Kumar N, Harjit-Singh, Dhaliwal HS, Balyan HS, Gupta PK (2000) Identification of eight chromosomes and a microsatellite marker on 1AS associated with QTL for grain weight in bread wheat. Theor Appl Genet 100:1290–1294
Verma V, Worland AJ, Sayers EJ, Fish L, Calligari PDS, Snape JW (2005) Identification and characterization of quantitative trait loci related to lodging resistance and associated traits in bread wheat. Plant Breed 124:234–241
Wang RX, Hai L, Zhang XY, You GX, Yan CS, Xiao SH (2009) QTL mapping for grain filling rate and yield-related traits in RILs of the Chinese winter wheat population Heshangmai × Yu8679. Theor Appl Genet 118:313–325
Wang JS, Lin WH, Wang H, Li LH, Wu J, Yan XM, Li XQ, Gao AN (2011a) QTL mapping of yield-related traits in the wheat germplasm 3228. Euphytica 177:277–292
Wang YY, Sun XY, Zhao Y, Kong FM, Guo Y, Zhang GZ, Pu YY, Wu K, Li SS (2011b) Enrichment of a common wheat genetic map and QTL mapping for fatty acid content in grain. Plant Sci 181:65–75
Wang L, Ge H, Hao C, Dong Y, Zhang X (2012) Identifying loci influencing 1,000-Kernel weight in wheat by microsatellite screening for evidence of selection during breeding. PLoS One 7(2):e29432. doi:10.1371/journal.pone.0029432
Xiao SH, He ZH (2003) Wheat yield and end use quality improvement in China (Chapter 13). In: Zhuang QS (ed) Chinese wheat improvement and pedigree analysis. China Agricultural Publish Press, China
Yano M, Sasaki T (1997) Genetic and molecular dissection of quantitative traits in rice. Plant Mol Biol 35:145–153
Yu JK, Dake TM, Singh S, Benscher D, Li W, Gill B, Sorrells ME (2004) Development and mapping of EST-derived simple sequence repeat markers for hexaploid wheat. Genome 47:805–818
Zhang DL, Hao CY, Wang LF, Zhang XY (2012) Identifying loci influencing grain number by microsatellite screening in bread wheat (Triticum aestivum L.). Planta 236:1507–1517
Zheng BS, Gouis JL, Leflon M, Rong WY, Laperche A, Brancourt-Hulmel M (2010) Using probe genotypes to dissect QTL × environment interactions for grain yield components in winter wheat. Theor Appl Genet 121:1501–1517
Acknowledgments
This research was supported by the National 863 Plans Projects of China (Grant No. 2011AA100103) and National Natural Science Foundation of China (Grant No. 30971765). The authors thank Prof. Sishen Li, College of Agronomy, Shandong Agricultural University, Taian, China, for kindly providing EST-SSR markers.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by J. Wang.
F. Cui and C. Zhao contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Cui, F., Zhao, C., Ding, A. et al. Construction of an integrative linkage map and QTL mapping of grain yield-related traits using three related wheat RIL populations. Theor Appl Genet 127, 659–675 (2014). https://doi.org/10.1007/s00122-013-2249-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-013-2249-8