
Abstract
Grain number (GN) is one of three major yield-related components in wheat. We used the Chinese wheat mini core collection to undertake a genome-wide association analysis of grain number using 531 SSR markers randomly located on all 21 chromosomes. Grain numbers of all accessions were measured in four trials, i.e. two environments in four growing seasons. Association analysis based on a mixed linear model (MLM) revealed that 27 SSR loci were significantly associated with mean GN (MGN) estimated by the best linear unbiased predictor (BLUP) method. These included numerous breeder favorable alleles with strong positive effects at 23 loci. Significant or extremely significant differences were detected on MGN between varieties conveying favored allele and varieties with other alleles. Moreover, statistical simulation showed that the favored alleles have additive genetic effects. Although modern varieties combined larger numbers of favored alleles, the numbers of favored alleles were not significantly different from those in landraces, especially those alleles contributing mostly to the phenotypic variation. These results indicate that there is still considerable genetic potential for use of markers for genome selection of GN for high yield in wheat.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Bread wheat (Triticum aestivum L.) is one of the most important food crops, feeding about 40 % of the world’s population (Gupta et al. 2008). Selection for high grain yield is an important objective in all wheat breeding programs. Grain yield is a complex trait and is usually controlled by quantitative trait locus (QTL). It is influenced by environmental factors, which make it difficult to be manipulated and improved in breeding programs. Improvement is therefore slow due to the lack of genetic information about the number, location and contribution of each QTL to the final expression of yield (Koebner and Snape 1999).
Grain yield can be dissected into a number of components. Grain number (GN), as one of three major components of yield in wheat, has steadily increased over the period. During the past decades, numerous studies on QTLs (or genes) for grain number have been reported in wheat. Araki et al. (1999) detected QTLs for spikelet number per spike (SNS) on chromosome 4AS. Kato et al. (2000) found a minor QTL for SNS on chromosome 5A. Li et al. (2002) detected QTLs for spike length (SL) on chromosomes 1AL, 1BS, 4AL and 7AL. QTLs for grain number per spike (GNS) were detected on 4A and 7D by Börner et al. (2002), by Huang et al. (2004) on 1D, 2A, 3D, 6A, 7A and 7D in an elite winter cultivar, by Narasimhamoorthy et al. (2006) on 3B and 3D, by Cuthbert et al. (2008) on 1A, 2D, 3B, 5A and 7A, and by Deng et al. (2011) on 4B. Kumar et al. (2007) found coincident QTLs for SNS on 2DS and 5AL, and QTLs for GNS on 1AL and 1BL in two populations. From the above analytical results, QTLs (or genes) for grain number were primarily located on chromosomes 1A, 1B, 1D, 2A, 2D, 3B, 3D, 4A, 5A, 6A, 7A and 7D.
All the above QTLs for grain number were detected by linkage mapping using F2, RIL or DH populations. With huge reductions in the cost of high-throughput genomic technologies (Atwell et al. 2010; Huang et al. 2010) and improvements in statistical methods (Zhu et al. 2008), association mapping (also named LD mapping) has become a reality in plants (Thornsberry et al. 2001). As an alternative to traditional linkage analysis, association mapping has gradually gained favor in plant genetic research because of several advantages: (1) association mapping reduces research time by using existing germplasm populations rather than segregating populations. (2) It can identify novel and superior alleles because more alleles are involved and their effects can be compared in the same diverse population. Associated markers can be used to assist breeders in introducing the best alleles into new varieties. (3) Using the historical recombination events over time, association mapping in a relatively small population of individuals can give a fine mapping resolution that requires very large populations in traditional biparental crosses. Association analysis can even detect associated genetic variation within a gene so as to prove its function (Thornsberry et al. 2001; Zhu et al. 2008; Cockram et al. 2010; Su et al. 2011). (4) Association mapping has lower costs in that only one set of genotypes is required to assess many different traits in many different environments, while replicating the phenotypic measurements to reduce environmental noise (Atwell et al. 2010). In wheat, Yao et al. (2009) detected loci contributing to spikelets per spike and grains per spike on chromosome 2A by association mapping using 125 markers, and demonstrated that association mapping can enhance QTL information and achieve higher resolution. Neumann et al. (2011) performed a genome-wide association mapping study in wheat considering 20 traits including GN and thousand kernel weight (TKW), and those loci provide opportunities for further wheat improvement, based on a marker approach. Wang et al. (2012) used association analysis to provide useful information for marker-assisted selection in breeding wheat for TKW. Each of these studies indicated that association analysis is an effective complementary approach to dissect important quantitative traits in crops compared with the traditional linkage mapping.
In this study, we used the diverse Chinese wheat mini core collection (MCC) (Hao et al. 2008) to undertake a genome-wide association analysis of grain number using 531 SSR markers randomly located on all 21 chromosomes. The purposes of the study were (1) to search for QTLs controlling grain number in a diverse germplasm gene pool, (2) to identify markers and wheat germplasm with favored alleles or genotypes for use in predictive molecular breeding, and (3) to consider the genetic mechanism of grain yield in bread wheat.
Materials and methods
Phenotypic assessment
A set of Chinese wheat MCC (Zhang et al. 2007; Hao et al. 2008, 2011) was chosen for genome-wide association of grain number (GN) with SSR markers. The MCC contains 262 wheat accessions, including 157 landraces, 88 modern varieties, and 17 introduced lines, representing 1 % of the national collection but more than 70 % of the entire genetic diversity (Hao et al. 2008). All accessions were planted in a randomized complete block design in four environments, viz. 2002, 2005 and 2006 in Luoyang, Henan province, and 2010 in Shunyi, Beijing (named GN02, GN05, GN06 and GN10, respectively). Each accession was planted in a 2 m two-row plot with 30 cm between rows, and 40 seeds planted per row. Ten plants from the middle of each plot were used in investigating GN and TKW (Wang et al. 2012). Mean values of GN and standard errors were analyzed by SPSS 16.0 (http://www.brothersoft.com/downloads/spss-16.html). The mixed mean GN (MGN) was estimated by the best linear unbiased predictor (BLUP) method according to Bernardo (1996a, b, c).
SSR genotyping
Genomic DNA was extracted from leaves of ten seedlings in each accession according to Sharp et al. (1989) and fingerprinted by PCR amplifications that identified alleles at 531 SSR loci. Genetic map positions for most of the markers (512 loci) can be found in Hao et al. (2011). The loci were distributed evenly across all 21 wheat chromosomes. The primer sequences and genetic locations of the loci were obtained from http://www.shigen.nig.ac.jp and http://wheat.pw.usda.gov, Röder et al. (1998) and Somers et al. (2004). The annealing temperature for each primer pair was obtained from Röder et al. (1998) and GrainGenes (http://wheat.pw.usda.gov). The amplified products were separated on an ABI 3730 DNA Analyzer (Applied Biosystems, Foster City, CA, USA) after the purification. Fragment sizes were determined using an internal size standard (GeneScan™-500 LIZ, Applied Biosystems), and the outputs were analyzed using GeneMapper software (http://www.appliedbiosystems.com.cn/). The minimum allelic frequency (MAF) was set as 0.05 for statistical analysis.
Association analysis
In order to define the degree of genetic covariance between pairs of individuals, a kinship (K) analysis was conducted on genotypic data with SPAGeDi software (Hardy and Vekemans 2002). Calculation of pairwise kinship coefficients was according to Loiselle et al. (1995) with 10,000 permutation tests. Negative values between individual pairs were then set to 0, as this indicated that they were less related than random individuals (Yu et al. 2006). To reduce the risk of false or spurious associations, population structure was estimated by STRUCTURE v2.2 software (Pritchard and Rosenberg 1999; Pritchard et al. 2000), based on 42 unlinked loci from both arms of each chromosome with a burn-in period equal to 50,000 iterations and a run of 500,000 replications of Markov Chain Monte Carlo (MCMC) after burn in (Wang et al. 2012).
The mixed linear model (MLM) module with Q + K of the TASSEL 2.1 software package (http://www2.maizegenetics.net/) (Bradbury et al. 2007; Zhang et al. 2010a, b) was used for genome-wide association of GN in each trial and MGN. The relative value of the favored allele for GN (R 2) was calculated according to the equation, R 2 = (SSA − f A × MSE)/SST where SSA indicated the sum of squares between groups of favored alleles and others, f A indicated the degrees of freedom of the group with the favored alleles, MSE indicated the error mean square, and SST indicated the sum of squares (Agrama et al. 2007; Zhang et al. 2010a, b).
Results
Phenotypic assessment
Grain number (GN) in the Chinese wheat MCC was determined over four growing seasons and two environments, including Luoyang, Henan Province, in 2002, 2005, 2006, and Shunyi, Beijing, in 2010. Grain number per spike showed high variation, ranging from 22.4 to 100.4 with an average of 49.8 (Table 1). There were significant correlations among environments, although there were also clear differences (Table 2). This indicated that the grain number had high heritability, but with obvious interactions with environment. Comparing landraces with modern varieties, there were minor differences in GN means in all environments, except GN-05 (Table 1). Mean grain number (MGN) of modern varieties, calculated with BLUP methods based on multiple environments, was not significantly different (P = 0.21) from that of landraces, suggesting that GN might not have undergone strong selection in breeding in comparison with 1,000-kernel weight (Wang et al. 2012).
Genome-wide association analysis of grain number
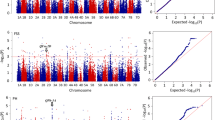
Population structure showed that the MCC entries comprised two sub-populations. One group was mainly the landraces, and the other included modern varieties and introduced lines (Wang et al. 2012). We therefore used the MLM model (Yu et al. 2006) to undertake marker/grain number association analysis. Thirty-two loci were significantly (P < 0.05) associated with MGN. Association analysis between 531 SSR markers and grain number measured in four individual environments was performed to further identify SSR loci significantly associated with phenotypic values in multiple environments. Among the 32 loci associated with MGN, four were detected in three environments, viz. Xgwm311-2A, Xgwm131-3B, Xcfd52-5D and Xcfe273-6A; six loci were detected in two environments; and 17 were detected in one environment. Five loci were not associated in any single environment (Fig. 1; Table S1).

Genome-wide association analysis of grain number per spike and SSR loci in the A (a), B (b) and D (c) genomes of wheat. GNs collected from four trials were used to estimate mean values (MGN). GN-02, GN-05, GN-06 and GN-10 are grain numbers for 2002, 2005, 2006 in Luoyang (Henan province) and 2010 in Shunyi (Beijing), respectively
Among the 27 loci associated with MGN in at least one trial, we found breeder-favored alleles with strong positive effects at 23 loci on 14 different chromosomes. Chromosome 3D had four loci, viz. Xgwm108, Xgwm2, Xcfd64 and Xbarc42. The genetic distance among these four loci is more than 10 cM, indicating they may not relate to a single yield gene. The allelic effect on MGN at each locus was estimated by ANOVA (SPSS16). Significant or extremely significant differences were detected on MGN between varieties conveying favored alleles and the varieties with other alleles. Two loci with the strongest effects were detected on chromosomes 3B (Xgwm131) and 6A (Xcfe273), which individually accounted for more than 10 % of the total variation (R 2 > 10 %) (Table 3).
Distribution of favored alleles at associated loci
Based on the mini core collection entries, the frequencies of favored alleles at each locus in the MCC were estimated for landraces and modern entries. The frequencies of five favored alleles were much higher in modern varieties than in the landraces and only one favored allele, Xgwm249 on 2A, was present as the favored allele in modern varieties (Fig. 2). The frequencies of favored alleles at the 23 loci in modern varieties were not significantly different from those in landraces. The favored alleles at eight loci did not increase in frequency in modern varieties compared with landraces, or even decreased. The frequencies of three favored alleles were higher than 50 % in modern varieties. However, two loci (Xgwm131 and Xcfe273) explaining the highest levels of phenotypic variation were at low frequency in modern varieties (Table 3; Fig. 2).

Pairwise comparisons of favored alleles between landraces and modern varieties at 23 loci in the Chinese wheat MCC
Positive selection of favored alleles at key loci was also clearly implicated by changes in number and frequency. Modern varieties tended to have greater numbers of favored alleles than landraces (Fig. 3a), indicating that in modern breeding there was an accumulation of favored alleles in selected varieties. The best modern variety (GN 61) had 13 favored alleles at 23 marker loci, whereas the best landrace (GN 60) had 10. These results illustrate the reliability of identifying favored alleles. However, no modern cultivar had favored alleles at all 23 loci, indicating further opportunities for improvement of GN by marker-assisted selection. Importantly, the favored alleles explaining the highest amounts of phenotypic variation (Xgwm311 500 , Xgwm131 110 and Xcfe273 306 ) did not have high frequencies in modern varieties; indeed, in some instances the frequencies were lower.

Comparison of frequencies (a) and phenotypic values (b) of varieties containing different numbers of favored alleles at 23 loci between landraces and modern varieties in the Chinese wheat MCC
We also analyzed grain numbers of varieties containing different numbers of favored alleles in landraces and in modern varieties. The phenotypic values of varieties containing more favored alleles were much higher than those possessing less favored alleles in both sets of accessions (Fig. 3b).
Relationship of GN and TKW
In our group about identifying loci influencing 1,000-kernel weight in wheat, phenotyping and association study of TKW were analyzed (Wang et al. 2012). So in this study, we further analyzed the relationship between GN and TKW.
In order to avoid the effect of population structure, all accessions were divided into landraces and modern varieties for evaluating the relationship between grain number (GN) and thousand kernel weight (TKW). While there was no significant correlation within landraces (Fig. 4a), an extremely significant positive correlation occurred between GN and TKW in modern varieties (Fig. 4b). Varieties from the 1960s showed lower GN (<53) and TKW (<40 g), those from the 1970s showed higher GN (>53) and lower TKW (<40 g), those from the 1980s showed higher GN (>53) and TKW (>40 g). We also analyzed the relationship of favored alleles between GN and TKW, and similar results were obtained (Fig. 4c, d). Varieties from the 1960s contained less favored alleles for GN (<7) and TKW (<9), varieties from the 1970s contained more favored alleles for GN (>7), but less for TKW (<9), and those after the 1980s contained more favored alleles for both GN (>7) and TKW (>9).

Relation between GN and TKW in landraces (a, c) and modern varieties (b, d). Modern varieties from different periods are labeled by different colored dots, corresponding to the 1940s (dark green circles), 1950s (medium green circles), 1960s (light green circles), 1970s (light orange circles), 1980s (medium orange circles) and 1990s (dark orange circles)
We selected 11 significantly associated loci explaining high levels of phenotypic variation (5 loci with R 2 > 5 % for GN and 6 loci with R 2 > 10 % for TKW (Wang et al. 2012)) to evaluate the phenotypic effect of favored alleles on GN and TKW (Fig. 5). The favored alleles at six loci (Xcfa223, Xgwm156, Xcfa2257, Xgwm2, Xgwm131 and Xwmc304) showed significant positive effects for both GN and TKW, accounting for 55 % of the analyzed loci; and favored alleles at only two loci (Xwmc17 and Xgwm311) showing an inverse effect for GN and TKW, accounting for 18 % of the loci, but the effect was not significant.

The phenotypic values of Favored alleles at 11 associated loci explaining high levels of phenotypic variation. Asterisk Instances where the phenotypic effects of Favored alleles on TKW and GN were significantly higher than the phenotypic effects for Others
Discussion
Advantages of association mapping of complex traits
Finding genetic variation underlying complex traits in crops not only provides insights into genetic pathways, but also provides targets for marker-assisted selection in breeding (Clark 2010). Linkage mapping in biparental crosses has been the most commonly employed method to identify genetic marker: phenotype associations. To date, many plant genes have been identified (Harjes et al. 2008) and cloned based on the linkage mapping, especially in Arabidopsis thaliana and Oryza sativa. Association mapping (also known as linkage disequilibrium mapping) is an alternative approach that captures multiple historical recombination events among a selected panel or population of unrelated individuals (Myles et al. 2009). Any candidate markers associated with agronomic and environmentally adaptive traits in plants were recently identified by this method (Wilson et al. 2004; Zhang et al. 2005; Breseghello and Sorrells 2006; Cockram et al. 2010; Huang et al. 2010).
An important advantage of association mapping, as mentioned and reviewed many times (e.g. Thornsberry et al. 2001; Zhu et al. 2008; Atwell et al. 2010; Clark 2010; Huang et al. 2010), is that association mapping is highly efficient in detecting multiple QTLs in the same genetic network at the same time, and provides opportunities to understand the pleiotropic effects of some chromosome regions, QTLs or genes. The populations used can be so diverse that most of the variation related to many target traits at different loci can be included and detected in the same mapping population. Results from the present study confirm the high efficiency of association mapping in QTL detection. We detected more than 100 QTLs related to grain number using a population of 262 individuals. Previously reported QTLs related to grain number based on linkage mapping in biparental populations were not more than 10 (Huang et al. 2004; Wang et al. 2009). We compared the associated QTLs with the reported QTLs according to genetic distances of the markers, and found there was high consistency between linkage analysis and association mapping. Associations of candidate markers with traits in this study were also detected in previous linkage studies (Table 3). Clearly, some novel markers and QTLs were also detected in this study, such as Xbarc42 and Xcfe172 on 3D, Xcfd52 on 5D and Xcfe273 on 6A.
Another advantage of association mapping relative to linkage mapping is that many variations at one locus may be present, enabling identification of the best of several alleles rather than the better of two. Moreover, association approaches are amenable to high-throughput genomics that can be used to characterize all of the genes in a genome (Buckler and Thornsberry 2002). In this way varieties or individuals with the most useful alleles can be identified. Moreover, association analysis allows detection of QTLs for a wide array of different traits in the same population and so permits the discovery of pleiotropic regions. In this study, we found that Xwmc304 was associated with grain number. Wang et al. (2012) detected this marker was also associated with TKW. Importantly, Xwmc304 126 was not only a favored allele for grain number, but was also favored for TKW. Both grain number and TKW are important components of grain yield. For a long time it was believed that these traits were negatively correlated, i.e. small-seeded varieties normally produce more seeds. However, alleles such as Xwmc304 126 can be favored since they improve both traits.
Thus, whole genome association can provide a profile of chromosome regions or loci contributing to yield, their relative contributions to the phenotypic variation of target traits, and the relative phenotypic effects of different alleles. It identifies the candidate regions or markers that should be considered in breeding programs and in dissecting genetic variation related to target traits.
Frequencies and genetically additive effects of favored alleles indicate potential for yield increases by selection of loci associated with GN
Improvement in grain productivity can be achieved by increasing both grain weight and grain number. TKW increased from a mean 31.5 g in the 1940s to 44.64 g in the 2000s, with a 2.19 g increase in each decade (Zhang et al., data not shown). A large difference in TKW was found between landraces and modern varieties in the Chinese wheat MCC (Wang et al. 2012). Meanwhile the increase of grain number was relatively slow and only minor differences existed between the landraces and modern varieties in the MCC. Our results indicate that there are five favored alleles, whose frequencies were much higher in modern varieties than in the landraces (Fig. 2). The frequencies of most of the favored alleles in modern varieties were not significantly different from those in landraces, and favored allele at eight loci did not increase in frequency in modern varieties relative to landraces; some even decreased. Moreover, the favored alleles contributing most to the phenotypic variation occurred at low frequencies in modern varieties. Donmez et al. (2001) suggested that the number of kernels per spike was more amenable to genetic improvement than kernel weight. There seems to be potential for improving yield by selection for grain number.
Molecular breeding by design aims to select the most valuable genotypes or alleles and to combine them in developing a desired variety. Wang et al. (2012) detected linear correlations between TKW and favored alleles. We also found linear correlations between grain number and favored alleles in this study. The evidence indicates that there are additive effects of QTLs or genes; and effects that should not be ignored in practice and breeders could improve grain number by combining more favored alleles. Frisch et al. (2010) found that predictions based on 50 chosen genes were as accurate as predictions based on 5,000 random genes. Therefore, it is feasible to increase grain number in wheat by marker-assisted selection.
Genetic basis of the relationship between GN and TKW in Chinese wheat
Grain number and grain weight are important aspects of the domestication syndrome, which partly explain the process of domestication of crop plants. Traditionally, there was a negative correlation between these traits, evident from repeated observations that small-seeded plants normally produce more seeds. In wheat, this negative correlation also exists (Jia 1984; Gaju et al. 2009). However, some recent studies showed there was no correlation between the two traits (Wang et al. 2001; Brancourt-Hulme et al. 2003), and sometimes even showed positive correlation (Aycicek and Yildirim 2006). Clearly, the relationship between the two traits depends on the actual experimental materials. In the present study, we found that there was no correlation between the two traits in Chinese landraces, but there was a significantly positive correlation in modern varieties. We concluded that selection played a major role in this difference. For landraces in China, natural selection made them produce more spikelets, larger grain numbers and lower TKW. However, during modern wheat breeding for high yield, artificial selection played a major role. Many studies found that grain weight per spike was a leading contributor to improved yield, and TKW was the main component of grain weight per spike. Therefore, the main focus was on TKW which underwent the greatest response among all yield components (Hu and Zhao 1995). The trend in grain number per spike has changed in different production areas and different periods. Study of varieties from the 1980s showed that changes in grain number per spike among varieties were quite small (Ting 1979; Qian et al. 1989), whereas studies after 1990 (Tian 1991; Hu and Zhao 1995) showed an increasing trend in grain number, and even more so after 2000 (Zhang et al., data not shown).
It is normally assumed that genotypic correlations between complicated traits such as yield and its component traits arise from pleiotropy. Pleiotropic effects of QTL and epistatic loci appeared to be responsible for the negative correlation between KW and GN. In this study, we undertook a genome-wide association analysis of GN using the same 531 SSR markers as Wang et al. (2012). Among the associated markers, only 30 loci were simultaneously associated with GN and TKW, accounting for 17 % of all associated loci, and most of the associated loci were associated with only one of traits. The favored alleles at these loci should increase the phenotypic value of one trait, without negatively affecting the phenotypic value of the other. In other words, there was a significant proportion of identified QTL affecting GN, but not affecting TKW. Thus, selection of these QTLs was likely a major factor in changing the relationship between GN and TKW over time. More detailed information about the loci involved in different yield components and their genetic relationships will be helpful in improving yield potential by deliberately choosing these loci for marker-assisted selection.
Abbreviations
- GN:
-
Grain number
- MLM:
-
Mixed linear model
- BLUP:
-
Best linear unbiased predictor
- MGN:
-
Mean grain number
- SNS:
-
Spikelet number per spike
- SL:
-
Spike length
- GNS:
-
Grain number per spike
- TKW:
-
Thousand kernel weight
- MCC:
-
Mini core collection
References
Agrama HA, Eizenga GC, Yan W (2007) Association mapping of yield and its components in rice cultivars. Mol Breeding 19:341–356
Araki E, Miura H, Sawada S (1999) Identification of genetic loci affecting amylose content and agronomic traits on chromosome 4A of wheat. Theor Appl Genet 98:977–984
Atwell S, Huang YS, Vilhjálmsson BJ, Willems G, Horton M, Li Y, Meng D, Platt A, Tarone AM, Hu T, Jiang R, Muliyati NW, Zhang X, Amer MA, Baxter I, Benjamin B, Chory J, Dean C, Debieu M, Meaux J, Ecker JR, Faure N, Kniskern JM, Jones Jonathan DG, Michael T, Nemri A, Roux F, Salt DE, Tang C, Todesco M, Brian TM, Weigel D, Marjoram P, Borevitz JO, Bergelson J, Nordborg M (2010) Genome-wide association study of 107 phenotypes in Arabidopsis thaliana inbred lines. Nature 465:627–631
Aycicek M, Yildirim T (2006) Path coefficient analysis of yield and yield components in bread wheat (Triticum aestivum L.) genotypes. Pak J Bot 38:417–424
Bernardo R (1996a) Test cross additive and dominance effects in best linear unbiased prediction of maize single-cross performance. Theor Appl Genet 93:1098–1102
Bernardo R (1996b) Marker-based estimate of identity by descent and alikeness in state among maize inbreds. Theor Appl Genet 93:262–267
Bernardo R (1996c) Best linear unbiased prediction of maize single-cross performance. Crop Sci 36:50–56
Börner A, Schumann E, Fürste A, Cöster H, Leithold B, Röder MS, Weber WE (2002) Mapping of quantitative trait loci determining agronomic important characters in hexaploid wheat. Theor Appl Genet 105:921–936
Bradbury PJ, Zhang ZW, Kroon DE, Casstevens TM, Ramdoss Y, Buckler ES (2007) TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23:2633–2635
Brancourt-Hulme M, Doussinault G, Lecomte C, Bérard P, Le Buanec B, Trottet M (2003) Genetic improvement of agronomic traits of winter wheat cultivars released in France from 1946 to 1992. Crop Sci 43:37–45
Breseghello F, Sorrells ME (2006) Association mapping of kernel size and milling quality in wheat (Triticum aestivum L.) cultivars. Genetics 172:1165–1177
Buckler ES, Thornsberry JM (2002) Plant molecular diversity and applications to genomics. Curr Opin Plant Biol 5:107–111
Clark RM (2010) Genome-wide association studies coming of age in rice. Nat Genet 42:926–927
Cockram J, White J, Zuluaga DL, Smith D, Comadran J, Macaulay M, Luo ZW, Kearsey MJ, Werner P, Harrap D, Tapsell C, Liu H, Hedley PE, Stein N, Schulte D, Steuernagel B, Marshall DF, Thomas WTB, Ramsay L, Mackay L, Balding DJ (2010) Genome-wide association mapping to candidate polymorphism resolution in the unsequenced barley genome. Proc Natl Accad Sci USA 107:21611–21616
Cuthbert JL, Somers DJ, Brǔlé-Babel AL, Brown PD, Crow GH (2008) Molecular mapping of quantitative trait loci for yield and yield components in spring wheat (Triticum aestivum L.). Theor Appl Genet 117:595–608
Deng SM, Wu XR, Wu YY, Zhou RH, Wang HG, Jia JZ, Liu SB (2011) Characterization and precise mapping of a QTL increasing spike number with pleiotropic effects in wheat. Theor Appl Genet 122:281–289
Donmez E, Sears RG, Shroyer JP, Paulsen GM (2001) Genetic gain in yield attributes of winter wheat in the great plains. Crop Sci 41:1412–1419
Frisch M, Thiemann A, Fu J, Schrag TA, Scholten S, Melchinger AE (2010) Transcriptome-based distance measures for grouping of germplasm and prediction of hybrid performance in maize. Theor Appl Genet 120:709–720
Gaju O, Reynolds MP, Sparkes DL, Foulkes MJ (2009) Relationships between large-spike phenotype, grain number, and yield potential in spring wheat. Crop Sci 49:961–973
Gupta PK, Mir RR, Mohan A, Kumar J (2008) Wheat genomics: present status and future prospects. Int J Plant Genomics 2008:896451
Hai L, Guo HJ, Wagner C, Xiao SH, Friedt W (2008) Genomic regions for yield and yield parameters in Chinese winter wheat (Triticum aestivum L.) genotypes tested under varying environments correspond to QTL in widely different wheat materials. Plant Sci 175:226–232
Hao CY, Dong YC, Wang LF, You GX, Zhang HN, Ge HM, Jia JZ, Zhang XY (2008) Genetic diversity and construction of core collection in Chinese wheat genetic resources. Chin Sci Bull 53:1518–1526
Hao CY, Wang LF, Ge HM, Dong YC, Zhang XY (2011) Genetic diversity and linkage disequilibrium in Chinese bread wheat (Triticum aestivum L.) revealed by SSR markers. PLoS One 6(2):e17279
Hardy OJ, Vekemans X (2002) SPAGeDi: a versatile computer program to analyze spatial genetic structure at the individual or population levels. Mol Ecol Notes 2:618–620
Harjes CE, Rocheford TR, Bai L, Brutnell TP, Bermudez Kandianis C, Sowinski SG, Stapleton AE, Vallabhaneni R, Williams M, Wurtzel ET, Yan JB, Buckler ES (2008) Natural genetic variation in lycopene epsilon cyclase tapped for maize biofortification. Science 319:330–333
Hu YJ, Zhao TF (1995) Studies on the effect of grain weight in breeding of high-yield wheat. Acta Agron Sin 21:671–678
Huang XQ, Kempf H, Ganal MW, Röder MS (2004) Advanced backcross QTL analysis in progenies derived from a cross between a German elite winter wheat variety and a synthetic wheat (Triticum aestivum L.). Theor Appl Genet 109:933–943
Huang XH, Wei XH, Sang T, Zhao Q, Feng Q, Zhao Y, Li CY, Zhu CR, Lu TT, Zhang ZW, Li M, Fan DL, Guo YL, Wang AH, Wang L, Deng LW, Li WJ, Lu YQ, Weng QJ, Liu KY, Huang T, Zhou TY, Jing YF, Li W, Lin Z, Buckler ES, Qian Q, Zhang QF, Li JY, Han B (2010) Genome-wide association studies of 14 agronomic traits in rice landraces. Nat Genet 42:961–965
Jia JZ (1984) The statistical analysis for correlation factors in kernel weight and plant characters in wheat (T. aestivum L.). Acta Agron Sin 10:201–205
Kato K, Miura H, Sawada S (2000) Mapping QTLs controlling grain yield and its components on chromosome 5A of wheat. Theor Appl Genet 101:1114–1121
Koebner RMD, Snape JW (1999) Actual and potential contributions of biotechnology to wheat breeding. In: Satorre EH, Slafer GA (eds) Wheat: ecology and physiology of yield determination, Food Products Press, p 441–460
Kumar N, Kulwal PL, Balyan HS, Gupta PK (2007) QTL mapping for yield and yield contributing traits in two mapping populations of bread wheat. Mol Breeding 19:163–177
Li WL, Nelson JC, Chu CY, Shi LH, Huang SH, Liu DJ (2002) Chromosomal locations and genetic relationship of tiller and spike characters in wheat. Euphytica 125:357–366
Li SS, Jia JZ, Wei XY, Zhang XC, Li LZ, Chen HM, Fan YD, Sun HY, Zhao XH, Lei TD, Xu YF, Jiang FS, Wang HG, Li LH (2007) A intervarietal genetic map and QTL analysis for yield traits in wheat. Mol Breeding 20:67–178
Loiselle BA, Sork VL, Nason J, Graham C (1995) Spatial genetic structure of a tropical understory shrub, Psychotria officinalis (Rubiaceae). Am J Bot 82:1420–1425
Marza F, Bai GH, Carver BF, Zhou WC (2006) Quantitative trait loci for yield and related traits in the wheat population Ning7840 × Clark. Theor Appl Genet 112:688–698
Myles S, Peiffer J, Brown PJ (2009) Association mapping: critical considerations shift from genotyping to experimental design. Plant Cell 21:2194–2202
Narasimhamoorthy B, Gill BS, Fritz AK, Nelson JC, Brown-Guedira GL (2006) Advanced backcross QTL analysis of a hard winter wheat × synthetic wheat population. Theor Appl Genet 112:787–796
Neumann K, Kobiljski B, Denčić S, Varshney RK, Börner A (2011) Genome-wide association mapping: a case study in bread wheat (Triticum aestivum L.). Mol Breeding 27:37–58
Peng JH, Ronin Y, Fahima T, Röder MS, Li YC, Nevo E, Korol A (2003) Domestication quantitative trait loci in Triticum dicoccoides, the progenitor of wheat. Proc Natl Acad Sci USA 100:2489–2494
Pritchard JK, Rosenberg NA (1999) Use of unlinked genetic markers to detect population stratification in association chain will tend to get stuck moving among very similar studies. Am J Hum Genet 65:220–228
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Qian MM, Sun HW, Song CH, Yang XM (1989) Study on evolution of agronomic characters in northern winter wheat region. Chin Seeds 1:3–5
Röder MS, Korzun V, Wendehake K, Plaschke J, Tixier MH, Leroy P, Ganal MW (1998) A microsatellite map of wheat. Genetics 149:2007–2023
Sharp PJ, Chao S, Desai S, Gale MD (1989) The isolation, characterization and application in Triticeae of a set of wheat RFLP probes identifying each homoeologous chromosome arm. Theor Appl Genet 78:342–348
Somers DJ, Isaac P, Edwards K (2004) A high-density microsatellite consensus map for bread wheat (Triticum aestivum L.). Theor Appl Genet 109:1105–1114
Su ZQ, Hao CY, Wang LF, Dong YC, Zhang XY (2011) Identification and development of a functional marker of TaGW2 associated with grain weight in bread wheat (Triticum aestivum L.). Theor Appl Genet 122:211–223
Thornsberry JM, Goodman MM, Doebley J, Kresovich S, Nielsen D, Buckler ES (2001) Dwarf8 polymorphisms associate with variation in flowering time. Nat Genet 28:286–289
Tian XM (1991) Studies on evolution and its tendency of agronomic character of winter wheat during cultivar alternation in Xinjiang. Acta Agron Sin 17:297–303
Ting SK (1979) Investigation on ways of selecting wheat varieties with a yield over 1,500 jin per mu. Sci Agri Sin 3:8–16
Wang ZL, Cao WX, Dai TB (2001) Genotypic differences in formation of kernel number per spike and analysis of improvement approaches in wheat. Acta Agron Sin 27:236–242
Wang RX, Hai L, Zhang XY, You GX, Yan CS, Xiao SH (2009) QTL mapping for grain filling rate and yield-related traits in RILs of the Chinese winter wheat population Heshangmai × Yu8679. Theor Appl Genet 118:313–325
Wang LF, Ge HM, Hao CY, Dong YS, Zhang XY (2012) Identifying loci influencing 1,000-kernel weight in wheat by microsatellite screening for evidence of selection during breeding. PLoS One 7(2):e29432. doi:10.1371/journal.pone.0029432
Wilson LM, Whitt SR, Ibanez AM, Rocheford TR, Goodman MM, Buckler ES (2004) Dissection of maize kernel composition and starch production by candidate gene association. Plant Cell 16:2719–2733
Yao J, Wang L, Liu L, Zhao C, Zheng Y (2009) Association mapping of agronomic traits on chromosome 2A of wheat. Genetica 137:67–75
Yu JM, Pressoir G, Briggs WH, Bi IV, Yamasaki M, Doebley JF, Mcmullen MD, Gaut B, Nielsen DM, Holland JB, Kresovich S, Buckler ES (2006) A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat Genet 38:203–208
Zhang N, Xu Y, Akash M, McCouch S, Oard JH (2005) Identification of candidate markers associated with agronomic traits in rice using discriminant analysis. Theor Appl Genet 110:721–729
Zhang XY, Tong YP, You GX, Hao CY, Ge HM, Wang LF, Li B, Dong YS, Li ZS (2007) Hitchhiking effect mapping: a new approach for discovering agronomically important genes. Agri Sci China 6:255–264
Zhang LY, Liu DC, Guo XL, Yang WL, Sun JZ, Wang DW, Zhang AM (2010a) Genomic distribution of quantitative trait loci for yield and yield-related traits in common wheat. J Int Plant Biol 52:996–1007
Zhang ZW, Ersoz E, Lai CQ, Fodhunter RJ, Tiwari HK, Gore MA, Bradbury PJ, Yu JM, Arnett DK, Ordovas JM, Buckler ES (2010b) Mixed linear model approach adapted for genome-wide association studies. Nat Genet 42:355–360
Zhu C, Gore M, Buckler E, Yu J (2008) Status and prospects of association mapping in plants. Plant Genome 1:5–20
Acknowledgments
The authors are grateful to HN Zhang, YH Tian, J Lin and YJ Wang for excellent genotyping and phenotyping of the mini core collection. We also gratefully acknowledge help from Prof. Robert A McIntosh, University of Sydney, with English editing. This work was supported by the Chinese Ministry of Science and Technology (2010CB125900) and Chinese Agricultural Research System, Ministry of Agriculture (CARS-3-1-2) and the National High Technology Research and Development Program of China (2006AA10Z1F2).
Author information
Authors and Affiliations
Corresponding author
Additional information
D. Zhang and C. Hao contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Zhang, D., Hao, C., Wang, L. et al. Identifying loci influencing grain number by microsatellite screening in bread wheat (Triticum aestivum L.). Planta 236, 1507–1517 (2012). https://doi.org/10.1007/s00425-012-1708-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00425-012-1708-9