
Abstract
Grain protein content (GPC) and gluten quality are the most important factors determining the end-use quality of wheat for pasta-making. Both GPC and gluten quality are considered to be polygenic traits influenced by environmental factors and other agricultural practices. Two related F8:9 recombinant inbred line (RIL) populations were generated to localise genetic factors controlling seven quality traits: GPC, wet gluten content (WGC), flour whiteness (FW), kernel hardness (KH), water absorption (Abs), dough development time (DDT) and dough stability time (DST). These lines were derived by crossing Weimai 8 and Jimai 20 (WJ) and by crossing Weimai 8 and Yannong 19 (WY). In total, WJ comprised 485 lines, while WY comprised 229 lines. Data on these seven quality traits were collected from each line in five different environments. Up to 85 putative QTLs for the seven traits were detected in WJ and 65 putative QTLs were detected in WY. Of these QTLs, 31 QTLs (36.47%) were detected in at least two trials in WJ, while 24 QTLs (36.92%) were detected in at least two trials in WY. Three QTLs from WJ and 25 from WY accounted for more than 10% of the phenotypic variance. The total 150 QTLs were spread throughout all 21 wheat chromosomes. Of these, at least thirteen pairwise were common to both populations, accounting for 20.00 and 15.29% of the total QTLs in WJ and WY, respectively. A major QTL for GPC, accounting for 53.04% of the phenotypic variation, was detected on chromosome 5A. A major QTL for WGC also shared this interval, explained more than 36% of the phenotypic variation, and was significant in two environments. Though co-located QTLs were common, every trait had its unique control mechanism, even for two closely related traits. Due to the different sizes of the two line populations, we also assessed the effects of population size on the efficiency and precision of QTL detection. In sum, this study will enhance our understanding of the genetic basis of these seven pivotal quality traits and facilitate the breeding of improved wheat varieties.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Breeding new varieties of wheat with improved end-use quality is a key aim of wheat breeding-programs. The end-use quality of wheat is dependent on a large complex of genes that are greatly influenced by environment conditions. Grain protein content (GPC), wet gluten content (WGC), kernel hardness (KH), flour whiteness (FW), water absorption (Abs), dough development time (DDT) and dough stability time (DST) are all important grain quality traits in both durum and bread wheat. Kernel hardness and FW affect milling quality and are closely related to processing quality. It is well known that WGC, Abs, DDT and DST are also important quality traits strongly correlated with GPC. Grain protein content is one of the most important factors affecting pasta-making (Matsuo et al. 1982; Autran et al. 1996) and bread-making quality, and is also important to human nutrition. Many recent reports (Joppa and Cantrell 1990; Sourdille et al. 1999; Zanetti et al. 1999; Perretant et al. 2000; Blanco et al. 1996, 2002, 2006) concluded that genetic factors impacting GPC in both cultivars and wild wheat were distributed over all 21 wheat chromosomes. Chee et al. (2001) detected a high grain protein QTL, QGpc.ndsu.6Bb, from Triticum turgidum L. var. dicoccoides, and deduced that the high GPC locus was insensitive to environmental conditions. The hardness (Ha) locus on chromosome 5D is the main determinant of grain texture in hexaploid wheat, and this locus also controlled the production of puroindoline proteins (Morris 2002). Recently, Chantret et al. (2004, 2005) sequenced the Ha locus. The recent emphasis on end-use quality has increased the economic value of these traits (Dohlman and Hoffman 2000).
Population size has a great effect on the estimation of QTL number and genetic effects (Sourdille et al. 1996; Beavis 1998; Mackay 2001; Schön et al. 2004; Vales et al. 2005; Zou et al. 2005; Buckler et al. 2009). The precision and efficiency of QTL detection is enhanced by combining more than two related populations (Kumar et al. 2007; Ma et al. 2007; Buckler et al. 2009; Uga et al. 2010). In the present study, we performed QTL detection for seven traits based on the recombination of two related line populations, one of which was a large population with up to 485 lines (WJ), while the other had a smaller, but still substantial, 229 lines (WY). The objectives of this study were to (i) accurately identify the genetic factors affecting wheat quality, (ii) specify the genetic relationships among the seven quality traits at the QTL level, and (iii) assess the effect of combining two related populations of different sizes on the efficiency and precision of QTL detection.
Materials and methods
Experimental populations and their evaluation
Two F8:9 recombinant inbred line (RIL) populations derived from crosses between three common Chinese wheat varieties, i.e., between Weimai 8 and Jimai 20 (WJ) and between Weimai 8 and Yannong 19 (WY), comprising 485 and 229 lines, respectively, were used in the present study. The three parental lines differed markedly in many morphological and quality traits. Weimai 8 is a large-spike wheat of the ideotype model and was released by Weifang Municipal Academy of Agricultural Sciences, Shandong, China in 2003; Jimai 20 and Yannong 19, two superior quality wheat varieties, are multi-spike types, and they were released by the Crop Research Institute, Shandong Academy of Agricultural Sciences, China in 2003, and by Yantai Municipal Academy of Agricultural Sciences, Shandong, China in 2001, respectively. In addition, the common parent Weimai 8 is a 1BL/1RS translocation line whereas the other two parents have the common 1B chromosome. The parents together with the RILs were evaluated in five environments (E1–E5) in Shandong province, China: Taian in 2007–2008 (E1), Taian in 2008–2009 (E2), Taian in 2009–2010 (E3), Zaozhuang in 2009–2010 (E4), and Jining in 2009–2010 (E5). A two-row plot with rows 2 m long and 30.0 cm apart was used, and 50 seeds were planted in each row. Normal agricultural practices were applied for disease and weed control. The lines were harvested individually at maturity, and naturally air dried. The seven quality traits, grain protein content (GPC), wet gluten content (WGC), kernel hardness (KH), flour whiteness (FW), water absorption (Abs), dough development time (DDT) and dough stability time (DST), were measured by near-infrared reflectance spectroscopy (NIRS) on a Perten DA-7200 instrument (Perten Instruments, Huddinge, Sweden) and expressed on a 14% moisture basis. The measurements were calibrated using calibration samples. Li et al. (2009) confirmed the correlation between NIRS and standard methods for WGC and Abs. There were no significant differences in results between NIRS and traditional methods. Indeed, NIRS determinations were highly correlated with traditional methods. Again, previous studies have demonstrated the importance of NIRS on wheat quality traits, especially grain protein content (flour protein content) and kernel hardness (Sourdille et al. 1996; Perretant et al. 2000; Blanco et al. 2002; Nelson et al. 2006; Kuchel et al. 2006; Suprayogi et al. 2009; Sun et al. 2010). Furthermore, Mann et al. (2009) determined moisture content of the grain and flour samples by NIRS. So that NIRS is a powerful method for measuring the quality traits of wheat.
Analysis of molecular and biochemical markers
Leaves were taken from all RILs and the parents for DNA extraction following the procedure described by Stein et al. (2001), but with the minor modification of using 70% ethanol as the washing solution. Various molecular markers, including G-SSR, EST-SSR, ISSR, STS, SRAP and RAPD were used to genotype the three parents and the RILs. Relevant information regarding G-SSR markers, including BARC, CFA, CFD, CFT, GWM, GDM, GPW, WMC and PSP codes, as well as PCR-based STS markers of the MAG codes, were taken from the GrainGenes Web site (http://wheat.pw.usda.gov). Relevant information about EST-SSR markers prefixed CFE, KSUM and CNL were publicly available (http://wheat.pw.usda.gov/ITMI/EST-SSR/). The EST-SSR markers of SWES and WW codes were developed and kindly provided by Professor Sishen Li, College of Agronomy, Shandong Agricultural University, China. The EST-SSR markers with the prefixes CWEM, EDM and CWM were published in reference articles by Peng and Lapitan (2005), Mullan et al. (2005) and Gao et al. (2004), respectively. The ISSR markers were developed by the University of British Columbia Biotechnology Laboratory (UBCBL) (Nagaoka and Ogihara 1997). Relevant information about chromosome 1RS-specific markers of rye were detailed by Zhao et al. (2009), and functional markers were provided by Liu et al. (2008) and Liang et al. (2010). The different high molecular weight glutenin subunits (HMW-GS) at Glu-a1, Glu-b1 and Glu-d1 between the parents were detected and used as biochemical markers.
Each PCR reaction for G-SSR, EST-SSR and PCR-based STS markers was conducted in a total volume of 25 μL in a TakaRa PCR thermal cycler or in a Bio-Rad 9600 thermal cycler. PCR was compounded according to the procedure described by Röder et al. (1998). Amplifications were performed using a touchdown PCR protocol as detailed by Hao et al. (2008). The PCR reaction volume and PCR protocol for SRAP and ISSR markers followed the proportions and the procedure detailed by Li et al. (2007), and RAPD markers were amplified using the method of Suenaga et al. (2005). The types of high molecular weight glutenin subunits (HMW-GS) were detected by using sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) (Singh and Shepherd 1991). Markers of BARC, CFA, CFD, GWM, GDM and WMC codes were screened against the nullisomic-tetrasomic stocks of Chinese Spring (CSNT) to assign them to chromosomes where possible.
Construction of the genetic linkage map
Linkage groups were constructed by MAPMAKER 3.0 (Lander et al. 1987). First, the ‘‘ANCHOR’’ command was used to locate marker loci to chromosomes based on the CSNT identification and the public genetic maps in GrainGenes 2.0 (http://wheat.pw.usda.gov/GG2/index.shtml). Then, the assignment of the remaining loci to chromosomes was made using the ‘‘ASSIGN’’ command at a LOD score of 3.0. Based on the linkage group defined above, JoinMap version 3.0 (Biometris, Wageningen, The Netherlands, http://www.joinmap.nl), was used to construct the linkage map, and centimorgan units were calculated using the Kosambi mapping function (Kosambi 1944).
Data analysis and QTL mapping
The trait data were analysed using SPSS13.0 (SPSS, Chicago, USA). The observed phenotypic values obtained from each environment (E1–E5), and the pooled data collected from the average of the five environments (E6) were used for QTL mapping analyses. Inclusive composite interval mapping by IciMapping 3.0 was used based on stepwise regression of simultaneous consideration of all marker information (http://www.isbreeding.net/). The walking speed for all QTLs was 1.0 cM. The threshold LOD scores were calculated using 1,000 permutations, and a genome-wide error rate of 0.10 (suggestive) and 0.05 (significant). However, we ignored the QTL with a LOD value of <3.0 to make the QTL reported herein authentic and reliable.
Results
Analysis of phenotypic data and correlations among the seven traits
All seven traits varied widely among the individual lines, showed transgressive segregation, and were normally distributed (Table 1). Phenotypic correlations between pairs of the seven quality traits are listed in Fig. 1a, b. The distribution of correlation coefficients in the two populations was similar. In both WJ and WY, higher positive correlation coefficients were observed between GPC and WGC, WGC and Abs, KH and Abs, KH and DDT, KH and DST, and as well between DDT and DST. Higher negative correlation coefficients were observed between GPC and KH, GPC and DST, WGC and DDT, WGC and DST, FW and KH, FW and Abs, and between GPC and DDT. In both populations, the highest positive correlation coefficients were found between DDT and DST, while the highest negative correlation coefficients were found between FW and Abs. The trait pairs Abs/DDT and GPC/Abs exhibited significant positive correlations coefficients in both WJ and WY line populations. However, the positive correlation coefficients differed significantly between the two populations as well between the different environments. Correlations between the trait pairs Abs/DST, FW/DST and FW/DDT were greatly influenced by the environment, and the coefficients were greatly different between the two populations.

Correlation coefficient between pairs of the seven quality traits in the WJ (a) and WY (b) populations and the number of pleiotropic QTL (c)
Construction of genetic linkage maps
The genetic map constructed based on the WJ population included 344 loci distributed over all wheat chromosomes and spanned 2855.5 cM, with average density of one marker per 8.30 cM. There were six linkage gaps with distances >50 cM. Marker distribution ranged from 45 on chromosome 4A to 3 on chromosomes 4D and 7D. The WY population was used to establish a genetic map consisting of 358 loci distributed in 27 linkage groups because of six linkage gaps. The map covered 3010.70 cM of the whole genome with an average distance of 8.41 cM between the adjacent loci. The number of markers per chromosome ranged from 40 on chromosome 1B to 3 on chromosomes 3D. The two linkage maps contained 69 common loci. The chromosomal locations and the orders of the markers in the two maps were in general agreement with published reports in GrainGenes 2.0 (http://wheat.pw.usda.gov/GG2/index.shtml). Positions of the loci common were approximately in accordance between the two maps. Functional markers and biochemical markers were accurately mapped to their corresponding chromosomes. The recommended interval length for genome-wide QTL scanning is less than 10 cM (Doerge 2002). Thus, both maps were suitable for genome-wide QTL scanning. The two linkage maps along with seven spike-related traits will be reported in another paper.
QTL mapping in the WJ and WY population
A total of 85 additive QTLs were detected for the seven traits in the WJ line population, and they were distributed over all 21 wheat chromosomes except 5D. In total, 65 additive QTLs were identified for the seven traits in WY. Again, these QTLs were assigned to all 21 wheat chromosomes except 3D and 4D.
Grain protein content
Overall, in the six trials, QTL mapping analysis produced a total of 9 and 10 putative additive QTLs for GPC in WJ and WY (Tables 2, 3; Figs. 2, 3), respectively. For the WJ lines, one each was located on chromosomes 2B, 3A, 4D, 5B, 7A and 7B, and three each on 4A. These QTLs individually explained 3.06–9.83% of the phenotypic variation. Among these, QGpc.WJ-7A.3 was verified in three environments and explained 4.94–9.83% of the phenotypic variance, while QGpc.WJ-2B.2, QGpc.WJ-4D.2 and QGpc.WJ-5B.2 were identified in two environments. The remaining five QTLs could be detected only in one environment. Of the 10 QTLs found in the WY lines, one each was detected on 1A, 1B, 2A, 2D, 3A, 4B, 5A, 5D, 6B and 7D. Each WY line QTL individually explained 6.32–53.04% of the phenotypic variation. The QTL, QGpcWY-2D.3, was identified in three environments and demonstrated an additive enhancing effect from Yannong 19. Both QGpc.WY-1B.2 and QGpc.WY-3A.2 were verified in two trials and the remaining QTLs showed environment-specific traits. Of all the QTLs isolated, QGpc.WY-5A.1 accounted for the highest phenotypic variation (53.04%).

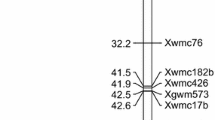
Partial genetic linkage map showing QTLs detected in more than two trials for the seven traits and QTLs explaining the phenotypic variation of more than 10% in only one trial in WJ. The vertical white line indicates the maker interval of the corresponding putative additive QTL. The chromosome length per cM is defined as 0.2 cm. For abbreviations and QTL symbols, see Table 2

Partial genetic linkage map showing QTLs detected in more than two trials for the seven traits and QTLs explaining the phenotypic variation of more than 10% in only one trial in WY. The vertical black line indicates the maker interval of the corresponding putative additive QTL. The chromosome length per cM is defined as 0.2 cm. For abbreviations and QTL symbols, see Table 2
Wet gluten content
In all of six trials, seven additive QTLs for WGC were detected in WJ, while eight additive QTLs were detected in WY lines (Tables 2, 3; Figs. 2, 3). In WJ lines, these QTLs were located on chromosomes 1A, 1D, 2B (2 QTLs), 4A (2 QTLs) and 7A. In WY lines, the eight QTLs were distributed to 1A (2 QTLs), 1B, 2A, 2D, 5A, 7A and 7D. The QTL, QWgc.WJ-1D.3, was identified in three environments and accounted for 4.64–5.39% of the phenotypic variation. The QTL, QWgc.WJ-7B.1, explained the highest level of the phenotypic variation for WGC in WJ at 7.15%. Four major QTLs influencing WGC were detected in WY. Of these, QWgc.WY-2D.5 was identified reproducibly in five trials and QWgc.WY-1B.3 was detected in three, and accounted for 9.85–25.39 and 7.45–12.63% of the phenotypic variation, respectively. The QTL, QWgc.WY-5A.2, was detected in E1 and E2, and it accounted for 28.37–36.76% of the phenotypic variance. Finally, QWgc.WY-2A.1 exerted significant effects on phenotype in only one environment where it accounted for 14.02% of the phenotypic variation.
Flour whiteness
Qualitative trait loci mapping analysis produced a total of 17 putative additive QTLs for flour whiteness in WJ (Table 2; Fig. 2) and 13 in WY (Table 3; Fig. 3). The 17 QTLs in WJ covered 10 wheat chromosomes: 1A, 1B (2 QTLs), 2B (2 QTLs), 2D, 3B, 4A (3 QTLs), 5B (2 QTLs), 6A, 6D (3 QTLs) and 7D. The 13 QTLs in WY distributed to chromosomes 1A, 1B (2 QTLs), 1D, 2A, 2B, 2D, 3B, 5B, 6A, 7B (2 QTLs) and 7D. The QTL, QFw.WJ-3B.4, was detected in four environments (E1, E2, E5 and E6) and accounted for 3.28–7.08% of the phenotypic variance. The major QTL, QFw.WY-5B.3, was identified in E3, E4 and E6, and explained 10.07–13.55% of the phenotypic variance. The QTLs, QFw.WJ-2D.3 and QFw.WJ-6A.3, were both detected in two trials and explained 3.46–9.10 and 4.03–8.46% of the phenotypic variance, respectively. Seven QTLs were found in two trials that explained 3.10–3.18 (QFw.WJ-2B.2), 3.41–4.30 (QFw.WJ-4A.2), 4.46–5.19 (QFw.WJ-5B.2a), 8.33–10.68 (QFw.WJ-5B.2b), 4.22–5.23 (QFw.WJ-6D.2), 5.71–9.34 (QFw.WY-1A.2) and 9.16–11.30% (QFw.WY-7D.2) of the phenotypic variance. The remaining QTLs were identified in only one trial.
Kernel hardness
For KH, 16 additive QTLs were detected in WJ lines and 14 additive QTLs were detected WY lines, each accounting for 2.25–8.33% of the phenotypic variance in WJ lines and 4.39–12.21% of the phenotypic variance in WY lines (Tables 2, 3; Figs. 2, 3). Together, these QTLs covered all 21 wheat chromosomes except 1A, 3D, 5A, 5D, 6A and 6B. All 30 KH QTLs identified demonstrated significant additive effects. In WJ, the QTL, QKh.WJ-7A.5, was identified in five trials (all except E1), and explained 3.98–8.33% of the phenotypic variance. The QTL, QKh.WJ-3B.3, was detected in three trials. In WY, QKh.WY-2B.3 explained up to 10.41–12.21% of the phenotypic variation for KH and was detected in three trials. Additive effects were as high as 10.63, 9.70 and 9.85% in the three trials, respectively. Another major QTL, QKh.WY-4A.3, was detected in three environments, E3, E5 and E6, and explained 7.36–10.69% of the phenotypic variance. The additive effects were 7.00 (E3), 5.74 (E5) and 4.37% (E6).
Water absorption
Fourteen putative additive QTLs were identified for Abs in WJ in all of six trials (Table 2; Fig. 2). Of these QTLs, one each was distributed on chromosome 2A, 2D and 3B, two each on 2B, 4A, 6A and 6D, and three each on 5B. However, no QTLs for Abs were found that individually accounted for more than 10% of the phenotypic variance in WJ. Of these 14 QTLs, five (QAbs.WJ-2A.2, QAbs.WJ-2B.2, QAbs.WJ-3B.2, QAbs.WJ-5B.2a and QAbs.WJ-5B.2b) were involved in two of the six trials and one (QAbs.WJ-6A.3) was detected in three trials. The QTL, QAbs.WJ-2D.4, was detected in four trials that explained 2.33–5.49% of the phenotypic variance. The remaining seven QTLs showed environment-specific effects that could be detected only in one environment. Eight putative additive QTLs for Abs, accounting for 5.03–16.19% of the phenotypic variance, were detected in WY (Table 3; Fig. 3). They distributed on seven chromosomes, two each on 7D, and one each on 1D, 2B, 4B, 5B, 5D and 6A. Of these eight QTLs, five were found that individually accounted for more than 10% of the phenotypic variance. The QTL QAbs.WY-1D.3 accounted for 7.21–16.19% of the phenotypic variance, and was identified in E3, E5 and E6. Similarly, QAbs.WY-2B.3 explained 7.17–13.44% of the phenotypic variation. The QTL, QAbs.WY-5B.2, was detected in two trials and accounted for 10.13–10.26% of the phenotypic variation. Both QAbs.WY-4B.1 and QAbs.WY-7D.1a were detected in only one trial, accounting for 12.47 and 12.12% of the phenotypic variance, respectively.
Dough development time
In total, 13 significant additive QTLs for DDT were detected in WJ (Table 2; Fig. 2). They were located on chromosomes 1A, 1D, 2A, 3A, 3B, 3D, 4A (2 QTLs), 5A, 5B (2 QTLs), 6B and 7A. These QTLs individually explained 2.47–10.05% of the phenotypic variance. Of these, QDdt.WJ-5B.1 accounted for 10.05% of the phenotypic variance. Three QTLs, QDdt.WJ-1A.2, QDdt.WJ-3B.2 and QDdt.WJ-5B.2, were detected in two of the six trials and QDdt.WJ-7A.3 was detected in three, but each exhibited only moderate additive effects. In WY (Table 3; Fig. 3), five putative additive QTLs for DDT were distributing on chromosomes 1B (2 QTLs), 3B, 6D and 7A. The QTL, QDdt.WY-1B.5, was detected in five trials (all except E4), accounting for 5.22–16.97% of the phenotypic variance. The QTL, QDdt.WY-1B.2, was verified in two trials and QDdt.WY-3B.3 and QDdt.WY-7A.3 were both detected in three trials, accounting for 6.65–6.71, 5.05–11.82 and 6.11–6.77% of the phenotypic variance, respectively.
Dough stability time
Qualitative trait loci mapping analysis produced a total of nine putative additive QTLs for DST in WJ and seven in WY (Tables 2, 3; Figs. 2, 3). They were assigned to chromosomes 1A, 1D, 2B, 3B (2 QTLs), 4D, 5A, 5B and 7A in WJ, and to 1B (2 QTLs), 3B, 4B, 7A, 7B and 7D in WY. Of the nine WJ QTLs, four (QDst.WJ-1A.2, QDst.WJ-3B.2, QDst.WJ-5B.2 and QDst.WJ-7A.2) were detected in two trials, accounting for 3.95–8.36, 2.75–2.88, 12.85–13.21 and 2.59–3.41% of the phenotypic variance, respectively. The remaining QTLs were identified in one trial only. The QTL, QDst.WJ-5B.2, had the highest LOD values of 10.01 and 10.96. Three significant QTLs, QDst.WY-1B.4, QDst.WY-3B.2 and QDst.WY-7A.3, were verified in at least two trials and accounted for 5.42–18.09, 10.89–12.84 and 3.86–12.96% of the phenotypic variance, respectively. The remaining QTLs showed significance in only one of the six trials, and individually explained less than 10% of phenotypic variance.
Discussion
Positive contributions of parental alleles and effect of environment on quality traits
In this study, the phenotypic values of Weimai 8 were lower than those of Jimai 20 and Yannong 19 for the three traits KH, DDT and DST, but the three parents contributed positive alleles for all traits (Tables 2, 3). For these three traits, many more positive alleles were donated by Jimai 20 and Yannong 19 than by Weimai 8. For GPC, however, six positive alleles originating from Weimai 8 were isolated in WJ and six positive Weimai 8 alleles were isolated in WY lines. In contrast, only three positive alleles for GPC originated from Jimai 20 in WJ and only four positive alleles in WY originated from Yannong 19. The allele that enhanced GPC the most was the major QTL QGpc.WY-5A.1 contributed by Weimai 8. For WGC, the positive alleles of five WJ QTLs and three WY QTLs originated from Weimai 8, while only two positive alleles in the WJ line originated from Jimai 20 and only five positive alleles in the WY line originated from Yannong 19. For FW, only four positive WJ alleles and seven positive WY alleles came from Weimai 8. In contrast, 13 positive WJ alleles and six positive WY alleles came from Jimai 20 and Yannong 19. For Abs, 11 of the 14 positive WJ alleles were contributed by Weimai 8, while only three positive alleles were contributed by Jimai 20, in WJ. In the WY lines, only three positive alleles came from Weimai 8 while five from Yannong 19. Thus, the contributions of the parental lines to the positive alleles vary significantly between these seven qualitative traits.
Four of 9 QTLs for GPC, 2 of 7 QTLs for WGC, 8 of 17 QTLs for FW, 3 of 16 QTLs for KH, 7 of 14 QTLs for Abs, 4 of 13 QTLs for DDT and 4 of 9 QTLs for DST in WJ, and 3 of 10 QTLs for GPC, 3 of 8 QTLs for WGC, 3 of 13 QTLs for FW, 5 of 14 QTLs for KH, 3 of 8 QTLs for Abs, 4 of 5 QTLs for DDT and 3 of 7 QTLs for DST were detected in at least two trials in the WY line population. Most QTLs showed environment-specific such that they could be detected in only one environment. Thus, those QTLs that are influenced significantly by the environment can only be detected in QTL studies conducted in a range of environments. Other QTLs accounting for higher phenotypic variance were usually detected in two environments or more, so they can be considered ‘stable’ QTLs and may prove most valuable in marker-assisted selection (MAS) for improvement of wheat varieties.
Common QTLs resolved in both the populations
Qualitative trait loci detection based on a single mapping population usually results in only a limited number of QTLs and results are generally not conclusive. With the rapid development of molecular marker technology, additional research on QTL effects in more than one different or related genetic background is both feasible and warranted. Turner et al. (2004) conducted QTL analysis to study the genetic basis of grain protein levels and grain texture using RILs and two recombinant substitution line (RSL) populations. Kulwal et al. (2005) used two independent RIL populations to conduct QTL analysis on grain protein content, but no QTLs were common between the two populations. Ma et al. (2007) identified a great number of common QTL for spike-related traits using recombinant inbred lines (RILs) and RILs-derived IF2 populations. Buckler et al. (2009) utilized NAM (Nested Association Mapping population) comprising 5,000 RILs, to dissect QTLs for flowering time in maize, and also found evidence for numerous small-effect QTLs shared among families. Five mapping populations were involved in a report of Uga et al. (2010) for comprehensively QTL detection in rice.
In this paper, two related RIL populations were applied to dissect QTLs for seven quality traits. Based on common markers in the two genetic maps, comparisons of congruent QTLs were conduced (Table 4). At least 13 pairwise common QTLs were identified in the two populations, although most of them were detected in different environments and had different additive effects, accounting for 15.29 and 20.00% of the phenotypic variation in WJ and WY, respectively. Six of the 13 QTLs showed consistent additive effects with the common parent Weimai 8 alleles, whether negative or positive, simultaneously in the two populations. As is well known, a reducing or enhancing effect is not absolute but relative to the effect of the two parental alleles, so the remaining seven QTLs can still be regarded as congruent QTLs. Five QTLs, QFw.WJ-3B.4, QFw.WJ-5B.2b, QKh.WJ-4A.1b, QAbs.WJ-5B.1 and QDst.WJ-3B.1 were pleiotropic or co-located QTLs, and they were confirmed in WY lines with higher phenotypic variation (>10%). The precise prediction and definition of common QTLs in the two populations was hampered by the limited number of common loci in the two genetic maps, although the positions of most QTLs for the same trait identified in the two populations were highly congruent. The results show that QTLs from the common parent in the two related populations can be detected repeatedly to a certain extent, and the comparable QTLs are authentic.
QTL clusters and potential pleiotropic effects
As might be predicted from the significant correlations among the traits, many pleiotropic or co-located QTLs between pairs of the seven traits were detected (Fig. 1c). In addition, the trends in the numbers of co-located QTLs for pairwise traits were in approximate in agreement with their phenotypic correlation coefficients. There was at least one co-located QTL for each paired combination of the seven quality traits in both WJ and WY, with the exception of GPC/FW in WJ, GPC/DDT, WGC/DDT and Abs/DTT in WY. In this paper, 85 QTLs distributed over all 21 chromosomes except 5D in the WJ lines and 65 QTLs in the WY line were located on all 21 chromosomes except 3D and 4D. Of these QTLs, up to 19 loci in WJ and 20 loci in WY shared common intervals for two traits or more. In WJ, 6 of 16 QTLs for KH were located to intervals similar to those for GPC and Abs, six QTLs for FW shared common intervals with Abs, and five QTLs for DDT were co-located with QTLs for DST. In WY, the four traits FW, KH, DDT and DST were detected in the interval Xmag972.2–Xme3em2.7, 11 of 13 QTLs for FW were located to intervals similar to those for KH, and five QTLs for GPC co-located with QTLs for WGC, in accordance with the higher phenotypic correlations. These results confirm that we identified the marker intervals containing genes controlling different quality traits.
Comparison of the present study with previous researches
Previous studies have demonstrated the importance of grain protein content on wheat quality (Joppa et al. 1997; Cantrell and Joppa 1991; Harjit-Singh et al. 2001; Chee et al. 2001; Blanco et al. 1996, 2002, 2006; Prasad et al. 2003; Olmos et al. 2003; Turner et al. 2004; Kulwal et al. 2005; Li et al. 2009; Liang et al. 2010), and many studies have concluded that factors influencing protein concentration in cultivars and wild wheat were located on chromosome 6B (Joppa et al. 1997; Cantrell and Joppa 1991; Blanco et al. 2002; Olmos et al. 2003). However, no QTL for GPC locating to 6B was detected in WJ or WY. A major QTL, QGpc.WY-5A.1, for GPC on chromosome 5A, explaining 53.04% of the phenotypic variation, was close to a QTL detected by Blanco et al. (2002). This QTL, which could be result in higher WGC, was positively influenced by GPC, and both traits shared a common locus. The QTL, QWgc.WY-5A.2, shared a common fragment with QGpc.WY-5A.1 and contributed higher phenotypic variation to WGC (with positive effect from Weimai 8). Both DDT and DST are also important bread-making quality traits and they are strongly associated with the content of gluten. Kuchel et al. (2006) detected a major QTL for DDT on chromosome 1A and QTLs for DST on chromosomes 1A and 1B. The QTLs, QDdt.WY-3B.3 and QDst.WY-3B.2, were detected within a common interval and exhibited higher phenotypic variation.
Kernel hardness is crucial to the end-use quality of any wheat variety (Pomeranz and Williams 1990). It affects milling yield and the size and shape of the flour particles. Previous studies had indicated that chromosome 5D harboured important factors affecting KH (Law et al. 1978; Aitken 1993). Law et al. (1978) concluded that the Ha locus on the short arm of 5D was the mainly factor affecting KH. Nevertheless, no QTL for KH was detected on 5D in WJ or WY. Only one QTL detected on 5B corresponded to a QTL reported by Li et al. (2009). In this report, a major QTL for KH, QKh.WJ-7A.5, was reproducibly detected in five trials within the interval Xgwm473–Xedm16.1, with the positive effect from Jimai 20.
Water absorption is also an important quality trait. High Abs can increase the output of bread per unit of flour, increase the softness of bread, and prolong freshness. Only a few studies have documented QTL analysis for Abs (Kuchel et al. 2006; Li et al. 2009). Li et al. (2009) detected a major QTL for Abs on the short arm of chromosome 5D. Significant QTL associated with Abs were detected on chromosomes 1A and 2D (Kuchel et al. 2006). In this study, a major QTL for Abs was detected on 5B in both WJ and WY, indicating an authentic QTL. For flour whiteness, three congruent QTLs were identified in the two populations and QFw.WJ-3B.4 was identified in four environments.
Effect of population size on QTL detection
The limited population sizes can lead to underestimation of QTL number, overestimation of QTL effects, and failure to quantify QTL interactions (Vales et al. 2005). Beavis (1998) suggested that even 200 individuals might be too few for reliable QTL detection. Buckler et al. (2009) utilized NAM comprising 5000 RILs to dissect QTLs for flowering time in maize and concluded that, with large enough samples, additive QTL models can accurately predict phenotype. False positive QTLs might be caused by parental sharing when the RIL population was not large enough to permit completely random mating (Zou et al. 2005). Schön et al. (2004) exploited a large experimental population, 976 F5 maize test cross progenies, for QTL detection, and found a large effect of sample size on power and QTL detection as well as on accuracy and precision of QTL estimates.
Due to the significant differences in population size between WJ and WY, we evaluated the effect of population size on accuracy and precision of QTL estimates, although differences in genetic backgrounds exist in the two populations. Based on the data of Tables 2, 3 and 5, we speculate that (i) it is difficult to detect minor QTL using a small population, (ii) the limited population sizes can lead to underestimation of QTL number and sometimes be trait-dependent, and (iii) QTL effects are likely apt to be overestimated with small populations.
References
Aitken KS (1993) Genetic analysis of grain protein content in wheat. PhD Thesis, University of East Anglia, Norwich, UK
Autran JC, Abecassis J, Feillet P (1996) Statistical evaluation of different technological and biochemical tests for quality assessment in durum wheat. Cereal Chem 63:390–394
Beavis WB (1998) QTL analyses: power, precision, and accuracy. In: Patterson AH (ed) Molecular dissection of complex traits. CRC Press, Boca Raton
Blanco A, De Giovanni C, Laddomada B, Sciancalepore A, Simeone R, Devos KM, Gale MD (1996) Quantitative trait loci influencing grain protein content in tetraploid wheats. Plant Breed 115:10–316
Blanco A, Pasqualone A, Troccoli A, DiFonzo N, Simeone R (2002) Detection of grain protein content QTL across environments in tetraploid wheats. Plant Mol Biol 48:615–623
Blanco A, Simeone R, Gadaleta A (2006) Detection of QTL for grain protein content in durum wheat. Theor Appl Genet 112:1195–1204
Branlard G, Bernard S, Boeuf C, Bernard M (1999) Detection of QTL for bread making quality in wheat using molecular markers. In: Scarascia Mugnozza GT, Porceddu E, Pagnotta MA (eds) Genetics and breeding for crop quality and resistance. Kluwer, Dordrecht, pp 361–366
Buckler ES, Holland JB, Acharya CB, Brown PJ, Browne C, Ersoz E, Flint-Garcia S, Garcia A, Glaubitz JC, Goodman MM, Harjes C, Guill K, Kroom DE, Larsson S, Lepak NK, Li HH, Mitchell SE, Pressoin G, Peiffer JA, Rosas MO, Rocheford TR, Romay MC, Romero S, Salvo S, Villeda HS, Sliva HSD, Sun Q, Tian F, Upadyayula N, Ware D, Yates H, Yu J, Zhang Z, Kresovich S, McMullen MD (2009) The genetic architecture of maize flowering time. Science 325:714–718
Cantrell RG, Joppa LR (1991) Genetic analysis of quantitative traits in wild emmer (Triticum turgidum L. var dicoccoides). Crop Sci 31:645–649
Chantret N, Cenci A, Sabot F, Anderson O (2004) Sequencing of the Triticum monococcum hardness locus reveals good microcolinearity with rice. Mol Genet Genomics 271:377–386
Chantret N, Salse J, Sabot F, Rahman S, Bellec A, Laubin B, Dubois I, Dossat C, Sourdille P, Joudrier P, Gautier MF, Cattolico L, Bechert M, Aubourg S, Weissenbach J, Caboche M, Bernard M, Leroy P, Chalhoub B (2005) Molecular basis of evolutionary events that shaped the hardness locus in diploid and polyploidy wheat species (Triticum and Aegilops). Plant Cell 17:1033–1045
Chee PW, Elias EM, Anderson JA, Kianian SF (2001) Evaluaion of a high protein QTL from Triticum turgidum L.v ar. dicoccoides in an adapted durum wheat background. Crop Sci 41:295–301
Doerge RW (2002) Multifactorial genetics: mapping and analysis of quantitative trait locus in experimental populations. Nat Rev 3:43–52
Dohlman E, Hoffman L (2000) The new agricultural trade negotiations: background and issues for the U.S. wheat sector. Wheat yearbook. Economic Research Service, USDA
Gao LF, Jing RL, Huo NX, Li Y, Li XP, Zhou RH, Chang XP, Tang JF, Ma ZY, Jia JZ (2004) One hundred and one new microsatellite loci derived from ESTs (EST-SSR) in bread wheat. Theor Appl Genet 108:1392–1400
Hao YF, Liu AF, Wang YH, Feng DS, Gao JR, Li XF, Liu SB, Wang HG (2008) Pm23: a new allele of Pm4 located on chromosome 2AL in wheat. Theor Appl Genet 117:1205–1212
Harjit-Singh Prasad M, Varshney RK, Roy KJ, Balyan HS, Dhaliwal HS, Gupta PK (2001) STMS markers for grain protein content and their validation using near-isogenic lines in bread wheat. Plant Breed 120:273–278
Joppa LR, Cantrell RG (1990) Chromosomal location of genes for grain protein content of wild tetraploid wheat. Crop Sci 30:1059–1064
Joppa LR, Du C, Hart GE, Hareland GA (1997) Mapping a QTL for grain protein in tetraploid wheat (Triticum turgidum L.) using a population of recombinant inbred chromosome lines. Crop Sci 37:1586–1589
Kosambi DD (1944) The estimation of map distances from recombination values. Annu Eugen 12:172–175
Kuchel H, Landridge P, Mosinek L, Williams K, Jefferies SP (2006) The genetic control of milling yield, dough rheology and baking quality of wheat. Theor Appl Genet 112:1487–1495
Kulwal P, Kumar N, Kumar A, Gupta RK, Balyan HS, Gupta PK (2005) Gene networks in hexaploid wheat: interacting quantitative trait loci for grain protein content. Funct Integr Genomics 5:254–259
Kumar N, Kulwal PL, Balyan HS, Gupta PK (2007) QTL mapping for yield and yield contributing traits in two mapping populations of bread wheat. Mol Breeding 19:163–177
Lander ES, Green P, Abrahamson J, Barlow A, Daly MJ, Lincoln SE, Newburg L (1987) MAPMAKER: an interactive computer package for constructing primary genetic linkage maps of experimental and natural populations. Genomics 1:174–181
Law CN, Young CF, Brown JWS, Snape JW, Worland AJ (1978) The study of grain protein control in wheat using whole chromosome substitution lines. In: Seed protein improvement by nuclear techniques. International Atomic Energy Agency, Vienna, pp 483–502
Li SS, Jia JZ, Wei XY, Zhang XC, Li LZ, Chen HM, Fan YD, Sun HY, Zhao XH, Lei TD, Xu YF, Jiang FS, Wang HG, Li LH (2007) A intervarietal genetic map and QTL analysis for yield traits in wheat. Mol Breeding 20:167–178
Li Y, Song Y, Zhou R, Branland Jia J (2009) Detection of QTLs for bread-making quality in wheat using a recombinant inbred line population. Plant Breed 128:235–243
Liang D, Tang JW, Peña RJ, Singh R, He XY, Shen XY, Yao DN, Xia XH, He ZH (2010) Characterization of CIMMYT bread wheats for highand low-molecular weight glutenin subunits and other quality-related genes with SDS-PAGE, RP-HPLC and molecular markers. Euphytica 172:235–250
Liu SX, Chao SM, Anderson JA (2008) New DNA markers for high molecular weight glutenin subunits in wheat. Theor Appl Genet 118:177–185
Ma ZQ, Zhao DM, Zhang CQ, Zhang ZZ, Xue SL, Lin F, Kong ZX, Tian DG, Luo QY (2007) Molecular genetic analysis of five spike-related traits in wheat using RIL and immortalized F2 populations. Mol Gen Genomics 277:31–42
Mackay TFC (2001) The genetic architecture of quantitative traits. Annu Rev Genet 35:303–339
Mann G, Diffey S, Cullis B, Azanza F, Martin D, Kelly A, McIntyre L, Schmidt A, Ma WJ, Nath Z, Kutty I, Leyne PE, Rampling L, Quail KJ, Morell MK (2009) Genetic control of wheat quality: interactions between chromosomal regions determining protein content and composition, dough rheology, and sponge and dough baking properties. Theor Appl Genet 118:1519–1537
Matsuo RR, Dexter JE, Kosmolak FG, Leisle D (1982) Statistical evaluation of tests for assessing paghetti-making quality of durum wheat. Cereal Chem 59:222–228
Morris CF (2002) Puroindolines: the molecular genetic basis of wheat grain hardness. Plant Mol Biol 48:633–647
Mullan DJ, Platteter A, Teakle NL, Appels R, Colmer TD, Anderson JM, Francki MG (2005) EST-derived SSR markers from defined regions of the wheat genome to identify Lophopyrum elongatum specific loci. Genome 48:811–822
Nagaoka T, Ogihara Y (1997) Applicability of inter-simple sequence repeat polymorphisms in wheat for use as DNA markers in comparison to RFLP and RAPD markers. Theor Appl Genet 94:597–602
Nelson JC, Andreescu C, Breseghello F, Finney PL, Gualberto DG, Bergman CJ, Peña RJ, Perretant MR, Leroy P, Qualset CO, Sorrells ME (2006) Quantitative trait locus analysis of wheat quality traits. Euphytica 149:145–159
Olmos S, Distelfeld A, Chicaiza O, Schlatter AR, Fahima T, Echenique V, Dubcovsky J (2003) Precise mapping of a locus affecting grain protein content in durum wheat. Theor Appl Genet 107:1243–1251
Peng JH, Lapitan NLV (2005) Characterization of EST-derived microsatellites in the wheat genome and development of eSSR markers. Funct Integr Genomics 5:80–96
Perretant MR, Cadalen T, Charmet G, Sourdlle P, Nicolas P, Boeuf C, Tixier MH, Branlard G, Bernard S, Bernard M (2000) QTL analysis of bread-making quality in wheat using a doubled haploid population. Theor Appl Genet 100:1167–1175
Pomeranz Y, Williams PC (1990) Wheat hardness: its genetic, structural and biochemical background, measurement and significance. In: Pomeranz Y (ed) Advances in cereal science and technology, vol 10. American Association of Cereal Chemists, St Paul, pp 471–544
Prasad M, Kumar N, Kulwal PL, Röder MS, Balyan HS, Dhaliwal HS, Gupta PK (2003) QTL analysis for grain protein content using SSR markers and validation studies using NILs in bread wheat. Theor Appl Genet 106:659–667
Röder MS, Korzun V, Wendehake K, Plaschke J, Tixier M-H, Leroy P, Ganal MW (1998) A microsatellite map of wheat. Genetics 149:2007–2023
Schön CC, Utz HF, Groh S, Truberg B, Openshaw S, Melchinger AE (2004) Quantitative trait locus mapping based on resampling in a vast maize testcross experiment and its relevance to quantitative genetics for complex traits. Genetics 167:485–498
Singh NK, Shepherd KW (1991) A simplified SDS-PAGE procedure for separation LMW subunits of glutenin. J Cereal Sci 14:203–208
Sourdille P, Perretant MR, Charmet G, Leroy P, Gautier MF, Joudrier P, Nelson JC, Sorrells ME, Bernard M (1996) Linkage between RFLP markers and gene affecting kernel hardness in wheat. Theor Appl Genet 93:580–586
Sourdille P, Perretant MR, Charmet G, Cadalen T, Tixier MH, Joudrier P, Gautier MF, Branlard G, Bernard S, Boeuf C, Bernard M (1999) Detection of QTL for bread making quality in wheat using molecular markers. In: Scarascia Mugnozza GT, Porceddu E, Pagnotta MA (eds) Genetics and breeding for crop quality and resistance. Kluwer, Dordrecht, pp 361–366
Stein N, Herren G, Keller B (2001) A new DNA extraction method for high-throughout marker analysis in a large-genome species such as Triticum aestivum. Plant Breed 120:354–356
Suenaga K, Khairallah M, William HM, Hoisington DA (2005) A new intervarietal linkage map and its application for quantitative trait locus analysis of ‘‘gigas’’ features in bread wheat. Genome 48:65–75
Sun XC, Marza F, Ma HX, Carver Brett F, Bai GH (2010) Mapping quantitative trait loci for quality factors in an inter-class cross of US and Chinese wheat. Theor Appl Genet 120:1041–1051
Suprayogi Y, Pozniak CJ, Clarke FR, Clarke JM, Knox RE, Singh AK (2009) Identification and validation of quantitative trait loci for grain protein concentration in adapted Canadian durum wheat populations. Theor Appl Genet 119:437–448
Turner AS, Bradburne RP, Fish L, Snap JW (2004) New quantitative trait loci influencing grain texture and protein content in bread wheat. J Cereal Sci 40:51–60
Uga Y, Siangliw M, Nagamine T, Ohsama R, Fujimura T, Fukuta Y (2010) Comparative mapping of QTLs determining glume, pistil and stamen sizes in cultivated rice (Oryza sativa L.). Plant Breed 129:657–669
Vales MI, Schön CC, Capettini F, Chen XM, Corey AE, Mather DE, Mundt CC, Richardson KL, Sandoval-Islas JS, Utz HF, Hayes PM (2005) Effect of population size on the estimation of QTL: a test using resistance to barley stripe rust. Theor Appl Genet 111:1260–1270
Zanetti S, Keller M, Winzeler M, Saurer W, Keller B, Messmer M (1999) QTL for quality parameters for bread-making quality in a segregating wheat by spelt population. In: Scarascia Mugnozza GT, Porceddu E, Pagnotta MA (eds) Genetics and breeding for crop quality and resistance. Kluwer, Dordrecht, pp 357–360
Zhao CH, Cui F, Zong H, Wang YH, Bao YG, Hao YF, Du B, Wang HG (2009) Transmission of the Chromosome 1R in winter wheat germplasm Aimengniu and its derivatives revealed by molecular markers. Agric Sci China 8(6):652–657
Zou F, Gelfond JAL, Airey DC, Lu L, Manly KF, Williams RW, hreadgill DW (2005) Quantitative trait locus analysis using ecombinant inbred intercross (RIX): theoretical and empirical considerations. Genetics 170:1299–1311
Acknowledgements
This research was supported by the National Basic Research Program of China (973 Program, 2006CB101700). The author thanks Sishen Li, College of Agronomy, Shandong Agricultural University, Taian, China, for kindly providing EST-SSR markers.
Author information
Authors and Affiliations
Corresponding author
Additional information
J. Li, F. Cui, A. Ding and C. Zhao contributed equally to this work.
Rights and permissions
About this article
Cite this article
Li, J., Cui, F., Ding, Am. et al. QTL detection of seven quality traits in wheat using two related recombinant inbred line populations. Euphytica 183, 207–226 (2012). https://doi.org/10.1007/s10681-011-0448-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10681-011-0448-4