
Abstract
A set of 142 winter wheat recombinant inbred lines (RILs) deriving from the cross Heshangmai × Yu8679 were tried in four ecological environments during the seasons 2006 and 2007. Nine agronomic traits comprising mean grain filling rate (GFRmean), maximum grain filling rate (GFRmax), grain filling duration (GFD), grain number per ear (GNE), grain weight per ear (GWE), flowering time (FT), maturation time (MT), plant height (PHT) and thousand grain weight (TGW) were evaluated in Beijing (2006 and 2007), Chengdu (2007) and Hefei (2007). A genetic map comprising 173 SSR markers and two EST markers was generated. Based on the genetic map and phenotypic data, quantitative trait loci (QTL) were mapped for these agronomic traits. A total of 99 putative QTLs were identified for the nine traits over four environments except GFD, PHT and MT, measured in two environments (BJ07 and CD07), respectively. Of the QTL detected, 17 for GFRmean, 16 for GFRmax, 21 for TGW and 10 for GWE involving the chromosomes 1A, 1B, 2A, 2D, 3A, 3B, 3D, 4A, 4D, 5A, 5B, 6D and 7D were identified. Moreover, 13 genomic regions showing pleiotropic effects were detected in chromosomes 1A, 1B, 1D, 2A, 2B, 2D, 3A, 3B, 4B, 4D, 5B, 6D and 7D; these QTL revealing pleiotropic effects may be informative for a better understanding of the genetic basis of grain filling rate and other yield-related traits, and represent potential targets for multi-trait marker aided selection in wheat.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Grain filling is a crucial and dynamic process of wheat growth. Its duration and rate determine the individual grain size, grain weight and as a result, the economic yield of the crop (Li and Pan 2005). Grain filling duration (GFD) is much influenced by temperature, particularly in the presence of stress (Pinthus and Shalom 1978; Wiegand and Cuellar 1981; Knott and Gebeyehou 1987), while grain filling rate (GFR) appears to be largely under genetic control (Nass and Reisser 1975; Van Sanford 1985). Moreover, since wheat is harvested at the beginning of summer and some other crops are planted in succession in China, duration was not randomly prolonged in order to achieve high yield altogether; therefore, the selection of genotypes with high GFR appears to be a successful strategy for increasing grain yield.
The physiological mechanisms by which crops regulate GFR have been widely explored (Egli et al. 1989; Jenner and Rathjen 1972; Manness 1989), but little is known until now about the genetics and loci affecting this complex trait. Availability of molecular maps enables to dissect complex quantitative traits into component loci and study their relative effects on a specific trait in a segregating population by QTL analysis (Röder et al. 1998). Up to now, QTL for grain yield and yield components such as grain weight per ear (GWE), grain numbers per ear (GNE) and thousand grain weight (TGW), etc., have been previously reported in wheat by several studies (Sourdille et al. 2003; Groos et al. 2003; Börner et al. 2002; Huang et al. 2003, 2004, 2006; Kumar et al. 2006; Quarrie et al. 2005), whereas only one paper on QTL for GFR has been reported by Kirigwi et al. (2007), in which a QTL controlling GFR was located on chromosome 4A in wheat.
Since final yield and yield components represent the results of various biochemical and physiological processes, a genetic analysis based trait such as yield may not shed much light on the genetics of the early determinants of yield and/or yield-limiting processes (Cui et al. 2003). To break through the current plateau affecting yield potential of wheat, it will be necessary to add physiological traits to the morphological ones currently identified as limiting yield. Generally, winter wheat originating from the north of China possesses the characteristic of fast grain filling, which forms the physiological foundation of larger grains and tolerance against stress, but the genetic basis of this ideotype is not understood clearly. Thus, our objectives were to detect QTL associated with GFR and yield-related agronomic traits in four distinct ecological environments, to provide molecular markers linked to these QTL, and to characterize the environmental stability of any identified GFR QTL.
Materials and methods
Plant materials
The mapping population comprised 142 F7 to F9 recombinant inbred lines (RILs) bred by single seed descent from the cross HSM × Y8679. HSM is a Chinese hard red local variety with a lower TGW (25 g) originating from Low and Middle Yantze Valley, winter wheat region, while Y8679 is a Chinese winter cultivar with a higher TGW (65 g) from Yellow and Huai River Valley, winter wheat region. Besides, the differences in TGW, Y8679 shows a higher GFR, a larger spike and grain size, but a lower plant height (PHT) compared to HSM.
Field trials
During the 2005/2006 and 2006/2007 winter wheat seasons, field trials were carried out at the experimental station of the Institute of Crop Science, Chinese Academy of Agricultural Sciences, Beijing (latitude 39º53′ N, longitude 117º24′ E, with an average rainfall of 628.9 mm per year, an average temperature of 11.6°C per year and average sunlight of 2,662 h per year). In 2006/2007, two further field trials were performed, one located at the experimental station of the Institute of Crop Research, Sichuan Province Academy of Agricultural Sciences, Chengdu (30º40′ N, 104º04′ E, with an average rainfall of 1,100 mm per year, an average temperature of 15.9°C per year and an average sunlight of 1,227 h per year), and the second one located at the experimental station of the Institute of Crop Research, Anhui Province Academy of Agricultural Sciences, Hefei (31º58′ N, 117º24′ E, with an average rainfall of 950 mm per year, an average temperature of 15.5°C per year and an average sunlight of 2,100 h per year). All the RILs, along with the two parents in Beijing comprised four-row plots with a length of 1 m and 25-cm spacing, whereas all lines and parental lines were grown in two-row plots with a length of 1 m and 25-cm spacing in Chengdu and Hefei. The field management followed common practice for wheat production.
Trait evaluation
A total of nine traits, comprising GFRmean, GFRmax, GFD, TGW, GWE, GNE, PHT, MT and FT were evaluated in the present study described as follows:
(1) Measurement of GFRmean and GFRmax: the flowering time of each line was recorded and at least 50 simultaneously flowering spikes per lines were tagged. Five tagged spikes from each line were sampled in a 5-day interval starting from anthesis up to maturity. The grains were separated from the glumes, held at 105°C for 10 min, and then at 70°C until reaching a constant weight. At this stage, the number of the total grains was counted and their weight was recorded. The grain filling process was adjusted using the logistic growth equation as described by Mo (1992): Y = K/(1 + ae −bt) (I), where Y is the mean weight per grain (mg), t is number of days post anthesis and K is the maximum theoretic weight calculated by the following formula: K = ((Y 2 2−(Y 1 + Y 3)–2Y 1 Y 2 Y 3))/(Y 2 2–Y 1 Y 3), where Y 1, Y 2 and Y 3 are the weight per grain at, respectively, 5, 20 and 35 days after anthesis, while a and b are coefficients determined by regression (Mo 1992). The calculation of GFRmax was performed as follows: first, the second derivative of formula (I) was calculated by deriving the formula (I) into d 2 Y/dt 2 = (Kabe –bt(abe –bt–b))/(1 + ae –bt)3. Setting d 2Y/dt2 = 0, t max will be equal to lna/b. Finally, if t treated as t max, GFRmax can be calculated by using the formula (II): GFRmax = dY/dT = Kabe −bt/(1 + ae –bt)2. The active GFD was defined as days corresponding to the period when Y was between 5% (t 1 = (ln a – 1.1317)/b) to 99% (t 2 = (ln a + 2.1972)/b) of K. GFRmean was also calculated by the ratio: W max/GFD, where W max is the maximum weight during GFD. (2) TGW (g) was measured as the average weight of two independent samples of 1,000 grains from each plot. (3) GNE was measured as the average number of filled spikelets per ear based on ten ears. (4) GWE (g) was determined as the average weight of bulked harvested grains per ear from ten ears. (5) PHT (cm) was measured in centimeters from the soil surface to the tip of the tallest ear excluding the awns. (6) MT was determined by the day on which the peduncles of 75% of the plants in the plots were no longer green.
SSR fingerprinting
From the genetic map of Röder et al. (1998), Gupta et al. (2002), and the GrainGenes database (http://wheat.pw.usda.gov/ggpages/maps), 1,132 SSR loci were assayed for information on the parental lines, along with ten EST markers, and those which were polymorphic following this test were used to genotype the RIL population. Genomic DNA was isolated from fresh young leaf tissue of both parents (Y8679 and HSM) and the 142 RILs using CTAB procedure. PCR reactions were performed on a programmable thermal controller (PTC-100, MJ Research Inc., MA USA) in a total volume of 15 uL, containing 1× buffer, 1.8 mmol L−1 MgCl2, 0.25 mmol L−1 dNTPs, 0.24 μmol L−1 of each primer, 1 U Taq-polymerase and 50 ng genomic DNA as template. After an initial denaturing step for 5 min at 95°C, 33 cycles were performed for 50 s at 94°C, 55 s at either 50°C, 55°C or 60°C (depending on the different primer pairs), 60 s at 72°C, followed by a final extension step of 10 min at 72°C. Amplicons were separated by 6% (w/v) denaturing polyacrylamide gels and visualized by silver staining (Bassam et al. 1991).
Linkage map and QTL analysis
Linkage analysis was performed using MAPMAKER/EXP version 3.0b (Lander et al. 1987). The command ‘compare and order’ was used to generate the linkage groups, which were assigned to chromosomes based on the microsatellite consensus map of Somers et al. (2004). The ‘Kosambi’ mapping function (Kosambi 1944) was used to transform recombination frequencies into centiMorgan (cM) map distances. QTL analysis was performed using composite interval mapping (CIM) method (Zeng 1994). Automatic cofactor selection by a forward/backward regression (forward P < 0.01, backward P < 0.01) was performed with Windows QTL Cartographer version 2.5. The step size chosen for all QTL was 2 cM. A QTL was claimed to be significant at a LOD value of 2.5 (The LOD value between 2.0 and 3.0 was claimed significant for the location of QTL; if the value was too high, the QTL with small effects have not been detected, whereas, if the value was too low, some fake QTL may be thought the real QTL. For complex quantitative traits, it is necessary to use the relative low LOD value to find all the QTL loci).
Statistical analysis
Statistical analysis was carried out using the SAS V8.0 statistics package (SAS Institute Inc., 1999). Correlation analysis between pairs of traits was calculated using ‘proc corr’ procedure and single marker regression analysis by ‘proc anova’. Heritability (h 2) was calculated using the ‘proc varcomp’ procedure, by h 2 = σ 2g /(σ 2g + σ 2ge /n + σ 2e /rn), where σ 2g is the genetic variance, σ 2ge is the genetic-by environment interaction variance, σ 2e is the error variance, n represent the number of environments and r the number of replicates per line tested in each environment.
Results
Linkage map of the RIL population HSM × Y8679
Of the 1,132 SSR and ten EST markers, 186 (16.29%) turned out to be polymorphic between the parents and therefore were used to genotype the RIL population. Finally, a total of 173 SSR and two EST markers were mapped generating 27 linkage groups. The linkage map reveals a total length of 1584.6 cM with an average interval length of 9.3 cM. The number of loci per linkage group ranged from 3 (chromosome 3A) to 18 (chromosome 2A) (Fig. 1).


A genetic map derived from the RIL population bred from the cross ‘HSM × Y8679’, showing the location of putative QTL. The lengths of the bars indicated the map distances, are shown in centiMorgans and were calculated using the Kosambi (1944) mapping function. Supported intervals for QTL are indicated by vertical bars, the length of the bar show a one-LOD confidence interval. LOD max is pointed by a triangle. Abbreviations for traits: GFD grain filling duration, GFR max maximum grain filling rate, GFR mean mean grain filling rate, MT maturation time, FT flowering time, GNE grain number per ear, GWE grain weight per ear, PHT plant height, TGW thousand grain weight
Phenotypic variation and correlation analysis
Parental phenotypic variation and the distribution among RILs for GWE, GNE, TGW, PHT, FT, MT, GFRmean, GFRmax and GFD in four different environments are shown in Table 1. Regarding all four environments, the measured target traits varied in the RIL population following a continuous distribution representing a normal phenotypic segregation for QTL mapping. Moreover, all traits except PHT were higher in parent Y8679 compared to HSM (Table 1). The estimated heritability varied between 16.6% (GFD) and 84.6% (TGW). Pairwise correlation coefficients between the nine traits are given in Table 2. GFRmean was correlated with all the traits except FT and GNE. Significant positive correlations were observed between GFRmean and GWE, TGW as well as GFRmax, respectively; furthermore, a positive correlation can also be observed between GWE and GNE; between FT and MT. The strongest positive correlation was observed between GFRmean and TGW with correlation coefficient r = 0.85 (P < 0.0001). The correlation between GFRmax and GFRmean was also highly significant (r = 0.84, P < 0.0001), whereas weak negative correlations were found between GFRmean and PHT (r = −0.21, P < 0.05) as well as FT (r = −0.10, P > 0.05), respectively.
QTL analysis
Putative QTL detected in each environment are listed in Table 3 and their locations are shown in Fig. 1. GFRmean, GFRmax, GWE, GNE, TGW and FT were analysed in all four environments (BJ06, BJ07, CD07 and HF07), whereas PHT, GFD and MT were only analysed in BJ06 and HF07. Across all the traits and environments, 99 putative QTL are identified, located on all of the wheat chromosomes except chromosomes 6A, 7A and 7B.
Mean grain filling rate
In total, 17 QTL were mapped on chromosomes 1A, 1B, 2A, 3A, 3B, 3D, 4D, 5B and 6D. Among them, four, five, three and five QTL were detected in BJ06, BJ07, CD07 and HF07, accounted for phenotypic variations of GFRmean by 8.72–15.99, 7.17–10.90, 7.24–13.77 and 7.27–20.83% for single QTL in the respective environment. Across all four environments no common QTL was identified; however two QTL on chromosomes 1B (QGfrmean.nfcri-1B) and 2A (QGfrmean.nfcri-2A) were detected in three environments and three QTL on chromosomes 3B (QGfrmean.nfcri-3B), 5B (QGfrmean.nfcri-5B) and 6D (QGfrmean.nfcri-6D) were detected in two environments, respectively. For example, QTL (QGfrmean.nfcri-1B) with the positive alleles derived from Y8679, was identified in BJ06, BJ07 and HF07, and explained 15.99, 10.65 and 7.27% of the phenotypic variation of the trait in the respective environment.
Maximum grain filling rate
Sixteen QTL for GFRmax were identified over the four environments, of which, four, five, two and five QTL were detected in BJ06, BJ07, CD07 and HF07, respectively, accounted for phenotypic variations of GFRmax by 6.31–15.95% for single QTL. These QTL were located on chromosomes 1A, 1B, 2A, 2D, 3B, 4D, 5B and 7D. Most of the positive alleles were contributed by Y8679, although the positive allele for QGfr max .nfcri-4D originated from HSM. One common QTL QGfr max .nfcri-2A for GFRmax was identified on chromosome 2A across all the four environments, but contributed only 7.46, 6.46, 6.31 and 8.90% of the phenotypic variation in BJ06, BJ07, CD07 and HF07, respectively. QTL QGfr max .nfcri-1B on chromosome 1B was found in three environments (BJ06, BJ07 and HF07), whereas two QTL, QGfr max .nfcri-3B on chromosome 3B and QGfr max .nfcri-7D on chromosome 7D, were detected in two environments (BJ07 and HF07), respectively.
Thousand grain weight
Twenty-one QTL controlling TGW on chromosomes 1B, 2A, 2D, 3B, 4A, 4D, 5A, 6D and 7D were identified across the four environments, and three, two, nine and seven QTL were detected in BJ06, BJ07, CD07 and HF07, respectively, explaining 9.80–13.96, 7.70–16.80, 4.36–10.47 and 5.75–13.96% of the phenotypic variation in the respective environment. Two common QTL (QTgw.nfcri-1B and QTgw.nfcri-2A) were found across all four environments, of which both positive alleles are contributed by Y8679, explaining more than 10.0 and 12.0% of the phenotypic variation of TGW, respectively. Furthermore, on the chromosome 3B, two QTL accounting for nearly 7.0 or 6.0% of the phenotypic variation of TGW were identified in CD07 and HF07.
Grain filling duration
Six QTL involving chromosomes 1A, 3B, 5D and 6D were identified for GFD across two environments (BJ07 and CD07). All positive alleles of the detected QTL were contributed by Y8679 except one QTL (QGfd.nfcri-5D) detected in CD07. Two common QTL, QGfd.nfcri-1A and QGfd.nfcri-3B, were simultaneously expressed in both BJ07 and CD07. The former explained 10.31% of the phenotypic variation in BJ07, and 6.68% in CD07, while the latter accounted for, respectively, 9.67 and 15.72%.
Grain weight per ear
Ten GWE QTL were mapped on chromosomes 1A, 2A, 3B, 4B, 4D and 6B. Among these QTL detected, one was identified in BJ06 and HF07, whereas three and five QTL were found in BJ07 and CD07, respectively. The proportion of the phenotypic variation explained by each of these QTL in turn ranged from 5.93 to 24.06%. Although none was expressed in all four environments, the QTL QGwe.nfcri-2A was identified in BJ07 (7.30%) and CD07 (6.42%). One QTL, QGwe.nfcri-3B, was found in three environments (BJ07, CD07 and HF07), which contributed 16.26, 24.06 and 9.86% of the phenotypic variation of GWE in BJ07, CD07 and HF07, respectively.
Grain number per ear
Eight QTL for GNE were mapped on chromosomes 1D, 2D, 2A, 3A, 4D and 6D, and were responsible for, respectively, 11.34–13.13, 7.43–10.77, 6.87–8.95 and 15.82% of the trait variation in each of the four environments. On the short arm of chromosome 4D, one QTL QGne.nfcri-4D with the positive alleles derived from Y8679 was identified in both BJ07 and CD07, which explained around 7.0% of the phenotypic variation for GNE in each environment. Another QTL QGne.nfcri-2D on chromosome 2D was expressed in BJ06 (11.34%) and CD07 (7.13%).
Plant height
A total of six QTL for PHT were identified on chromosomes 1D, 2D, 3D and 4D in two environments (BJ07 and CD07). In each environment, three QTL were detected and explained 5.83–13.12 and 6.70–25.24% of the phenotypic variation in respective location for single QTL. Two common QTL (QPht.nfcri-1D and QPht.nfcri-4D) with the positive allele contributed by HSM were identified. In each environment, QPht.nfcri-1D explained around 6.0% of the phenotypic variation of PHT, whereas QPht.nfcri-4D explained 13.12 and 25.24% of the phenotypic variation of PHT, respectively.
Flowering time
Eight QTL for FT were identified on chromosomes 1B, 2B, 3B, 5D and 6D, of which two, one, three and two were expressed in, respectively, BJ06, BJ07, CD07 and HF07. But none occurred in all four environments. QTL QFt.nfcri-1B with the positive allele from Y8679 was identified in three environments (BJ06, BJ07 and CD07) and explained 15.77, 10.14 and 11.63% of the phenotypic variation of FT in the respective environment.
Maturation time
Of the seven MT QTL identified, four expressed in BJ07 accounted for 7.14–9.61% of the phenotypic variation, and mapped on chromosomes 1B, 2A, 4B and 6D. Three QTL were found on chromosomes 1B, 3B and 5D in CD07, which accounted for 8.28–10.77% of the phenotypic variation of MT. One common QTL QMt.nfcri-1B with the positive allele derived from Y8679 contributed around 10.0% of the phenotypic variation of MT over the two environments (BJ07 and CD07).
Single marker regression and alternative allele effects analysis
A single marker regression analysis, based on RILs means across three environments (BJ07, CD07 and HF07), was conducted to validate the QTL identified by CIM method. Environment BJ06 was excluded because the missing data for grain filling rate of 26 lines, which had seldom plants because of frozen weather, and the QTL location for grain filling rate was just based on 116 lines in BJ06 and the QTL location for grain filling rate of BJ07, CD07 and HF07 were based on all the 142 lines. The markers closely associated with TGW and GFRmean are listed in Table 4. The three SSR markers identified in this way were xwmc419, xgwm359 and xbarc113, located on, respectively, chromosomes 1B, 2A and 3B. The phenotypic variation could be explained by these markers ranged from 14.58% (xbarc113) to 24.07% (xwmc419) for TGW and from 14.41% (xbarc113) to 20.39% (xgwm359) for GFRmean, respectively.
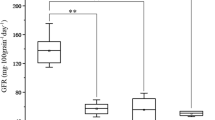
Alternative allele effects in the QTL regions on chromosomes 1B, 2A and 3B for TGW and GFRmean are shown in Fig. 2. RILs carrying all the positive alleles from Y8679 on 1B, 2A and 3B (AAA) showed the highest TGW and GFRmean, with an average value of 44.4 g for TGW and 1.27 mg grain−1 day−1 for GFRmean, whereas those RILs carrying the opposite alleles from HSM (aaa) have the lowest TGW and GFRmean, with an average value of 32.2 g for TGW and 0.99 mg grain−1 day−1 for GFRmean. Furthermore, there were significant differences between the RILs possessing two positive alleles (AAa, AaA, aAA) for TGW and GFRmean (39.8 g and 1.15 mg grain−1 day−1) in comparison to one positive allele (Aaa, aAa, aaA; with 34.4 g, 0.98 mg grain−1 day−1). The lines carrying two positive alleles derived from Y8679 showed a higher TGW of 5.4 g and a higher GFRmean of 0.17 mg grain−1 day−1 compared to those only with one positive allele from the same parental line Y8679.

Boxplot distributions of lines possessing alternative alleles in the QTL genomic regions on chromosomes 1B, 2A and 3B for TGW and GFRmean. Data were based on means over three experiments (BJ07, CD07 and HF07). Genotypes were classified by the allelic state at xwmc419 (1B), xgwm359 (2A) and xbarc113 (3B) linked to the corresponding QTL, respectively. Twenty-two lines were not classified because of missing marker values. ‘A’ represents alleles contributed by parental line ‘Y8679’, while ‘a’ represents alleles contributed by ‘HSM’; solid lines, median value; individual data points falling outside the confidence interval indicated by black circles
Discussion
In general, the process of grain filling is regulated by both GFR and GFD (Gebeyehou et al. 1982; Darroch and Backer 1990; Cross 1975; Wang et al. 1999); however, their relative contribution remains controversial. Mashiringwani and Schweppenhauser (1992) reported that genotypic differences in grain yield of wheat were due to differences in GFR. Further on, Nass and Reisser (1975) have observed that genetic differences in final grain weight were related to differences in GFR rather than GFD. A lack of relationship between the final grain yield and GFD has been discussed by Van Sanford (1985) as well as Bruckner and Frohberg (1987). In contrast, Gebeyehou et al. (1982) found that grain yield was strongly correlated with GFD. In the present study, the heritability of GFD was lower than that of the other eight traits analysed, and thus is a character much influenced by some, or all, environmental variables, including temperature, light intensity and moisture availability, as also suggested by Nass and Reisser (1975). In contrast, the heritability of GFR was high both in our experiments and those of Mou and Kronstad (1994). As GFRmean is strongly correlated with GFRmax, TGW and GWE, while in contrast, GFD is correlated with neither TGW nor GWE. Therefore, it could be concluded that more progress towards wheat yield improvement should be achievable by focusing on GFR rather than on GFD.
QTL for grain filling rate and yield related traits
As reported previously by numerous studies, complex traits like yield and yield-related traits showing continuous agronomic variation usually were governed by a number of genes known as quantitative trait loci (QTL), which are highly affected by environmental conditions (Börner et al. 2002; Marza et al. 2006; Thomson et al. 2003). In the present study, numerous QTL for GFRmean, GFRmax, GFD, TGW, GWE, GNE, FT, MT and PHT have been detected in four environments with QTL numbers varying from one to nine. Concerning the four environments, the average temperature, sunlight and rainfall, etc. were very different from each other, which might be the reason why only a few major large-effects QTL were detected across all four environments. The present results confirmed the assumption by Paterson et al. (1991) that the QTL analysis conducted in a single environment was likely to underestimate the number of QTL for a certain trait and that was the reason why a QTL analysis should be done across several environments.
GFRmean and GFRmax are the two most important parameters associated with grain filling process, which determine the final wheat production (Yang and Zhang 2006). Kirigwi et al. (2007) identified a QTL controlling GFR within the marker interval xwmc89-xwmc420 on chromosome 4A in a distinct RIL population. This QTL is different from our results; however, a QTL (QTgw.nfcri-4A) for TGW was located in the comparable region of our RIL population, indicating that this is a genomic region which seems to contain important loci or genes affecting grain yield.
Regarding QTL for TGW identified in our population, the same loci have previously been reported by several authors. For example, both Börner et al. (2002) and Peng et al. (2003) have detected the same QTL for TGW linked to the marker xgwm498 on chromosome 1B, and this SSR locus was closely linked to the marker xwmc419 (Somers et al. 2004), which in our RIL population was associated with QTgw.nfcri-1B. Similarly, on chromosome 2A, in the interval xbarc1165-xbarc124, the detected QTL QTgw.nfcri-2A for TGW identified in our population seemed to correspond with the QTL previously detected by Campbell et al. (1999), which was linked to the marker xcdo456B, 2 cM distant from xcdo456B according to our genetic map. Furthermore, QTgw.nfcri-3B found in the present study, closely linked to the marker xgwm533 seemed to correspond to the QTL for TGW described by Groos et al. (2003). This QTL was linked to the marker xcfd79, which mapped in a distance of 7 cM from xgwm533 according to the consensus map of Somers et al. (2004).
Some of the eight QTL for GNE identified here have probably also been located in other populations. For example, QGne.nfcri-2D and QGne.nfcri-4D were located in the same genomic regions as reported by Börner et al. (2002), Marza et al. (2006) and McCartney et al. (2005). For GWE, out of nine QTL identified on chromosomes 1A, 2A, 3B, 4D and 6B, some have been previously reported. For example, QGwe.nfcri-2A appears to be coincident with the locus described by Li et al. (2007), and QGwe.nfcri-3B coincident QTL QTgw.nfcri-3B detected in our study seemed to correspond to QTL for GWE detected by Groos et al. (2003).
In addition, FT is positively correlated with MT (r = 0.68, P < 0.0001) in our population; as expected, two coincided loci were detected for these two traits. For example, the QTL QFt.nfcri-1B and QFt.nfcri-2B were found in the same genomic region with QMt.nfcri-1B and QMt.nfcri-2B for MT. One of them, QTL QFt.nfcri-1B was located in the same region reported by Marza et al. (2006); on the other hand, QFt.nfcri-2B located in the interval xbarc373-xbarc160 seemed to correspond to the interval xrz444-xcdo405 of the QTL for FT as reported by Börner et al. (2002). Among the six QTL for GFD detected in our study, QGfd.nfcri-1A and QGfd.nfcri-3B are probably coincident with the QTL for GFD described by Börner et al. (2002). Furthermore, the QTL (QGfd.nfcri-1B) were linked to the marker xgwm11 on chromosome 1B and QTL (QGfd.nfcri-5A) linked to the marker xgwm293 on chromosome 5A had been reported by Yang et al. (2002).
QTL for PHT have been identified in almost every wheat chromosome. In the present study, four QTL for PHT were identified in our population on chromosomes 1D, 2D, 3D and 4D. Particularly, QPht.nfcri-1D and QPht.nfcri-4D were detected in both trial sites. Moreover, QPht.nfcri-2D, linked to the marker xgwm296, may correspond to the QTL for PHT likely previously reported by Börner et al. (2002); QPht.nfcri-4D, located within the marker interval xwmc617-xwmc48, seemed to correspond to the QTL for PHT found by McCartney et al. (2005).
Pleiotropic effects and QTL pyramiding for breeding
Out of 99 putative QTL detected for grain filling rate and yield-related traits, a large number of QTL showed relatively small effects and only a few major large-effects QTL were identified in the present study. Thus presents a serious challenge for using single QTL with small effect for MAS, and therefore we have paid more attention to explore those stable QTL detected in three or four environments and those QTL with pleiotropic effects. So a QTL pyramiding scheme seemed to be useful for high-yield breeding based on the idea of efficiently accumulating beneficial QTL by using MAS.
In the present study, 13 loci distributed on chromosomes 1A, 1B, 1D, 2A, 2B, 2D, 3A, 3B, 4B, 4D, 5B, 6D and 7D are detected revealing pleiotropic effects. Particularly, three QTL for GFRmean located on the chromosomes 1B (QGfrmean.nfcri-1B), 2A (QGfrmean.nfcri-2A) and 3B (QGfrmean.nfcri-3B) were simultaneously associated with several traits such as GFRmax, GWE and TGW, and accounted for a substantial proportion of the phenotypic variation. Moreover, these three QTL have been confirmed by the single marker regression analysis (Table 4). Focusing on TGW and GFRmean, the analysis of effects of alternative alleles indicated that the more positive alleles of xwmc419 (1B), xgwm359 (2A) and xbarc113 (3B) contributed by the parental line Y8679 the higher TGW and GFRmean. Furthermore, there were significant differences between the RILs possessing two positive alleles (AAa, AaA, aAA) compared to one positive allele (Aaa, aAa, aaA). In summary, all these facts indicated that these three QTL revealing pleiotropic effects seemed to be effective and useful for MAS breeding.
Further study
The grain filling process is important not only for the accumulation of grain weight, but also for the determination of grain size. We are therefore seeking to explore the relationship between grain filling rate and grain size, by conducting a QTL mapping for grain size (grain length, grain width, grain thickness). Moreover, grain filling is a process of metabolism of carbohydrate and starch accumulation with 33 enzymes involved. Among them, sucrose synthase (SuSase), ADP glucose pyrophosphorylase (ADPGase), starch synthase (SSase) and starch branch enzyme (SBE) play important roles in starch biosynthesis and accumulation (Yang et al. 2003; Hurkman et al. 2003; Zhao et al. 2005). Of interest would be to attempt to correlate the activity of these key enzymes with grain filling rate, which could be achieved by a time-related QTL mapping exercise. A comparison of such time-related QTL and GFR QTL could serve to clarify the relationship between the enzymes and GFR. Moreover, considering the time-consuming measurement of GFR, the coincident loci controlling these traits will provide valuable information for the selection of genotypes with high GFR by the easier measurement of the activity of the enzymes at a certain stage during the grain filling period of wheat. Further on, the same assay could then also be applied for a search for novel alleles in wheat germplasm.
Abbreviations
- CIM:
-
Composite interval mapping
- IMTI:
-
International triticeae mapping initiative
- MAS:
-
Marker aided selection
- GFRmean :
-
Mean grain filling rate
- GFRmax :
-
Maximum grain filling rate
- GFD:
-
Grain filling duration
- GNE:
-
Grain numbers per ear
- GWE:
-
Grain weight per ear
- FT:
-
Flowering time
- MT:
-
Maturation time
- PHT:
-
Plant height
- TGW:
-
Thousand grain weight
- QTL:
-
Quantitative trait loci
- RILs:
-
Recombinant inbred lines
- SSR:
-
Simple sequence repeat
- EST:
-
Expressed sequence tag
- Y8679:
-
Yu8679
- HSM:
-
Heshangmai
- BJ:
-
Beijing
- CD:
-
Chengdu
- HF:
-
Hefei
References
Bassam BJ, Caetano AG, Gresshoff PM (1991) Fast and sensitive silver staining of DNA in polyacrylamide gels. Anal Biochem 196:80–83
Börner A, Schumann E, Furste A, Coster H, Leithold B, Röder MS, Weber WE (2002) Mapping of quantitative trait loci determining agronomic important characters in hexaploid wheat (Triticum aestivum L.). Theor Appl Genet 105:921–936
Bruckner PL, Frohberg RC (1987) Rate and duration of grain fill in spring wheat. Crop Sci 27:451–455
Campbell KG, Bergman CJ, Gualberto DG, Anderson JA, Giroux MJ, Hareland G, Fulcher RG, Sorrells ME, Finney PL (1999) Quantitative trait loci associated with kernel traits in a soft × hard wheat cross. Crop Sci 39:1184–1195
Cross HZ (1975) Diallel analysis of duration and rate of grain filling of seven inbred lines of corn. Crop Sci 15:532–535
Cui KH, Peng SB, Xing YZ, Yu SB, Xu CG, Zhang Q (2003) Molecular dissection of the genetic relationships of source, sink and transport tissue with yield traits in rice. Theor Appl Genet 106:649–658
Darroch BA, Backer RJ (1990) Grain filling in three spring wheat genotypes: statistical analysiss. Crop Sci 30:525–529
Egli DE, Ramseur EL, Yu ZW, Sullivan CH (1989) Source-sink alterations affect the number of cells in soybean cotyledons. Crop Sci 29:732–735
Gebeyehou G, Knott DR, Baker RJ (1982) Rate and duration of grain filling in durum wheat cultivars. Crop Sci 22:337–340
Groos C, Robert N, Bervas E, Charmet G (2003) Genetic analysis of grain protein-content, grain yield and thousand-kernel weight in bread wheat. Theor Appl Genet 106:1032–1040
Gupta PK, Balyan HS, Edwards KJ, Isaac P, Korzun V, Röder MS, Gautier MF, Joudrier P, Schlatter AR, Dubcovsky J, DelaPena RC, Khairallah M, Penner G, Hayden MJ, Sharp P, Kellar B, Wang RCC, Hardouin JP, Jack P, Leroy P (2002) Genetic mapping of 66 new microsatellite (SSR) loci in bread wheat. Theor Appl Genet 105:413–422
Huang XQ, Coster H, Ganal MW, Röder MS (2003) Advanced backcross QTL analysis for the identification of quantitative trait loci alleles from wild relatives of wheat (Triticum astivum L.). Theor Appl Genet 106:1379–1389
Huang XQ, Kempf H, Ganal MW, Röder MS (2004) Advance backcross QTL analysis in progenies derived from a cross between a German elite winter wheat variety and a synthetic wheat (Triticum aestivum L.). Theor Appl Genet 109:933–943
Huang XQ, Cloutier S, Lycar L, Radovanovic N, Humphreys DG, Noll JS, Somers DJ, Brown PD (2006) Molecular detection of QTL for agronomic and quality traits in a doubled haploid population derived from two Canadian wheats (Triticum aestivum L.). Theor Appl Genet 113:753–766
Hurkman WJ, McCue KF, Altenbach SB, Korn A, Tanaka CK, Kothari KM, Johnson EL, Bechtel DB, Wilson JD, Anderson OD, DuPont FM (2003) Effect of temperature on expression of genes encoding enzymes for starch biosynthesis in developing wheat endosperm. Plant Sci 164:873–881
Jenner CF, Rathjen AJ (1972) Limitations to the accumulation of starch in the developing wheat grain. Ann Bot 36:743–754
Kirigwi FM, Ginkel MV, Guedira GB, Gill BS, Paulsen GM, Fritz AK (2007) Markers associated with a QTL for grain yield in wheat under drought. Mol Breed 20:401–413
Knott DR, Gebeyehou G (1987) Relationship between the lengths of the vegetative and grain filling periods and the agronomic characters in three durum wheat crosses. Crop Sci 27:857–860
Kosambi DD (1944) The estimation of map distances from recombination values. Ann Eugen 12:172–175
Kumar N, Kulwal PL, Gaur A, Tyagi AK, Khurana JP, Khurana P, Balyan HS, Gupta PK (2006) QTL analysis for grain weight in common wheat. Euphytica 151:135–144
Lander ES, Green P, Abrahamson J, Barlow A, Daly MJ, Lincoln SE, Newburg L (1987) MAPMAKER: an interactive computer package for constructing primary genetic linkage maps of experimental and natural populations. Genomics 1:174–181
Li SS, Jia JZ, Wei XY, Zhang XC, Li LZ, Chen HM, Fan YD, Sun HY, Zhao XH, Lei TD, Xu YF, Jiang FS, Wang HG, Li LH (2007) A intervarietal genetic map and QTL analysis for yield traits in wheat. Mol Breed 20:167–178
Li XJ, Pan ZD (2005) A study on the grain filling characteristic of different weight wheat. Rev China Agri Sci Tech 7:26–30
Manness NO (1989) High temperature limitation for enzymatic sugar conversion in wheat kernels. Wheat, Barley and Triticale Abstract 6, 422
Marza F, Bai GH, Carver BF, Zhou WC (2006) Quantitative trait loci for yield and related traits in the wheat population Ning 7840 × Clark. Theor Appl Genet 112:688–689
Mashiringwani NA, Schweppenhauser MA (1992) Phenotypic characters associated with yield adaptation of wheat to a range of temperature conditions. Field Crops Res 29:69–77
McCartney CA, Somers DJ, Humphreys DG, Lukow O, Ames N, Noll J, Cloutier S, McCallum BD (2005) Mapping quantitative trait loci controlling agronomic traits in the spring wheat cross RL4452 × ‘AC Domain’. Genome 48:870–883
Mo HD (1992) Agricultural experimentation, 2nd edn. Shanghai Sci. & Tech Press, Shanghai, pp 22–25
Mou B, Kronstad WE (1994) Duration and rate of grain filling in selected winter wheat populations: I. Inherit Crop Sci 34:833–837
Nass HG, Reisser B (1975) Grain filling period and grain yield relationships in spring wheat. Can J Plant Sci 55:673–678
Paterson AH, Damon S, Hewitt JD, Zamir D, Rabinowitch HD, Lincoln SE, Lander ES, Tanksley SD (1991) Mendelian factors underlying quantitative traits in tomato: comparison across species, generations, and environments. Genetics 127:181–197
Peng JH, Ronin Y, Fahima T, Röder MS, Li YC, Nevo E, Korol A (2003) Domestication quantitative trait loci in Triticum dicoccoides, the progenitor of wheat. Proc Natl Acad Sci USA 100:2489–2494
Pinthus MJ, Shalom YS (1978) Dry matter accumulation in the grains of wheat (Triticum aestivum L.) cultivars differing in grain weight. Ann Bot 42:469–471
Quarrie SA, Steed A, Calestani C, Semikhodskii A, Lebreton C, Chinoy C, Steele N, Pljevljakusic D, Waterman E, Weyen J, Schondelmaier J, Habash DZ, Farmer P, Saker L, Clarkson DT, Abugalieva A, Yessimbekova M, Turuspekov Y, Abugalieva S, Tuberosa R, Sanguineti MC, Hollington PA, Aragues R, Royo A, Dodig D (2005) A high-density genetic map of hexaploid wheat (Triticum aestivum L.) from the cross Chinese Spring × SQ1 and its use to compare QTLs for grain yield across a range of environments. Theor Appl Genet 110:865–880
Röder MS, Korzun V, Wendehake K, Plaschke J, Tixier MH, Leroy P, Ganal MW (1998) A microsatellite map of wheat. Genetics 149:2007–2023
Somers DJ, Isaac P, Edwards K (2004) A high-density microsatellite consensus map for bread wheat (Triticum aestivum L.). Theor Appl Genet 109:1105–1114
Sourdille P, Cadalen T, Guyomarch H, Snape JW, Perretant MR, Charmet G, Boeuf C, Bernard S, Bernard M (2003) An update of the Courtot × Chinese spring intervarietal molecular marker linkage map for the QTL detection of agronomic traits in wheat. Theor Appl Genet 106:530–538
Thomson MJ, Tai TH, McClung AM, Lai XH, Hinga ME, Lobos KB, Xu Y, Martinez CP, McCouch SR (2003) Mapping quantitative trait loci for yield, yield components and morphological traits in an advanced backcross population between Oryza rufipogon and the Oryza sativa cultivar Jefferson. Theor Appl Genet 107:479–493
Van Sanford DA (1985) Variation in kernel growth characters among soft red winter wheats. Crop Sci 25:626–630
Wang GL, Kang MS, Moreno O (1999) Genetic analyses of grain-filling rate and duration in maize. Field Crops Res 61:211–222
Wiegand CL, Cuellar JA (1981) Duration of grain filling and kernal weight as affected by temperature. Crop Sci 21:95–101
Yang JC, Zhang JH (2006) Grain filling of cereals under soil drying. New Phytol 169:223–236
Yang J, Sears RG, Gill BS, Paulsen GM (2002) Quantitative and molecular characterization of heat tolerance in hexaploid wheat. Euphytica 126:275–282
Yang JC, Zhang JH, Wang ZQ, Zhu QS, Liu LJ (2003) Activities of enzymes involved in sucrose-to-starch metabolism in rice grains subjected to water stress during filling. Field Crops Res 81:69–81
Zeng ZB (1994) Precision mapping of quantitative trait loci. Genetics 136:1457–1468
Zhao BH, Zhang WJ, Wang ZQ, Zhu QS, Yang JC (2005) Changes in activitis of the key enzymes related to starch synthesis in rice grains during grain filling and their relationships with the filling rate and cooking quality. Agri Sci China 4:26–33
Acknowledgments
This study was supported by the research project 06-02-03B granted by Chinese Ministry of Agriculture. We would like to thank researchers Zhonghu He and Xianchun Xia (Chinese Academy of Agricultural Sciences) for providing the SSR markers. We are grateful to researcher Ling Wu (Sichuan Province Academy of Agricultural Sciences) and researcher Rui Wang (Anhui Province Academy of Agricultural Sciences) for field evaluations in Chengdu and Hefei, respectively. We thank Dr. Carola Wagner (Department of Plant Breeding, Justus-Liebig-University Giessen, Germany) for improving the English language of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by C. Hackett.
Rights and permissions
About this article
Cite this article
Wang, R.X., Hai, L., Zhang, X.Y. et al. QTL mapping for grain filling rate and yield-related traits in RILs of the Chinese winter wheat population Heshangmai × Yu8679. Theor Appl Genet 118, 313–325 (2009). https://doi.org/10.1007/s00122-008-0901-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-008-0901-5