
Abstract
A population of 96 doubled haploid lines (DHLs) was prepared from F1 plants of the hexaploid wheat cross Chinese Spring × SQ1 (a high abscisic acid-expressing breeding line) and was mapped with 567 RFLP, AFLP, SSR, morphological and biochemical markers covering all 21 chromosomes, with a total map length of 3,522 cM. Although the map lengths for each genome were very similar, the D genome had only half the markers of the other two genomes. The map was used to identify quantitative trait loci (QTLs) for yield and yield components from a combination of 24 site × treatment × year combinations, including nutrient stress, drought stress and salt stress treatments. Although yield QTLs were widely distributed around the genome, 17 clusters of yield QTLs from five or more trials were identified: two on group 1 chromosomes, one each on group 2 and group 3, five on group 4, four on group 5, one on group 6 and three on group 7. The strongest yield QTL effects were on chromosomes 7AL and 7BL, due mainly to variation in grain numbers per ear. Three of the yield QTL clusters were largely site-specific, while four clusters were largely associated with one or other of the stress treatments. Three of the yield QTL clusters were coincident with the dwarfing gene Rht-B1 on 4BS and with the vernalisation genes Vrn-A1 on 5AL and Vrn-D1 on 5DL. Yields of each DHL were calculated for trial mean yields of 6 g plant−1 and 2 g plant−1 (equivalent to about 8 t ha−1 and 2.5 t ha−1, respectively), representing optimum and moderately stressed conditions. Analyses of these yield estimates using interval mapping confirmed the group-7 effects on yield and, at 2 g plant−1, identified two additional major yield QTLs on chromosomes 1D and 5A. Many of the yield QTL clusters corresponded with QTLs already reported in wheat and, on the basis of comparative genetics, also in rice. The implications of these results for improving wheat yield stability are discussed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The advent of molecular markers has revolutionised the genetic analysis of complex traits, and reports on the location of quantitative trait loci (QTLs) for yield for many cereal crops are commonplace [see Li et al. (2001) for rice; Thomas (2003) for barley; Khavkin and Coe (1997) for maize] . However, QTL analysis of quantitative traits in bread wheat has in part been hampered by its large genome size, estimated to be around 14,500 Mbp/1C (Bennett et al. 1982), with the large majority of this DNA being repetitive sequences. Thus, many markers are required to cover the whole genome adequately. In addition, because of the relatively recent origin of the species (Huang et al. 2002), hexaploid wheat also suffers from relatively low levels of polymorphism (Chao et al. 1989). In consequence, detailed genetic maps of the whole genome are much more difficult to achieve than for most other crop species.
A few genetic maps covering all of the hexaploid wheat genome have been reported (Liu and Tsunewaki 1991; Gale et al. 1995; Cadalen et al. 1997; Röder et al. 1998; Messmer et al. 1999; Chalmers et al. 2001; Groos et al. 2003; Paillard et al. 2003; for reviews, see Langridge et al. 2001 and Hart 2001), the most detailed of which is the International Triticeae Mapping Initiative (ITMI) wheat map from the cross Synthetic W7984 × Opata 85 (Nelson et al. 1995a, b, c; Van Deynze et al. 1995; Marino et al. 1996). To increase the levels of polymorphism, several of these maps have been made by crossing relatively distantly related parental lines (Gale et al. 1995; Röder et al. 1998; Messmer et al. 1999). The number of whole genome maps developed from crosses between wheat cultivars or breeding lines is more limited.
Grain yield is a particularly complex trait, being the end product of many processes in the plant and, in consequence, is very environmentally dependent. Therefore, yield usually has a low heritability. A detailed genetic map of wheat would allow the expression of this complex trait to be broken down into its genetic components by QTL analysis. Each of these genetic components could in turn be associated with particular yield components—ears per plant, grains per ear and thousand grain weight (TGW). The low heritability of yield would be revealed as variability in the location of yield QTLs from experiment to experiment or treatment to treatment. Thus, in a particular genetic background, QTL analysis would allow the presence of QTLs to be identified which are environmentally relatively stable, thereby providing the breeder with targets for marker-assisted variety improvement.
We are aware of only a very few reports of QTL analysis of grain yield covering the whole genome of hexaploid wheat (Ayala et al. 2002; Börner et al. 2002; Byrne et al. 2002; Groos et al. 2003). The most detailed analysis presented to date has been by Börner et al. (2002) for the ITMI wheat mapping population, who identified QTLs for yield components across a wide range of sites and years. Genetic control of tiller number per plant has also been examined in the ITMI population by Li et al. (2002), and grain size in wheat by Ammiraju et al. (2001), although only a few of the associated simple sequence repeats (SSRs) were mapped.
We present here a new genetic map for hexaploid wheat and describe its use to compare the distribution of QTLs for grain yield in a population of doubled haploid lines (DHLs) compiled from 24 site × treatment × year experiments. Because yield is so environmentally dependent, and its genetic control is, therefore, variable from one occasion to the next, the intention was to build up a picture of regions of the wheat genome where QTLs for yield were repeatedly present and to relate these QTLs to particular yield components. In this way, future work could be focused more effectively on these regions of consistency. Although one of the parents used for this mapping population (Chinese Spring) is unsuitable agronomically, it is well characterised genetically, and many aneuploid stocks are available to help assign chromosomal locations to molecular markers.
Materials and methods
Genetic stocks
The parents chosen for the mapping population were hexaploid wheat (Triticum aestivum L.) genotypes Chinese Spring (CS) and SQ1, a high abscisic acid-expressing breeding line (line no. 25/3/4/2/1) extracted from the F7 population of the spring wheat cross Highbury × TW269/9/3/4 (Quarrie 1981; 1987a, b). A population of DHLs was prepared from F1 plants using the maize pollination method (Laurie and Reymondie 1991). Of the original 152 DHLs, 96 were selected at random to construct the genetic map, but as one line (CSDH77) subsequently became sterile, 95 DHLs were ultimately used for most marker and all phenotype analyses. The mapping population of 95 DHLs and the parents are available from the John Innes Centre, Norwich (contact person: Prof. John Snape, e-mail: john.snape@bbsrc.ac.uk).
Molecular marker characterisation and map construction
Leaves were taken from the DHLs and the parents for DNA extraction using a CTAB method (Murray and Thompson 1980). For restriction fragment length polymorphism (RFLP) analysis, DNA was restricted with the enzymes EcoRI, EcoRV, DraI and HindIII, separated on agarose gels and blotted onto Hybond N+ membranes essentially as described by Sharp et al. (1988). Probe labelling with [32P] and filter hybridisation were also carried out essentially as described by Sharp et al. (1988). Probes for RFLP analysis came largely from the collections of the John Innes Centre (prefixes: psb, psr). Other RFLP probes came from the Institute for Resistance Genetics, Grünbach (prefix: mwg), University of Cornell, Ithaca (prefixes: bcd, wg), Prof. M. Hughes, Newcastle University (prefix: blt), Dr. J. Mundy, Carlsberg Research Centre, Copenhagen (prefix: rab), Prof. R. Quatrano, University of North Carolina, Chapel Hill (prefix: rsq).
Wheat microsatellites (SSRs), which originated from the collections of the John Innes Centre (prefix: psp), Institut für Pflanzengenetik und Kulturpflanzen-forschung, Gatersleben (prefix: gwm), Wheat Microsatellite Consortium (Agrogene-Consortium; prefix: wmc), duPont (prefix: dupw; Eujayl et al. 2002) and R. Ward (prefix: barc), were analysed at the John Innes Centre with the parents and DHLs using the protocol of Bryan et al. (1997) or at Saaten-Union Resistenzlabor GmbH by standard amplification protocols, with one of the SSR primers labelled with FAM, HEX or NED for separation and visualisation using an ABI 3100 capillary sequencer (Applied Biosystems, Foster City, Calif.). SSR probes with the prefix SU are proprietary probes.
Screening of the parents and the DHLs for amplified fragment length polymorphism (AFLPs) was carried out according to Vos et al. (1995) using the four-base cutter Mse1 and six-base cutter Pst1 (prefix: m). In addition, the population segregated qualitatively for several morphological and biochemical markers (the dwarfing gene Rht-B1, scored using the gibberellin insensitivity test; Vrn-A1, for vernalisation sensitivity; an iodine-binding isozyme, ibf-D1; seed glutenin and gliadin storage proteins, Glu and Gli). Markers were also screened on the nullisomic-tetrasomic stocks of CS to assign them to chromosomes, where possible.
A genetic map was constructed using mapmaker macintosh ver. 2.0 [kindly supplied by S. Tingey, duPont de Nemours, and based on the algorithms of mapmaker (Lander et al. 1987)] using the Kosambi (Kosambi 1944) mapping function. Previously mapped RFLP and SSR markers from Gale et al. (1995), GrainGenes (http://wheat.pw.usda.gov/GG2/index.shtml) and R. Appels (unpublished consensus map information) were used as anchors within linkage groups, and the best order of other markers within groups was determined using the compare command. Several AFLP markers were rejected either because of severe segregation distortion or because their insertion into the linkage group considerably extended the map length.
Using the initial marker orders within linkage groups given by mapmaker macintosh , the dataset of scores was examined for potential errors as suggested by the presence of implied adjacent recombination events. These scores were reassessed and suspicious scores eliminated. A final map was then constructed with mapmaker macintosh using the corrected dataset and with anchor markers oriented within linkage groups according to existing wheat map information.
Yield trials
Full experimental details of each field trial will be given elsewhere. Because of variability in plant establishment for some lines in some trials and variation in sowing density between trials, data are generally presented here as yield per plant.
Norwich, UK
The DHLs were trialled in 1994, 1997 and 1998. Seeds were sown in micro-plots at 5-cm spacings in 1.2-m-long rows spaced 15 cm apart. In 1994, lines were trialled in two replicates of a single row per DHL in a high-fertility soil, with the addition of fertiliser and in a low-fertility soil without the addition of fertiliser. In 1997 and 1998, DHLs were trialled in two-row plots in the low-fertility soil in three replicates with three nitrogen treatments of 45 (low), 90 (medium) and 170 (high) units ha−1 of N, using a latin square design. Seeds were sown in mid-March, and normal agricultural practices were applied for disease and weed control. Supplementary water was given by overhead irrigation when necessary to maintain adequate soil water contents.
Zajecar, Serbia
Plants were trialled in 1999–2000 and 2000–2001 at the Agricultural Research Institute-Serbia, Centre for Agricultural and Technological Research (CATR), situated in the drought-prone Timok region of southeastern Serbia. The DHLs were grown in a fertile soil in two treatments (rainfed and irrigated) with two replicate plots of three 1-m long rows at 20-cm spacings, with typically 50–70 plants per row reaching maturity. Irrigated plants were watered by hand once soil water contents had declined to less than 70% of field capacity. Seeds were sown in late October in 1999 and mid-November in 2000. The year 2000 was particularly dry, though 2001 was wetter than usual.
Almaty, Kazakhstan
The DHLs were trialled in widely-spaced (80 cm), single-row plots of ten plants at 15-cm spacings with three replicates in fields at an experimental farm of the Kazak Research Institute of Agriculture, Almaty region, Kazakhstan. The DHLs were tested in a low-salinity soil under irrigated and non-irrigated conditions in 1998, 1999 and 2000. Irrigation was given by temporarily flooding the site. Seeds were sown on 2 April in 1998 and 2000 and on 16 April in 1999. The 2000 season was particularly severe with high temperatures during grain filling, and a small number of DHLs produced ears but failed to produce any seeds. These DHLs were included in the analyses with yields of 0 g plant−1.
Zaragoza, Spain
A high-fertility site at the Unidad de Suelos y Riegos, Servicio Investigación Agroalimentaria was used to trial the DHLs for two seasons. This site was equipped with trickle irrigation to provide either fresh or saline water to the plots (Isla et al. 1997), which consisted of six 1.4-m-long rows with 20-cm spacings. Each DHL was represented by one row, with two replications in both a control (electrical conductivity, EC=1.5 dS m−1) and two saline (medium, EC=8.2 dS m−1; high, EC=16.4 dS m−1) irrigation treatments. Seeds were sown in November 1998 and 1999, and the saline irrigation treatments started in February. In the 1999–2000 trial, seed yields were determined from representative ears, and ears per plant were not determined. However, under the high-saline treatment, plants were usually uni-culm.
From all of the experiments, except Norwich 1994 and Zaragoza 1999, the yield components ears per plant (not in Zaragoza), grains per ear and TGW were recorded on representative plants.
Statistical analyses
Initially, individual plot yield data along the beds were examined for spatial heterogeneity around sites. There was no evidence in these trials for unusual trends within the beds. However, although genotypes were grouped according to expected height category, height trends along the beds were not always uniform. Therefore, yield data were tested for near-neighbour effect, using the mean height of the two adjacent genotypes as the independent variable in linear regressions against yield. Where regressions were significant, yield data were adjusted by adding the genotype regression residual to the plot mean yield.
Standard statistical analyses were performed using either Microsoft excel 98 (ver. 8.0), for Chi-square analysis, correlation and linear regression analysis, and anova or qstats (from the qtlcartographer suite, see below) for testing normality and identifying outliers. The presence of QTLs was determined with qtlcartographer (Basten et al. 1996) ver. 1.16c (March 2002) for Macintosh using either linear regression with lrmapqtl to identify individual markers significantly associated with trait variation, or interval mapping (zmapqtl option 3).
Results
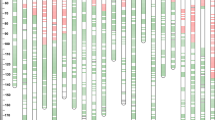
The genetic map (Fig. 1) consists of 567 markers assigned to 21 linkage groups, giving a total map length of 3,521.7 cM, with an average chromosome length of 168 cM [range: 93.0 cM (chromosome 1D) to 264.2 cM (chromosome 5D)]. In nearly every case, the order of anchor markers within linkage groups from previously published information was also the most likely order of markers identified by mapmaker macintosh. The approximate positions of centromeres were determined by comparison with the John Innes Centre (JIC) wheat consensus map (Gale et al. 1995). From the JIC consensus map, the CS × SQ1 map was estimated to provide effective coverage of 85–90% of the wheat genome.


a, b Genetic map of Chinese Spring (CS) × SQ1 derived from 96 DHLs, with the positions of marker loci on chromosomes shown as horizontal bars. Loci named in blue are RFLP markers, loci in red are PCR-based markers, loci in green are morphological, physiological and isozyme markers, loci in black are AFLP markers. Map distances are shown in centiMorgans and were calculated using the Kosambi (1944) mapping function. Underlined markers were assigned to chromosomes using CS nullisomic-tetrasomic stocks. Markers shown in bold were mapped in multiple genomes and are joined by dashed lines. Markers in parenthesis were excluded from the QTL analyses. Locations of major genes Rht-B1 and Vrn-A1 are shown as thick red and green horizontal bars, respectively. Chromosome segments shown in white and black indicate regions of significant segregation distortion (white—CS alleles in excess; black—SQ1 alleles in excess). The translocated segments of chromosomes 4A and 5AL are indicated: diagonal shading 4AL-5AL, dark hatching 4AL-4AS, pale hatching 7BL-4AS. Black ovals Estimated location of centromeres (based mainly on Gale et al. 1995)
The map length was divided approximately equally amongst the three genomes: 1,148.0 cM, 1,204.8 cM and 1,168.9 cM for the A, B and D genomes, respectively. However, the distribution of markers amongst the genomes was not uniform, with twice as many polymorphic markers mapping to the A and B genomes as to the D genome: 224, 228 and 115 markers for the A, B and D genomes, respectively. The majority of markers were SSRs (242), with 194 AFLPs, 123 RFLPs and eight other morphological and biochemical loci.
The distribution of markers within chromosomes was not uniform, with gaps of more than 40 cM on chromosomes 2D, 3A, 3D, 4D, 5D, 6A, 6D and 7B and concentrations of markers around the centromere particularly noticeable on chromosomes 2A, 5A, 6A and 6B. Duplicate or triplicate loci were identified by 37 probes/primers (markers in bold connected by dashed lines in Fig. 1), and these allowed relative alignments of the three genomes to be made. A further four loci on the chromosome 4A ‘long’ arm had duplications on the short arms of the group-7 chromosomes (Xpsr160, Xpsr392, Xpsr490 and Xwmc283), thereby confirming the 7BS translocation to chromosome 4A (Devos et al. 1995; Nelson et al. 1995a). Two loci on the group-4 chromosomes (Xgwm6 and Xgwm269) also had duplications on chromosomes 5AL and 5DL, confirming the 5AL/4AL-4AS translocation and rearrangement events (Devos et al. 1995; Nelson et al. 1995a). Markers shown in parenthesis in Fig. 1 were subsequently deleted from the map to remove duplications and provide more uniform spacing between markers for subsequent genetic analysis in this work and other investigations (manuscripts in preparation).
Regions of significant segregation distortion (shown as white or black boxes within chromosomes in Fig. 1) were found on chromosomes 1AL, 3BS, 4AL, 4AS, 4DS/L, 5AS, 6AS and 7BL. Four regions had an excess of alleles from CS and four from SQ1. The frequency of DHLs having parental (non-recombinant) chromosomes varied inversely with chromosome length, with 19 lines (20%) per chromosome of approximate length 150 cM.
QTLs for grain yield and yield components
Although the parents of the mapping population were originally chosen because of large differences in the drought-induced accumulation of the stress hormone abscisic acid (Quarrie et al. 1994), the progeny of the cross are very diverse for many other traits. In particular, the DHLs differ in several traits that are known to be associated with yield—such as flowering date (typically a range of 2–3 weeks under field conditions), vernalisation and day-length sensitivity—height (which ranges from 40 cm to 140 cm in the field and up to 180 cm under glasshouse conditions), awn length (which ranges from no awns to long awns) and variation in spikelet density, ear length and colour. In the highest-yielding UK trial (Norwich, high nitrogen, 1998), with a sowing density of 133 plants m−2, the yield of the best DHL from a spring sowing was equivalent to 10.0 t ha−1, and the worst, 2.9 t ha−1.
Yield trials were carried out over a wide range of conditions, including temperatures and photoperiods for which the DHLs were not necessarily well adapted and with or without the imposition of a range of stress treatments. In consequence, mean yields for the trials varied considerably (Table 1). Under non-stressed UK conditions, average yields for the spring-sown DHLs were the equivalent of 4–6 t ha−1 under the sowing densities used. Yields were particularly low under the severe nutrient deficiency conditions of the Norwich 1994 trial, the droughted treatment in Kazakhstan in 2000 and the high-salinity treatments of the Zaragoza trials.
When the small plot size of each trial is taken into consideration, the heritabilities on a genotype mean basis were relatively high, varying among trials from 25% to 98% (Table 1). In general, heritabilities were lower under stressed conditions.
Although many of the yield data have also been analysed using interval and composite interval mapping software (qtlcartographer zmapqtl options 3 and 6, results not presented), for simplicity and to facilitate comparison amongst experiments the results are presented here using the marker-by-marker linear regression analysis option of qtlcartographer (lrmapqtl), which gives results very similar to those for one-way anova and t-tests of allele classes at each marker locus.
The combined results for all 24 site × treatment × year combinations are shown in Fig. 2. For each significant (P<0.05) event at one or more markers, the vertical bars extend either side of the significant markers half-way to the first non-significant adjacent marker, so the lengths of bars are not a measure of the confidence interval. The thickness of the bars is proportional to the level of significance for the most significant marker in the cluster.

a, b Location of all QTLs for yield significant at P<0.05 using single marker analysis (lrmapqtl). QTLs are shown as vertical bars adjacent to chromosomes, with their lengths determined by the number of significant loci associated with a particular QTL (see text for description). QTLs obtained under non-stressed treatments (irrigated or good agronomy) are shown with solid bars; QTLs obtained under mild and severe stress treatments are shown with dashed and dotted lines, respectively. The level of significance of the most significant marker associated with a particular QTL is proportional to the thickness of the bar. Bars to the left of chromosomes indicate beneficial alleles from SQ1; those to the right of chromosomes indicate beneficial alleles from CS. Clusters of five or more QTLs coincident at one or more markers are identified by brackets and are detailed in Table 2. The yield component(s) (when measured) most associated with each QTL is shown at the top of each bar (Ee, ears per plant; Gg, grains per ear; Tt, thousand grain weight). Uppercase letters indicate significant association with the yield component; lowercase letters indicate yield components not reaching significance at P<0.05. ? indicates that no yield component was obviously associated with the yield QTL. Other details as in Fig. 1
One or more QTLs for yield were identified on every chromosome, with chromosome 5B having the most (26) and chromosome 6D the least (3). Except for chromosome 3D, which had 20 significant QTLs, the D genome in general had fewer yield QTLs than the A and B genomes, which may reflect the much lower density of markers obtained for the D genome of this cross. The group-5 chromosomes gave the largest number of yield QTLs (67) and the group-6 chromosomes the least (23). Genes for yield were dispersed among the parents, although SQ1 alleles did account for almost 60% of the advantageous alleles at QTLs (187 and 135 for SQ1 and CS, respectively). High-yield alleles were predominantly from CS on the group-1 chromosomes and from SQ1 on the group-3 chromosomes. The greater number of advantageous alleles for yield QTLs for SQ1 was consistent with the yield characteristics of the parents. In most trials, SQ1 gave higher yields than CS (data not presented).
Despite the ubiquity of yield QTLs around the genome, several clusters of QTLs could be identified. Clusters of five or more QTLs are identified in Fig. 2 and listed in Table 2. Seventeen QTL clusters were distributed among each of the chromosome groups, although the group-4 chromosomes had the most QTL clusters. Of these yield QTL clusters, high yield in four of them was due predominantly to CS alleles and high yield in seven of them was due almost exclusively to SQ1 alleles; in the six remaining clusters, high yield was equally divided between CS and SQ1 alleles.
The clusters with the highest numbers of yield QTLs were the two occurring distal on the group-7 long arms, 7AL and 7BL, both of which occurred in 11 of the 24 trials. As the SSR locus from the wmc273 primers maps in the middle of the QTL clusters on both chromosomes, it is very likely that QTL effects here are due to homoeologous genes on 7AL and 7BL, with the high-yield alleles being contributed by SQ1 on 7AL and by CS on 7BL. Three other pairs of QTL clusters, possibly due to homoeologous genes on two of the genomes, were on 4BS/L and 4DL (associated with loci for SSR gwm165), 5AL and 5DL (coincident with the vernalisation genes, Vrn-A1 and Vrn-D1) and proximal on 5BS and 5DS.
Because of the wide range of yields among the trials, the sizes of allele effects at each QTL region (Table 2) are expressed in relation to the average yield across all DHLs for the trial, and these ranged from differences of around 12% to over 21% between the allele classes. The highest differences were for the homoeologous regions on chromosome 5BS/L and 5DS/L where the QTLs in both clusters were expressed predominantly in data from the Zaragoza salt-tolerance trials. However, as mean yields under the saline treatments were relatively low and the nature of the salt response was characteristically more variable than responses to the other stress treatments, larger differences among yields of the DHLs would be needed to reach statistical significance. The QTL region with the highest relative additive effect across a wide range of environments and treatments was on chromosome 7AL, with a yield difference between alleles of over 18% in favour of SQ1. For the high-yielding trials, this would represent a yield advantage for the SQ1 allele of 0.5 t ha−1.
In addition to the Zaragoza yield QTL clusters on chromosomes 5B and 5D, the only other region with a site-specific QTL cluster was on chromosome 4AS, which was specific to the Norwich trials. The yield QTL regions on chromosomes 1AL, 4BS/L, 4DL and 5DL were largely associated with one or other of the stress treatments.
The association of yield per plant with its components ears per plant, grain numbers per ear and TGW was highly significant (P<0.001) in a large majority of the trials, with correlations that were typically around 0.5–0.7 with each yield component. Exceptions for ears per plant were for N7C (see Table 2 for code) and N7M in Norwich, 1997, where correlations with yield were not significant. The correlation with grains per ear was not significant for K9C, and significant at only P<0.05 for K0S. With TGW, the correlations were not significant for Z0C and significant at only P<0.05 for N8L.
Consequently, the large majority of yield QTLs were coincident with QTLs for one or more yield components. Figure 2 and Table 2 show the yield components that were largely responsible for each of the significant yield QTLs. Most of the yield QTL clusters were coincident with significant effects (P<0.05, uppercase letters in Fig. 2) for more than one yield component, depending upon the trial. However, three yield QTL clusters were coincident with significant effects of, essentially, only a single yield component: TGW on chromosome 6BL and grain numbers per ear on the group-7 long arms.
Yield QTL variation across environments
Responses of CSDH lines to the environments were studied using the graphical analysis of Finlay and Wilkinson (1963) in which the yield of individual DHLs was compared with the trial mean yields. The DHLs showed large variation in their yield responses to the environment, with a threefold range in regression slopes between the extremes, CSDH146 and CSDH40 (Fig. 3). Linear regressions were significant at P<0.05 for every DHL. These linear regressions were used to calculate yields for each DHL at site mean yields of 6 g plant−1 and 2 g plant−1 (equivalent to about 8 t ha−1 and 2.5 t ha−1, respectively), which represent optimal conditions and a moderately severe stress. QTL analysis was carried out using both qtlcartographer lrmapqtl single-marker analysis and the zmapqtl module with option 3 for interval mapping, and the results for interval mapping compared in Fig. 4, which shows the QTL LR (likelihood ratio) output along each chromosome. Yield stability linear regressions for each DHL were also calculated with data for the two lowest yielding and two highest yielding trials deleted (to reduce bias due to leverage at the extremes) and yield data for another trial included (Zajecar, rainfed 2002; Dodig and Quarrie unpublished data). Yields for each DHL at mean yields of 6 g plant−1 and 2 g plant−1 were again calculated and the QTL analysis repeated. The two sets of analyses gave almost identical results in terms of the location of yield QTLs and their significance levels (data not presented).

A Finlay and Wilkinson (1963) plot of the two DHLs [line 146 (solid line and filled circles) and line 40 (dashed lineand open circles)] contrasting most in their yields across the 24 site × treatment × year trials. The fitted lines for linear regressions are shown

Interval mapping of QTL likelihood ratios (LR) for yields calculated at 6 g plant−1 (solid lines) and 2 g plant−1 (dotted lines). The direction of the QTL effect is indicated according to the orientation on the chromosome (left SQ1, right CS). The maximum level of significance of individual markers associated with QTLs, according to lrmapqtl, is shown: **P<0.01, ****P<0.0001. Brackets indicate the position of yield QTL clusters from Fig. 2. Other details as in Fig. 1
Under optimal conditions (6 g plant−1), highly significant (P<0.01) single-marker associations with yield were identified at five locations (2BS, 3BL, 4AL, 5BL and 7BL), although only the QTL on 7BL reached a LOD value greater than 2.0 (LR: 9.21) from interval mapping (Fig. 4). This QTL, near Xwmc273.2, had by far the strongest effect on yield (additive effect: 0.45 g plant−1) and was coincident with the yield QTL cluster identified from the individual trials. Another optimal yield QTL coincident with those from individual trials was on 2BS. At 2 g plant−1, the analysis gave seven highly significant (P<0.01) single-marker associations with yield (1DS, 3BS, 4AS, 4BL, 5AS, 7AL and 7BL), with those on 1DS, 3BS, 4AS, 5AS and 7AL exceeding a LOD score of 2.0 for interval mapping (Fig. 4). The QTLs on 4AS, 4BL, 7AL and 7BL were coincident with yield QTL clusters from individual trials, and the QTL on 7AL gave the largest additive effect (0.15 g plant−1).
The genotype of individual CSDH lines was examined at the five most significant QTL regions identified under optimal (6 g plant−1) and stressed conditions (2 g plant−1). CSDH146 gave the highest yields in several high-yielding individual trials (N8H, N8M, S9C) and was the highest yielding DHL at a site mean yield of 6 g plant−1 (9.0 g plant−1). This DHL also had the high-yield allele at each of the five QTLs under optimum conditions (SQ1 at 2BS, 3BL, 5BL; CS at 4AL and 7BL). Under stressed conditions (2 g plant−1), five DHLs among the six highest yielding DHLs (CSDH10, -18, -58, -90 and -96, with yields calculated to range from 2.4 g plant−1 to 2.9 g plant−1) had the high-yield allele at the five most significant locations for yield (SQ1 at 3BS, 4AS, 4BS, 5AS, 7AL; CS at 1DS).
Discussion
The genetic map for CS × SQ1 was very similar in length to those reported for the ITMI (Synthetic W7984 × Opata 85) map (3,551 cM; Nelson et al. 1995a, b, c; Van Deynze et al. 1995; Marino et al. 1996), CS × Synthetic map (2,830 cM; Gale et al. 1995), Arina × Forno map (3,086 cM; Paillard et al. 2003) and three maps described by Chalmers et al. (2001) of 3,164–4,110 cM. Chromosome lengths were similar to those reported for the ITMI and CS × Synthetic maps (Hart 2001; reviewed in Langridge et al. 2001). As with the other hexaploid wheat maps, the D genome gave the lowest number of polymorphic markers, although the map length of the D genome was similar to those of the A and B genomes. Clustering of markers around the centromeres is a common feature of wheat maps (see Chao et al. 1989; Gale et al. 1995; Blanco et al. 1998; Peng et al. 2000) because of reduced recombination in the proximal region of chromosome arms.
Although the relative order of markers within linkage groups is largely in agreement with other published mapping information, an anomalous locus on the 7B translocation segment of chromosome 4A remains unresolved. RFLP probe PSR490, a cDNA clone for sucrose synthase (Ss1), had loci on 7AS and 7BS but also on 4AL, despite careful checking of the marker scores and the testing of alternative map positions. Another RFLP probe (ABC465), which mapped on the 7BS-translocated chromosome 4A segment, has also been reported to map to the short arm of 7B (Chalmers et al. 2001). Chromosome 7BS on our map was definitely shorter than 7AS and 7DS. These results could be explained by an ancestral region of duplication on the group-7 short arms, with only part of the duplication on 7BS being translocated to 4A.
Although several regions of the genetic map showed significant segregation distortion, these were restricted to only 17 of 567 markers (3.0%) and were distributed among eight regions of the genome. In contrast, 27% of the markers mapped by Cadalen et al. (1997) deviated significantly from the 1:1 ratio in their mapping population (which was derived from anther culture), even though one of their parents was also CS. Notably, two major regions of segregation distortion around the centromeres of chromosomes 4A and 4D in our map are probably in locations equivalent to major regions of segregation distortion in the CS × Courtot map (no markers were located on 4D in that map). Cadalen et al. (1997) suggest that regions of segregation distortion may identify regions of the genome involved in the control of androgenic capability. Both populations were generated using chromosome doubling techniques to produce doubled haploids. The much smaller proportion of distorted segregations in our population implies that the maize pollination method has advantages over anther culture, as the former is associated with reduced androgenic influence on segregation ratios that were a feature of the CS × Courtot doubled haploids.
When we used the map to study the genetic control of yield a large number of regions of the wheat genome were revealed that regulate yield, depending on the site, treatment and year, with clustering of yield QTLs in several regions. The similar widespread occurrence of yield QTLs from occasion to occasion has also been noted in barley (Thomas 2003). The two yield QTL clusters on the group-7 long arms were particularly striking as they occurred in almost one-half of all the site × treatment × year trials (11 of 24 for each QTL cluster). Taken together these QTLs were equivalent to a yield advantage for the SQ1 genotype on 7AL and the CS genotype on 7BL of about 1 t ha−1 under favourable agronomic conditions. Both QTL regions were also identified using interval mapping of yields calculated for trial yields of 2 g plant−1 and 6 g plant−1. According to zmapqtl option 3, the 7AL QTL accounted for 22% of the variance at 2 g plant−1, and the 7BL QTL for 35% of the variance at 6 g plant−1.
Several of the QTL clusters coincided with yield or yield component QTLs previously identified in wheat. Two of the yield QTL regions in the CS × SQ1 population coincided with major genes mapped in our DHL population regulating plant height (Rht-B1 on 4BS) and vernalisation sensitivity (Vrn-A1 on 5AL). The majority of the QTLs on the 4BS/L cluster were associated with variation in grain number per ear. This would be expected as a pleiotropic consequence of the dwarfing gene Rht-B1 (Gale and Youssefian 1985). A third region of yield QTLs on 5DL is coincident with the predicted location of the vernalisation gene Vrn-D1, about 3.4 cM proximal of Xgwm292 (Snape et al. 2001).
A third set of major genes influencing environmental adaptability are the photoperiod sensitivity genes, Ppd, on chromosomes 2BS and 2DS. Ppd-B1 has been mapped 6.6 cM distal of Xgwm429 (equivalent to the location of m86p65.1 on 2BS in the CS × SQ1 map) and Ppd-D1 14.4 cM proximal of Xgwm261 (Butterworth 2000). Markers associated with variation in flowering date under different photoperiods have also been identified in the CS × SQ1 population at Xm86p65.1 on 2BS and Xwmc112 on 2DS (Quarrie and Melchiorre unpublished results), so it is likely that CS and SQ1 possess different alleles of both Ppd-B1 and Ppd-D1. The location of Ppd-B1 was coincident with another cluster of yield QTLs that centered on Xm86p65.1 (Fig. 2, Table 2). The Rht, Vrn and Ppd genes have been the focus of breeding for many years as a means to reduce the height of the crop and to fit plants better to their environments by flowering at the appropriate time, so the frequent association between these genes and QTLs for yield is not surprising. QTLs for yield have previously been identified on chromosomes 2B and 5B of bread wheat (Byrne et al. 2002), although the location on the linkage groups of these yield QTLs under high temperature was not specified.
The presence of two probably homoeologous regions proximal on chromsomes 5B and 5D (Fig. 2) controlling yield under conditions of salinity suggests the presence of one or more genes controlling salt tolerance. For some years the group-5 chromosomes have been regarded to carry genes for abiotic stress resistance (Sutka and Snape 1989; Manyowa and Miller 1991; Cattivelli et al. 2002) , including salt tolerance (Forster et al. 1988; Mahmood and Quarrie 1993). With the advent of molecular marker technologies, QTLs for salt tolerance have since been located on the group-5 chromosomes of both wheat (Semikhodskii et al. 1997) and barley (Mano and Takeda 1997; Pakniyat et al. 1997). In wheat, using an earlier version of the CS × SQ1 genetic map, Semikhodskii et al. (1997) identified a significant QTL for plant survival at Xpsr128 on chromosome 5A that was homoeologous to the two yield clusters on chromosomes 5B and 5D associated with the salinity trials in Spain. Pakniyat et al. (1997) studied salt tolerance in barley genotypes containing different alleles of the breviaristatum mutant (ari-e) affecting height and found a significant variation in shoot dry weight accumulation under salinity. The ari-e gene maps about 2 cM from the centromere on the short arm of 5H, corresponding well with the 5B and 5D clusters of yield QTLs in wheat.
Several other clusters of yield QTLs have also been identified previously in wheat, either controlling yield itself or a yield component. Groos et al. (2003) identified nine significant QTLs for yield in the bread wheat cross Renan × Recital, six of which were coincident with yield QTL clusters in the CS × SQ1 cross, either directly or at homoeologous loci. On the group 4s, yield QTLs found by Groos et al. (2003) on 4AL at Xcdo545 and centromeric on 4B near Xgwm113 (map alignments from Röder et al. 1998) corresponded to yield QTL clusters on 4AL (Xgwm160) and 4BS/L (Xgwm513) in our mapping population (Fig. 2, Table 2). Two homoeologous yield QTLs on chromosomes 5A and 5B of Renan × Recital corresponded with the locations of the vernalisation genes Vrn-A1 and Vrn-B1 (map alignments from Chalmers et al. 2001) already referred to above. Two 7DL yield QTLs near Pch1 in the Renan × Recital population were homoeologous to the two yield QTL clusters distal on 7AL and 7BL (map alignment from Gale et al. 1995). A QTL for grain number per ear (the yield component contributing to yield QTLs on 7AL and 7BL in the CS × SQ1 cross) was identified by Börner et al. (2003) to be distal on 7DL in the ITMI wheat mapping population. This yield component QTL was closest to Xfba204, which is in a homoeologous position to our 7AL and 7BL QTLs clustered around the Xwmc273 loci (map alignment from Röder et al. 1998). The ITMI population also gave a QTL on 7AL for the yield component spikelets per ear (a component of grains per ear) centered on Xfba97 (Li et al. 2002). This QTL is very close to the yield QTL cluster at Xgwm332 on 7AL in the CS × SQ1 population associated with grains per ear (map alignment from Röder et al. 1998). An effect of the group-7 long arm on yield has also been recorded using a 7DL.7Ag translocation line (Singh et al. 1998).
The yield QTL cluster on the chromosome 6B centromeric region (Fig. 2, Table 2) was in a homoeologous position to a yield QTL region identified by Blanco et al. (2002) that is near Xrsq805 on 6A. Blanco et al. (2002) also identified the yield QTL near the centromere on 4B. Our yield QTL cluster on 6B was due largely to variation in TGW. An association between an inter(I)-SSR marker on wheat 6BL and TGW has also recently been reported (Ammiraju et al. 2001). A yield QTL in the centromeric region of 6B was also found by Ayala et al. (2002) using the ITMI population. Another yield QTL identified by Ayala et al. (2002) at XksuG59 on 3DL was within the region of the yield QTL cluster on 3DL in our population (map alignment from Röder et al. 1998).
The yield QTLs identified by both single-marker analysis and interval mapping on 1D and distal on 5AS (Fig. 4) for a site mean yield of 2 g plant−1 do not appear to correspond to any yield or yield component QTLs previously published for wheat. These QTLs are unlikely to be statistical artifacts as they were present using both single-marker analysis and interval mapping with both the full set of site × treatment × year yield data as well as with the reduced set with extreme yielding sites excluded. Furthermore, as well as several QTLs for yield, individual yield components were frequently associated significantly with centromeric markers on 1D as well as those distal on 5AS (Xwg232.5): five occasions on 1D (mainly grains per ear) and ten occasions on 5AS (mainly grains per ear and TGW).
It is likely that the genes for two of our yield QTL clusters have already been cloned as the Rht-B1 and Vrn-A1 genes (Peng et al. 1999; Yan et al. 2003, respectively). Both of these genes were cloned using comparative genetics with rice at some stage to help locate the sequences. Therefore, the equivalent location in rice of each yield QTL cluster was determined (Table 2). This was achieved using a rice-wheat comparative database of RFLP markers mapped into both species (available at http://jic-bioinfo.bbsrc.ac.uk/cereals/wheat-rice.html, Quarrie et al. unpublished data). This database contains the predicted chromosomal location of around 500 loci in rice and the Triticeae. Only the yield QTL clusters on the long arms of chromosomes 4B and 4D could not be clearly assigned a probable homologous location in rice. These locations in rice were compared with the yield QTLs identified in rice by Li et al. (2001). Assuming three of our QTLs correspond to the Rht-B1, Vrn-A1 and Vrn-D1 genes, ten of the remaining 14 yield QTLs correspond with yield QTLs identified by Li et al. (2001) (Table 2).
The yield component most frequently associated with yield QTL clusters was grain numbers per ear (G), and the least frequent yield component was ears per plant (E). Every yield QTL should be the consequence of variation in one or more of the yield components E, G and T (Fig. 2, Table 2). Several yield QTL clusters were associated with all three yield components (1BL, 4AL, 5AL, 5DL, Table 2), depending on the trial. As each yield component is determined at a different phase of development of the plant (Slafer 2003), these particular yield QTL clusters are likely to be associated with plant biomass production, as a relatively vigorous plant is likely to have a better yield whichever yield component(s) determine(s) yield in a particular trial. This was true for three of these four yield QTL regions: 4AL, 5AL and 5DL. For these regions, biomass at harvest was significantly associated with the same markers as those for yield in each of the trials comprising the QTL clusters (data not presented). In the yield cluster on 1BL, biomass was not associated with these markers, although it was not measured in all trials within the QTL cluster.
The wide diversity of yield QTLs from trial to trial is an illustration of the large genotype × environment interaction that is usually found for yield. Reynolds et al. (2002) concluded that the phase of wheat most sensitive to environmental factors causing genotype × environment interaction is generally the phase of spike primordial growth–i.e. the phase during which the potential number of grains per ear is determined. In this regard, it is worth noting that the three regions of the genome showing the biggest contrast between yield QTLs at site yields of 2 g plant−1 and 6 g plant−1 (1DS/L, 5AS and 7AL, Fig. 4) were all regions of significant variation in the yield component grains per ear (G): four of five trials at 1DS/L, six of ten trials at 5AS and eight out of eight trials in which yield components were measured at 7AL.
Although neither of the parents of our mapping population is a commercial wheat variety, the locations of yield QTL clusters we report here could usefully be studied for allelic variation among breeding germplasm, as illustrated by Quarrie et al. (2003). Targeted recombination of genotypes containing the most beneficial alleles for yield at these locations could provide genotypes with improved yields in a commercial breeding programme.
Conclusion
We have presented here a genetic map for a new bread wheat population comprising DHLs that are particularly diverse phenotypically. As the large majority of the genome is covered with markers, the population is ideal for QTL analysis of a wide range of traits. The compilation of yield QTLs from 24 site × treatment × year trial combinations has shown, on the one hand, the ubiquity of yield QTLs around the genome, but on the other hand, the presence of several clusters of QTLs. A few of these correspond with major genes already known to be important in determining yield, and other QTL clusters correspond with regions of the genome already shown by others to contain QTLs for yield or yield components. Many of the QTL clusters also correspond with regions of the rice genome regulating yield—a useful prerequisite for any future strategies to clone the genes responsible. Our results suggest it may be possible to improve yield stability by combining yield QTLs expressed under contrasting yielding environments. In particular, combining the CS allele on 7BL and the SQ1 allele on 7AL into wheat genetic backgrounds having both low-yield alleles may increase yield potential (7BL) as well as increase yield stability (7AL). In future publications we shall dissect further the processes likely to be regulating yield at particular QTLs.
References
Ammiraju JSS, Dholakia BB, Santra DK, Singh H, Lagu MD, Tamhankar SA, Dhaliwal HS, Rao VS, Gupta VS, Ranjekar PK (2001) Identification of inter simple sequence repeat (ISSR) markers associated with seed size in wheat. Theor Appl Genet 102:726–732
Ayala L, Henry M, van Ginkel M, Singh R, Keller B, Khairallah M (2002) Identification of QTLs for BYDV tolerance in bread wheat. Euphytica 128:249–259
Basten CJ, Zeng Z-B, Weir BS (1996) qtlcartographer: a suite of programs for mapping quantitative trait loci. Abstracts to Plant Genome IV. Academic Press, San Diego, p 108
Bennett MD, Smith JB, Heslop-Harrison JS (1982) Nuclear DNA amounts in angiosperms. Proc R Soc London Ser B 216:179–199
Blanco A, Bellomo MP, Cenci A, De Giovanni C, D’Ovidio R, Iacono E, Laddomada B, Pagnotta MA, Porceddu E, Sciancalepore A, Simeone R, Tanzarella OA (1998) A genetic linkage map of durum wheat. Theor Appl Genet 97:721–728
Blanco A, Pasqualone A, Troccoli A, Di Fonzo N, Simeone R (2002) Detection of grain protein content QTLs across environments in tetraploid wheats. Plant Mol Biol 48:615–623
Börner A, Schumann E, Furste A, Coster H, Leithold B, Röder MS, Weber WE (2002) Mapping of quantitative trait loci determining agronomic important characters in hexaploid wheat (Triticum aestivum L.). Theor Appl Genet 105:921–936
Bryan GJ, Collins AJ, Stephenson P, Orry A, Smith JB, Gale MD (1997) Isolation and characterisation of microsatellites from hexaploid bread wheat. Theor Appl Genet 96:557–563
Butterworth KJ (2000) Flowering time genes of wheat and their influence on environmental adaptability. PhD thesis, University of East Anglia
Byrne PF, Butler JD, Anderson GR, Haley SD (2002) QTL’s for agronomic and morphological traits in a spring wheat population derived from a cross of heat tolerant and heat sensitive lines (poster). In: Plant, Animal and Microbe Genomes X Conf. San Diego, Calif.
Cadalen T, Boeuf C, Bernard S, Bernard M (1997) An intervarietal molecular marker map in Triticum aestivum L. Em. Thell. and comparison with a map from a wide cross. Theor Appl Genet 94:367–377
Cattivelli L, Baldi P, Crosatti C, Di Fonzo N, Faccioli P, Grossi M, Mastrangelo AM, Pecchioni N, Stanca AM (2002) Chromosome regions and stress-related sequences involved in resistance to abiotic stress in Triticeae. Plant Mol Biol 48:649–665
Chalmers KJ, Campbell AW, Kretschmer J, Karakousis A, Henschke PH, Pierens S, Harker N, Pallotta M, Cornish GB, Shariflou MR, Rampling LR, McLauchlan A, Daggard G, Sharp PJ, Holton TA, Sutherland MW, Appels R, Langridge P (2001) Construction of three linkage maps in bread wheat, Triticum aestivum. Aust J Agric Res 52:1089–1119
Chao S, Sharp PJ, Worland AJ, Warham EJ, Koebner RMD, Gale MD (1989) RFLP-based genetic maps of wheat homoeologous group 7 chromosomes. Theor Appl Genet 78:495–504
Devos KM, Dubcovsky J, Dvorak J, Chinoy CN, Gale MD (1995) Structural evolution of wheat chromosomes 4A, 5A, and 7B and its impact on recombination. Theor Appl Genet 91:282–288
Eujayl I, Sorrells ME, Baum M, Wolters P, Powell W (2002) Isolation of EST-derived microsatellite markers for genotyping the A and B genomes of wheat. Theor Appl Genet 104:399–407
Finlay KW, Wilkinson GN (1963) The analysis of adaptation in a plant-breeding programme. Aust J Agric Res 14:342–354
Forster BP, Miller TE, Law CN (1988) Salt tolerance of two wheat- Agropyron junceum disomic addition lines. Genome 30:559–564
Franckowiak J (1997) Revised linkage maps for morphological markers in barley, Hordeum vulgare. Barley Genet Newsl 26:9–21
Gale MD, Youssefian S (1985) Dwarfing genes in wheat. In: Russell GE (ed) Progress in plant breeding. J Butterworth and Co, London, pp 1–35
Gale MD, Atkinson MD, Chinoy CN, Harcourt RL, Jia J, Li QY, Devos KM (1995) Genetic maps of hexaploid wheat. In: Li ZS, Xin ZY (eds) Proc 8th Int Wheat Genetics Symp. Agric Scientech Press, Beijing, pp 29–40/Genome 37:871–875
Groos C, Robert N, Bervas E, Charmet G (2003) Genetic analysis of grain protein-content, grain yield and thousand-kernel weight in bread wheat. Theor Appl Genet 106:1032–1040
Hart GE (2001) Molecular-marker maps of the cultivated wheats and other Triticum species. In: Phillips RL, Vasil IK (eds) DNA-based markers in plants. Kluwer, Dordrecht, pp 421–441
Huang S, Sirikhachornkit A, Su X, Faris J, Gill B, Haselkorn R, Gornicki P (2002) Genes encoding plastid acetyl-CoA carboxylase and 3-phosphoglycerate kinase of the Triticum/Aegilops complex and the evolutionary history of polyploid wheat. Proc Natl Acad Sci USA 99:8133–8138
Isla R, Royo A, Aragues R (1997) Field screening of barley cultivars to soil salinity using a sprinkler and a drip irrigation system. Plant Soil 197:105–117
Khavkin E, Coe E (1997) Mapped genomic locations for developmental functions and QTLs reflect concerted groups in maize (Zea mays L.). Theor Appl Genet 95:343–352
Kosambi DD (1944) The estimation of map distances from recombination values. Ann Eugen 12:172–175
Lander ES, Green P, Abrahamson J, Barlow A, Daly MJ, Lincoln SE, Newburg L (1987) mapmaker: an interactive computer package for constructing primary genetic linkage maps of experimental and natural populations. Genomics 1:174–181
Langridge P, Lagudah ES, Holton TA, Appels R, Sharp PJ, Chalmers KJ (2001) Trends in genetic and genome analysis in wheat: a review. Aust J Agric Res 52:1043–1077
Laurie DA, Reymondie S (1991) High frequencies of fertilization and haploid seedling production in crosses between commercial hexaploid wheat varieties and maize. Plant Breed 106:182–189
Li WL, Nelson JC, Chu CY, Shi LH, Huang SH, Liu DJ (2002) Chromosomal locations and genetic relationships of tiller and spike characters in wheat. Euphytica 125:357–366
Li Z-K, Luo LJ, Mei HW, Wang DL, Shu QY, Tabien R, Zhong DB, Ying CS, Stansel JW, Khush GS, Paterson AH (2001) Overdominant epistatic loci are the primary genetic basis of inbreeding depression and heterosis in rice. I. Biomass and grain yield. Genetics 158:1737–1753
Liu Y-G, Tsunewaki K (1991) Restriction fragment length polymorphism (RFLP) analysis in wheat. II. Linkage maps of the RFLP sites in common wheat. Jpn J Genet 66:617–633
Mahmood A, Quarrie SA (1993) Effects of salinity on growth, ionic relations and physiological traits of wheat, disomic addition lines from Thinopyrum bessarabicum and two amphiploids. Plant Breed 110:265–276
Mano Y, Takeda K (1997) Mapping quantitative trait loci for salt tolerance at germination and the seedling stage in barley (Hordeum vulgare L.). Euphytica 94:263–272
Manyowa NM, Miller TE (1991) The genetics of tolerance to high mineral concentrations in the tribe Triticeae—a review and update. Euphytica 57:175–185
Marino CL, Nelson JC, Lu YH, Sorrells ME, Leroy P, Tuleen NA, Lopes CR, Hart GE (1996) Molecular genetic maps of the group 6 chromosomes of hexaploid wheat (Triticum aestivum L. em. Thell). Genome 39:359–366
Messmer MM, Keller M, Zanetti S, Keller B (1999) Genetic linkage map of a wheat × spelt cross. Theor Appl Genet 98:1163–1170
Murray MG, Thompson WF (1980) The isolation of high molecular weight plant DNA. Nucleic Acids Res 8:4321–4325
Nelson JC, Sorrells ME, Van Deynze AE, Lu YH, Atkinson M, Bernard M, Leroy P, Faris JD, Anderson JA (1995a) Molecular mapping of wheat—major genes and rearrangements in homoeologous groups 4, 5, and 7. Genetics 141:721–731
Nelson JC, Van Deynze AE, Autrique E, Sorrells ME, Lu YH, Merlino M, Atkinson M, Leroy P (1995b) Molecular mapping of wheat. Homoeologous group 2. Genome 38:516–524
Nelson JC, Van Deynze AE, Autrique E, Sorrells ME, Lu YH, Negre S, Bernard M, Leroy P (1995c) Molecular mapping of wheat. Homoeologous group 3. Genome 38:525–533
Paillard S, Schnurbusch T, Winzeler M, Messmer M, Sourdille P, Abderhalden O, Keller B, Schachermayr G (2003) An integrative genetic linkage map of winter wheat (Triticum aestivum L.). Theor Appl Genet 107:1235–1242
Pakniyat H, Handley LL, Thomas WTB, Connolly T, Macaulay M, Caligari PDS, B.P. Forster BP (1997) Comparison of shoot dry weight, Na+ content and δ13 C values of ari-e and other semi-dwarf barley mutants under salt-stress. Euphytica 94:7–14
Peng J, Richards DE, Hartley NM, Murphy GP, Devos KM, Flintham JE, Beales J, Fish LJ, Worland AL, Pelica F, Sudhakar D, Christou P, Snape JW, Gale MD, Harberd NP (1999) ’Green revolution’ genes encode mutant gibberellin response modulators. Nature 400:256–261
Peng J, Korol AB, Fahima T, Röder MS, Ronin YI, Li YC, Nevo E (2000) Molecular genetic maps in wild Emmer wheat, Triticum dicoccoides: genome-wide coverage, massive negative interference, and putative quasi-linkage. Genome Res 10:1509–1531
Quarrie SA (1981) Genetic variability and heritability of drought-induced abscisic acid accumulation in spring wheat. Plant Cell Environ 4:147–151
Quarrie SA (1987a) Evaluation of the influence of a metabolic character on drought resistance exemplified by studies on abscisic acid in wheat and maize. In: Monti L, Porceddu E (eds) Drought resistance in plants: physiological and genetic aspects. Commission of the European Communities, Luxembourg, pp 111–129
Quarrie SA (1987b) Use of genotypes differing in endogenous abscisic acid levels in studies on physiology and development. In: Hoad GV, Lenton JR, Jackson MB, Atkin RK (eds) Hormone action in plant development: a critical appraisal. Butterworths, London, pp 89–105
Quarrie SA, Gulli M, Calestani C, Steed A, Marmiroli N (1994) Location of a gene regulating drought-induced abscisic acid production on the long arm of chromosome 5A of wheat. Theor Appl Genet 89:794–800
Quarrie SA, Dodig D, Pekic S, Kirby J, Kobiljski B (2003) Prospects for marker-assisted selection of improved drought responses in wheat. Bulg J Plant Physiol Special Issue 2003, pp 83–95
Reynolds MP, Trethowan R, Crossa J, Vargas M, Sayre KD (2002) Physiological factors associated with genotype by environment interaction in wheat. Field Crops Res 75:139–160
Röder MS, Korzun V, Wendehake K, Plaschke J, Tixier M-H, Leroy P, Ganal MW (1998) A microsatellite map of wheat. Genetics 149:2007–2023
Semikhodskii AG, Quarrie SA, Snape JW (1997) Mapping quantitative trait loci for salinity responses in wheat. In: Jevtic S, Pekic S (eds) Drought and plant production, vol. 2. Proc Int Symp. Agricultural Research Institute Serbia, Belgrade, pp 83–92
Sharp PJ, Kreis M, Shewry PR, Gale MD (1988) Location of B-amylase sequences in wheat and its relatives. Theor Appl Genet 75:286–290
Singh RP, Huerta-Espino S, Rajaram S, Crossa J (1998) Agronomic effects from chromosome translocations 7DL.7Ag and 1BL.1RS in spring wheat. Crop Sci 38:27–33
Slafer GA (2003) Genetic basis of yield as viewed from a crop physiologist’s perspective. Ann Appl Biol 142:117–128
Snape JW, Sarma R, Quarrie SA, Fish L, Galiba G, Sutka J (2001) Mapping genes for flowering time and frost tolerance in cereals using precise genetic stocks. Euphytica 120:309–315
Sutka J, Snape JW (1989) Location of a gene for frost resistance on chromosome 5A of wheat. Euphytica 42:41–44
Thomas WTB (2003) Prospects for molecular breeding of barley. Ann Appl Biol 142:1–12
Van Deynze AE, Dubcovsky J, Gill KS, Nelson JC, Sorrells ME, Dvorak J, Gill BS, Lagudah ES, McCouch SR, Appels R (1995) Molecular-genetic maps for group 1 chromosomes of Triticeae species and their relation to chromosomes in rice and oat. Genome 38:45–59
Vos P, Hogers R, Bleeker M, Reijans M, van de Lee T, Hornes M, Frijters A, Pot J, Peleman J, Kuiper M, Zabeau M (1995) AFLP: a new technique for DNA fingerprinting. Nucleic Acids Res 23:4407–4414
Yan L, Loukoianov A, Tranquilli G, Helguera M, Fahima T, Dubcovsky J (2003) Positional cloning of the wheat vernalization gene VRN1. Proc Natl Acad Sci USA 100:6263–6268
Acknowledgements
The authors acknowledge the following funding sources that allowed aspects of this research to be carried out: molecular marker work from the BC-MURST (Italy) programme (C. Calestani), EU FP4 Biotechnology programme for the EGRAM project (E. Waterman) and EU FP5 Sustainable agriculture SUSTAIN project no. QLK5-CT-2001-01461 (D. Z. Habash, J. Weyen, J. Schondelmaier); molecular marker work and field trials from the BBSRC RASP (Resource Allocation and Stress in Plants) programme (C. Chinoy, P. Farmer, L. Saker, D.T. Clarkson), EU INTAS programme (A. Abugalieva, M. Yessimbekova, Y. Turuspekov, S. Abugalieva, R. Tuberosa, M.-C. Sanguineti) and EU INCO-DC programme (N. Steele, P. Hollington, R. Aragüés, A. Royo). D. Dodig acknowledges financial support from the Serbian Ministry of Science and Technology. The John Innes Centre is supported by a grant-in-aid from the Biotechnological and Biological Sciences Research Council. The helpful comments on this manuscript of Professor John Snape are gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by P. Langridge
Rights and permissions
About this article
Cite this article
Quarrie, S.A., Steed, A., Calestani, C. et al. A high-density genetic map of hexaploid wheat (Triticum aestivum L.) from the cross Chinese Spring × SQ1 and its use to compare QTLs for grain yield across a range of environments. Theor Appl Genet 110, 865–880 (2005). https://doi.org/10.1007/s00122-004-1902-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-004-1902-7