
Abstract
Background and Objective
Denosumab (XGEVA®; AMG 162) is a fully human IgG2 monoclonal antibody, which binds to the receptor activator of nuclear factor K-B ligand (RANKL) and prevents terminal differentiation, activation and survival of osteoclasts. We aimed to characterize the population pharmacokinetics of denosumab in patients with advanced solid tumours and bone metastases.
Methods
A total of 14 228 free serum concentrations of denosumab from 1076 subjects (495 healthy subjects and 581 advanced cancer patients with solid tumours and bone metastases) included in 14 clinical studies were pooled. Denosumab was administered as either single intravenous (n= 36), single subcutaneous (n= 490) or multiple subcutaneous doses (n = 550) ranging from 30 to 180 mg (or from 0.01 to 3 mg/kg) and was given every 4 or 12 weeks for up to 3 years. An open two-compartment pharmacokinetic model with first-order absorption, linear distribution to a peripheral compartment, linear clearance and quasi-steady-state approximation of the target-mediated drug disposition was used to describe denosumab pharmacokinetics, using NONMEM Version 7.1.0 software. The influence of covariates (body weight, age, race, tumour type) was investigated using the full model approach. Model evaluation was performed through visual predictive checks. Model-based simulations were conducted to explore the role of covariates on denosumab serum concentrations and inferred RANKL occupancy.
Results
After subcutaneous administration, the dose-independent bioavailability and mean absorption half-life of denosumab were estimated to be 61% and 2.7 days, respectively. The central volume of distribution and linear clearance were 2.62L/66kg and 3.25mL/h/66kg, respectively. Clearance and volume parameters were proportional to body weight. Assuming 1:1 denosumab-RANKL binding, the baseline RANKL level, quasi-steady-state constant and RANKL degradation rate were inferred to be 4.46 nmol/L, 208ng/mL and 0.00116 h-1, respectively. Between-subject variability in model parameters was moderate. Following 120 mg dosing every 4 weeks, the inferred RANKL occupancy at steady state exceeded 97% during the entire dosing interval in more than 95% of subjects, regardless of the patient covariates.
Conclusions
The integration of pharmacokinetic data from 14 clinical studies demonstrated denosumab RANKL-mediated pharmacokinetics. Pharmacokinetics-based dosage adjustments on the basis of body weight, age, race and tumour type are not necessary in patients with bone metastases from solid tumours.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The most common metastatic site for many types of cancer, such as breast and prostate cancer, is bone.[1] These metastases can result in substantial morbidity, including pathological fractures, spinal cord compression and the need to undergo radiation or surgery of the bone.[2–4] These events are collectively referred to as ‘skeletal-related events’ (SREs). Metastatic tumour cells in bone secrete cytokines and growth factors, which induce osteoblasts to release the receptor activator of nuclear factor κ-B ligand (RANKL), a tumour necrosis factor (TNF) superfamily protein, which plays a critical role in bone remodelling.[5] RANKL binds to its receptor, RANK, on osteoclast precursors and mature osteoclasts to promote terminal differentiation, activation and survival of osteoclasts, the sole cell type responsible for bone resorption.[6–8] Inhibition of RANKL activity is a therapeutic target for treatment of several bone disorders associated with increased bone resorption, such as prevention of SREs in patients with bone metastases from solid tumours.
Denosumab (AMG 162, XGEVA®) is a fully human IgG2 monoclonal antibody with high affinity (dissociation constant [KD] 3 × 10−12 mol/L) and specificity for RANKL.[9] Denosumab is highly specific because it binds only to RANKL and not to other members of the TNF family, such as TNFα, TNFβ, TNF-related apoptosis-inducing ligand (TRAIL) or CD40 ligand.[9] Like native osteoprotegerin (a decoy receptor for RANKL, which is also produced by osteoblasts),[7,10,11] denosumab neutralizes the activity of human membrane-bound or soluble RANKL by blocking binding to RANK and thus prevents terminal differentiation, activation and survival of osteoclasts, thereby reducing bone resorption, tumour-induced bone destruction and SREs.
Several phase I and phase II studies have determined the pharmacological activity and tolerability of denosumab, as well as its pharmacokinetics and pharmacodynamics when administered to cancer patients as a single intravenous dose, a single subcutaneous dose or multiple subcutaneous doses ranging from 30 to 180 mg (or from 0.01 to 3 mg/kg) and given every 4 or 12 weeks for up to 3 years.[12–15] Furthermore, results from pivotal randomized double-blinded phase III studies demonstrated that denosumab was superior to zoledronic acid in suppressing bone resorption and in delaying or preventing SREs in patients with breast cancer, prostate cancer and other solid tumours with bone metastases, and that it was well tolerated.[16–18] Currently, denosumab 120 mg administered subcutaneously every 4 weeks is approved in the US, EU and other countries for prevention of SREs in patients with bone metastases from solid tumours.
According to non-compartmental analyses, denosumab exposure after subcutaneous administration ranges from 36% to 78% relative to intravenous dosing. Denosumab is slowly absorbed after subcutaneous administration, with peak serum concentration (Cmax) values generally reached within 4 weeks post-dose and a small volume of distribution after intravenous dosing, indicative of limited extravascular distribution. Approximately 2-fold accumulation of denosumab in serum was observed with repeated doses of 120 mg administered every month, which reached steady-state exposure within 4–5 months and maintained that exposure with continued dosing. After the last administered dose, serum denosumab levels declined over a period of 4–5 months, with a mean half-life of approximately 25–30 days. As observed for other monoclonal antibodies, denosumab exhibits dose-dependent, parallel linear and nonlinear elimination over a wide dose range when administered by intravenous and subcutaneous routes, and moderate between-subject pharmacokinetic variability. However, approximately dose-proportional increases in exposure are observed for doses ≥60 mg. In addition, there was no evidence of time-dependent pharmacokinetics after repeated denosumab dosing under the regimens examined for up to 4 years of exposure.[19] Reversibility of pharmacodynamic effects (based on serum bone turnover markers) was observed.[14]
Furthermore, a population pharmacokinetic analysis of denosumab[19] in healthy subjects and women with osteopenia or osteoporosis demonstrated that an open two-compartment pharmacokinetic model with linear distribution to the peripheral compartment, parallel linear and RANKL-mediated elimination, and first-order absorption following subcutaneous administration, was suitable to describe the time course of free denosumab serum concentrations following different intravenous and subcutaneous dosing schedules. In that analysis, the clinical impact of changes in body weight, race and age on denosumab exposure were found to be limited, leading to a recommendation for no dose adjustments when administering denosumab 60 mg subcutaneously every 6 months to postmenopausal woman with osteoporosis.
In the present population analysis, the pharmacokinetics of denosumab were evaluated in patients with bone metastases from solid tumours by pooling full concentration-time profiles from healthy subjects and cancer patients enrolled in phase I studies with sparse observations collected from cancer patients with bone metastases enrolled in proof-of-concept and registration studies. The primary objectives of the population pharmacokinetic analysis of denosumab were three-fold: (i) to characterize the pharmacokinetic profile of denosumab after intravenous and subcutaneous administration; (ii) to quantify the degree of between-subject variability of denosumab pharmacokinetic parameters in patients with bone metastases from solid tumours; and (iii) to evaluate patient-related covariates as potential sources of variability in the pharmacokinetics of denosumab.
Methods
Clinical Data
This population pharmacokinetic analysis pooled data from 14 clinical studies of denosumab, which included 14 228 free denosumab serum concentrations from 1076 subjects (495 healthy subjects and 581 patients with bone metastases from solid tumours). The relevant characteristics of each clinical study used for the current analyses are summarized in table I. Additional details of these clinical trials are reported elsewhere.[12–18,20–21] In these studies, denosumab was administered as single intravenous (n = 36), single subcutaneous (n = 490) or multiple subcutaneous doses (n = 550), which ranged from 30 to 180 mg (or from 0.01 to 3 mg/kg) and were given every 4 weeks or every 12 weeks for up to 3 years. All studies were sponsored by Amgen Inc., conducted in accordance with the principles for human experimentation as defined in the Declaration of Helsinki and the International Conference on Harmonization Good Clinical Practice guidelines, and approved by the respective institutional review boards. Informed consent was obtained from each subject after they were told about the potential risks and benefits, as well as the investigational nature of the study.

Summary of clinical studies of denosumab
Bioanalysis
For the clinical studies used in this analysis, free serum denosumab concentrations were determined using a validated enzyme linked immunosorbent assay (ELISA). Microtitre plates coated with osteoprotegerin ligand (OPGL, Part #890705; R&D Systems, Minneapolis, MN, USA) were used to capture denosumab from serum. After sample incubation and washing steps, OPGL conjugated to horseradish peroxidase (Part #890841; R&D Systems) was used to detect denosumab bound to the plate. The validated assay range was from 0.8 to 35 ng/mL. The intra-assay and inter-assay variations were <6% and <9%, respectively.
Software
Nonlinear mixed effects modelling for the population pharmacokinetic analysis of denosumab was performed using NONMEM® Version 7.1.0 (Icon Development Solutions, Ellicott City, MD, USA) with a GFortran 4.4 compiler. The ADVAN13 subroutine and the first-order conditional estimation method with interaction (FOCEI) were used for the key intermediate model analysis. The stochastic approximation expectation maximization (SAEM) method was used to provide the parameter estimation of the final model. After 1000 iterations, the objective function value (OFV) was obtained by running the iterative-two-stage (ITS) method with the final parameter estimates. Graphical data visualization, evaluation of NONMEM® outputs, construction of goodness-of-fit plots and graphical model comparisons were conducted using S-Plus Version 8.0.4 (TIBCO Software Inc., Palo Alto, CA, USA).
Structural Model
The structural pharmacokinetic model of denosumab, displayed in figure 1, is based on the concept of target-mediated drug disposition (TMDD)[22] and is consistent with the structural model used to analyse the denosumab free serum concentration in women with osteopenia or osteoporosis.[19] The subcutaneous absorption of denosumab was represented by the first-order absorption rate constant (ka). Absolute bioavailability (F) was estimated by simultaneously analysing the data obtained after subcutaneous and intravenous administration. After an intravenous bolus or subcutaneous absorption, free denosumab was distributed into the central compartment, with the volume of distribution represented by V1. The nonspecific distribution from the central compartment into the peripheral compartment was characterized by intercompartmental clearance (Q), and the peripheral volume of distribution was represented by V2. The free denosumab in the central compartment was assumed to be eliminated by a linear pathway, quantified by CLlin, or by binding to RANKL (R) following a second-order rate constant (kon). The denosumab-RANKL complex (RC) that was formed was dissociated according to a first-order rate constant (koff) and generated free denosumab and RANKL, or was eliminated through a first-order process, characterized by the rate constant kint. RANKL was also assumed to be produced following a zero-order process, characterized by ksyn, and degraded by a first-order process, with the elimination rate constant represented by kdeg. Since RANKL is produced as a membrane-bound protein, which is cleaved into a soluble form by ectodomain shedding,[23] both kint and kdeg are hybrid constants. Actually, kint characterizes both the internalization rate of the denosumab : membrane-bound RANKL complex and the degradation rate of the denosumab : soluble RANKL complex. Similarly, kdeg characterizes the degradation rate of the soluble and membrane-bound RANKL. In this TMDD model, the nonlinearity of denosumab pharmacokinetics was explained by the limited RANKL in the central compartment at a given time relative to the free denosumab concentration.

Denosumab pharmacokinetic model. D, C, and P represent the depot, central and peripheral compartments, respectively. Absorption following subcutaneous (SC) administration follows a first-order process (absorption rate constant [ka]) with bioavailability represented by F. Non-specific distribution from the central compartment (V1) to the peripheral compartment (V2) is linear with intercompartmental clearance (Q). Elimination of the drug from the central compartment is described using parallel linear clearance (CLlin) and quasi-steady-state approximation of target-mediated drug disposition. The R and RC compartments represent the total receptor activator of nuclear factor κ-B ligand (RANKL) and denosumab-RANKL complex, respectively. Target turnover is described by the first-order degradation rate constant (kdeg) and the zero-order synthesis rate (ksyn), where the baseline RANKL level (R0) is equal to ksyn/kdeg. The denosumab-RANKL binding affinity is estimated as the quasi-steady-state constant (Kss), calculated as Kss = (koff + kint)/kon, where koff is the first-order dissociation rate constant, kint is the first-order elimination rate constant, and kon is the second-order association rate constant of the drug-target complex. IV = intravenous. [Reproduced from Sutjandra et al. (figure 1),[19] with permission from Adis (© Adis Data Information BV 2012. All rights reserved).]
As the denosumab-RANKL association rate was much faster than the elimination rates of denosumab (CLlin/V1) and the denosumab-RANKL complex (kint), and the binding of denosumab to RANKL can be assumed to be nearly irreversible (KD = 3 × 10−12 mol/L), the full TMDD model described above became overparameterized and numerical difficulties precluded obtaining accurate estimations of the binding constants kon and koff. Therefore, the quasi-steady-state approximation of the TMDD model, which assumes that the concentrations of free denosumab, RANKL and denosumab-RANKL complex were at steady state, was used.[22,24–26] The denosumab-RANKL binding affinity, defined as koff/kon, was equivalent to KD and could not be estimated independently. Rather, the quasi-steady-state constant (Kss) defined as KD + kint/kon, was estimated directly from the data. The equations used to describe the system were as follows (equations 1–7):
where AD denotes the amount of denosumab in the subcutaneous depot compartment available for absorption, and AC and AP refer to the amount of free denosumab in the central and peripheral compartments, respectively. C is the free denosumab concentration in the central compartment, and t is time in hours. The total amount of denosumab (Atot) in serum was introduced as the sum of AC and RC • V1. In addition, CLtot is denosumab total clearance, and Rtot is the total RANKL level (including both free RANKL and RANKL bound to denosumab), which at steady state (Rtot,ss) can be explicitly determined by equation 8:
The initial conditions of this differential equation system were set as (equation 9):
Statistical Model
Between-subject variability in pharmacokinetic parameters was initially assumed to follow an independent log-normal distribution, and the correlations between random effects were explored and incorporated into the model if deemed necessary. As concentrations are distributed log-normally rather than normally, the ‘transformation of both sides’ approach was applied to describe the residual error.[27] In addition, the exploratory analysis performed with phase I data, further confirmed by previous population pharmacokinetic analysis,[19] demonstrated that the magnitude of the residual variability in the log domain was greatest for low concentrations. In order to account for this complexity, the following residual error model implemented in the log-transformed concentration scale described different variances for low and high concentrations according to equation 10:
where Cij is the ith observed serum concentration of denosumab in the jth subject; Ĉij is the corresponding model-predicted concentration; ɛij is a random, independent, normally distributed variable with a mean of 0 and variance of 1; σL and σH represent the approximate coefficient of variations for low and high denosumab serum concentrations, respectively; and C50 is the estimated denosumab serum concentration where the coefficient of variation of the residual variability is equal to the mean between σL and σH. In addition, to account for the differences between the intensive samples in phase I studies and the relatively sparse samples in phase II and III studies, σH was estimated separately as σH,Ph1 and σH,Ph2/3, respectively.
Covariate Analysis
It is well known that the distribution and elimination of monoclonal antibodies are often proportional to body weight,[28,29] therefore CLlin, V1, Q and V2 were scaled by body weight (normalized for the typical body weight of women with postmenopausal osteoporosis [66 kg], to be consistent with the previously published analysis) using a power function. In addition, the covariates included in the analysis were age, sex, race (Caucasian, Hispanic, Black, Asian and other) and disease status (healthy subjects versus cancer subjects, who were also stratified by tumour type as subjects with breast cancer, prostate cancer, other solid tumours or giant cell tumour of the bone). Since it is generally accepted that monoclonal antibodies are eliminated by catabolism and/or receptor-mediated processes and not by hepatic phase 1 or phase 2 metabolism, markers of hepatic function were not evaluated as potential covariates in the current analysis.[30,31] Similarly, markers of renal function were not investigated as potential covariates, as the high molecular weight of denosumab (144.7 kD) precludes its elimination through renal excretion, and a renal impairment study[32] has shown no effect of renal insufficiency on the pharmacokinetics of denosumab.
Empirical Bayes Estimates (EBEs) of the individual model parameters were computed and used to identify potential correlations with patient covariates. A full model approach[33] was used for the covariate analysis and the inferences about statistical significance of model parameters were based on the resulting parameter estimates and measures of estimation precision (asymptotic standard errors). If the magnitude of the change of the parameter due to a covariate’s influence was less than 20% over the range of values that were evaluated, the covariate factor was not considered to be clinically relevant. The effects of continuous covariates were modelled multiplicatively using normalized power models, whereas the effects of categorical covariates were modelled multiplicatively using a similar notation (equation 11):
where the typical value of a model parameter (TVP) is described as a function of M individual continuous covariates (covm, m = 1,..,M) and P individual categorical (0 or 1) covariates (covM + p, p = 1,..,P), θn is the estimated typical model parameter value with covariates equal to the reference covariate values (covm = refm, covM + p = 0), and θ(n + m) and θ(n + M + p) are estimated parameters that describe the magnitude of the covariate-parameter relationships.
Model Evaluation
A visual predictive check (VPC)[34] was performed as a model validation technique. The denosumab serum concentration-time profile was simulated for 1000 virtual subjects. Specific covariates for virtual subjects were sampled from the original dataset. Descriptive statistics of simulated denosumab serum concentrations were then graphically compared with observed denosumab serum concentrations. From this evaluation, an assessment of model adequacy was made.
Model-Based Simulations
In order to compare the time course of denosumab serum concentrations after subcutaneous administration of 2 mg/kg or 120 mg doses every 4 weeks, the final estimates of the fixed and random effects from the denosumab population pharmacokinetic model were used to simulate the time course of denosumab serum concentrations following six doses administered to 1000 virtual subjects. Individual body weights were obtained by resampling from the body weights of cancer patients included in this study. The 5th, 50th and 95th percentiles of the denosumab concentrations over time were compared across the two dosing regimens. Additionally, the time course of denosumab serum concentrations was simulated for a monthly subcutaneous dose of 120 mg administered to Caucasian, Hispanic and Black subjects. The denosumab pharmacokinetics were then compared among races. Similarly, the time course of denosumab serum concentrations for a typical 66 kg patient was simulated for a monthly subcutaneous dose of 120 mg, and the denosumab pharmacokinetic profile was compared among the cancer types in order to evaluate the effect of the disease status. The effect of body weight, age, race and tumour type on the time course of serum denosumab concentrations and inferred RANKL inhibition was also computed for typical subjects covering the entire distribution of each covariate. The determination of the fraction of RANKL neutralized by denosumab was computed as RC/Rtot, and was a function of the free denosumab serum concentration.
Results
The quasi-steady-state TMDD model that was developed provided a reasonable fit to the denosumab pharmacokinetic data gathered from up to 3 years of treatment. Simplifying the quasi-steady-state parameterization of the TMDD model to the Michaelis-Menten model parameterization resulted in a significant increase in the minimum OFV (ΔMOFV = 5063). Therefore, the quasi-steady-state assumption was selected for further model development. In addition, the power coefficients associated with the body weight effects on CLlin, V1, Q and V2 were estimated to be 0.849, 0.758, 1.14 and 1.05, respectively. Since these values were very similar to 1 and their estimation yielded a minor change in the OFV (ΔMOFV = −10.231), fixing the body weight exponents to 1 was deemed appropriate. If the exponents were assumed to be different from 1, the values of the pharmacokinetic parameters differed by less than 20% in more than 95% of subjects, compared with fixing the exponents to 1. Furthermore, the inclusion of the race effect on CLlin and V1 was significant on the basis of the 95% confidence interval for the point estimates (ΔMOFV = −62.496, df = 2, p < 0.001), although only a 1% reduction of between-subject variability for those parameters was observed. Hispanic subjects had 27% faster CLlin relative to Caucasian, Asian and Black subjects. On the other hand, inclusion of the tumour type on CLlin was associated with a 4% reduction of between-subject variability on CLlin (ΔMOFV = −133.519, df = 4, p < 0.001). Subjects with solid tumours had 10–37% faster linear clearance compared with the rates in healthy subjects. As in women with osteopenia or osteoporosis,[19] ka decreased as a function of age and remained relatively constant for subjects older than 53 years (ΔMOFV = −79.227, df = 2, p < 0.001); however, only an 8% reduction of between-subject variability in ka was observed when the age effect was incorporated into the model.
No other evaluated covariates were found to significantly improve the goodness of fit. An off-diagonal element for the covariance between CLlin and V1 random effects was also estimated. In addition, as the phase I data consisted of intensive concentration-time profiles whereas the majority of the phase II/III data were sparse, estimating σH separately for phase I studies (σH,Ph1) and phase II/III studies (σH,Ph2/3) was associated with substantial model improvement (ΔMOFV = −6760) and reflected the difference in residual variability across study types, as previously reported in other meta-analyses.[35,36]
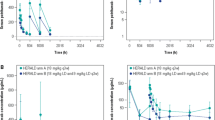
Table II displays the parameter estimates of the final population pharmacokinetic model using the SAEM method. Both fixed and random effects were precisely estimated, with relative standard errors (RSEs) of less than 8%. Overall, population and individual model predictions adequately described the observed data, and goodness-of-fit plots revealed a random normal scatter around the line of identity with no apparent trend in the conditional weighted residuals over the range of concentrations and time that were evaluated (figure 2). Histograms of individual random effects on parameters showed approximately normal distribution, and no significant correlation between random effects was discernible (data not shown), other than CLlin vs V1. The VPC figures (figure 3) indicate excellent predictive ability of the model to describe denosumab concentrations following a single subcutaneous dose of 120 mg (figure 3a) and multiple monthly subcutaneous doses of 120 mg (figure 3b). Overall, the model appeared to adequately characterize the nonlinear pharmacokinetics of denosumab and was suitable to explore any covariate effects on the denosumab serum concentration-time course through model-based simulations.

Estimated population pharmacokinetic parameters of denosumab

Goodness-of-fit plots: (a) observed vs population-predicted concentrations; (b) observed vs individual-predicted concentrations; (c) conditional weighted residuals vs population-predicted concentrations; (d) conditional weighted residuals vs time.

Model evaluation, showing concentration-time profiles of denosumab following subcutaneous administration of (a) a single 120 mg dose and (b) 120 mg every 4 weeks. The light grey shaded areas indicate the 5th and 95th percentiles of the simulated concentrations.
The magnitude of the effects of body weight, race and tumour type on the time course of denosumab serum concentrations and the estimated RANKL inhibition are shown in figure 4. Figure 4a and 4b indicate the similarity of the denosumab pharmacokinetic profile and the time course of RANKL inhibition across different body weights. While the denosumab area under the concentration-time curve (AUC) values at steady state for typical 45 kg and 120 kg subjects were 43% higher and 47% lower, respectively, than the denosumab AUC for a typical 66 kg subject, the estimated RANKL inhibition AUC values at steady state for 45 kg and 120 kg subjects were only 0.21% higher and 0.63% lower, respectively, than the target occupancy AUC at steady state for the typical 66 kg subject. A comparison of the denosumab serum concentration-time course after administration of a 120 mg or 2 mg/kg dose was conducted to assess the effects of body weight on exposure for the fixed dosing regimen. Figure 4e and 4f show the similarity of the median and 90% prediction interval of the simulated denosumab serum concentration-time profile and the estimated time course of RANKL inhibition for both dosing regimens administered to cancer patients with a body weight range from 38 to 174 kg, respectively. Finally, after adjustment for differences in body weight, the effects of race (figure 4a and 4b) and tumour type (figure 4c and 4d) on denosumab disposition were limited. Actually, the estimated RANKL inhibition at steady state following 120 mg administered subcutaneously every month was greater than 97% during the entire dosing interval in more than 95% of subjects, regardless of the patient covariate values (figure 4b and 4d).

Model simulation and target occupancy: (a–d) simulation of the effects of body weight, race and tumour type on the pharmacokinetics of denosumab 120 mg every 4 weeks and on the receptor activator of nuclear factor κ-B ligand (RANKL) target occupancy. Graphs () and (b) show the effects of weight and race. Graphs (c) and (d) show the effects of weight and tumour type. Graphs (e) and (f) show simulations of the concentration-time profiles and RANKL target occupancy, respectively, after dosing of denosumab 120 mg every 4 weeks (q4wk) vs 2 mg/kg every 4 weeks (q4wk). PI = prediction interval.
Discussion
A primary goal of this analysis was to develop a population pharmacokinetic model to characterize the time course of denosumab absorption and disposition after intravenous and subcutaneous administration in patients with bone metastases from solid tumours. Consistent with the pharmacokinetics of other monoclonal antibodies and denosumab pharmacokinetics in women with osteopenia and osteoporosis,[19] an open two-compartment pharmacokinetic model with linear distribution to the peripheral compartment, parallel linear and target-mediated elimination, and first-order absorption following subcutaneous administration was suitable to describe the time course of denosumab serum concentrations in patients with bone metastases from solid tumours. The model that was developed represents the quasi-steady-state approximation of the general TMDD model[25] and was able to quantitatively characterize denosumab pharmacokinetics for different intravenous and subcutaneous dosing schedules administered to healthy subjects and advanced cancer patients. Rich pharmacokinetic information, including data from intravenous administration, obtained from healthy subjects, was previously published[19,20] and included in the present analysis to support the identification of the structural pharmacokinetic model parameters.
Bioavailability following subcutaneous administration of proteins is affected by the extent of absorption from the subcutaneous injection site and by pre-systemic catabolism. Accordingly, denosumab absolute bioavailability following subcutaneous administration in cancer patients with bone metastases was estimated to be 61%, which is also consistent with the relative exposure obtained from the non-compartmental analysis of the intravenous and subcutaneous data in Study 20010124,[20] the results of the previous population pharmacokinetics analysis of denosumab[19] and the bioavailability values reported for efalizumab, omalizumab, adalimumab and etanercept.[29,31,37] Furthermore, no evidence of dose-dependent bioavailability was observed for denosumab within the range of doses evaluated in the current and previous population pharmacokinetic analyses.[19]
The primary pathways for systemic absorption of monoclonal antibodies following subcutaneous administration include convective transport of the antibody through lymphatic vessels into the blood, and diffusion of the antibody across blood vessels distributed near the site of injection. As the flow rate of the lymphatic system is relatively slow and movement dependent, and because monoclonal antibodies are substantially large in comparison with cell membrane junctions, these proteins are absorbed at greatly varying rates across subjects and over relatively long periods of time (days or weeks) after drug administration. In humans, the typical absorption half-life (t½,ka = ln(2)/ka) for denosumab was estimated to be 2.7 days and was associated with large between-subject variability (51.5%). These values are consistent with the absorption rates reported previously for efalizumab, omalizumab and ustekinumab, which ranged from 1.4 to 3.6 days, with between-subject variability varying from 40% to 44%,[37] and are consistent with the results of the previous population pharmacokinetics analysis of denosumab in postmenopausal osteoporosis.[19] Exploratory analysis of ka versus dose did not reveal any trends and, therefore, ka was similar across the dose range that was evaluated. However, ka was associated with age and, on average, a 50% increase in age was associated with a 19% reduction in ka up to an age of 53 years. This relationship is physiologically plausible, since subcutaneous absorption depends on passive lymphatic transport, which in turn may depend on the activity level, which may correlate inversely with age. A decrease of the absorption rate with age has been observed for other biologics administered subcutaneously, such as erythropoiesis-stimulating agents, and has also been reported for denosumab in women with osteopenia or osteoporosis.[19,38,39] Model-based simulations indicated that, despite the differences in the absorption rates, the steady-state concentration-time profiles in young and old subjects were very similar (data not shown), suggesting limited clinical relevance of the effect of age on denosumab absorption in cancer patients.
V1 for IgG antibodies typically ranges from 2 to 3 L, with the volume of distribution at steady state, calculated as V1 + V2, ranging from 3.5 to 7 L, depending on the affinity for and capacity of binding to the ligand and its distribution.[40] V1 for denosumab was estimated to be 2.62 L, which was associated with moderate to large between-subject variability (48.5%). In addition, Black subjects have approximately 23% lower V1 values than non-Black subjects. Similar findings have been observed by Sutjandra et al. in postmenopausal women with osteopenia or osteoporosis treated with denosumab,[19] and for other monoclonal antibodies such as ustekinumab in patients with psoriasis, where V1 was found to be 11% smaller in non-Caucasian subjects than in Caucasian subjects.[37]
As for other monoclonal antibodies, the distribution of denosumab into the extravascular space may take place by convective transport from the blood to the interstitial space of the tissues or by receptor-mediated transcytosis. The alpha-phase half-life is approximately 13.6 hours, which is significantly shorter than the absorption half-life; therefore, it is not observed following subcutaneous administration because the slow absorption masks the distribution phase. At high concentrations (when the nonlinear target-mediated pathway is saturated), the denosumab beta-phase half-life is approximately 35.8 days. The volume of distribution at steady state (3.99 L/66 kg) was estimated to be similar to the plasma volume, as previously reported.[19] However, this value is slightly lower than the values reported for other monoclonal antibodies, which indicates the lack of extensive extravascular distribution of denosumab.[37] Nevertheless, the small volume of distribution does not preclude attainment of pharmacologically active concentrations, as is clearly demonstrated by the pharmacodynamics and efficacy of denosumab.[13,14,16–18] In cases where antibodies show high affinity and relatively high-capacity binding, as with denosumab, the true steady-state volume of distribution may be greater than the estimated distribution volume, especially if significant drug elimination occurs from the peripheral tissues.[30]
Like any other monoclonal antibody, denosumab exhibits a dual elimination process mediated by a nonspecific (linear) pathway and a RANKL-mediated pathway, which was implemented in the model as the quasi-steady-state approximation of the (nonlinear) target-mediated elimination process (figure 1). CLlin was estimated to be 3.25 mL/h/66 kg (between-subject variability 34.4%), which is about one third of the reported clearance for endogenous immunoglobulins (8.75–10 mL/h) and other monoclonal antibodies, but similar to the values reported for denosumab in women with osteopenia or osteoporosis.[30,37]
At monthly subcutaneous doses of 120 mg, the RANKL-mediated elimination pathway is fully saturated, since denosumab serum concentrations are much higher than 1872 ng/mL (9-fold the Kss value). At this dose level, denosumab pharmacokinetics are essentially linear, and denosumab disposition is mainly affected by the linear elimination pathway. Interestingly, after adjustment for body weight differences, CLlin in Hispanic subjects was 27% faster relative to the rates in Caucasian, Asian and Black subjects. However, given the small magnitude of the race effects on V1 and CLlin, it translated into only a slight effect (<15%) on denosumab serum concentrations at steady state and on total exposure (figure 4a). Similarly, a statistically significant association was found for subjects with solid tumours (breast, prostate, giant cell and other tumours), who had a 10–37% increase in CLlin relative to healthy volunteers. Again, these differences are small relative to the body weight effect and the inherent variability in denosumab pharmacokinetics. Therefore, the clinical relevance of race and tumour type on denosumab pharmacokinetics is expected to be limited, as the RANKL occupancy seems to be extensive during the entire dosing interval (figure 4b).
At very low concentrations, the nonlinearity in denosumab pharmacokinetics becomes more apparent, and total clearance is as fast as 85 mL/h at a concentration of 23 ng/mL when approximately 10% of the inferred RANKL is blocked. This rate is about 46% faster than the total clearance estimated for postmenopausal woman with osteoporosis at a similar concentration range.[19] Furthermore, the nonlinear elimination through the binding to RANKL reached half capacity at a concentration of 208 ng/mL, where the total clearance was then reduced to 49 mL/h, which is about 2.9-fold faster than the rate estimated for denosumab in postmenopausal woman with osteoporosis.[19] Following monthly subcutaneous 120 mg dosing, the RANKL-mediated elimination pathway is estimated to have >90% saturation during the time denosumab serum concentrations are higher than 1872 ng/mL (9-fold the estimated Kss value).
The apparent Kss value estimated from this analysis (1.44 nmol/L) is much higher than the in vitro KD estimated at 3.00 pmol/L but very similar to the half maximal inhibitory concentration for RANKL-induced formation of osteoclasts in murine macrophage precursor cells (1.64 nmol/L).[41] Part of this discrepancy might be explained by the quasi-steady-state assumption, where Kss is the sum of KD + kint/kon. However, this does not account for the three orders of magnitude difference between Kss and KD. Other factors might be potentially important to understand the in vivo-in vitro differences in KD. For instance, antibody concentrations in the interstitial fluid of the target tissue are likely to be substantially lower than antibody concentrations in serum, particularly if the small volume of distribution of denosumab relative to other monoclonal antibodies is considered. In this situation, the apparent KD would be over-estimated and will be closer to the hybrid parameter, Kss. On the other hand, simple calculations showed that an in vivo KD similar to the estimated in vitro KD would lead to maximal binding at all observed denosumab concentrations. In that situation, the concentration-time profile would not exhibit a clear non-linear pharmacokinetic profile, as was evidenced in the denosumab clinical studies. Nevertheless, the reasons why the in vivo and in vitro KD were different are still not well understood.
The typical value of the baseline RANKL level [R0] (4.46 nmol/L), assuming 1 : 1 binding, represented the steady-state total serum RANKL level and was associated with large between-subject variability (64.9%). In addition, the relative levels and extent of distribution of soluble and membrane-bound RANKL in both plasma and bone tissues are not known. The model assumes that RANKL distribution between plasma and bone tissues occurs instantaneously, but there are no data available to confirm or refute this hypothesis. Thus, the fast equilibrium assumption is acknowledged to be a limitation of the model. The inferred RANKL level is higher than values reported in the literature for healthy young women,[42] Chinese pre- and post-menopausal women,[43] osteoarthritic males[44] and multiple myeloma patients,[45] probably because of disease-related differences in RANKL expression in healthy populations and various disease settings. It is hypothesized that tumour cells in the bone lead to increased expression of RANKL on osteoblasts and their precursors. Excessive RANKL-induced osteoclast activity results in resorption and local bone destruction (with evidence of elevated levels of bone turnover markers), leading to SREs.[46]
Since the relative levels of soluble and membrane-bound RANKL are not known, the model assumes that kint and kdeg are hybrid elimination rates. According to this assumption, the apparent estimate of the half-life associated with the estimated kint (2.6 days) corresponds to a weighted average of the internalization of the denosumab : membrane-bound RANKL complex (a process that normally takes a few minutes) and the degradation of the denosumab : soluble RANKL complex (a process that normally takes about 3 weeks). As was observed in the population pharmacokinetic analysis in women with osteopenia or osteoporosis,[19] the elimination rate of the denosumab-RANKL complex (kint = 0.0112 h−1) was estimated to be much faster than the elimination rates of RANKL (kdeg = 0.00116 h−1) and denosumab (CLlin/V1 = 0.00124 h−1). One potential mechanism that might explain this phenomenon involves binding of the soluble antibody-antigen complex to Fcγ receptors on cells such monocytes and macrophages, which subsequently triggers internalization and catabolism of the complex.[37] If this elimination mechanism was present for denosumab, then it would be reasonable to anticipate that clearance of soluble denosumab-RANKL complex would be faster than that of free denosumab. A similar phenomenon has been reported for omalizumab (an anti-IgE monoclonal antibody); the apparent clearance of the omalizumab-IgE complex was 0.32 L/day and that of free omalizumab was 0.18 L/day.[47] The reasons behind this finding are not well understood, and it is unclear whether the estimated value reflects the actual complex elimination rate or difficulties in the estimation of this parameter in the absence of RANKL or denosumab-RANKL complex measurements.[25] Therefore, the values of kint, and also kdeg, should be interpreted with caution.
No evidence of time-dependent kinetics was found in the current analysis. Denosumab systemic exposure is consistent following repeated monthly subcutaneous administration of 120 mg (figure 3b). Consistent with the results of the non-compartmental analysis, simulated serum concentration-time profiles predicted 2.65- and 2.68-fold increases in the maximum and mean denosumab concentrations at steady state following monthly 120 mg subcutaneous dosing. The pharmacokinetics of denosumab were similar in healthy subjects and in patients with bone metastases from solid tumours, and no difference in R0 with respect to these two populations was evident. Notably, this finding does not imply that the relative levels of RANKL and osteoprotegerin would be the same across these two populations.
As body weight has been reported to be the most important demographic predictor of monoclonal antibody pharmacokinetics,[28,29] denosumab pharmacokinetic parameters were scaled by body weight, and simulations were conducted in order to assess whether body weight-based dosing was necessary or not. The typical pharmacokinetic profiles and their variability following denosumab 120 mg versus 2 mg/kg dosing were similar across the two dosing regimens (figure 4e), clearly indicating that there are no relevant differences in denosumab exposure and that dose adjustment based on body weight is not necessary. Model-based simulations revealed that following 120 mg dosing every 4 weeks, the inferred RANKL occupancy at steady state exceeded 97% during the entire dosing interval in more than 95% of subjects, independent of body weight, race (figure 4b) and disease type (figure 4d). Taken together, these data support the selection of a 120 mg dose administered monthly to prevent SREs in patients with bone metastases from solid tumours, and also explain the lack of clinically relevant effects of the patient covariates (such as body weight, race, age and type of tumour) affecting denosumab pharmacokinetics.
Conclusions
The integration of the pharmacokinetic data generated during denosumab development demonstrated that an open two-compartment pharmacokinetic model with linear distribution to the peripheral compartment, parallel linear and RANKL-mediated elimination, and first-order absorption following subcutaneous administration was suitable to describe the time course of denosumab serum concentrations following different intravenous and subcutaneous dosing schedules administered to patients with bone metastases from solid tumours. A monthly subcutaneous dose of denosumab 120 mg provides sufficient target coverage to prevent SREs in patients with bone metastases from solid tumours, regardless of the patient-specific covariates. In addition, given the moderate to large between-subject variability in denosumab pharmacokinetics, the clinical relevance of the effects of body weight, race, age and tumour type on pharmacokinetic parameters is likely to be limited, since a substantial overlap in simulated concentration-time profiles was observed and the inferred RANKL occupancy at steady state exceeded 97% during the entire dosing interval in more than 95% of subjects, regardless of body weight, age, sex, race and tumour type. Therefore, pharmacokinetically based dose adjustments on the basis of these patient covariates are not warranted for denosumab in patients with bone metastases from solid tumours.
References
Coleman RE. Clinical features of metastatic bone disease and risk of skeletal morbidity. Clin Cancer Res 2006; 12: 6243s–9s
Coleman RE. Skeletal complications of malignancy. Cancer 1997; 80: 1588–94
Coleman RE. Metastatic bone disease: clinical features, pathophysiology and treatment strategies. Cancer Treat Rev 2001; 27: 165–76
Abrahm JL, Banffy MB, Harris MB. Spinal cord compression in patients with advanced metastatic cancer: “all I care about is walking and living my life”. JAMA 2008; 299: 937–46
Anderson DM, Maraskovsky E, Billingsley WL, et al. A homologue of the TNF receptor and its ligand enhance T-cell growth and dendritic-cell function. Nature 1997; 390: 175–9
Burgess TL, Qian Y, Kaufman S, et al. The ligand for osteoprotegerin (OPGL) directly activates mature osteoclasts. J Cell Biol 1999; 145: 527–38
Lacey DL, Timms E, Tan HL, et al. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell 1998; 93: 165–76
Yasuda H, Shima N, Nakagawa N, et al. Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc Natl Acad Sci U S A 1998; 95: 3597–602
Elliott R, Kostenuik PJ, Chen C, et al. Denosumab is a selective inhibitor of human receptor activator of NF-KB ligand (RANKL) that blocks osteoclast formation and function [abstract no. P149]. Osteoporos Int 2007; 18: S54
Simonet WS, Lacey DL, Dunstan CR, et al. Osteoprotegerin: a novel secreted protein involved in the regulation of bone density. Cell 1997; 89: 309–19
Akatsu T, Murakami T, Nishikawa M, et al. Osteoclastogenesis inhibitory factor suppresses osteoclast survival by interfering in the interaction of stromal cells with osteoclast. Biochem Biophys Res Commun 1998; 250: 229–34
Fizazi K, Lipton A, Mariette X, et al. Randomized phase II trial of denosumab in patients with bone metastases from prostate cancer, breast cancer, or other neoplasms after intravenous bisphosphonates. J Clin Oncol 2009; 27: 1564–71
Lipton A, Steger GG, Figueroa J, et al. Extended efficacy and safety of denosumab in breast cancer patients with bone metastases not receiving prior bisphosphonate therapy. Clin Cancer Res 2008; 14: 6690–6
Body JJ, Facon T, Coleman RE, et al. A study of the biological receptor activator of nuclear factor-kappaB ligand inhibitor, denosumab, in patients with multiple myeloma or bone metastases from breast cancer. Clin Cancer Res 2006; 12: 1221–8
Yonemori K, Fujiwara Y, Minami H, et al. Phase 1 trial of denosumab safety, pharmacokinetics, and pharmacodynamics in Japanese women with breast cancer-related bone metastases. Cancer Sci 2008; 99: 1237–42
Stopeck AT, Lipton A, Body JJ, et al. Denosumab compared with zoledronic acid for the treatment of bone metastases in patients with advanced breast cancer: a randomized, double-blind study. J Clin Oncol 2010; 28: 5132–9
Henry DH, Costa L, Goldwasser F, et al. Randomized, double-blind study of denosumab versus zoledronic acid in the treatment of bone metastases in patients with advanced cancer (excluding breast and prostate cancer) or multiple myeloma. J Clin Oncol 2011; 29: 1125–32
Fizazi K, Carducci M, Smith M, et al. Denosumab versus zoledronic acid for treatment of bone metastases in men with castration-resistant prostate cancer: a randomised, double-blind study. Lancet 2011; 377: 813–22
Sutjandra L, Rodriguez RD, Doshi S, et al. Population pharmacokinetics meta-analysis of denosumab in healthy subjects and postmenopausal women with osteopenia or osteoporosis. Clin Pharmacokinet 2011; 50: 793–807
Bekker PJ, Holloway DL, Rasmussen AS, et al. A single-dose placebo-controlled study of AMG 162, a fully human monoclonal antibody to RANKL, in postmenopausal women. J Bone Miner Res 2004; 19: 1059–66
Thomas D, Henshaw R, Skubitz K, et al. Denosumab in patients with giant-cell tumour of bone: an open-label, phase 2 study. Lancet Oncol 2010; 11: 275–80
Mager DE, Jusko WJ. General pharmacokinetic model for drugs exhibiting target-mediated drug disposition. J Pharmacokinet Pharmacodyn 2001; 28: 507–32
Nakashima T, Kobayashi Y, Yamasaki S, et al. Protein expression and functional difference of membrane-bound and soluble receptor activator of NF-kappaB ligand: modulation of the expression by osteotropic factors and cytokines. Biochem Biophys Res Commun 2000; 275: 768–75
Mager DE, Krzyzanski W. Quasi-equilibrium pharmacokinetic model for drugs exhibiting target-mediated drug disposition. Pharm Res 2005; 22: 1589–96
Gibiansky L, Gibiansky E, Kakkar T, et al. Approximations of the target-mediated drug disposition model and identifiability of model parameters. J Pharmacokinet Pharmacodyn 2008; 35: 573–91
Ma P. Theoretical considerations of target-mediated drug disposition models: simplifications and approximations. Pharm Res 2012; 29: 866–82
Girard P. Data transformation and parameter transformations in NONMEM. Eleventh Meeting, Population Approach Group in Europe; 2002 Jun 6–7; Paris [online]. Available from URL: http://www.page-meeting.org/page/page2002/PascalGirardPage2002.pdf [Accessed 2012 Feb 15]
Mahmood I. Clinical pharmacology of therapeutic proteins. Rockville (MD): Pine House Publishers, 2006
Kuester K, Kloft C. Pharmacokinetics of monoclonal antibodies. In: Meibohm B, editor. Pharmacokinetics and pharmacodynamics of biotech drugs: principles and case studies in drug development. Weinheim: Wiley-VCH Verlag GmbH & Co. KGaA, 2006: 45–91
Wang W, Wang EQ, Balthasar JP. Monoclonal antibody pharmacokinetics and pharmacodynamics. Clin Pharmacol Ther 2008; 84: 548–58
Lobo ED, Hansen RJ, Balthasar JP. Antibody pharmacokinetics and pharmacodynamics. J Pharm Sci 2004; 93: 2645–68
Block G, Bone HG, Fang L, et al. A single dose study of denosumab in patients with various degrees of renal impairment [abstract no. 57]. J Amer Kidney Dis 2010; 55(4): B46
Gastonguay MR. Full covariate models as an alternative to methods relying on statistical significance for inferences about covariate effects: a review of methodology and 42 case studies. Twentieth Meeting, Population Approach Group in Europe; 2011 Jun 7–10; Athens [online]. Available from URL: http://www.page-meeting.org/pdf_assets/1694-GastonguayPAGE2011.pdf [Accessed 2012 Feb 16]
Yano Y, Beal SL, Sheiner LB. Evaluating pharmacokinetic/pharmacodynamic models using the posterior predictive check. J Pharmacokinet Pharmacodyn 2001; 28: 171–92
Pérez-Ruixo JJ, Piotrovskij V, Zhang S, et al. Population pharmacokinetics of tipifarnib in healthy subjects and adult cancer patients. Br J Clin Pharmacol 2006; 62: 81–96
Frame B, Koup J, Miller R, et al. Population pharmacokinetics of clinafloxacin in healthy volunteers and patients with infections: experience with heterogeneous pharmacokinetic data. Clin Pharmacokinet 2001; 40: 307–15
Dirks NL, Meibohm B. Population pharmacokinetics of therapeutic monoclonal antibodies. Clin Pharmacokinet 2010; 49: 633–59
Olsson-Gisleskog P, Jacqmin P, Pérez-Ruixo JJ. Population pharmacokinetics meta-analysis of recombinant human erythropoietin in healthy subjects. Clin Pharmacokinet 2007; 46: 159–73
Agoram BM, Martin SW, van der Graaf PH. The role of mechanism-based pharmacokinetic-pharmacodynamic (PK-PD) modelling in translational research of biologics. Drug Discov Today 2007; 12: 1018–24
Roskos LK, Davis CG, Schwab GM. The clinical pharmacology of therapeutic monoclonal antibodies. Drug Dev Res 2004; 61: 108–20
Kostenuik PJ, Nguyen HQ, McCabe J, et al. Denosumab, a fully human monoclonal antibody to RANKL, inhibits bone resorption and increases BMD in knock-in mice that express chimeric (murine/human) RANKL. J Bone Miner Res 2009; 24: 182–95
Hofbauer LC, Schoppet M, Schuller P, et al. Effects of oral contraceptives on circulating osteoprotegerin and soluble RANK ligand serum levels in healthy young women. Clin Endocrinol (Oxf) 2004; 60: 214–9
Liu JM, Zhao HY, Ning G, et al. Relationships between the changes of serum levels of OPG and RANKL with age, menopause, bone biochemical markers and bone mineral density in Chinese women aged 20–75. Calcif Tissue Int 2005; 76: 1–6
Findlay D, Chehade M, Tsangari H, et al. Circulating RANKL is inversely related to RANKL mRNA levels in bone in osteoarthritic males. Arthritis Res Ther 2008; 10: R2 [online]. Available from URL: http://arthritis-research.com/content/pdf/ar2348.pdf [Accessed 2011 Oct 6]
Marathe A, Peterson MC, Mager DE. Integrated cellular bone homeostasis model for denosumab pharmacodynamics in multiple myeloma patients. J Pharmacol Exp Ther 2008; 326: 555–62
Coleman RE, Major P, Lipton A, et al. Predictive value of bone resorption and formation markers in cancer patients with bone metastases receiving the bisphosphonate zoledronic acid. J Clin Oncol 2005; 23: 4925–35
Hayashi N, Tsukamoto Y, Sallas WM, et al. A mechanism-based binding model for the population pharmacokinetics and pharmacodynamics of omalizumab. Br J Clin Pharmacol 2007; 63: 548–61
Acknowledgements
Part of the content of this manuscript was presented at a poster podium session at the third American Conference on Pharmacometrics, San Diego, CA, USA, from 3 to 6 April 2011 (Gibiansky L, Sutjandra L, Doshi S, Zheng J, Sohn W, Peterson M, Jang G, Chow A, Pérez-Ruixo JJ. Population Pharmacokinetic Analysis of Denosumab in Advanced Cancer Patients with Solid Tumors).
The authors thank the thousands of patients, investigators, and medical, nursing and laboratory staff who participated in the clinical studies that were included in the present analysis; Mark Ma for coordinating the bioanalytical analysis for denosumab plasma concentrations; and Belén Valenzuela for the editorial comments provided during the preparation of the manuscript.
This study was sponsored by Amgen Inc., which was involved in the study design; the data collection, analysis, interpretation; the writing of the manuscript, and the decision to submit the manuscript for publication. Leonid Gibiansky was a consultant for Amgen Inc. and received consultation fees for contributing to the current analysis. Liviawati Sutjandra, Sameer Doshi, Jenny Zheng, Winnie Sohn, Graham Jang, Andrew Chow and Juan José Pérez-Ruixo were employees of Amgen Inc. and owned stock in Amgen Inc. at the time when the analysis was conducted. Mark Peterson is a former employee of Amgen Inc.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Gibiansky, L., Sutjandra, L., Doshi, S. et al. Population Pharmacokinetic Analysis of Denosumab in Patients with Bone Metastases from Solid Tumours. Clin Pharmacokinet 51, 247–260 (2012). https://doi.org/10.2165/11598090-000000000-00000
Published:
Issue Date:
DOI: https://doi.org/10.2165/11598090-000000000-00000