
Purpose
The aim of this study is to derive and evaluate an equilibrium model of a previously developed general pharmacokinetic model for drugs exhibiting target-mediated drug disposition (TMDD).
Methods
A quasi-equilibrium solution to the system of ordinary differential equations that describe the kinetics of TMDD was obtained. Computer simulations of the equilibrium model were carried out to generate plasma concentration-time profiles resulting from a large range of intravenous bolus doses. Additionally, the final model was fitted to previously published pharmacokinetic profiles of leukemia inhibitory factor (LIF), a cytokine that seems to exhibit TMDD, following intravenous administration of 12.5, 25, 100, 250, 500, or 750 μg/kg in sheep.
Results
Simulations show that pharmacokinetic profiles display steeper distribution phases for lower doses and similar terminal disposition phases, but with slight underestimation at early time points as theoretically expected. The final model well-described LIF pharmacokinetics, and the final parameters, which were estimated with relatively good precision, were in good agreement with literature values.
Conclusions
An equilibrium model of TMDD is developed that recapitulates the essential features of the full general model and eliminates the need for estimating drug-binding microconstants that are often difficult or impossible to identify from typical in vivo pharmacokinetic data.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The traditional paradigm of pharmacokinetic/pharmacodynamic (PK/PD) systems analysis typically involves a separation principle, whereby the time courses of plasma drug concentrations are modeled first and then fixed as driving functions in appropriate mechanism-based PD models (1,2). It is generally assumed that the amount of drug distribution to a biophase or drug interacting with a pharmacological target is negligible and does not affect PK profiles. Target-mediated drug disposition (TMDD) represents a distinct case wherein this assumption is invalid, and a significant proportion of the drug (relative to dose) is bound with high affinity to a receptor, enzyme, or transporter (3). Such interactions may result in nonlinear dose-dependent effects on drug disposition, with a decreasing apparent volume of distribution with increasing dose most commonly observed. When drug-target binding is also implicated in the elimination of the drug, such as receptor-mediated endocytosis, then concentration-dependent drug elimination may manifest as well (4).
A general PK model of TMDD has been described (5) and has since been utilized to characterize the PK of several compounds in humans and animals, including: bosentan and imirestat (5), interferon-β1a (6,7), vascular endothelial growth factor (8), warfarin (9), thrombopoietin (10), and leukemia inhibitory factor (LIF) (11). This modeling approach essentially integrates the thermodynamics of drug-target binding within the system of ordinary differential equations that describe drug disposition (see Theoretical), and drug-binding microconstants (kon and koff) are included in the model. However, these parameters are sometimes difficult to estimate from routine in vivo PK data. It has been shown, for example, that kon–koff parameters were not uniquely identifiable when the TMDD model was applied to abciximab PK/PD profiles, and a final stable model was achieved by assuming a quasi-equilibrium between free and bound drug concentrations (12,13). For the abciximab model, assuming a quasi-equilibrium state, along with some additional modifications, reduces one case of the TMDD model into one of the general nonlinear binding models originally described by Wagner (14). Although such a model was successfully applied to abciximab, these models do not allow for temporal changes in the homeostasis of the pharmacological target (e.g., production, degradation, and density up- and down-regulation). Drug exposure after relatively large doses or following multiple dosing may result in functional adaptation processes, and changes in receptor or enzyme kinetics may further impact the disposition of compounds exhibiting TMDD. Here we derive a quasi-equilibrium solution to the general TMDD model and evaluate its properties through computer simulations and nonlinear regression fitting to previously published LIF PK data resulting from the administration of a large range of intravenous (IV) doses in sheep (11).
Theoretical
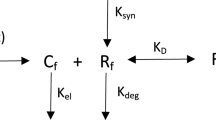
The details of the general pharmacokinetic model of TMDD have been introduced previously (5). As shown in Fig. 1, the key feature of this model responsible for observable nonlinear pharmacokinetic behavior is the saturable, high-affinity binding of the drug to its pharmacological target. Drug in the central compartment (C) binds at the second-order rate (kon) to free receptors (R) to form a drug receptor complex (RC). The RC complex may dissociate at the first-order rate (koff) or be internalized and degraded by the first-order rate process of endocytosis (kint). Free drug can also be directly eliminated at a first-order rate (kel) or be distributed to a nonspecific tissue-binding site (AT) by first-order processes (ktp and kpt). Free receptor can be synthesized at a zero-order rate (ksyn) and degraded at a first-order rate (kdeg). The input rate [In(t)] to the free drug compartment accounts for any process (IV bolus, zero-order infusion, first-order absorption, etc.) that may require additional model components. Free drug C, free receptor R, and drug-receptor complex RC are expressed in molar concentrations, whereas AT denotes amount (moles) of nonspecifically tissue bound or distributed drug. The model equations are as follows (5):
where Vc denotes the apparent volume of the free drug compartment. If the free drug is endogenously present (e.g., hormone or enzyme), then the initial conditions for the above system will be defined by the steady-state (baseline) values:
assuming that the free drug is administered as an IV bolus dose.

General pharmacokinetic model of target-mediated drug disposition. Adapted from Mager and Jusko (5). Symbols are defined in Theoretical.
Often, the binding of drug to free receptor and dissociation of the drug-receptor complex are of several orders of magnitude faster than the remaining system processes. Thus, equilibrium between the binding and dissociation is achieved almost instantly and observable data contain limited information about these processes. Consequently, the estimation of the parameters kon and koff becomes difficult, if not impossible. To resolve this problem, we additionally assume that free drug, free receptor, and drug-receptor complex are at quasi-equilibrium or rapid equilibrium (15):
where KD denotes the equilibrium dissociation constant. The quasi-equilibrium assumption [Eq. (6)] adds to the system [Eqs. (1)–(4)] and results in five equations describing four unknowns. To reduce the number of equations, one can add side by side Eq. (1) to Eq. (4) and Eq. (3) to Eq. (4) and introduce new system variables:
and
where Ctot can be interpreted as the total drug concentration and Rtot as the total receptor concentration. Then the system will be described by four variables C, Ctot, Rtot, and AT and the following four equations:
Derivations are provided in Appendix A. The initial conditions for Eqs. (8)–(10) are determined by the steady-state (baseline) values:
which are calculated in Appendix B and will be nonzero only if the endogenous production of free drug is present.
The assumption of fast drug-receptor binding allows the TMDD model to be simplified to its quasi-equilibrium form. However, the equilibrium model may contain parameters, which might not be identifiable from real data sets, and further simplifications should follow. If the receptor turnover is slow with respect to the experimental time scale, then one can postulate to let the ksyn and kdeg parameters to be small such that the baseline value for the free receptors R0 is constant:
This simplifies Eq. (10) to yield:
Also, the steady state for Rtot [Eq. (12c)] will be undetermined. Providing that there is no prior exposure of receptors to drug, the initial condition for Rtot should be:
If the receptors were partially occupied prior to the experiment, then Rtot(0) should become a model parameter because determination of the free receptor baseline values would not be possible.
Further simplifications can be made by assuming that the internalization rate is slow (kint ≈ 0). Then Eq. (14) implies that the total receptor concentration will remain constant (Rtot ≡ R0), and the model equations will consist of Eqs. (8), (9), and (11). Such a model has been introduced previously by Wagner to describe drug concentration in plasma that is eliminated by fast binding to tissue and a parallel first-order process as well as distributed to a peripheral compartment (14). In this terminology, our free receptor compartment represents the tissue, and Rtot is the maximum receptor (tissue) capacity. Additionally, the differential equation for the variable C was derived. We can obtain it by differentiating with respect to time the equilibrium Eq.(6) (see Appendix C):
with the initial condition C(0) = Dose/Vc. Thus, the quasi-equilibrium model with slow receptor turnover and internalization reduces to the Wagner nonlinear tissue-binding model that can be described by a single equation [Eq. (16)].
methods
Quasi-EquilibriumApproximation
The quasi-equilibrium assumption [Eq. (6)] is based on the observation that target-binding processes are much faster than all other processes described by the model. Comparison of the rates of change of the model variables requires a reference (time scale). From the point of view of observable data, the characteristic time (tchar) would be the length of the time interval between measurements. If the sampling times are of order of minutes, one should not expect to collect data containing information about processes that last seconds (e.g., receptor binding). A characteristic time for the binding process can be inferred from the receptor-binding rate constant, kon. If Cchar is the characteristic ligand concentration (for IV bolus drug administration, Cchar = Dose/Vc), then Ccharkon will be the first-order rate constant for the depletion of the receptor pool, and its reciprocal could be interpreted as the mean time a receptor resides in the receptor compartment prior to binding with its ligand:
The binding is fast if τB ≪ tchar, and hence the ratio τB/tchar ≡ ɛ ≪ 1. The time and time-dependent variables of the TMDD model can be scaled using tchar, τB, and other characteristic scales (e.g., a characteristic concentration of free receptors, Rchar), and the assumption of fast binding (ɛ→0) can be used to find 0th-order approximations of the model by the means of singular perturbation theory (16,17). Such a technique has been used to derive the equilibrium equation in enzyme kinetics (15). In our case, it will yield Eq. (6) along with the quasi-equilibrium model [Eqs. (8)–(10)]. The change of variables from C, R, and RC to C, Ctot, and Rtot in the singular perturbation analysis will be dictated by the presence of higher-order approximate solutions in equations for the 0th-order approximate equations which can be removed in the same way as the binding terms were disposed of for Eqs. (1)–(4) (see Appendix A). Presenting details of this procedure exceeds the scope of this work, but the general conclusion of accuracy of the approximation provided by the singular perturbation theory can be drawn. If the ratio τB/tchar is small, the approximation is usually accurate (assuming that the remaining scaling assumptions not discussed here hold). If τB/tchar ≈ 1 or τB is greater than tchar, then one might not observe good agreement between TMDD and equilibrium models. Because of the presence of the unknown parameters Vc and kon in the definition of τB, this criterion has limited practical value. However, if prior knowledge of such parameters is available, then an assessment can be made and ɛ may be a criterion for model selection. It should be noted that the error of the quasi-equilibrium approximation of the full model solution is O(ɛ), which can be interpreted as the error being proportional to ɛ without specifying how big the proportionality constant is. In practice, this precludes any definite values of ɛ that might serve as criteria except that they should be less than 1.
Another assumption for ensuring accuracy of the equilibrium model as required by the singular perturbation approach is:
which can be interpreted as the level of receptors available for binding must be comparable to or lower than the drug concentration [the symbol O(1) means that the left-hand side of Eq. (18) is bound by a constant]. If the free receptor concentration exceeds the drug concentration by an order of magnitude, then the accuracy of the quasi steady-state approximation might not be maintained and the full model should be used. Simulations will be performed where the differences between solutions of the equilibrium and full model with decreasing dose can be demonstrated.
Simulations
Computer simulations of the quasi-equilibrium model [Eqs. (8)–(12)] were conducted using the ADAPT II software program (18). To make a direct comparison with previous simulations of the general TMDD model (5), identical system parameters and conditions were selected. Two distinct cases of drug elimination were evaluated, where drug is eliminated from the central compartment alone (Case A, kel = 1 h−1, kint = 0) or in parallel with drug-target internalization (Case B, kel = 1 h−1, kint = 0.1 h−1). Nonspecific tissue binding (AT) was excluded (kpt = ktp = 0), and total receptor density was assumed to be constant (dRtot/dt = 0 assuming kint = kdeg). The remaining parameters were as follows: Rtot = 100 units, KD = 1 unit (for the general model, kon was 0.1 unit−1 h−1 and koff was 0.1 h−1), and Vc = 10 units. Escalating IV doses ranged from 100 to 4,000 units, and the initial condition for Eq. (8) was Ctot(0) = Dose/Vc [i.e., no endogenous drug is present in the system or In(t) = 0].
Data Analysis
Leukemia inhibitory factor plasma concentration-time profiles were obtained from a recent preclinical study conducted in sheep (11). Briefly, six groups of sheep (n = 2 per group) received single IV doses of recombinant human LIF of 12.5, 25, 100, 250, 500, or 750 μg/kg into the jugular vein. Blood samples (3 mL) were withdrawn from the indwelling jugular vein catheter at predose and at various times over 24 h. Plasma was separated and stored at −20°C until analyzed for immunoreactive LIF using a commercially available ELISA that was validated for use with sheep plasma (Quantikine; R&D Systems, Minneapolis, MN, USA). The intra- and interassay variability were less than 15%, and the limit of quantification was defined to be 50 pg/mL (from the lowest spiked plasma sample that demonstrated acceptable accuracy and precision).
Traditional methods of noncompartmental analysis (19) were applied to the PK profiles, revealing dose-dependent elimination (total clearance ranged from 5.18 to 1.09 mL/min/kg) and distribution (volume of the central compartment ranged from 68.9 to 39.1 mL/kg, although no clear trend was observed for the steady-state volume of distribution) as dose increased (11). After evaluating several nonlinear PK models, it was determined that the full TMDD model best characterized the data [Eqs. (1)–(4)]. Minor modifications to the general model included setting the first-order rate terms of nonspecific binding equal to one another (kpt = ktp = knsb) and allowing a separate total receptor density parameter for the largest IV dose (Rtotss2). No drug was detected in predose blood samples, and so initial conditions for Eqs. (1)–(4) are C(0) = Dose/Vc, AT(0) = 0, R(0) = Rtotss, and RC(0) = 0. The parameters ksyn and koff were fixed as secondary parameters according to the relationships: ksyn = kdegRtotss and koff = konKD, where the KD term was fixed to an in vitro literature estimate (0.1 nM). Final model parameters thus included knsb, kel, kdeg, kon, kint, Rtotss1, Rtotss2, and Vc, which were estimated using the maximum likelihood estimator in ADAPT II (nota bene: additional compartments and parameters were included to comodel PK profiles resulting from subcutaneous administration of LIF that are not considered in this present analysis). The variance model was defined as:
where σ is a variance model parameter to be estimated and y(t j ) represents model-predicted values of drug concentration.
In this study, the LIF concentration-time profiles were refitted with the quasi-equilibrium model [Eqs. (8)–(11)]. The previous minor modifications were included, namely, kpt = ktp = knsb and a separate Rtotss for the highest IV dose. The zero-order production of receptors (ksyn) was specified as a secondary parameter as before; however, the KD term was allowed to be estimated during the fitting process. Again, the maximum likelihood estimator in ADAPT II was used to estimate model parameters, along with the identical variance model [Eq. (19)].
Results and Discussion
Drugs that demonstrate TMDD produce complex nonlinear pharmacokinetic profiles that necessitate the incorporation of drug-target binding into models used to characterize the PK and sometimes PD properties of such compounds. Here we present a new modeling approach based on the quasi-equilibrium conditions of a general TMDD model (Fig. 1). The major advantage of this equilibrium model is that drug-binding microconstants (kon and koff), which are often difficult to estimate from typical in vivo PK experiments, are replaced by the equilibrium dissociation constant (KD).
A comparison of simulated pharmacokinetic profiles from the general and quasi-equilibrium TMDD models is shown in Fig. 2. The cases differ depending on whether drug elimination occurs only from the central compartment (Case A) or in parallel with drug-target complex internalization or metabolism (Case B). For both cases, there is good agreement between the models for relatively large doses, with only slight deviations at very early time points near the initial concentration and at inflection points around 5 h. In contrast, simulated data for low doses using the equilibrium model show systematic deviations from the general model over the entire time course. The immediate impact of this difference is difficult to interpret because error in measuring drug concentrations is not considered here. Regardless, the implementation of the equilibrium model clearly may be affected by the administered dose levels and the frequency of blood sampling, where the basic assumptions of quasi-equilibrium may not be satisfied.

Simulated concentration-time profiles for escalating intravenous doses (100, 1,000, 2,000, and 4,000 units) using target-mediated drug disposition models including binding microconstants [Eqs. (1)–(4); solid lines] or quasi-equilibrium conditions [Eqs. (8)–(11); dashed lines]. Cases correspond to elimination from the central compartment only (Case A) or in parallel with drug-target complex internalization/metabolism (Case B). Model parameterizations are defined in Methods.
If the free receptor concentration exceeds the drug concentration by an order of magnitude, then the accuracy of the quasi steady-state approximation might not be maintained and the full model should be used. This situation is depicted in Fig. 2 where the differences between solutions of the equilibrium and full model increase with decreasing dose. Assuming Rchar = Rtot, the ratios of Rchar/Cchar = 10 and 1 result in apparent differences, whereas the accuracy is acceptable for Rchar/Cchar < 1. For doses 100 and 1,000, the τB values were 1 and 0.1 h, which also contributed to observed discrepancies.
The quasi-equilibrium model was applied to LIF concentration-time profiles following escalating IV doses in sheep. The LIF data set was chosen because the general model was shown to well characterize the profiles previously, and the data seem to satisfy the above equilibrium criteria. Using the previously estimated parameter values for LIF and the molecular weight of 19,710 (11), for the lowest dose of 12.5 μg/kg, τB = 2.7 s, whereas the characteristic time for data sampling was tchar = 1 min. In addition, the initial concentration produced by the lowest dose was about 181 ng/mL, or approximately 9.18 nM, which exceeds the prior estimated Rtotss of 5.30 nM. The mean data and model-predicted profiles from simultaneously fitting the model to all of the IV data are shown in Fig. 3. The equilibrium model well captured the LIF pharmacokinetic data, and the predicted profiles are essentially identical to prior fittings using the general TMDD model [see Fig. 2A in Ref. (11)].

Plasma leukemia inhibitory factor (LIF) concentration-time profiles following intravenous administration in sheep. Lines represent predicted profiles from fitting the quasi-equilibrium target-mediated drug disposition model [Eqs. (8)–(11)] to all of the data simultaneously. Symbols represent previously reported mean data (11) and correspond to LIF doses of 12.5 (•), 25 (○), 100 (▾), 250 (▿), 500 (▪), and 750 (□) μg/kg (n = 2 per observation).
Final estimated pharmacokinetic parameters for LIF using either the general or quasi-equilibrium model are listed in Table I. Overall, there is good agreement between parameter estimates, with the exception of the equilibrium dissociation constant. The KD estimate from the equilibrium model (1.22 nM) is about 10-fold higher than the fixed value used for fitting the general TMDD model. Although the KD term was fixed in the previous analysis, the authors note that allowing both kon and koff to be estimated resulted in a similar KD value of around 0.1 nM, which is similar to in vitro measurements (11). There are several potential sources for the discrepancy in this parameter estimate. First, the prior analysis also included three additional PK profiles resulting from the subcutaneous administration of 10, 20, and 50 μg/kg of LIF. It is unclear whether these data influence KD estimates, but were excluded from this analysis to avoid any complexities associated with the subcutaneous absorption of therapeutic proteins. The second and more probable cause for disagreement in KD estimates is that the model may be insensitive to KD values. Simulations of TMDD models for two drugs, abciximab and tissue plasminogen activator, reveal that a 100-fold variation in the KD value had little effect on the resulting time course of plasma drug concentrations (13,20). A more complete sensitivity analysis of the equilibrium model is thus warranted and the subject of current study.
In summary, a new modeling approach for characterizing the pharmacokinetics of drugs showing target-mediated drug disposition has been developed based on quasi-equilibrium conditions of the general TMDD model. The equilibrium model eliminates the need to estimate the often problematic microconstants of drug-target binding, alternatively incorporating the equilibrium dissociation constant. Care must be taken to ensure that assumptions of equilibrium conditions have been satisfied, for which the newly described metrics of characteristic time (tchar) and ligand concentration (Cchar) may be calculated for verification. In contrast to prior equilibrium nonlinear protein-binding models, the kinetics of the pharmacological target may be included in the model, thereby removing the need to assume that total target density remains constant with drug exposure. This new quasi-equilibrium TMDD model retains the potential for exploiting the time course of pharmacological target occupancy in subsequent pharmacodynamic analyses of drug effects.
References
D. E. Mager E. Wyska W. J. Jusko (2003) ArticleTitleDiversity of mechanism-based pharmacodynamic models Drug Metab. Dispos. 31 510–518 Occurrence Handle10.1124/dmd.31.5.510 Occurrence Handle12695336
H. Derendorf B. Meibohm (1999) ArticleTitleModeling of pharmacokinetic/pharmacodynamic (PK/PD) relationships: concepts and perspectives Pharm. Res. 16 176–185 Occurrence Handle10.1023/A:1011907920641 Occurrence Handle10100300
G. Levy (1994) ArticleTitlePharmacologic target-mediated drug disposition Clin. Pharmacol. Ther. 56 248–252 Occurrence Handle7924119
E. D. Lobo R. J. Hansen J. P. Balthasar (2004) ArticleTitleAntibody pharmacokinetics and pharmacodynamics J. Pharm. Sci. 93 2645–2668 Occurrence Handle10.1002/jps.20178 Occurrence Handle15389672
D. E. Mager W. J. Jusko (2001) ArticleTitleGeneral pharmacokinetic model for drugs exhibiting target-mediated drug disposition J. Pharmacokinet. Pharmacodyn. 28 507–532 Occurrence Handle10.1023/A:1014414520282 Occurrence Handle11999290
D. E. Mager W. J. Jusko (2002) ArticleTitleReceptor-mediated pharmacokinetic/pharmacodynamic model of interferon-beta 1a in humans Pharm. Res. 19 1537–1543 Occurrence Handle10.1023/A:1020468902694 Occurrence Handle12425473
D. E. Mager B. Neuteboom C. Efthymiopoulos A. Munafo W. J. Jusko (2003) ArticleTitleReceptor-mediated pharmacokinetics and pharmacodynamics of interferon-beta1a in monkeys J. Pharmacol. Exp. Ther. 306 262–270 Occurrence Handle10.1124/jpet.103.049502 Occurrence Handle12660309
S. M. Eppler D. L. Combs T. D. Henry J. J. Lopez S. G. Ellis J.H. Yi B. H. Annex E. R. McCluskey T. F. Zioncheck (2002) ArticleTitleA target-mediated model to describe the pharmacokinetics and hemodynamic effects of recombinant human vascular endothelial growth factor in humans Clin. Pharmacol. Ther. 72 20–32 Occurrence Handle10.1067/mcp.2002.126179 Occurrence Handle12152001
G. Levy D. E. Mager W. K. Cheung W. J. Jusko (2003) ArticleTitleComparative pharmacokinetics of coumarin anticoagulants L: physiologic modeling of S-warfarin in rats and pharmacologic target-mediated warfarin disposition in man J. Pharm. Sci. 92 985–994 Occurrence Handle10.1002/jps.10345 Occurrence Handle12712418
F. Jin W. Krzyzanski (2004) ArticleTitlePharmacokinetic model of target-mediated disposition of thrombopoietin AAPS Pharm Sci 6 E9 Occurrence Handle10.1208/ps060109
A. M. Segrave D. E. Mager S. A. Charman G. A. Edwards C. J. Porter (2004) ArticleTitlePharmacokinetics of recombinant human leukemia inhibitory factor in sheep J. Pharmacol. Exp. Ther. 309 1085–1092 Occurrence Handle10.1124/jpet.103.063289 Occurrence Handle14872093
D. E. Mager M. A. Mascelli N. S. Kleiman D. J. Fitzgerald D. R. Abernethy (2004) ArticleTitleNonlinear binding models suggest that abciximab pharmacokinetics and pharmacodynamics are target-mediated Clin. Pharmacol. Ther. 75 88 Occurrence Handle10.1016/j.clpt.2003.11.338
D. E. Mager M. A. Mascelli N. S. Kleiman D. J. Fitzgerald D. R. Abernethy (2003) ArticleTitleSimultaneous modeling of abciximab plasma concentrations and ex vivo pharmacodynamics in patients undergoing coronary angioplasty J. Pharmacol. Exp. Ther. 307 969–976 Occurrence Handle10.1124/jpet.103.057299 Occurrence Handle14534354
J. G. Wagner ((1971)) A new generalized nonlinear pharmacokinetic model and its implications J. G. Wagner (Eds) Biopharmaceutics and Relevant Pharmacokinetics Drug Intelligence Publications Hamilton, IL 302–317
I. H. Segel (1975) Enzyme Kinetics. Behavior and Analysis of Rapid Equilibrium and Steady-State Enzyme Systems John Wiley & Sons New York
J. D. Murray (2002) Mathematical Biology Springer New York
C. C. Linand L. A. Segel (1974) Mathematics Applied to Deterministic Problems in the Natural Sciences Macmillan Publishing Co., Inc. New York
D. Z. D'Argenio A. Schumitzky (1997) ADAPT II User's Guide Biomedical Simulations Resource Los Angeles
M. Gibaldi D. Perrier (1982) Pharmacokinetics Marcel Dekker, Inc. New York
M. J. Kemme R. C. Schoemaker J. Burggraaf M. Linden Particlevan der M. Noordzij M. Moerland C. Kluft A. F. Cohen (2003) ArticleTitleEndothelial binding of recombinant tissue plasminogen activator: quantification in vivo using a recirculatory model J. Pharmacokinet. Pharmacodyn. 30 3–22 Occurrence Handle10.1023/A:1023293325245 Occurrence Handle12800805
Acknowledgments
The authors would like to thank Dr. S. A. Charman, Dr. C. J. H. Porter, and Dr. A. M. Segrave for providing the LIF PK data. This study was funded in part by Grant 57980 from the National Institute of General Medical Sciences, National Institutes of Health (for W. K.) and start-up funds (to D. E. M.) from the University at Buffalo, State University of New York.
Author information
Authors and Affiliations
Corresponding author
Appendices
Appendix A
Quasi-equilibrium equations for the TMDD model
Equations(7a) and (7b) define the variables Ctot and Rtot as the concentration sums of free and bound complexes. To derive the differential equations describing them, similar operations should be performed on the equations for the TMDD model. Adding Eq. (1) to Eq. (4) will result in:
The RC variable can be calculated from Eq. (7a), such that:
After substituting Eq. (A2) for RC in Eq. (A1), it can be rearranged to Eq. (8). Similarly, to obtain the differential equation for Rtot, one needs to add Eqs. (3) and (4) and eliminate the variables RC and R using Eq. (A2):
This procedure results in differential equations for Ctot and Rtot that contain the variable C which in turn can be calculated from the quasi-equilibrium Eq. (6) after substituting Eqs. (A2) and (A3) for RC and R:
Equation (A4) can be multiplied side by side by the denominator of the left-hand side and rearranged to the quadratic equation for C:
Because KD > 0 and Ctot ≥ 0, this equation has exactly one nonnegative solution:
which is Eq. (11). Equation (A6) is algebraically equivalent to Eq. (A4) under assumption C ≥ 0. The differential equation for AT in the equilibrium model is identical to Eq. (2).
Appendix B
Steady-state equations for the quasi-equilibrium TMDD model
To derive the steady-state equations for the quasi-equilibrium model, we will assume that the input rate is constant [In(t) ≡ In] and the nonspecific tissue-binding site (AT) is present (ktp, kpt > 0). The derivations will hold for receptor turnover with the synthesis rate ksyn > 0 and degradation or internalization (kint + kdeg > 0). Also, we consider the model with at least one clearance mechanism (kel + kint > 0) and the binding process always present (KD > 0). At steady state, there is no change in the model variables, and therefore, the time derivatives in Eqs. (8)–(10) can be set to 0 resulting in a system of four algebraic equations with four unknowns (Css, ATss, Ctotss, and Rtotss):
Equation (B4) is equivalent to the equilibrium Eq. (6) that can be rewritten as follows:
where the relationships defined by Eqs. (7a) and (7b) have been exploited. Equation (B2) allows one to express ATss as a function of Css:
Dividing Eq. (B2) by Vc and adding it to Eq. (B1), we cancel off the AT term and obtain:
If In = 0, and because Ctotss ≥ Css and ksyn > 0, then Eqs. (B3), (B5), and (B7) imply that kdeg > 0, and:
Later, we will assume that In > 0. Let us first consider the case where receptors are eliminated through the turnover process (kdeg > 0). Equation (B5) can be used to calculate Ctotss − Css and substitute for this term in Eq. (B3) yielding:
The only unknown now is Css. If kint = 0, then Css can be easily calculated from Eq. (B7):
If kint > 0, then calculation of the steady states becomes more troublesome. Css can be determined from Eq. (B3) if Eq. (B5) is used to replace Rtotss and Eq. (B7) to substitute for Ctotss − Css resulting in the following equation for Css:
After multiplying both sides of Eq. (B11) by kintCss, it can be rearranged to the equation for Css:
If kel = 0, then the positive solution exists only if ksyn > In:
If kel > 0, then the positive solution is:
Let us now consider the case kdeg = 0. Then the only clearance mechanism for bound receptors is the internalization process. If kint > 0, then Eq. (B3) implies:
and from Eq. (B7), it follows that for kel > 0 and In > ksyn:
Rtotss can be determined from Eq. (B8). If kel = 0, then after adding Eqs. (B3) and (B7), one can conclude that ksyn = In, thus implying:
Hence,
Inserting Rtotss from Eq. (B18) and Ctotss described by Eq.(B15) into Eq. (B5) yields an equation for Css:
which can be transformed to the following quadratic equation:
Equation (B21) has only one nonnegative solution:
This completes derivations of the steady-state solutions forthe quasi-equilibrium model, which are summarized in Table II.
Appendix C
Derivation of Wagner nonlinear tissue binding [Eq. (16)]
For derivation of Eq. (16), the best form of the equilibrium Eq. (6) is:
where Rtot is constant. One can differentiate both sides of Eq.(C1) and obtain:
Equation (8), with kint = 0, can be substituted in the left-hand side of Eq. (C2) resulting in:
Finally, one can solve Eq. (C3) for dC/dt and arrive at Eq. (16).
Rights and permissions
About this article
Cite this article
Mager, D.E., Krzyzanski, W. Quasi-Equilibrium Pharmacokinetic Model for Drugs Exhibiting Target-Mediated Drug Disposition. Pharm Res 22, 1589–1596 (2005). https://doi.org/10.1007/s11095-005-6650-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11095-005-6650-0