
Abstract
Osteoporosis is a chronic progressive disorder and is regarded as an important worldwide health issue. The development of novel treatments and the comparison of the effects of novel and existing treatments in osteoporosis are complicated by the difficulties of establishing drug effects on disease progression, as reflected in the slowly changing primary biomarker, bone mineral density. In recent years, research has considerably improved our understanding of the pathophysiology of osteoporosis. Specifically, various biomarkers have been identified that reflect bone physiology at the cellular level. These biomarkers mirror the dynamics of bone formation and degradation on a shorter timescale than bone mineral density as a composite measure. These markers can therefore, in principle, be used to characterize the underlying regulatory system and to quantify drug effects in osteoporosis.
Recently, the concept of disease system analysis has been proposed as a novel approach to characterize, in a strictly quantitative manner, drug effects on disease progression. This approach integrates physiology, disease progression and drug treatment in a comprehensive mechanism-based model, using dynamic information on a network of biomarkers. This review focuses on the use of disease system analysis for the characterization of drug effects on osteoporosis. It is concluded that, although the development of fully mechanistic disease system models may be practically impossible, parsimonious — but mechanism-based — disease system models may ultimately be used to adequately predict the long-term effects of drug treatment on clinical outcomes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Osteoporosis is a progressive disease, with fractures of the spine, hip or wrist as primary clinical manifestations. Most often, osteoporosis is diagnosed only after a fracture, and it is therefore referred to as a ‘silent’ disease. It is regarded as an important worldwide health issue because of the associated morbidity, mortality and costs resulting from a cumulative increase in fracture incidence and fracture risk associated with an increased life expectancy.[1–6] When considering prevalence, lifetime risk and socioeconomical impact, osteoporosis is placed alongside or even above other diseases such as breast cancer, cardiovascular disease and diabetes mellitus.[1,7,8]
The development of new treatments is challenging, as the trials needed to establish the prevention and treatment of osteoporosis require the inclusion of large numbers of patients and are long in duration. The former is due to statistical aspects of establishing antifracture benefit, and the latter is due to the slow progression of the disease, as reflected in the primary biomarker — bone mineral density (BMD) — required for registration. The biomarkers that change more rapidly — bone turnover markers (BTMs) — are considered secondary endpoints and are therefore not accepted by regulators as surrogate endpoints.
In the meantime, there is extensive knowledge about the physiology of bone and the underlying mechanisms of osteoporosis. Specifically, biomarkers reflecting different aspects and levels of bone physiology and the disease are at hand that enable comprehensive description of bone physiology and osteoporosis. Based on biomarker data and conceptual mathematical frameworks in the literature, the investigation of bone disease can be taken a step further. By combining the information from different sources within a disease system analysis (DSA) approach, while accepting that not all processes or information can or must be included, the observable results (e.g. marker responses) of the underlying disease system can be characterized. By identifying and capturing the essential time- and rate-limiting steps and the regulatory interactions within the system that can be supported by the available marker data, the important aspects of bone physiology are preserved. This perception forms the starting point for development of mechanism-based models of the disease, which integrate the pertinent information regarding bone physiology and osteoporosis into the description of the effects of current and upcoming treatments earlier and more adequately.
In this review, we discuss the pertinent information on the biology of bone physiology, bone disease (osteoporosis) and (drug) treatment. We emphasize the importance of these components in the transition from ‘simple’ models to comprehensive mechanism-based disease progression models utilizing the recently proposed concept of DSA.[9] In the end, the integration of these components can lead to an accurate prediction of the long-term clinical outcome fractures, from short- to medium-term observations. As there are many multifaceted aspects to this subject, the various components are discussed, with the intention being to highlight the wealth of information and to outline a novel approach to advance the understanding of osteoporosis and its treatments.
1. Introduction to Bone Physiology
1.1 Bone Structure
The skeletal system represents a major constituent of the human body and comprises the specialized connective tissues bone and cartilage. Bone is a dynamic organ, of which the composition is approximately 30% organic and 70% inorganic. The organic part, or the extracellular matrix of bone tissue, primarily holds type I collagen fibres, making up about 90% of its composition (osteoid matrix), in addition to proteoglycans and noncollagenous proteins. This combined protein matrix forms 98% of the organic constituents within bone. The remaining 2% is made up of bone cells, growth factors and cytokines, which play a vital role in bone homeostasis. The inorganic part of the bone tissue contains, for the most part, crystals of hydroxyapatite, an insoluble salt of calcium and phosphorus, which precipitates on the protein matrix, together forming the bone tissue.[10–15]
Two types of bone are distinguished, based on their structural, mechanical and metabolic function: cortical and trabecular. Cortical bone, making up around 80% of the skeleton by mass, forms the outer shell of bones. It consists of layers of bone (lamellae), which are organized around central canals in which blood vessels, nerves, connective tissue and lymphatic vessels are found (the Haversian system, also referred to as the osteon). Trabecular bone, representing only 20% of the skeletal mass but 80% of the bone surface, is found in the distal ends of long bones, in short bones, in the inner surfaces of flat bones and in irregular bones (e.g. vertebrae). Trabecular bone consists of an interconnected structure within which bone marrow is found.
As cortical bone is dense and compact calcified tissue, it has a high resistance to bending and torsion, and therefore provides mechanical strength and protection of vital internal organs and bone marrow. Furthermore, it forms the basis for muscle attachment, supporting locomotion. Trabecular bone is less dense and is composed of thin trabeculae forming a robust, 3-D framework with elastic properties, contributing to mechanical support, particularly in bones such as the vertebrae. Trabecular bone, in contrast to cortical bone, has a high turnover rate and therefore has a major function in metabolic processes by serving as a reservoir of calcium and phosphate for the maintenance of mineral homeostasis. Cortical bone has such metabolic function only in the situation of severe or prolonged mineral deficits.[10–15]
1.2 Bone Physiology
1.2.1 Bone Modelling and Remodelling
Bone modelling and remodelling are the main processes by which bone tissue continuously adapts and renews in reaction to metabolic and biomechanical stimuli. Bone modelling relates to the process of bone growth and adaptation, and controls the size, structure, quantity and shape of the skeletal structure. In this modelling state, the processes of resorption and formation of bone occur at different locations as bone grows and adapts. During the first two decades of life, it accounts for growth, and in the adult skeleton, it accounts for the healing of fractured bone and the increase in bone mass in reaction to biomechanical stress, such as extensive exercise.[13]
Bone remodelling refers to the process of bone renewal. In this process, new bone formation equals old bone resorption. Thereby, the action of bone cells is coupled, regulating the quality and the mechanical integrity of the skeleton during adult life.[13] The process of bone remodelling is the main controlling mechanism of calcium and phosphorus homeostasis. The bone cells that are active in remodelling (i.e. osteoblasts and osteoclasts) are temporally and spatially coupled, and closely collaborate within basic multicellular units (BMUs).[16] Bone modelling differs from remodelling in the sense that the bone cells responsible for bone formation and resorption can even be completely uncoupled in their activity.[17,18] Generally, if the removal of bone exceeds its formation, this results in the breakdown of bone. On the other hand, when the formation of bone structurally exceeds its removal, an increase in the bone mass is observed.
1.2.2 Bone Cells and Remodelling Stages
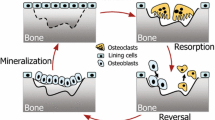
During a remodelling cycle, the turnover of bone is managed by a sequential process of the bone cells within a BMU under the control of various mechanical, systemic and local factors.[10,13,15,19–22] Osteoclasts are the bone cells responsible for bone removal, whereas osteoblasts form bone. Figure 1 presents the various phases during bone remodelling and includes the bone cells involved during the different stages of the remodelling process.

The bone remodelling cycle within the context of the basic multicellular unit depicted on a trabecular bone surface. Bone lining cells and osteoblastic stromal cells are involved in the activation phase of the osteoclastic cells and the initiation of a remodelling cycle. The arrows depict the collaborative pathways of the various cells. The osteoclasts remove bone during the resorption phase and arise from haematopoietic precursor cells. During the reversal phase, the bone surface is prepared for bone formation. Osteoclastic cells and regulatory factors signal the mesenchymal precursor cells to proliferate and differentiate into osteoblasts. These osteoblasts subsequently form bone matrix and finally become enclosed in bone to develop into osteocytes or transform into bone lining cells. (Adapted from Raisz,[19] with permission.)
The remodelling cycle begins with activation of a quiescent bone surface through a cascade of signals to osteoclastic precursors, which may be mediated by cells of the osteoblastic lineage. Bone-lining cells disappear from the bone surface, and haematopoietic precursor cells migrate to the exposed mineralized area and form osteoclasts that remove bone. During this resorption phase, the protein and mineral components of bone and various local paracrine and autocrine regulatory factors are released. This phase is followed by a reversal phase in which the bone surface is prepared for bone formation. Regulatory factors signal mesenchymal precursor cells to differentiate into osteoblasts, which are responsible for the production and maturation of the osteoid matrix. In this phase, osteoblasts synthesize and deposit collagen, and the initial mineralization of the formed osteoid matrix equals this collagen deposition. In a next step, the production of collagen decreases, and the secondary full mineralization of the matrix takes place.[10] In addition, the osteoblasts produce various regulatory factors, which are partly stored in the newly created bone for the future, to be released during subsequent remodelling cycles. Behind the osteoclasts that progress forward and continue the resorption of bone, new osteoblast teams are recruited to fill the site of resorption. The osteoclasts therefore consist of cells of different ages, and the successive groups of osteoblasts that build the bone are of the same age.[23] The resorption and reversal phases are relatively fast — in the order of weeks — compared with the slower process of formation, which can take several months.[10,13,15,19–22]
At the end of the formation phase, osteoblasts (i) enter apoptosis; (ii) become enclosed in the bone matrix, forming osteocytes; or (iii) stay on the bone surface to become bone-lining cells.[10,13,19–22] The osteocytes form a complex network within bone that probably functions as a mechanosensor, enabling a metabolic response to mechanical stimuli, or a lack thereof, by specifically recruiting osteoclasts to sites that need modelling or remodelling.[10,13,15,19–22,24]
It should be noted that, although the activation-resorption-formation cycle is similar for both types of bone, the organization of the BMU differs between trabecular and cortical bone. In cortical bone, the front (cutting cone) of the BMU forms a cylindrical canal by digging a tunnel (forward in the Haversian canal) through the bone, which is followed by osteoblasts that fill this tunnel (closing cone). For trabecular bone, the BMU progresses across the trabecular surface by digging a trench rather than a tunnel, and osteoblasts fill the resorption cavity. It has been shown that a BMU on a trabecular surface can be regarded as the lower half of a BMU within cortical bone. In cortical bone, an osteon is formed; in trabecular bone, a hemi-osteon is formed. The lifespan of a BMU is about 2–8 months.[10,25] (For an overview of the BMU and its progression on the trabecular surface or through cortical bone, see Parfitt.[23]) The remodelling process renews approximately 10% of bone each year, and the entire skeleton is renewed in approximately 7–10 years. Trabecular bone is metabolically more active and is remodelled at a higher rate (25% per year) than cortical bone (3% per year).[26] The higher turnover of trabecular bone is likely due to the fact that there is a large bone surface area, which is also in close contact with the bone marrow in which regulatory factors are present, whereas cortical bone is located more distantly from these factors.[10,26–29]
1.2.3 Signalling Pathway and Regulation of Remodelling: Mechanical, Systemic and Local Factors
Cumulative evidence shows that the osteoprotegerin (OPG)/receptor activator of NF-κB ligand (RANKL)/receptor activator of NF-κB (RANK) system is the major regulatory system in which the coordination of osteoclastogenesis and bone remodelling converges.[15,30–32] Figure 2 gives a schematic representation of this OPG/RANKL/RANK system. Cells of the osteoblastic lineage activate a remodelling cycle and initiate osteoclastogenesis, although the specific factors responsible for the activation have not yet been fully elucidated. Osteoblasts produce macrophage colony-stimulating factor (M-CSF, stimulating the c-fms receptor on osteoclasts) and bone marrow stromal cells, and osteoblasts express RANKL (a cytokine of the tumour necrosis factor family) on their surface, which binds to the RANK receptor on the surface of osteoclastic progenitors and osteoclasts. M-CSF and RANKL are both essential and sufficient for osteoclastogenesis, with RANKL being the primary driver of osteoclastogenesis, activation and survival of the osteoclast, and M-CSF being one of the potentiating factors.[33] Osteoblastic cells additionally secrete the soluble receptor OPG, which counteracts the differentiation and activation of osteoclasts by acting as a decoy receptor for RANKL. A decoy receptor recognizes and binds its ligand with high affinity and specificity but is structurally incapable of signalling.[34] The OPG/RANKL/RANK system is a fundamental system requiring the input of systemic hormones and local factors that regulate the expression of RANKL, RANK, OPG and M-CSF, which ultimately promotes the differentiation, migration and activation of osteoclasts.[15,20,30–32,35–38] In turn, osteoclastic activity regulates the differentiation and activation of osteoblastic cells through a variety of paracrine pathways, which include transforming growth factor-β, bone morphogenetic proteins, 1,25-dihydroxyvitamin D or calcitriol [1,25-(OH)2D], matrix metalloproteinases and bone lining cells. Overexpression of OPG or depletion of RANKL has been shown to induce osteopetrosis, and the reverse has been shown to induce osteoporosis in mice.[19,36,38] The finding of this OPG/RANKL/RANK mechanism has put a focus on the osteoblastic RANKL as a major set-point of osteoclastic formation, and the RANKL to OPG ratio as a gateway of osteoclastic activity.

Osteoclastogenesis as a result of the osteoprotegerin (OPG)/receptor activator of NF-κB ligand (RANKL)/receptor activator of NF-κB (RANK) system. Cells of the osteoblastic lineage initiate bone remodelling by contact with osteoclastic progenitors. Macrophage colony-stimulating factor (M-CSF) stimulates the c-fms receptor on osteoclasts. The preosteoclasts then present RANK, which is stimulated by RANKL, leading to proliferation and activation of osteoclasts. This interaction is blocked by OPG. Osteoclastogenesis is influenced by various systemic hormones and local factors such as cytokines, parathyroid hormone (PTH), vitamin D, calcitonin and prostaglandin E (PGE). (Adapted from Raisz,[19] with permission.)
Despite the crucial role of OPG/RANKL/RANK system in bone turnover, attempts to assess its status in patients have been relatively unsuccessful. This can be explained by a combination of technical difficulties with the current assays and discrepancies between serum concentrations and expression of RANKL and OPG in bone itself.[37] The latter may be caused by the technical difficulties with the assays but may also be related to the lack of specificity of these cytokines for bone, as they are also expressed in other tissues. (See Hofbauer et al.,[30] Lerner,[31] Kearns et al.[32] and Vega et al.[35] for extensive overviews of the current understanding and limitations regarding the OPG/RANKL/RANK system.) In conclusion, at present it is difficult, if not impossible, to assess the RANKL and OPG status in patients without invasive procedures such as bone biopsies.
As discussed, mechanical strain, systemic hormones and local factors exert an interrelated direct and/or indirect influence on the balance between RANKL and OPG in this specific regulatory mechanism underlying bone turnover. Osteocytes are thought to convert mechanical stimuli into metabolic responses, thereby organizing bone turnover under mechanical strain and immobilization.[13,22,30] Being descended from osteoblasts, they have similar paracrine capabilities. Increased mechanical strain inhibits RANKL and upregulates OPG, which decreases osteoclastic activity and therefore increases bone mass indirectly through the osteocytes.[10]
Various systemic hormones have an effect on bone turnover, which is mediated by local factors and probably converges at the level of RANKL and OPG. The primary systemic hormones include parathyroid hormone (PTH) as an important regulator of calcium homeostasis by stimulating bone resorption; the hormonally active metabolite of vitamin D, i.e. 1,25-(OH)2D; calcitonin with anabolic properties; thyroid hormones and glucocorticoids, which exert stimulatory and inhibitory effects on bone turnover; and estrogen, which controls osteoclast formation. Basically, PTH rapidly influences calcium concentrations through an increase in renal calcium reabsorption. Secondly, by increasing the transformation of 25-OH-vitamin D to its active metabolite, it indirectly leads to an increase in calcium concentrations.[10,13,15,39,40] (More elaborate discussions of the intriguingly complex action of PTH are available in the literature.[15,41–43])
Among the local paracrine and autocrine effectors are local cytokines and growth factors, which can act either as stimulators or as inhibitors of bone resorption or bone formation. Importantly, some of these factors act independently of the cytokines RANKL and OPG, and are necessary factors in the recruitment and differentiation of osteoblastic and osteoclastic cells. A subset of examples include members of the transforming growth factor-β family, tumour necrosis factor-α, various interleukins (e.g. interleukin−1, −4, −6, −7, −13, −17), insulin-like growth factors I and II, prostaglandin E2 and M-CSF.[15,19,31,32,40,44–47]
Clearly, the current description of the OPG/RANKL/RANK system is limited, and the importance and functioning of the various systemic hormones and local factors in relation to bone physiology is more complex. (For a more extensive outline on this multifaceted signalling pathway, including the mechanical, systemic and local factors, overviews have been presented by Seibel et al.,[15] Weitzmann and Pacifici,[20] Hofbauer et al.,[30] Lerner,[31] Kearns et al.,[32] Tanaka et al.,[36,48] Rogers and Eastell[37,49] and Hofbauer et al.[50–52])
Capturing these characteristics of the interaction between bone cells and including regulatory mechanisms in conceptual and mathematical frameworks have enhanced the understanding of the importance of these underlying systems in the description of bone physiology.[25,53–56]
2. Measuring Different Levels in the Dynamics of Bone Physiology
2.1 Bone Mineral Density (BMD) and Bone Turnover Markers (BTMs)
BMD presents the amount of bone mass (amount of minerals) per unit area or volume and provides a static measurement of skeletal status as it changes slowly over a period of months to years. Various methods are used to measure BMD at different skeletal sites, such as ultrasound, radiographic absorptiometry, dual photon absorptiometry, quantitative computed tomography (QCT), quantitative ultrasound and dual energy x-ray absorptiometry (DXA).[2,4,7,57] The gold standard for BMD measurements is DXA, which presents the bone mineral content per area (g/cm2). The spine (predominantly trabecular), hip (mixed trabecular/cortical) and wrist (predominantly cortical) are measured, as they represent the clinically most relevant areas for fractures.[4,57] A fracture of the hip is the site associated with the greatest morbidity, mortality and cost.[1,7]
BTMs present a dynamic expression of bone turnover, as they can reflect changes on a daily to weekly basis and thus on a substantially shorter timescale than BMD. A division into three BTM categories can be made, depending on their relation to the BMU in combination with bone: collagenous bone resorption markers, bone formation markers and markers of osteoclast regulatory proteins.[11,58,59] Table I and figure 3 present a summary overview of various markers of bone turnover in relation to their origin and function. The collagenous resorption markers are degradation products of bone collagen, and their concentrations reflect the rate of bone resorption.[15,61–63] Bone formation markers can be measures of the enzyme activity of osteoblasts, measures of the bone protein of which bone matrix is composed or measures of pro-collagen markers. The osteoclast regulatory proteins are divided into markers reflecting the rate of osteoclastogenesis and the osteoclast numbers. Finally, calcium (and phosphate) balance studies can present additional information on bone turnover and reflect the net index of the resorption and formation of bone in the whole body.[15,64] However, calcium balance studies are costly, time consuming and difficult to perform well.[49,65]


Biochemical markers of bone turnover in relation to their origination during bone remodelling. BSAP = bone-specific alkaline phosphatase; CTx = C-terminal cross-linked telopeptide of type I collagen; DPD = deoxypyridinoline; ICTP = C-telopeptide pyridinoline cross-links of type I collagen; NTx = N-terminal cross-linked telopeptide of type I collagen; OCN = osteocalcin; OPG = osteoprotegerin; PICP = C-terminal pro-peptide of type I collagen; PINP = procollagen type I N-propeptide; PYR = pyridinoline; RANK = receptor activator of NF-κB; RANKL = RANK ligand; TRAcP-5b = tartrate-resistant acid phosphatase. (Adapted from Leeming et al.,[11] with permission. © Springer-Verlag 2006.)
2.2 Variability in BMD and BTMs
Assessments of BMD and BTMs are subject to variability arising from both technical/analytical and biological sources.
The analytical or technical variability of BMD measures arises from the type of scanner used (DXA; e.g. lunar or hologic). Different scanners present slightly different outcomes, and the precision can vary because of device errors, technician variability, intra- and inter-observer variability and between-centre variations.[66–71] Nevertheless, the overall reproducibility of DXA measurements is satisfactory, as has been established with phantom measurements.[72,73] To overcome the issues of comparing differences in the numerical measures (g/cm2) for different types of scanners, each manufacturer provides a reference distribution for each device to internally normalize the individual measures. This presents the T-score, i.e. the number of standard deviation (SDs) above or below the mean reference value for young adults, which can be compared between scanners. In section 4.3, the use of the T-score in diagnosing osteoporosis and the points to consider when using these normalized scores are discussed.
Biological variability reflected in BMD measures results from the influence of seasonal variation, age, sex, demographics, recent fractures, drugs, disease, mobility and lifestyle, including food and vitamin D intake, and arises from the effects of these factors on the underlying physiological system.[11,26,58] The BMD precision error is often reported as a coefficient of variation (CV [%]) or as the SD (g/cm2). The short-term precision error for the lumbar spine and total hip is reported to be constant (0.010–0.15 g/cm2 [CV 1–1.5%] with 1 g/cm2 as the reference) over a wide clinical range.[74] This implies that the CV will increase with decreasing BMD measures. Therefore, it is best to base the smallest detectable differences in an individual on the SD.[73–75] It has also been shown that in older subjects, the measurement error increases for the spine and total hip.[73–75] The long-term precision error (SD) in a clinical study increased by 6.5–9.2% with each additional year of monitoring.[76]
BTMs exhibit substantial intra- and interindividual variability originating from analytical and biological sources. The analytical variability in BTM measures is determined by the marker itself (e.g. resorption markers are often a mixture of molecular entities), the assay used for a specific marker, the precision of the assay used, inter-laboratory variation, the mode (24-hour or second morning void for urinary markers) and timing of sampling (diurnal variation, menstrual rhythm), the type of sample taken (e.g. urine or serum) and the status of the subject (e.g. fasting, exercised). These are typically controllable factors, and they should thus be accounted for by investigators and laboratories — for example, by study design, specimen collection and use of standardized assays and working protocols.[77,78] The CVs for the inter- and intra-assay variability are typically within 10%.[11,15,58,79,80]
An important issue in measuring BTMs and comparing treatment effects between studies based on these markers lies in the fact that various assays, which are sometimes presented in different units (e.g. U/L or ng/L), exist for practically every marker. This has an influence on comparison of the results of ‘identical’ markers based on different assay methods. The markers of formation and resorption of bone can be measured by a variety of assays such as colourimetric, electrophoretic or precipitation techniques; high-performance liquid chromatography; or immunological assays such as enzyme immunoassay, immunoradiometric assay, radioimmunoassay, chemiluminescence immunoassays, electrochemiluminescence immunoassays and ELISA.[15,58] Furthermore, comparison of treatment effects based on the results of different markers is a challenge, as the markers present specific sensitivity for the change in bone turnover with a treatment. Overall, this leads to questions as to how to compare a treatment with itself (e.g. between different types of assay) and with other treatments (e.g. treatment-specific sensitivity of the marker) based on BTMs. To complicate matters further, relationships between markers and fractures have often been presented in relation to a specific assay. (See Cremers and Garnero,[59] Szulc and Delmas[81] and Garnero[82] for discussions on the markers of bone turnover and the challenges of their use in clinical practice and in clinical drug development.) As we will discuss later, a mechanistic platform model will aid in comparing disease and treatment effects based on these heterogeneous sets of markers, as it captures the underlying dynamics present in the data, independently of the measured markers and corresponding assays or reported assay units. Differences in the manner in which the markers are analysed in different studies can also be captured with such a mechanistic model. Furthermore, the previously mentioned differences in the numerical values produced with respect to the devices for measuring BMD can be incorporated into a platform model without relating them first to internal reference properties that are typical of a specific scanner.
The biological variation in the measures of BTMs arises from comparable factors, as mentioned previously for BMD. Besides the previously mentioned controllable factors, there are also uncontrollable factors of biological variability, such as age, sex, demographics, recent fractures, drugs, disease, mobility and lifestyle.[58,78] However, in contrast to BMD, the influence of several of these factors on BTM concentrations can be observed on a daily to monthly basis. In combination, these factors result in substantial amounts of intra- and interindividual variation in BTM.[83] Generally, the variability in the markers of bone turnover increases with advancing age and after menopause.[11,77,83–85] In paediatric to adolescent populations, the observed variability even dictates the characterization of different age categories to allow interpretation of bone marker values.[86]
An example of biological variation is the rhythms due to short- and long-term hormonal and seasonal differences. Interestingly, BMD decreases during winter and increases during summer, and an inverse relationship of this seasonal variation is observed for BTM concentrations. This is a direct result of a decrease in vitamin D concentrations and an increase in PTH concentrations during the winter.[26,58,87,88] Furthermore, influences of kidney function are relevant for renally cleared markers measured in urine and serum. With decreased creatinine filtration, markers corrected by urinary creatinine can present an apparent increase. On the other hand, decreased renal function might present an apparent decrease in urinary bone makers.[58,89]
The intraindividual biological variations of BTM for urinary and serum N-terminal cross-linked telopeptide of type I collagen (NTx) have been reported as being as large as 13.1% and 6.3%, respectively, within 3 days, and 15.6% and 6.3%, respectively, within 3 months. Others have reported intraindividual variability in the order of 5–27% in healthy and osteoporotic women for bone-specific alkaline phosphatase and osteocalcin determined during a period ranging from weeks to up to 2 years. Intraindividual variability of 9–48% for collagen cross-link immunoassays have been reported for comparable periods.[11,58,77,80,83,90]
In summary, BTMs are subject to substantial short- and long-term variability arising from controllable and uncontrollable (e.g. analytical and biological) variation, and for BMD, this variability appears to be somewhat smaller.[11,58,79,80,91]
When monitoring the individual effectiveness of treatment on BMD and BTMs, induced changes in these markers have to go beyond intraindividual variability. This means that the individual change in the marker — BTM or BMD — should be greater than the imprecision or noise of the measured value. This is called the least significant change.[58] Treatment generally induces changes within days to months in BTMs, and these changes typically range from 20% to 80%, depending on the choice of marker and treatment efficacy.[11,58,78,79,92,93] Intravenous zoledronic acid has presented a maximum decrease in the mean of the urinary resorption marker NTx at 1 week, and denosumab has shown decreases in concentrations of serum C-terminal cross-linked telopeptide of type I collagen (CTx) within 3 days.[94,95] For BMD, the changes range from 0% to 9% after a period of 1–3 years.[79,90] Interestingly, for the monoclonal antibody to RANKL (denosumab), significant increases in BMD have been observed within 1 month of treatment.[95–98]
Both BTMs and BMD are thus capable of detecting treatment efficacy, but BTMs present this information on a shorter timescale, which is also shown by the fact that changes in BTMs within 1–6 months correlate with BMD changes observed for up to 3 years.[11,58,77,79,99–103]
3. Imbalance and Activation in Bone Physiology
Bone turnover is perturbed in many diseases, each characterized by a specific pathophysiology. Examples of such metabolic bone diseases are Paget’s disease of bone, postmenopausal osteoporosis, hyperparathyroidism and hypoparathyroidism. However, external factors such as the use of corticosteroids, immobilization, and calcium and vitamin D deficiency are also well known as leading to altered bone turnover.[4,40,104] The change in the turnover arises from direct or indirect alterations at a certain stage in bone remodelling, which may result in an imbalance between resorption and formation and/or an increased activation frequency of the BMU.[1,13,14,31,32,36,105]
As bone remodelling is strictly regulated within a tightly coupled system, any local or systemic perturbations leading to uncoupling of osteoclastic and osteoblastic collaboration may substantially change bone mass. Figure 4 shows a comparison between normal turnover and high turnover of bone. Moreover, in disarranged regulation, any changes in osteoclastic activation or activity will be further attenuated by the different timescales on which osteoclasts and osteoblasts function. Osteoblasts will initially be unable to follow the increased resorption, which results in decreased bone mass. Clearly, trabecular bone, which has a higher turnover than that of cortical bone, is more sensitive to perturbations during remodelling.

Schematic representations of the effect on bone mass of normal and high bone turnover. During normal bone turnover, the amount of bone formed (F) equals the amount removed (R). During high turnover, the quantity of removed bone is not compensated for by the amount of bone that is formed, leading to a decrease in bone mass. (Adapted from Tanaka,[36] with permission. Copyright © 2007 S. Karger AG, Basel.)
4. Bone Pathology: Osteoporosis
Osteoporosis is a systemic skeletal disease characterized by low bone mass and microarchitectural deterioration of bone tissue, leading to enhanced bone fragility and a consequent increase in fracture risk.[57] Various forms of osteoporosis exist, which include postmenopausal osteoporosis, senile or age-related osteoporosis, idiopathic and juvenile osteoporosis, or osteoporosis as a result of other diseases (e.g. hypogonadism, endocrine states, diabetes), conditions (e.g. immobilization), deficiencies (e.g. vitamin D, calcium) or medications (e.g. corticosteroid use).[106,107]
4.1 OPG/RANKL/RANK System
The most common forms of osteoporosis are age-related osteoporosis in both women and men, and postmenopausal osteoporosis in women (figure 5). Alterations in the OPG/RANKL/RANK osteoclast regulatory system have been related to various metabolic bone diseases, including osteoporosis.[20,30–32] Loss of trabecular and cortical bone occurs as part of normal aging and affects both men and women from the fourth or fifth decade of life onward. In women, this bone loss presumably starts within 15 years preceding menopause and affects primarily trabecular bone, while cortical bone appears unaffected until menopause.[108] The slow bone loss relates to a combination of age-related mechanisms, including reduced bone formation, cumulative effects of calcium and vitamin D deficiency, decreased physical activity, and an age-related decrease in gonadal function.[1,13,20,108–120] For example, one factor that probably contributes to age-related bone loss is a deficiency in calcium and vitamin D, which can result in increased resorption of bone through the action of PTH. This secondary hyperparathyroidism stimulates increased calcium release from bone, an increase in BTMs and a subsequent decrease in bone mass.[31] Counterintuitively, serum OPG concentrations have been shown to increase with age in both women and men, but this is thought of as a protective compensatory mechanism, as the concentrations of bone markers have been negatively correlated to OPG concentrations.[32] This conflicting finding may also result from the fact that it is unclear whether serum concentrations of OPG accurately reflect the ratio of RANKL to OPG on the tissue level, where it is expected to have its effect.[32] In OPG-knockout mice, the level of osteopenia tends to increase with age, and increased concentrations of RANKL and decreased concentrations of OPG have been shown in normal aging mice.[32] Estrogen concentrations have been shown to be positively correlated with both OPG concentrations and BMD, and inversely correlated with markers of bone turnover.[11,19,20,30–32,37,50,121–123]

Bone mass dynamics during life. The bone mass increases during the initial decades of life. The attained peak bone mass is greater in men than in women. In both men and women, an age-related decrease in bone mass is observed. For women, this decrease is further attenuated by an accelerated phase of bone resorption during menopause.
At the time of menopause, women experience an additional transient period of accelerated bone loss when estrogen concentrations rapidly decrease. During this phase, increased concentrations of BTMs are observed, and these are correlated to decreasing BMD.[111,124–126] The onset of this postmenopausal phase lies between the ages of 50 and 65 years, and it presumably lasts for a period of 5–10 years and follows an exponential loss pattern in BMD.[13,108,109,127–133] During this period, trabecular bone is primarily affected because of its higher turnover and larger surface area compared with cortical bone, and the loss of trabecular mass appears to be a sensitive indicator of estrogen deficiency.[130] Estrogen deficiency in postmenopausal women has been shown to induce an increase in RANKL and a decrease in OPG, in addition to increased responsiveness of osteoclasts upon activation of the RANK receptor. Moreover, RANKL has been positively correlated with markers of bone turnover, and OPG has been shown to have an inverse relationship with these markers, although contradictory results have been published.[20,30–32,35,37]
After this accelerated phase, the slower phase of bone loss continues. During the postmenopausal phase, the majority of fractures are related mainly to trabecular bone, and they include acute vertebral compression fractures and distal forearm (Colles’) fractures.[7,129,134–137] In the long term, the age-related decrease in bone mass affects both trabecular and cortical bone, and hip fractures related to age-related bone loss are observed after the age of 70 years in both women and men.[7,138] The prevalence of osteoporosis and the lifetime probability of fractures beyond the age of 50 is about 3-fold higher in women than in men.[7]
4.2 Risk Factors for Osteoporosis and Osteoporotic Fractures
The major determinants of bone mass and the subsequent risk of osteoporosis and osteoporotic fractures are the peak bone mass accrued during the third decade of life and the subsequent rate of bone loss commencing after the fourth to fifth decades of life (figure 6). This makes women more prone to the effects of decreasing bone mass than men, as women have a lower peak bone mass and they experience accelerated bone loss after menopause.[1,139,140]

Major determinants of bone mass and the subsequent risk of osteoporosis and osteoporotic fractures. (Adapted from Riggs,[107] with permission from the BMJ Publishing Group.)
Various genetic and environmental factors determine the bone mass during life by influencing the accrued peak bone mass and the subsequent bone loss rate. These factors include modifiable risk factors and non-modifiable risk factors such as age, the menarche, (premature/early) menopause, a family history of osteoporosis/fractures, previous fragility fractures, low bodyweight, life expectancy, nutritional status, physical activity/immobility, lifestyle, race, sex, smoking, excessive alcohol (ethanol) consumption, and various medications and diseases.[4,7,57,133,136,138] In combination, these independent risk factors determine bone strength and the risk of osteoporotic fractures.
The total fracture risk is determined by the bone strength in addition to the frequency and direction of falls and the individual contribution of each risk factor to the overall fracture risk.[1,7,19,136,138,141]
Considering osteoporosis, one should therefore clearly distinguish between the diagnosis of osteoporosis and the associated fracture risk assessment.[4,57,65,142]
The WHO endorses a tool that takes into account all of these factors to predict the 10-year risk of osteoporosis fracture in men and women: FRAX®. A link to this tool and other programs that calculate cost-effectiveness and quality of life can be found on the website of the International Osteoporosis Foundation.[143] It should be noted that the links provide no quantitative insight into the contribution of each separate risk factor on which the calculations are based.
4.3 Diagnosis and Whole Bone Strength
BMD is the best single predictor of bone strength, and the diagnosis of osteoporosis is based on this confirmatory measurement, which accounts for 60–85% of bone strength.[2,4,57,144–146] Based on the deviation of an individual BMD measurement from that of the young healthy adult female mean (T-score), four categories are defined by the WHO (table II).[57] These categories are considered arbitrary cutoff values, as they heavily depend on the reference population, e.g. race, sex, demographics.[4,138,147–158]

Although the BMD is a strong predictor of bone strength, a low BMD alone accounts for only up to 44% of the fracture risk.[159] As discussed previously, independent risk factors (mostly extraskeletal factors) then provide complementary information on the individual risk of osteoporosis and osteoporotic fractures. As the total fracture risk reduction cannot be explained by BMD alone, the term ‘bone quality’ was designated to capture other skeletal properties that contribute to the whole bone strength.[160] These include the shape, geometry, microarchitecture, bone tissue composition, mineralization, microdamage and the rate of bone turnover.[1,161–165]
The term ‘bone quality’ is currently under discussion, as (i) BMD and bone quality are practically inseparable because BMD is an aggregate measure of bone (e.g. it includes the mineralization of bone); and (ii) the definition of bone quality is imprecise and difficult to measure and, at present, it is therefore difficult to translate bone quality into antifracture efficacy. (See Sievänen et al.[160] and Jarvinen et al.[166] for discussions on bone strength and bone quality semantics.) Nevertheless, the terms ‘bone strength’ and ‘bone quality’ aid the discussion on the complex properties of bone that contribute to the probability of fractures. More recently, advances in this field have been made by assessing the mechanical competence (strength) of bone by finite element modelling based on 3-D micro-computed tomography (micro-CT) images and high-resolution peripheral QCT (HR-pQCT) data.[167–169] An alternate view to the discussion on bone quality has been presented by Hernandez and Keaveny.[170] They argue that changes in bone quality should be looked at in a more clinically meaningful manner by quantifying the biomechanical effects of these changes rather than trying to resolve the semantics on bone quality. They state: “Because a clinical fracture is ultimately a biomechanical event, it follows then that any clinically relevant modification of bone quality must change bone mechanical performance (strength) relative to bone mass (density)”. A more comprehensive framework for quantifying the biomechanical effects is proposed, which enables evaluation of the relationship between bone mechanical performance and bone density. Furthermore, the hierarchical nature of the framework enables characterization of the scale at which the clinically relevant changes in bone quality occur, rather than looking at separate physical levels in bone quality. The previously discussed HR-pQCT technique would fit into this framework by presenting information on a specific level of bone biomechanics. Hernandez[161] also presented this framework to investigate how bone turnover could independently influence fracture risk by altering both bone biomechanics (strength) and bone mass (density).
Biochemical markers of bone provide easily accessible information on aspects of bone quality, such as the rate of bone turnover, and they become increasingly more important as supplementary and even independent predictors of BMD and fracture risk.[11,58,59,77–79,171–183] Furthermore, these markers present the response to treatment and patient compliance on a shorter timescale than that of BMD.[11,58,59,77,92,93,140,180,184–186] These markers cannot diagnose osteoporosis, because there is a broad overlap between healthy and diseased populations. The current markers also do not discern between trabecular and cortical bone loss.[1,4,57,142]
Other measures that provide information on parts of the bone quality framework include quantitative assessment of trabecular and cortical bone structure, the activation frequency and the remodelling imbalance. These include histomorphometric techniques, such as HR-pQCT, which present specific information on cortical and trabecular bone characteristics at the cellular and tissue level by using invasive bone biopsies and various imaging techniques to provide 3-D images of bone.[13,14,59,105,187–191] Interestingly, BTMs such as osteocalcin, N-terminal pro-peptide of type I procollagen and tartrate-resistant acid phosphatase have been linked to histomorphometric measurements and can thus be regarded as presenting an insight into the actual turnover and true quality of bone.[58,77,167]
4.4 BTMs, BMD and Fractures
BMD measurements present the surrogate marker for the diagnosis and management of osteoporosis and for the development of new treatments in this area. As discussed previously, low BMD has a strong relationship to the risk of fracture, and this measurement alone explains a substantial part, but not all components, of whole bone strength and fracture risk.[180,192] Single BTMs or combinations of BTMs have been shown to be related independently to the risk of fractures, to predict changes in BMD and to reflect the efficacy and mechanism of action of different treatments.[11,26,91,99–103,117,171,175,193–212] It has been shown that combining a clinical risk factor with low hip BMD and high levels of bone resorption improved the predictive value of the relative risk of fracture from 1.8–2.8 to 5.8. Combining high levels of bone resorption with BMD or a history of previous fracture results in a 70–100% higher 10-year probability of hip fracture than is associated with low BMD alone.[115,203,213] The levels of these markers semi-quantitatively reflect the actual bone turnover, and early changes in BTM are able to explain equal or even larger proportions of antifracture efficacy compared with changes in BMD.[15,171,175,178,181,214]
Of interest in the development of new treatment modalities is the shorter timescale with which BTMs change with respect to BMD and antifracture efficacy. The changes in BTMs can be assessed faster and may consequently be better drug development tools, with respect to antifracture efficacy, than slow changing BMD. As discussed previously, the combination of the information in BTMs and BMD has been shown to more accurately predict the risk of fracture than either marker alone. As bone strength comprises various skeletal characteristics besides bone density alone, combinations of markers at various stages of bone remodelling may present a more comprehensive insight into the total or true quality of bone. (See Seibel,[58,77] Cremers and Garnero[59] and Szulc and Delmas[81] for overviews of the applicability of biochemical markers of bone turnover.)
The US FDA and the European Medicines Agency distinguish between agents used in the treatment or prevention of osteoporosis.[65,215,216] Prevention relates to the avoidance of bone loss during the first years of menopause, while treatment refers to the reduction of fracture risk. BMD is regarded as the primary outcome marker for drugs developed for prevention of osteoporosis. For treatment of osteoporosis, the primary outcome is the reduction in vertebral and non-vertebral fractures in addition to proof of an increase in BMD.[7,65,142,215–217] Trials for prevention of osteoporosis require durations of 2 years, while those for treatment of osteoporosis must be of 3 years’ duration, with an additional requirement to include thousands of subjects to be able to establish antifracture efficacy. Although BTMs have been shown to exhibit potential for use as surrogate markers, they are regarded as secondary outcomes and are only approved as such by these regulatory agencies. An important shortcoming lies in the fact that these markers are not disease-specific and reflect every change in bone metabolism. Nevertheless, in combination with BMD, they provide a substantial body of information on treatment efficacy at different timescales in the development of treatments for osteoporosis.[11,58,77] In the development of new dose regimens for treatments such as risedronate, ibandronate and zoledronic acid, the combination of BTMs and BMD has been used to achieve effective dose regimens. For risedronate and ibandronate, these markers have been accepted by regulatory agencies as proof of efficacy for new dose regimens in addition to previously established antifracture efficacy.[59,218–230]
4.5 Treatment Options and Emerging Treatments
It is possible to distinguish between different treatments based on differences in their mechanisms of action, specific sites and modes of action, routes of administration (e.g. oral, intravenous, nasal spray), dosage regimens, and degrees of efficacy and effectiveness.[7,231–233]
Most agents decrease bone resorption (antiresorptive or anticatabolic treatment), some increase bone formation (anabolic treatment), and others have been reported to do both.[234] The nature of the treatment effect is reflected in the dynamics of BTMs. Figure 7 presents a schematic overview of these dynamics. Antiresorptive treatments result in a decrease in markers of bone resorption, which is followed by a decrease in formation markers due to coupling of the osteoblastic and osteoclastic actions. Anabolic treatments present an increase in markers of bone formation, which is followed by an increase in markers of bone resorption. During the period between the onset of the effect on the initial bone marker and the subsequent change in the second bone marker, bone mass increases. According to the bone markers, the effect of anabolic treatments appears to wear off after a period of time.[1,41] Figure 8 illustrates the change in BMD with antiresorptive and anabolic treatments. The antiresorptive effect (representing, for instance, estrogens, bisphosphonates and calcitonin) on BMD levels off after a period of time. The anabolic effect (representing, for instance, fluoride and intermittent PTH) on BMD initially continues to increase beyond the pronounced anabolic window (the first 6–12 months of treatment). This might relate to the fact that beyond this period, bone formation is still more greatly increased than bone resorption.[42,237] Therefore, the continuous increase in BMD with decreasing values of BTM is probably a result not just of secondary mineralization but also of a continued relative increase in bone formation.[235]

Schematic overview of the dynamics of formation and resorption markers after treatment that stimulates bone formation (parathyroid hormone; PTH) and after treatment with antiresorptive oral bisphosphonates. (Adapted from Cremers and Garnero,[59] with permission from Adis, a Wolters Kluwer business [© Adis Data Information BV (2006). All rights reserved.])

In general, antiresorptive treatments include hormone replacement therapy, bisphosphonates, selective estrogen-receptor modulators and calcitonin. The anabolic treatments are represented by intermittent PTH and its analogues, and sodium fluoride. The latter treatment has been abandoned, as it was shown to actually increase the risk of non-vertebral fractures.[233] Strontium ranelate has been shown to slightly increase bone formation and to slightly reduce bone resorption, thereby presenting a treatment that demonstrates both treatment actions. (For an overview of these pharmacological treatments, see Sambrook and Cooper,[1] Kanis et al.[7] and Borges and Bilezikian.[238]) Calcium and vitamin D derivates are important elements that positively influence bone homeostasis by supplementing any nutritional deficiencies in the elderly.[7] Table III presents an overview of the (pharmacological) interventions with references to background information.

Possible (pharmacological) interventions in osteoporosis
Emerging antiresorptive treatments include novel agents that are currently under development, such as cathepsin K inhibitors and a fully monoclonal antibody to RANKL, called denosumab, which have different mechanisms of action than the existing treatments. Cathepsin K inhibitors selectively decrease osteoclast-mediated bone resorption by inhibiting the enzyme required for the breakdown of type I collagen, presumably without altering bone formation. Denosumab blocks the binding of RANKL to RANK.[96,231,232] Sclerostin antibodies are another promising group of biological therapies, which have been shown to exert anabolic properties and to increase bone mass.[1,327]
Discussion of the complex matter of the exact biological mechanism of action of these agents is beyond the scope of this article, but it is acknowledged that these mechanisms might determine the relationship to outcome. From a conceptual and mathematical perspective, it is more important that there are agents available with different mechanisms, specific sites and modes of action, and degrees of efficacy and effectiveness, which result in qualitatively and quantitatively different dynamics of the biochemical markers of bone turnover.
The heterogeneity in the dynamics of the markers of bone turnover reflects the differences observed in BMD. For example, an increase in BMD results from a net positive bone-forming index, which can result from an increase in bone formation, a decrease in bone resorption or a combination of both. Each of these treatment effects is reflected by different bone marker dynamics (figures 7 and 8). To reflect and compare the complex mechanisms of action, combined use of these markers in drug development is thus warranted.[59] To compare treatments on a common basis, conceptual and mathematical models describing these combined dynamics can play a crucial role, as described in sections 5–7.
The development of new treatment regimens, such as monthly and yearly administration of agents (e.g. zoledronic acid[343]), is facilitated by the use of BTMs, as they provide a direct insight into bone dynamics and the related treatment effects. Combinations (figure 9) or sequential use of treatments with similar antiresorptive or opposite mechanisms of action require use of the markers to understand the complex dynamics that lead to an increase in BMD and ultimately a reduction in fracture risk.[77,237,308,309,344,345]

Changes in (a) markers of bone formation and (b) markers of bone resorption after treatment that stimulates bone formation (parathyroid hormone), after treatment with the antiresorptive bisphosphonate alendronate, and after a combination of both. Markers of both bone formation (procollagen type I N-propeptide [PINP]) and resorption (C-terminal cross-linked telopeptide of type I collagen [CTx]) changed after treatment. Parathyroid hormone increases the formation marker first and then the resorption marker. Alendronate decreases the resorption first and subsequently also the formation of bone. The combined treatment leads to a decrease in markers of bone formation and resorption. The bars represent the interquartile range. (Reproduced from Black et al.,[344] with permission. Copyright © [2003] Massachusetts Medical Society. All rights reserved.)
Finally, the site and mode of action of treatments and specific pharmacokinetic properties lead to differences in the duration of the effect on BTMs and BMD after withdrawal of treatment, perhaps indicative of disease modification or at least suggesting an appropriate duration of the effect.[98,185,310,346–352]
5. Modelling of Osteoporosis
In the area of osteoporosis, various conceptual, mathematical, statistical and epidemiological models have been established that have provided valuable insights into the complex area of bone research. In particular, conceptual models have provided an important basis for the understanding of the complex coupled interaction between the osteoclastic and osteoblastic actions within the BMU, and the influence on bone mass.[16–18,353] The models can be divided into those based on the theory of bone physiology and those based on the available markers and clinical outcome data.
5.1 Theory-Based Models in Bone Biology
The concepts of bone biology have been formalized into mathematical models incorporating the inherent dynamics of biological systems to deterministically describe the time trend in bone mass.[25,55,56,354–367] These models incorporate and explain the influence of various factors, such as age, peak bone mass, drug treatment and (patho-)physiological processes, on BMD. Many of the models are presented with a focus on the BMU concept and relate the system to mechanical and metabolic influences. Another group of conceptual mathematical models exist, which pursue a more mechanistic approach by incorporating these influences, with a direct focus on the dynamics and homeostatic regulation of the coupled interaction of the osteoclasts and osteoblasts within the BMU.[53,368–374] As such, the effects are described theoretically at the cellular level of the underlying physiological system. Figure 10 depicts the conceptual model proposed by Lemaire et al.,[53] which specifically incorporates the interaction between osteoblasts and osteoclasts.

Diagram of the model proposed by Lemaire et al.[53] to capture the interaction between osteoclasts (OCs) and osteoblasts (OBs). OPG = osteoprotegerin; PTH = parathyroid hormone; RANK = receptor activator of NF-κB; RANKL = RANK ligand; TGF-β = transforming growth factor-β. (Reproduced from Lemaire et al.,[53] with permission. Copyright [2004], with permission from Elsevier.)
The aforementioned conceptual and mathematical models present powerful frameworks for understanding the complex dynamic interactions and the resulting outcome of the process of bone remodelling in bone physiology. Moreover, the models are able to capture, explain and mimic the characteristics of bone biology, metabolic bone diseases and various treatment conditions, as discussed in the literature. A disadvantage of these methods lies in the fact that the inherent variability that exists in biological systems is generally not accounted for.
5.2 Data-Based Models in Bone Biology
The models based on the biological markers and clinical outcomes can be divided into statistical/epidemiological models that describe the correlation between markers of bone pathophysiology and clinical outcome, and models that describe the dynamics of various bone markers and clinical outcomes.
The statistical and epidemiological models have provided valuable information on the correlation, predictive value and interrelated time courses of various BTMs, BMD and clinical outcomes. Furthermore, this area of research has provided insight into the influences of various factors, such as age, lifestyle, and menopause, on these parameters, and has made it possible to evaluate, statistically confirm and compare the effects of different treatments.[7,11,91,101,102,115–119,134,136,137,154,157,173,177–179,181,192,196,198,204,205,213,214,375–377]
Nevertheless, these methods cannot be used for quantitative dynamic prediction of treatment efficacy in the area of drug development, because the effect of treatment is normally captured only as a categorical covariate without considering or quantifying the underlying temporal changes inherent in the biological system.
The models describing the dynamics of various bone markers based on data reflecting bone biology consist of (i) models that aim to extract quantitative measures of bone turnover; and (ii) models that describe the dynamics and relationships at the level of BTM, BMD and clinical outcomes. The first collection of models presents quantitative measures of bone turnover and bone biology, by using techniques such as tetracycline labelling with invasive histomorphometric measurements (bone biopsies) and noninvasive techniques such as tracer kinetics of labelled calcium and fluoride (radioisotope techniques and positron emission tomography).[13,62,63,169,189,359,378–384] Specific aspects of the bone quality can also be measured by assessing the mechanical competence (strength) of the bone by finite element modelling based on 3-D micro-CT imaging and HR-pQCT data.[167–169] All of the above methods specifically present intrinsic measures of bone turnover, such as the activation frequency of BMUs and osteoblastic and osteoclastic activity, and are able to characterize the underlying bone biology at the tissue and cellular level.[189,385] Generally, these models provide exceptional information on the underlying biology of bone; however, most of the methods cannot be applied on a large scale because of technical and ethical considerations, and they provide information only on specific parts of the skeleton.
The second collection of models is in a field of research that describes the dynamics of various bone markers, based on data reflecting bone biology, and is based on population pharmacokinetic-pharmacodynamic and disease progression modelling, also referred to as the ‘population approach’.[386–391] The value of this mathematical approach lies in the fact that the dynamics of BTM, BMD and clinical outcome can be dynamically linked even though the changes occur at vastly different rates, and that time-variant changes in the course of the disease can be explicitly taken into consideration in a mathematical structure describing different stages of metabolic bone diseases.[9,230] Various examples exist where the pharmacokinetics, BTM and BMD are linked within such a mathematical framework.[54,392–400] Figure 11 presents a schematic depiction of a population pharmacokinetic-pharmacodynamic model of ibandronate.

Schematic presentation of a population pharmacokinetic-pharmacodynamic model applied in the development of intermittent oral and intravenous ibandronate regimens. Ae = amount excreted in urine; CL R = renal clearance; Dose = ibandronate dose; k D = uCTx degradation rate constant; k S = uCTx formation rate; uCTx = urinary C-terminal cross-linked telopeptide of type I collagen; V x = volume of distribution in compartment x. (Reproduced from Pillai et al.,[392] with permission.)
Furthermore, this approach has been applied by the FDA to quantitatively link BMD to clinical fracture risks.[401] In addition, it has been used to dynamically describe the influence of seasonal variation on BMD and to establish new treatment schedules for bisphosphonates such as ibandronate.[226,230,394,398,402] This method provides a basis for the prediction and extrapolation of treatment effects, as the dynamics in the markers at various levels are considered in relation to the variability that exists between and within individuals, and it is therefore discussed further in sections 6 and 7.
6. Integration of Knowledge into Osteoporosis Research
6.1 Rationale for Combining Sources of Information
As presented, there is extensive knowledge on the physiology of bone, and several conceptual, statistical and mathematical approaches exist that capture various aspects of this biological system, each starting from either a theoretical or a data-driven point of view. Valuable information for understanding the complex area of bone biology and pathophysiology has been obtained by conceptualizing, utilizing and combining the available data that reflect the various levels of bone biology at different time stages of bone disease. The knowledge about bone physiology continues to increase, and different sources of information will continue to emerge. As a result, the conceptual, mathematical and statistical models will change accordingly.
None of the aforementioned approaches comprehensively integrates the conceptual and mathematical knowledge about the underlying regulatory system during the time course of osteoporosis, and the information about this system that is contained in the various BTMs and BMD measurements. As there is understanding of the pathophysiology of osteoporosis and the effect of various treatments, in addition to well established short- and long-term markers reflecting this disease, this combined knowledge can be integrated into a framework model with a mechanistic basis, which can ultimately be used to quantify clinical efficacy and optimize drug development in this area.[9]
Again, it should be noted that in order to follow this approach, one should recognize the fact that not all processes or information can or must be included to describe the observable results (e.g. markers) arising from the underlying complexity of the system, as long as the important aspects of bone physiology are maintained. Integration of knowledge on each regulatory process would result in a systems-biology approach, which would inherently lack the opportunity to comprehensively describe individual variability.[403] When pursuing more comprehensive models, one needs to identify which factors are likely to be predictive and what adjustments to the conceptual structures are needed to capture the relevant (or supported) rate-limiting steps. Obviously, this depends on the mathematical structures chosen, the aim of the model, the available information on the time course of biomarker dynamics, the available covariates and the disease status of the patients (e.g. early or late postmenopausal). Therefore, the identification of the factors and the necessary model adjustments need to be evaluated on an individual basis.
An integrated approach may allow for differentiation between current drug classes and novel agents with different mechanisms of action. Through this differentiation, a quantitative prediction of the final clinical outcomes of these novel agents can ultimately be obtained early in clinical development. Furthermore, development of new treatment regimens can be supported, and the effect on clinical outcomes of combinations or sequential use of treatments can be predicted.
6.2 Population Pharmacokinetic-Pharmacodynamic and Disease Progression Modelling
As discussed previously, an approach that allows for comprehensive integration of the knowledge of the disease system, individual treatment efficacy and the measures of bone turnover and bone density is in the area of population pharmacokinetics-pharmacodynamics and disease progression modelling. This methodology is based on nonlinear mixed-effects analysis, enabling identification and quantification of pharmacokinetic and pharmacodynamic responses to treatment within populations by estimating typical population parameters in addition to describing random effects.[404] It aims to comprehensively describe the dynamics of exposure and the related response of a treatment in relation to (patho-)physiological processes, while at the same time characterizing the interindividual variability in the drug and system-specific parameters, and the residual variability in the response. Furthermore, it aims to identify covariates that explain parts of the variability that exists between and within individuals. As such, this population approach characterizes individual profiles in relation to a typical population trend.
In recent years, population modelling has evolved from an empirical descriptive approach into a scientific means of identifying and quantifying the time course of drug effects, while considering the complex physiological and pathophysiological processes and mechanisms on the causal path between drug administration and effect.[388,389] A pertinent feature of this so-called mechanism-based pharmacokinetic-pharmacodynamic modelling is the separation of drug-specific and biological system-specific characteristics, which has been made possible by the availability of relevant biomarkers and the increase in knowledge about biological systems. This separation between the drug and the system is crucial for the prediction and extrapolation of treatment effects.[389] More recently, the effects of drugs on disease processes and disease progression has become of interest to account for the time-variant changes pertinent to biological systems that are under the influence of disease. This additional level of pharmacokinetic-pharmacodynamic modelling is of particular importance when considering disease-modifying treatment effects, as these agents specifically target the underlying time course of the disease.[9,389,405] So far, the population approach has been widely used for characterization and prediction of pharmacokinetic-pharmacodynamic responses and disease progression in relation to treatment in both clinical and drug-development settings.[386,406–409]
7. Population Modelling in Osteoporosis: Towards Disease System Analysis
An integrative approach to the conceptual and mathematical models and known statistical correlations allows for a mechanism-based description of osteoporosis, and presumably other bone diseases, without the need to incorporate each minute process of bone physiology. This approach can further optimize the use of various short- and long-term markers during drug development and in clinical practice. Such an approach, which incorporates the various levels and timescales of the disease and drug action by combining the available knowledge and data, is referred to as DSA.[9]
The population approach has been applied in the area of osteoporosis to dynamically link BTMs, most often resorption markers, to BMD and the latter to clinical fractures, albeit in a descriptive manner.[401] A more mechanistic approach to modelling of bisphosphonate treatment has been proposed, which accounts for a surface and a deep bone compartment in the pharmacokinetics of pamidronate in relation to the dynamics of a resorption marker.[394] A related example is presented for ibandronate, where the pharmacokinetics are coupled with CTx measurements through a bone compartment, and for a cathepsin K inhibitor, where the contributions of cortical and trabecular bone are accounted for in the production of CTx and NTx fragments during treatment.[392,393,410] Nevertheless, given the levels of knowledge about the system and the available data, this could conceptually be taken a step further by also incorporating markers of bone formation to mechanistically reflect the observed time delay between the onset of the treatment effect in resorption markers and formation markers caused by the coupled action of the osteoclastic and osteoblastic cells. Furthermore, the short-term treatment effect characterized from the resorption and formation markers could be used to translate these effects into long-term BMD changes. Ultimately, the fracture risk could be incorporated as an additional, clinically relevant, although not physiological marker.
From a more mechanistic point of view, the underlying regulatory system of bone physiology could be taken into consideration even more specifically, as the markers reflect explicit parts and levels of the system (e.g. BTMs), or derivatives of this system (e.g. BMD). The markers present information on distinct timescales about parts of the source system, which can be extracted when considering the concepts of bone pathophysiology. In this manner, a more accurate description of bone biology and treatment action could be achieved.
In view of the complex homeostatic control mechanisms in osteoporosis and the multitude of biomarkers that reflect its disease status, a DSA approach would require a cascade of compartments.[9] Lemaire et al.[53] and Komarova et al.[371] have presented multidimensional models that could serve as a basis for a DSA approach, to which BTMs and BMD measures could be linked.
7.1 Bone Physiology and Treatment Mechanism of Action
A benefit of a comprehensive platform model, incorporating the understanding of bone physiology and the information present in the markers at different stages and levels, is that it would allow for comparison of various treatments on a common basis during the time course of osteoporosis. When comparing treatments that decrease the resorption of bone with those that increase the formation of bone or treatments that induce both, different dynamics of the various bone resorption and formation markers are observed, each ultimately leading to an increase in BMD. Each treatment induces these effects through different sites and modes of action within the system and with different characteristic dynamics. For example, such a model would allow for comparison of teriparatide (which increases bone formation, which is followed by an increase in bone resorption) with the converse action of antiresorptive agents (see figure 7). Peterson and Riggs[54,411] have provided an example of a conceptual physiological model, in which bone physiology based on the OPG/RANKL/RANG regulatory system of Lemaire et al.[53] and calcium homeostasis by Raposo et al.[64] is combined, which enables comparison of these kinds of treatment effects on a common basis.
As the increase in BMD is, at least partly, dependent on the time-window between the changes in the formation and resorption markers, a combined approach could allow for characterization of this transient effect window, allowing for better understanding and more accurate prediction of the (individual) increases in BMD. This could be of great value when considering emerging treatments such as cathepsin K inhibitors, which decrease bone resorption while presumably exerting only modest effects on bone formation, and denosumab, which specifically interacts with the osteoblastic-osteoclastic regulatory system.[95,97,232]
A DSA approach offers the advantage that it brings all observations on biomarkers and BMD under a common denominator, leading to a definite conclusion on the treatment effect in terms of ‘disease modifying’ versus ‘symptomatic’, as well as ‘disease-dependent’ versus ‘disease-independent’.[9] This is important because analysis of the treatment effect on individual biomarkers may lead to different conclusions, depending on the choice of the biomarker. For instance, this can be observed in the investigation by Greenspan et al.,[198] where the effect of bisphosphonate treatment was apparently symptomatic when considering the dynamics of BTMs. Although the dynamics of both bone formation markers and bone resorption markers appear symptomatic, they have different dynamics after termination of the treatment. This is because the main action of bisphosphonates is inhibition of osteoclast-mediated bone resorption and because of the specific pharmacokinetics of bisphosphonates. Ultimately, the treatment effect on BMD dynamics appears to be disease-modifying.[9] We have recently made a similar observation in a DSA analysis of the effect of tibolone in combination with calcium on a variety of biomarkers and BMD (Post TM et al., unpublished data).
7.2 Extrapolation and Prediction
Another advantage of pursuing this comprehensive platform model using the population approach is that it characterizes the dynamics of a population, while identifying and explaining the diverse sources of variability. For instance, the seasonal influence on BTMs and BMD can be accounted for in a dynamic mechanistic fashion, and variation in stature, bodyweight and body composition, which correlate with BMD measures, can be taken into account to describe parts of the between- and within-subject variabilities. Eventually, by characterizing and quantifying these covariates and risk factors, and by accounting for inter- and intra-assay variation, extrapolation and prediction of various treatment effects along the trajectory of osteoporosis become feasible. The population approach allows an integrated mechanism-based model to leverage parameter information on specific parts of the system in a population analysis, where individual studies may lack information on specific components such as the disease, drug action or covariates.
From a drug-development perspective, establishing models that are capable of extrapolating and predicting treatment effects offers clear advantages in selecting effective compounds with the right dose, effective dosage regimens for the right population, a shorter timeframe and a reduced number of subjects for clinical trials. In addition, it would present an opportunity to take the best compound into full development based on quantitative differentiation between novel and existing drug classes, thus considering different mechanisms of action. Finally, it would allow prediction, comparison and selection of the efficacy of different treatment regimens for various mechanisms of action. Given the 2- to 5-year duration of clinical trials and the substantial number of subjects needed to establish antifracture risk, the efficiency of drug development in the area of osteoporosis would greatly benefit from predictive models for existing and novel compounds early in their development.
From a regulatory perspective, although these models cannot replace the actual trial, they may enable quantitative comparison between novel and existing drug classes to provide comprehensive supplementary guidance for assessment of treatment efficacy. From a clinical perspective, individual extrapolation would offer a tool for accurate estimation of individual treatment effectiveness, with prediction of its effects on BMD and perhaps the fracture risk, dose and treatment adjustments, treatment combinations and monitoring compliance. Nevertheless, one should bear in mind that knowledge about the system, and the information on relevant parts of the system and the available treatment effects, continue to increase and, inherently, the disease models should change accordingly to mirror important advances. Furthermore, established disease systems should be qualified extensively before they are applied in a regulatory setting, and especially in a clinical setting, in order to avoid misinterpretation of treatment effects.
8. Conclusion
In studying disease and treatment effects in the area of osteoporosis, an integrative approach is advocated in which the concept of DSA is applied. This concept combines the interplay between various levels of bone (patho-)physiology and relevant biomarkers in a dynamic quantitative framework. Depending on the quality and completeness of the data, this would allow for a mechanistic model capable of quantitative assessment of treatment efficacy at the various levels of bone physiology and at different stages of pathophysiology on a universal basis. An important characteristic of this approach lies in the identification and characterization of the essential time and rate-limiting steps in the regulatory system, based on the available literature data and conceptual mathematical models. The important aspects of bone physiology should be preserved, while avoiding a full physiologically based model incorporating each known process. This optimization of the use of information will bring the investigation of osteoporosis a step further by stepping away from ‘simple’ fragmented models, which may miss out on certain aspects that are relevant to the disease and treatment effects under investigation.
Ultimately, this should lead to a practical quantitative tool, which enables translation of short-term effects into long-term predictions in clinical and drug-development settings, and differentiation in efficacy between different drug classes. Moreover, with continuing advances in knowledge about the system, and with a multitude of markers becoming available that reflect bone physiology at various levels, such a platform model could be expanded, thereby increasing the translational power of such a platform model to compare, predict and extrapolate treatment effects with different mechanisms of action. Although development of fully mechanistic disease system models may be practically impossible, it is concluded that parsimonious — but mechanism-based — disease system models may ultimately be used to adequately predict the long-term effects of drug treatment on the clinical outcome: fractures.
References
Sambrook P, Cooper C. Osteoporosis. Lancet 2006; 367: 2010–18
Cummings SR, Bates D, Black DM. Clinical use of bone densitometry: scientific review. JAMA 2002; 288: 1889–97
Melton III LJ. The prevalence of osteoporosis: gender and racial comparison. Calcif Tissue Int 2001; 69: 179–81
WHO. Prevention and management of osteoporosis [technical report series no. 921]. Geneva: WHO, 2003 [online]. Available from URL: http://whqlibdoc.who.int/trs/WHO_TRS_921.pdf [Accessed 2009 Nov 13]
van Geel AC, Geusens PP, Nagtzaam IF, et al. Timing and risk factors for clinical fractures among postmenopausal women: a 5-year prospective study. BMC Med 2006; 4: 24
van Geel TA, Geusens PP, Nagtzaam IF, et al. Risk factors for clinical fractures among postmenopausal women: a 10-year prospective study. Menopause Int 2007; 13: 110–5
Kanis JA, Burlet N, Cooper C, et al. European guidance for the diagnosis and management of osteoporosis in postmenopausal women. Osteoporos Int 2008; 19: 399–428
Seeman E. Unmet needs in fracture prevention: new European guidelines for the investigation and registration of therapeutic agents. Osteoporos Int 2007; 18: 569–73
Post TM, Freijer JI, DeJongh J, et al. Disease system analysis: basic disease progression models in degenerative disease. Pharm Res 2005; 22: 1038–49
Hadjidakis DJ, Androulakis II. Bone remodeling. Ann N Y Acad Sci 2006; 1092: 385–96
Leeming DJ, Alexandersen P, Karsdal MA, et al. An update on biomarkers of bone turnover and their utility in biomedical research and clinical practice. Eur J Clin Pharmacol 2006; 62: 781–92
Dogan E, Posaci C. Monitoring hormone replacement therapy by biochemical markers of bone metabolism in menopausal women. Postgrad Med J 2002; 78: 727–31
Compston JE. Sex steroids and bone. Physiol Rev 2001; 81: 419–47
Seeman E, Delmas PD. Bone quality: the material and structural basis of bone strength and fragility. N Engl J Med 2006; 354: 2250–61
Seibel MJ, Robins SP, Bilezikian JP. Dynamics of bone and cartilage metabolism: principles and clinical applications. 2nd ed. San Diego (CA): Academic Press, 2006
Frost HM. The mechanostat: a proposed pathogenic mechanism of osteoporoses and the bone mass effects of mechanical and nonmechanical agents. Bone Miner 1987; 2: 73–85
Frost HM. Skeletal structural adaptations to mechanical usage (SATMU): 1. Redefining Wolff’s law: the bone modeling problem. Anat Rec 1990; 226: 403–13
Frost HM. Skeletal structural adaptations to mechanical usage (SATMU): 2. Redefining Wolff’s law: the remodeling problem. Anat Rec 1990; 226: 414–22
Raisz LG. Pathogenesis of osteoporosis: concepts, conflicts, and prospects. J Clin Invest 2005; 115: 3318–25
Weitzmann MN, Pacifici R. Estrogen deficiency and bone loss: an inflammatory tale. J Clin Invest 2006; 116: 1186–94
Rosen CJ. Pathogenesis of osteoporosis. Baillieres Best Pract Res Clin Endocrinol Metab 2000; 14: 181–93
Robling AG, Castillo AB, Turner CH. Biomechanical and molecular regulation of bone remodeling. Annu Rev Biomed Eng 2006; 8: 455–98
Parfitt AM. Osteonal and hemi-osteonal remodeling: the spatial and temporal framework for signal traffic in adult human bone. J Cell Biochem 1994; 55: 273–86
Hazenberg JG, Taylor D, Lee TC. The role of osteocytes and bone microstructure in preventing osteoporotic fractures. Osteoporos Int 2007; 18: 1–8
Hernandez CJ, Beaupre GS, Carter DR. A model of mechanobiologic and metabolic influences on bone adaptation. J Rehabil Res Dev 2000; 37: 235–44
Watts NB. Clinical utility of biochemical markers of bone remodeling. Clin Chem 1999; 45: 1359–68
Compston J. Local biosynthesis of sex steroids in bone. J Clin Endocrinol Metab 2002; 87: 5398–400
Compston JE. Bone marrow and bone: a functional unit. J Endocrinol 2002; 173: 387–94
Eriksen EF, Eghbali-Fatourechi GZ, Khosla S. Remodeling and vascular spaces in bone. J Bone Miner Res 2007; 22: 1–6
Hofbauer LC, Kuhne CA, Viereck V. The OPG/RANKL/RANK system in metabolic bone diseases. J Musculoskelet Neuronal Interact 2004; 4: 268–75
Lerner UH. Bone remodeling in post-menopausal osteoporosis. J Dent Res 2006; 85: 584–95
Kearns AE, Khosla S, Kostenuik PJ. Receptor activator of nuclear factor kappaB ligand and osteoprotegerin regulation of bone remodeling in health and disease. Endocr Rev 2008; 29: 155–92
Boyle WJ, Simonet WS, Lacey DL. Osteoclast differentiation and activation. Nature 2003; 423: 337–42
Colotta F, Re F, Muzio M, et al. Interleukin-1 type II receptor: a decoy target for IL-1 that is regulated by IL-4. Science 1993; 261: 472–5
Vega D, Maalouf NM, Sakhaee K. The role of receptor activator of nuclear factor-kappaB (RANK)/RANK ligand/osteoprotegerin: clinical implications. J Clin Endocrinol Metab 2007; 92: 4514–21
Tanaka S. Signaling axis in osteoclast biology and therapeutic targeting in the RANKL/RANK/OPG system. Am J Nephrol 2007; 27: 466–78
Rogers A, Eastell R. Circulating osteoprotegerin and receptor activator for nuclear factor kappaB ligand: clinical utility in metabolic bone disease assessment. J Clin Endocrinol Metab 2005; 90: 6323–31
Khosla S. Minireview: the OPG/RANKL/RANK system. Endocrinology 2001; 142: 5050–5
Jilka RL. Biology of the basic multicellular unit and the pathophysiology of osteoporosis. Med Pediatr Oncol 2003; 41: 182–5
Raisz LG. Physiology and pathophysiology of bone remodeling. Clin Chem 1999; 45: 1353–8
Canalis E, Giustina A, Bilezikian JP. Mechanisms of anabolic therapies for osteoporosis. N Engl J Med 2007; 357: 905–16
Rubin MR, Bilezikian JP. The anabolic effects of parathyroid hormone therapy. Clin Geriatr Med 2003; 19: 415–32
Rubin MR, Cremers S, Dempster DW, et al. PTH increases bone turnover in hypoparathyroidism [abstract no. F485]. 30th Annual Meeting, American Society for Bone and Mineral Research; 2008 Sep 12–16; Montreal (QC) [online]. Available from URL: http://www.abstractsonline.com/viewer/?mkey=DCB70C83-5B38-431A-B0E1-9221D66718D0 [Accessed 2009 Nov 13]
Teitelbaum SL. Bone resorption by osteoclasts. Science 2000; 289: 1504–8
Zaidi M, Blair HC, Moonga BS, et al. Osteoclastogenesis, bone resorption, and osteoclast-based therapeutics. J Bone Miner Res 2003; 18: 599–609
Schoppet M, Preissner KT, Hofbauer LC. RANK ligand and osteoprotegerin: paracrine regulators of bone metabolism and vascular function. Arterioscler Thromb Vasc Biol 2002; 22: 549–53
Sattler AM, Schoppet M, Schaefer JR, et al. Novel aspects on RANK ligand and osteoprotegerin in osteoporosis and vascular disease. Calcif Tissue Int 2004; 74: 103–6
Tanaka S, Nakamura I, Inoue J, et al. Signal transduction pathways regulating osteoclast differentiation and function. J Bone Miner Metab 2003; 21: 123–33
Eastell R. Role of oestrogen in the regulation of bone turnover at the menarche. J Endocrinol 2005; 185: 223–34
Hofbauer LC, Heufelder AE. Role of receptor activator of nuclear factor-kappaB ligand and osteoprotegerin in bone cell biology. J Mol Med 2001; 79: 243–53
Hofbauer LC, Khosla S, Dunstan CR, et al. The roles of osteoprotegerin and osteoprotegerin ligand in the paracrine regulation of bone resorption. J Bone Miner Res 2000; 15: 2–12
Hofbauer LC, Schoppet M. Clinical implications of the osteoprotegerin/RANKL/RANK system for bone and vascular diseases. JAMA 2004; 292: 490–5
Lemaire V, Tobin FL, Greller LD, et al. Modeling the interactions between osteoblast and osteoclast activities in bone remodeling. J Theor Biol 2004; 229: 293–309 [online]. Available from URL: http://www.sciencedirect.com/science/journal/00225193 [Accessed 2009 Nov 17]
Peterson MC, Riggs MM. Calcium homeostasis and bone remodeling: development of an integrated model for evaluation and simulation of therapeutic responses to bone-related therapies [abstract no. 1218]. 16th Meeting, Population Approach Group in Europe; 2007 Jun 13–15; Copenhagen [online]. Available from URL: http://www.page-meeting.org/default.asp?abstract=1218 [Accessed 2009 Nov 13]
Turner CH. Homeostatic control of bone structure: an application of feedback theory. Bone 1991; 12: 203–17
Turner CH. Toward a mathematical description of bone biology: the principle of cellular accommodation. Calcif Tissue Int 1999; 65: 466–71
WHO. Assessment of fracture risk and its application to screening for postmenopausal osteoporosis [technical report series no. 843]. Geneva: WHO, 1994
Seibel MJ. Biochemical markers of bone turnover: part I. Biochemistry and variability. Clin Biochem Rev 2005; 26: 97–122
Cremers S, Garnero P. Biochemical markers of bone turnover in the clinical development of drugs for osteoporosis and metastatic bone disease: potential uses and pitfalls. Drugs 2006; 66: 2031–58
Henriksen K, Tanko LB, Qvist P, et al. Assessment of osteoclast number and function: application in the development of new and improved treatment modalities for bone diseases. Osteoporos Int 2007; 18: 681–5
Arlot M, Meunier PJ, Boivin G, et al. Differential effects of teriparatide and alendronate on bone remodeling in postmenopausal women assessed by histomorphometric parameters. J Bone Miner Res 2005; 20: 1244–53
Charles P, Eriksen EF, Mosekilde L, et al. Bone turnover and balance evaluated by a combined calcium balance and 47calcium kinetic study and dynamic histomorphometry. Metabolism 1987; 36: 1118–24
Eastell R, Delmas PD, Hodgson SF, et al. Bone formation rate in older normal women: concurrent assessment with bone histomorphometry, calcium kinetics, and biochemical markers. J Clin Endocrinol Metab 1988; 67: 741–8
Raposo JF, Sobrinho LG, Ferreira HG. A minimal mathematical model of calcium homeostasis. J Clin Endocrinol Metab 2002; 87: 4330–40
Division of Metabolism and Endocrine Drug Products, US FDA. Guidelines for preclinical and clinical evaluation of agents used in the prevention or treatment of postmenopausal osteoporosis. Rockville (MD): FDA, 1994
Truscott JG, Simpson DS, Fordham JN. A suggested methodology for the construction of national bone densitometry reference ranges: 1372 Caucasian women from four UK sites. Br J Radiol 1997; 70: 1245–51
Truscott JG. Reference data for ultrasonic bone measurement: variation with age in 2087 Caucasian women aged 16-93 years. Br J Radiol 1997; 70: 1010–6
El-Maghraoui A, Achemlal L, Bezza A. Monitoring of dual-energy X-ray absorptiometry measurement in clinical practice. J Clin Densitom 2006; 9: 281–6
El-Maghraoui A, Do Santos Zounon AA, Jroundi I, et al. Reproducibility of bone mineral density measurements using dual X-ray absorptiometry in daily clinical practice. Osteoporos Int 2005; 16: 1742–8
Louis O, Verlinde S, Thomas M, et al. Between-centre variability versus variability over time in DXA whole body measurements evaluated using a whole body phantom. Eur J Radiol 2006; 58: 431–4
Blank RD, Malone DG, Christian RC, et al. Patient variables impact lumbar spine dual energy X-ray absorptiometry precision. Osteoporos Int 2006; 17: 768–74
Shepherd JA, Lu Y, Wilson K, et al. Cross-calibration and minimum precision standards for dual-energy X-ray absorptiometry: the 2005 ISCD official positions. J Clin Densitom 2006; 9: 31–6
Lodder MC, Lems WF, Ader HJ, et al. Reproducibility of bone mineral density measurement in daily practice. Ann Rheum Dis 2004; 63: 285–9
Wong JC, Griffiths MR. Precision of bone densitometry measurements: when is change true change and does it vary across bone density values? Australas Radiol 2003; 47: 236–9
Ravaud P, Reny JL, Giraudeau B, et al. Individual smallest detectable difference in bone mineral density measurements. J Bone Miner Res 1999; 14: 1449–56
Sadatsafavi M, Moayyeri A, Wang L, et al. Heteroscedastic regression analysis of factors affecting BMD monitoring. J Bone Miner Res 2008; 23: 1842–9
Seibel MJ. Biochemical markers of bone turnover: part II. Clinical applications in the management of osteoporosis. Clin Biochem Rev 2006; 27: 123–38
Delmas PD, Eastell R, Garnero P, et al. The use of biochemical markers of bone turnover in osteoporosis. Committee of Scientific Advisors of the International Osteoporosis Foundation. Osteoporos Int 2000; 11 Suppl. 6: S2–17
Miller PD, Baran DT, Bilezikian JP, et al. Practical clinical application of biochemical markers of bone turnover: consensus of an expert panel. J Clin Densitom 1999; 2: 323–42
Eastell R, Mallinak N, Weiss S, et al. Biological variability of serum and urinary N-telopeptides of type I collagen in postmenopausal women. J Bone Miner Res 2000; 15: 594–8
Szulc P, Delmas PD. Biochemical markers of bone turnover: potential use in the investigation and management of postmenopausal osteoporosis. Osteoporos Int 2008; 19: 1683–704
Garnero P. New biochemical markers of bone turnover. IBMS BoneKEy 2008 Mar; 5(3): 84–102
Hannon R, Eastell R. Preanalytical variability of biochemical markers of bone turnover. Osteoporos Int 2000; 11 Suppl. 6: S30–44
Ebeling PR, Atley LM, Guthrie JR, et al. Bone turnover markers and bone density across the menopausal transition. J Clin Endocrinol Metab 1996; 81: 3366–71
Garnero P, Mulleman D, Munoz F, et al. Long-term variability of markers of bone turnover in postmenopausal women and implications for their clinical use: the OFELY study. J Bone Miner Res 2003; 18: 1789–94
Yang L, Grey V. Pediatric reference intervals for bone markers. Clin Biochem 2006; 39: 561–8
Woitge HW, Knothe A, Witte K, et al. Circaannual rhythms and interactions of vitamin D metabolites, parathyroid hormone, and biochemical markers of skeletal homeostasis: a prospective study. J Bone Miner Res 2000; 15: 2443–50
Rapuri PB, Kinyamu HK, Gallagher JC, et al. Seasonal changes in calciotropic hormones, bone markers, and bone mineral density in elderly women. J Clin Endocrinol Metab 2002; 87: 2024–32
Hamano T, Fujii N, Nagasawa Y, et al. Serum NTX is a practical marker for assessing antiresorptive therapy for glucocorticoid treated patients with chronic kidney disease. Bone 2006; 39: 1067–72
Hannon R, Blumsohn A, Naylor K, et al. Response of biochemical markers of bone turnover to hormone replacement therapy: impact of biological variability. J Bone Miner Res 1998; 13: 1124–33
Looker AC, Bauer DC, Chesnut III CH, et al. Clinical use of biochemical markers of bone remodeling: current status and future directions. Osteoporos Int 2000; 11: 467–80
Delmas PD. Markers of bone turnover for monitoring treatment of osteoporosis with antiresorptive drugs. Osteoporos Int 2000; 11 Suppl. 6: S66–76
Delmas PD, Hardy P, Garnero P, et al. Monitoring individual response to hormone replacement therapy with bone markers. Bone 2000; 26: 553–60
Saag K, Lindsay R, Kriegman A, et al. A single zoledronic acid infusion reduces bone resorption markers more rapidly than weekly oral alendronate in postmenopausal women with low bone mineral density. Bone 2007; 40: 1238–43
McClung MR, Lewiecki EM, Cohen SB, et al. Denosumab in postmenopausal women with low bone mineral density. N Engl J Med 2006; 354: 821–31
Bone HG, Bolognese MA, Yuen CK, et al. Effects of denosumab on bone mineral density and bone turnover in postmenopausal women. J Clin Endocrinol Metab 2008; 93: 2149–57
Lewiecki EM, Miller PD, McClung MR, et al. Two-year treatment with denosumab (AMG 162) in a randomized phase 2 study of postmenopausal women with low BMD. J Bone Miner Res 2007; 22: 1832–41
Miller PD, Bolognese MA, Lewiecki EM, et al. Effect of denosumab on bone density and turnover in postmenopausal women with low bone mass after long-term continued, discontinued, and restarting of therapy: a randomized blinded phase 2 clinical trial. Bone 2008; 43: 222–9
Dobnig H, Sipos A, Jiang Y, et al. Early changes in biochemical markers of bone formation correlate with improvements in bone structure during teriparatide therapy. J Clin Endocrinol Metab 2005; 90: 3970–7
Grados F, Brazier M, Kamel S, et al. Prediction of bone mass density variation by bone remodeling markers in postmenopausal women with vitamin D insufficiency treated with calcium and vitamin D supplementation. J Clin Endocrinol Metab 2003; 88: 5175–9
Greenspan SL, Resnick NM, Parker RA. Early changes in biochemical markers of bone turnover are associated with long-term changes in bone mineral density in elderly women on alendronate, hormone replacement therapy, or combination therapy: a three-year, double-blind, placebo-controlled, randomized clinical trial. J Clin Endocrinol Metab 2005; 90: 2762–7
Greenspan SL, Rosen HN, Parker RA. Early changes in serum N-telopeptide and C-telopeptide cross-linked collagen type 1 predict long-term response to alendronate therapy in elderly women. J Clin Endocrinol Metab 2000; 85: 3537–40
Greenspan SL, Parker RA, Ferguson L, et al. Early changes in biochemical markers of bone turnover predict the long-term response to alendronate therapy in representative elderly women: a randomized clinical trial. J Bone Miner Res 1998; 13: 1431–8
Shoback D. Update in osteoporosis and metabolic bone disorders. J Clin Endocrinol Metab 2007; 92: 747–53
Seeman E. Bone quality: the material and structural basis of bone strength. J Bone Miner Metab 2008; 26: 1–8
Riggs BL, Melton III LJ. Involutional osteoporosis. N Engl J Med 1986; 314: 1676–86
Riggs BL. Overview of osteoporosis. West J Med 1991; 154: 63–77
Arlot ME, Sornay-Rendu E, Garnero P, et al. Apparent pre- and postmenopausal bone loss evaluated by DXA at different skeletal sites in women: the OFELY cohort. J Bone Miner Res 1997; 12: 683–90
Riggs BL. Endocrine causes of age-related bone loss and osteoporosis. Novartis Found Symp 2002; 242: 247–59; discussion 260-4
Recker R, Lappe J, Davies K, et al. Characterization of perimenopausal bone loss: a prospective study. J Bone Miner Res 2000; 15: 1965–73
Recker R, Lappe J, Davies KM, et al. Bone remodeling increases substantially in the years after menopause and remains increased in older osteoporosis patients. J Bone Miner Res 2004; 19: 1628–33
Becker C. Pathophysiology and clinical manifestations of osteoporosis. Clin Cornerstone 2006; 8: 19–27
Balasch J. Sex steroids and bone: current perspectives. Hum Reprod Update 2003; 9: 207–22
Riggs BL. Role of the vitamin D-endocrine system in the pathophysiology of postmenopausal osteoporosis. J Cell Biochem 2003; 88: 209–15
Garnero P, Delmas PD. Contribution of bone mineral density and bone turnover markers to the estimation of risk of osteoporotic fracture in postmenopausal women. J Musculoskelet Neuronal Interact 2004; 4: 50–63
Garnero P, Delmas PD. Noninvasive techniques for assessing skeletal changes in inflammatory arthritis: bone biomarkers. Curr Opin Rheumatol 2004; 16: 428–34
Garnero P, Delmas PD. Biochemical markers of bone turnover: applications for osteoporosis. Endocrinol Metab Clin North Am 1998; 27: 303–23
Garnero P. Biomarkers for osteoporosis management: utility in diagnosis, fracture risk prediction and therapy monitoring. Mol Diagn Ther 2008; 12: 157–70
Garnero P, Dargent-Molina P, Hans D, et al. Do markers of bone resorption add to bone mineral density and ultrasonographic heel measurement for the prediction of hip fracture in elderly women? The EPIDOS prospective study. Osteoporos Int 1998; 8: 563–9
Lips P, Hosking D, Lippuner K, et al. The prevalence of vitamin D inadequacy amongst women with osteoporosis: an international epidemiological investigation. J Intern Med 2006; 260: 245–54
Rogers A, Saleh G, Hannon RA, et al. Circulating estradiol and osteoprotegerin as determinants of bone turnover and bone density in postmenopausal women. J Clin Endocrinol Metab 2002; 87: 4470–75
Zallone A. Direct and indirect estrogen actions on osteoblasts and osteoclasts. Ann N Y Acad Sci 2006; 1068: 173–9
Fitzpatrick LA. Estrogen therapy for postmenopausal osteoporosis. Arq Bras Endocrinol Metabol 2006; 50: 705–19
Ross PD, Knowlton W. Rapid bone loss is associated with increased levels of biochemical markers. J Bone Miner Res 1998; 13: 297–302
Reeve J, Walton J, Russell LJ, et al. Determinants of the first decade of bone loss after menopause at spine, hip and radius. QJM 1999; 92: 261–73
Finkelstein JS, Brockwell SE, Mehta V, et al. Bone mineral density changes during the menopause transition in a multiethnic cohort of women. J Clin Endocrinol Metab 2008; 93: 861–8
Greer W, Smith R, Shipman AJ. A multi-exponential model of postmenopausal decline in vertebral bone mineral density: a new approach to the BMD reference range. J Clin Densitom 2003; 6: 113–24
Riggs BL, Khosla S, Atkinson EJ, et al. Evidence that type I osteoporosis results from enhanced responsiveness of bone to estrogen deficiency. Osteoporos Int 2003; 14: 728–33
Riggs BL, Khosla S, Melton III LJ, et al. A unitary model for involutional osteoporosis: estrogen deficiency causes both type I and type II osteoporosis in postmenopausal women and contributes to bone loss in aging men. J Bone Miner Res 1998; 13: 763–73
Gallagher JC. Effect of early menopause on bone mineral density and fractures. Menopause 2007; 14: 567–71
Chapurlat RD, Garnero P, Sornay-Rendu E, et al. Longitudinal study of bone loss in pre- and perimenopausal women: evidence for bone loss in perimenopausal women. Osteoporos Int 2000; 11: 493–8
Pi YZ, Wu XP, Liu SP, et al. Age-related changes in bone biochemical markers and their relationship with bone mineral density in normal Chinese women. J Bone Miner Metab 2006; 24: 380–5
Picard D, Imbach A, Couturier M, et al. Longitudinal study of bone density and its determinants in women in peri- or early menopause. Calcif Tissue Int 2000; 67: 356–60
Kanis JA, Johnell O, Oden A, et al. Ten-year risk of osteoporotic fracture and the effect of risk factors on screening strategies. Bone 2002; 30: 251–8
Kanis JA. Assessing the risk of vertebral osteoporosis. Singapore Med J 2002; 43: 100–5
Kanis JA, Borgstrom F, De Laet C, et al. Assessment of fracture risk. Osteoporos Int 2005; 16: 581–9
Kanis JA, Oden A, Johnell O, et al. The use of clinical risk factors enhances the performance of BMD in the prediction of hip and osteoporotic fractures in men and women. Osteoporos Int 2007; 18: 1033–46
Kanis JA. Diagnosis of osteoporosis and assessment of fracture risk. Lancet 2002; 359: 1929–36
Hansen MA, Overgaard K, Riis BJ, et al. Role of peak bone mass and bone loss in postmenopausal osteoporosis: 12 year study. BMJ 1991; 303: 961–4
Riis BJ, Hansen MA, Jensen AM, et al. Low bone mass and fast rate of bone loss at menopause: equal risk factors for future fracture: a 15-year follow-up study. Bone 1996; 19: 9–12
Chen YT, Miller PD, Barrett-Connor E, et al. An approach for identifying postmenopausal women age 50–64 years at increased short-term risk for osteoporotic fracture. Osteoporos Int 2007; 18: 1287–96
WHO. Guidelines for preclinical evaluation and clinical trials in osteoporosis. Geneva: WHO, 1998 [online]. Available from URL: http://whqlibdoc.who.int/publications/1998/9241545224_eng.pdf [Accessed 2009 Nov 13]
International Osteoporosis Foundation. Health economics [online]. Available from URL: http://www.iofbonehealth.org/health-professionals/health-econo mics.html [Accessed 2009 Nov 19]
Ammann P, Rizzoli R. Bone strength and its determinants. Osteoporos Int 2003; 14 Suppl. 3: S13–8
Schoenau E, Land C, Stabrey A, et al. The bone mass concept: problems in short stature. Eur J Endocrinol 2004; 151 Suppl. 1: S87–91
Schoenau E, Saggese G, Peter F, et al. From bone biology to bone analysis. Horm Res 2004; 61: 257–69
Lu Y, Genant HK, Shepherd J, et al. Classification of osteoporosis based on bone mineral densities. J Bone Miner Res 2001; 16: 901–10
Singer A. Osteoporosis diagnosis and screening. Clin Cornerstone 2006; 8: 9–18
Kanis JA, Delmas P, Burckhardt P, et al. Guidelines for diagnosis and management of osteoporosis. The European Foundation for Osteoporosis and Bone Disease. Osteoporos Int 1997; 7: 390–406
Kanis JA, Black D, Cooper C, et al. A new approach to the development of assessment guidelines for osteoporosis. Osteoporos Int 2002; 13: 527–36
Binkley N, Kiebzak GM, Lewiecki EM, et al. Recalculation of the NHANES database SD improves T-score agreement and reduces osteoporosis prevalence. J Bone Miner Res 2005; 20: 195–201
Binkley N, Bilezikian JP, Kendler DL, et al. Official positions of the International Society for Clinical Densitometry and executive summary of the 2005 Position Development Conference. J Clin Densitom 2006; 9: 4–14
Binkley NC, Schmeer P, Wasnich RD, et al. What are the criteria by which a densitometric diagnosis of osteoporosis can be made in males and non-Caucasians? J Clin Densitom 2002; 5 Suppl.: S19–27
Havill LM, Mahaney MC, Binkley L, et al. Effects of genes, sex, age, and activity on BMC, bone size, and areal and volumetric BMD. J Bone Miner Res 2007; 22: 737–46
Kiebzak GM, Binkley N, Lewiecki EM, et al. Diagnostic agreement at the total hip using different DXA systems and the NHANES III database. J Clin Densitom 2007; 10: 132–7
Leib ES, Lewiecki EM, Binkley N, et al. Official positions of the International Society for Clinical Densitometry. South Med J 2004; 97: 107–10
Lofman O, Larsson L, Toss G. Bone mineral density in diagnosis of osteoporosis: reference population, definition of peak bone mass, and measured site determine prevalence. J Clin Densitom 2000; 3: 177–86
Leib ES, Lenchik L, Bilezikian JP, et al. Position statements of the International Society for Clinical Densitometry: methodology. J Clin Densitom 2002; 5 Suppl.: S5–10
Stone KL, Seeley DG, Lui LY, et al. BMD at multiple sites and risk of fracture of multiple types: long-term results from the Study of Osteoporotic Fractures. J Bone Miner Res 2003; 18: 1947–54
Sievänen H, Kannus P, Jarvinen TL. Bone quality: an empty term. PLoS Med 2007; 4: e27
Hernandez CJ. How can bone turnover modify bone strength independent of bone mass? Bone 2008; 42: 1014–20
Bouxsein ML. Mechanisms of osteoporosis therapy: a bone strength perspective. Clin Cornerstone 2003; Suppl. 2: S13–21
Bouxsein ML. Bone quality: where do we go from here? Osteoporos Int 2003; 14 Suppl. 5: S118–27
Boivin G, Meunier PJ. Changes in bone remodeling rate influence the degree of mineralization of bone. Connect Tissue Res 2002; 43: 535–7
Compston J. Bone quality: what is it and how is it measured? Arq Bras Endocrinol Metabol 2006; 50: 579–85
Jarvinen TL, Kannus P, Sievänen H. Bone quality: emperor’s new clothes. J Musculoskelet Neuronal Interact 2008; 8: 2–9
Cohen A, Recker RR, Dempster D, et al. Marked abnormalities in cortical and trabecular microarchitecture and strength in premenopausal women with idiopathic osteoporosis: a bone biopsy study [abstract no. M310]. 30th Annual Meeting, American Society for Bone and Mineral Research; 2008 Sep 12–16; Montreal (QC) [online]. Available from URL: http://www.abstractsonline.com/viewer/?mkey=DCB70C83-5B38-431A-B0E1-9221D66718D0 [Accessed 2009 Nov 13]
Boutroy S, Van RB, Sornay-Rendu E, et al. Finite element analysis based on in vivo HR-pQCT images of the distal radius is associated with wrist fracture in postmenopausal women. J Bone Miner Res 2008; 23: 392–9
Muller R. Long-term prediction of three-dimensional bone architecture in simulations of pre-, peri- and post-menopausal microstructural bone remodeling. Osteoporos Int 2005; 16 Suppl. 2: S25–35
Hernandez CJ, Keaveny TM. A biomechanical perspective on bone quality. Bone 2006; 39: 1173–81
Ross PD. Predicting bone loss and fracture risk with biochemical markers: a review. J Clin Densitom 1999; 2: 285–94
Delmas PD. Treatment of postmenopausal osteoporosis. Lancet 2002; 359: 2018–26
Delmas PD, Licata AA, Reginster JY, et al. Fracture risk reduction during treatment with teriparatide is independent of pretreatment bone turnover. Bone 2006; 39: 237–43
Bjarnason NH, Christiansen C. Early response in biochemical markers predicts long-term response in bone mass during hormone replacement therapy in early postmenopausal women. Bone 2000; 26: 561–9
Bjarnason NH, Sarkar S, Duong T, et al. Six and twelve month changes in bone turnover are related to reduction in vertebral fracture risk during 3 years of raloxifene treatment in postmenopausal osteoporosis. Osteoporos Int 2001; 12: 922–30
Bauer DC, Garnero P, Bilezikian JP, et al. Short-term changes in bone turnover markers and bone mineral density response to parathyroid hormone in postmenopausal women with osteoporosis. J Clin Endocrinol Metab 2006; 91: 1370–5
Bauer DC, Garnero P, Hochberg MC, et al. Pretreatment levels of bone turnover and the antifracture efficacy of alendronate: the Fracture Intervention Trial. J Bone Miner Res 2006; 21: 292–9
Bauer DC, Black DM, Garnero P, et al. Change in bone turnover and hip, non-spine, and vertebral fracture in alendronate-treated women: the Fracture Intervention Trial. J Bone Miner Res 2004; 19: 1250–8
Bauer DC, Sklarin PM, Stone KL, et al. Biochemical markers of bone turnover and prediction of hip bone loss in older women: the study of osteoporotic fractures. J Bone Miner Res 1999; 14: 1404–10
Riggs BL, Melton III LJ. Bone turnover matters: the raloxifene treatment paradox of dramatic decreases in vertebral fractures without commensurate increases in bone density. J Bone Miner Res 2002; 17: 11–4
Eastell R, Barton I, Hannon RA, et al. Relationship of early changes in bone resorption to the reduction in fracture risk with risedronate. J Bone Miner Res 2003; 18: 1051–6
Eastell R, Hannon RA. Biomarkers of bone health and osteoporosis risk. Proc Nutr Soc 2008; 67: 157–62
Reginster JY, Collette J, Neuprez A, et al. Role of biochemical markers of bone turnover as prognostic indicator of successful osteoporosis therapy. Bone 2008; 42: 832–6
Delmas PD, Vrijens B, Eastell R, et al. Effect of monitoring bone turnover markers on persistence with risedronate treatment of postmenopausal osteoporosis. J Clin Endocrinol Metab 2007; 92: 1296–304
Sornay-Rendu E, Garnero P, Munoz F, et al. Effect of withdrawal of hormone replacement therapy on bone mass and bone turnover: the OFELY study. Bone 2003; 33: 159–66
Sornay-Rendu E, Munoz F, Garnero P, et al. Identification of osteopenic women at high risk of fracture: the OFELY study. J Bone Miner Res 2005; 20: 1813–9
Boivin G, Meunier PJ. The degree of mineralization of bone tissue measured by computerized quantitative contact microradiography. Calcif Tissue Int 2002; 70: 503–11
Chavassieux P, Seeman E, Delmas PD. Insights into material and structural basis of bone fragility from diseases associated with fractures: how determinants of the biomechanical properties of bone are compromised by disease. Endocr Rev 2007; 28: 151–64
Recker RR, Weinstein RS, Chesnut III CH, et al. Histomorphometric evaluation of daily and intermittent oral ibandronate in women with postmenopausal osteoporosis: results from the BONE study. Osteoporos Int 2004; 15: 231–7
Zoehrer R, Roschger P, Paschalis EP, et al. Effects of 3- and 5-year treatment with risedronate on bone mineralization density distribution in triple biopsies of the iliac crest in postmenopausal women. J Bone Miner Res 2006; 21: 1106–12
Seeman E. Bone quality. Osteoporos Int 2003; 14 Suppl. 5: S3–7
Delmas PD, Seeman E. Changes in bone mineral density explain little of the reduction in vertebral or nonvertebral fracture risk with anti-resorptive therapy. Bone 2004; 34: 599–604
Sarkar S, Reginster JY, Crans GG, et al. Relationship between changes in biochemical markers of bone turnover and BMD to predict vertebral fracture risk. J Bone Miner Res 2004; 19: 394–401
Sarkar S, Mitlak BH, Wong M, et al. Relationships between bone mineral density and incident vertebral fracture risk with raloxifene therapy. J Bone Miner Res 2002; 17: 1–10
Melton III LJ, Khosla S, Atkinson EJ, et al. Relationship of bone turnover to bone density and fractures. J Bone Miner Res 1997; 12: 1083–91
Garnero P, Sornay-Rendu E, Duboeuf F, et al. Markers of bone turnover predict postmenopausal forearm bone loss over 4 years: the OFELY study. J Bone Miner Res 1999; 14: 1614–21
Greenspan SL, Bone HG, Ettinger MP, et al. Effect of recombinant human parathyroid hormone (1–84) on vertebral fracture and bone mineral density in postmenopausal women with osteoporosis: a randomized trial. Ann Intern Med 2007; 146: 326–39
Greenspan SL, Emkey RD, Bone HG, et al. Significant differential effects of alendronate, estrogen, or combination therapy on the rate of bone loss after discontinuation of treatment of postmenopausal osteoporosis: a randomized, double-blind, placebo-controlled trial. Ann Intern Med 2002; 137: 875–83
Greenspan SL, Harris ST, Bone H, et al. Bisphosphonates: safety and efficacy in the treatment and prevention of osteoporosis. Am Fam Physician 2000; 61: 2731–6
Hochberg MC, Greenspan S, Wasnich RD, et al. Changes in bone density and turnover explain the reductions in incidence of nonvertebral fractures that occur during treatment with antiresorptive agents. J Clin Endocrinol Metab 2002; 87: 1586–92
Iki M, Morita A, Ikeda Y, et al. Biochemical markers of bone turnover may predict progression to osteoporosis in osteopenic women: the JPOS cohort study. J Bone Miner Metab 2007; 25: 122–9
Iki M, Morita A, Ikeda Y, et al. Biochemical markers of bone turnover predict bone loss in perimenopausal women but not in postmenopausal women: the Japanese Population-based Osteoporosis (JPOS) cohort study. Osteoporos Int 2006; 17: 1086–95
Johnell O, Oden A, De Laet C, et al. Biochemical indices of bone turnover and the assessment of fracture probability. Osteoporos Int 2002; 13: 523–6
Lenora J, Ivaska KK, Obrant KJ, et al. Prediction of bone loss using biochemical markers of bone turnover. Osteoporos Int 2007; 18: 1297–305
Lofman O, Magnusson P, Toss G, et al. Common biochemical markers of bone turnover predict future bone loss: a 5-year follow-up study. Clin Chim Acta 2005; 356: 67–75
Lukaszkiewicz J, Karczmarewics E, Marowska J, et al. Bone turnover rate in postmenopausal women: bimodal distribution? J Clin Densitom 2001; 4: 343–52
Marcus R, Holloway L, Wells B, et al. The relationship of biochemical markers of bone turnover to bone density changes in postmenopausal women: results from the Postmenopausal Estrogen/Progestin Interventions (PEPI) trial. J Bone Miner Res 1999; 14: 1583–95
McClung MR, San MJ, Miller PD, et al. Opposite bone remodeling effects of teriparatide and alendronate in increasing bone mass. Arch Intern Med 2005; 165: 1762–8
Rogers A, Hannon RA, Eastell R. Biochemical markers as predictors of rates of bone loss after menopause. J Bone Miner Res 2000; 15: 1398–404
Watts NB, Cooper C, Lindsay R, et al. Relationship between changes in bone mineral density and vertebral fracture risk associated with risedronate: greater increases in bone mineral density do not relate to greater decreases in fracture risk. J Clin Densitom 2004; 7: 255–61
Rosenbrock H, Seifert-Klauss V, Kaspar S, et al. Changes of biochemical bone markers during the menopausal transition. Clin Chem Lab Med 2002; 40: 143–51
Garnero P, Hausherr E, Chapuy MC, et al. Markers of bone resorption predict hip fracture in elderly women: the EPIDOS prospective study. J Bone Miner Res 1996; 11: 1531–8
Garnero P, Sornay-Rendu E, Claustrat B, et al. Biochemical markers of bone turnover, endogenous hormones and the risk of fractures in postmenopausal women: the OFELY study. J Bone Miner Res 2000; 15: 1526–36
Eastell R, Delmas PD. How to interpret surrogate markers of efficacy in osteoporosis. J Bone Miner Res 2005; 20: 1261–2
Committee For Medicinal Products For Human Use, European Medicines Agency. Note for guidance on postmenopausal osteoporosis in women [doc. ref. CPMP/EWP/552/95 Rev. 1]. London: European Medicines Agency, 2001
Committee For Medicinal Products For Human Use, European Medicines Agency. Draft guideline on the evaluation of new medicinal products in the treatment of primary osteoporosis [doc. ref. CPMP/EWP/552/95 Rev. 2]. London: European Medicines Agency; 2005 Dec 14 [online]. Available from URL: http://www.emea.europa.eu/pdfs/human/ewp/055295en.pdf [Accessed 2009Nov 13]
Kanis JA, Torgerson D, Cooper C. Comparison of the European and USA practice guidelines for osteoporosis. Trends Endocrinol Metab 2000; 11: 28–32
Miller PD, McClung MR, Macovei L, et al. Monthly oral ibandronate therapy in postmenopausal osteoporosis: 1-year results from the MOBILE study. J Bone Miner Res 2005; 20: 1315–22
Miller PD, Epstein S, Sedarati F, et al. Once-monthly oral ibandronate compared with weekly oral alendronate in postmenopausal osteoporosis: results from the head-to-head MOTION study. Curr Med Res Opin 2008; 24: 207–13
Miller PD. Optimizing the management of postmenopausal osteoporosis with bisphosphonates: the emerging role of intermittent therapy. Clin Ther 2005; 27: 361–76
Pyon EY. Once-monthly ibandronate for postmenopausal osteoporosis: review of a new dosing regimen. Clin Ther 2006; 28: 475–90
Chesnut III CH, Skag A, Christiansen C, et al. Effects of oral ibandronate administered daily or intermittently on fracture risk in postmenopausal osteoporosis. J Bone Miner Res 2004; 19: 1241–9
Delmas PD, Recker RR, Chesnut III CH, et al. Daily and intermittent oral ibandronate normalize bone turnover and provide significant reduction in vertebral fracture risk: results from the BONE study. Osteoporos Int 2004; 15: 792–8
Reginster JY, Adami S, Lakatos P, et al. Efficacy and tolerability of oncemonthly oral ibandronate in postmenopausal osteoporosis: 2 year results from the MOBILE study. Ann Rheum Dis 2006; 65: 654–61
Reginster JY, Felsenberg D, Cooper C, et al. A new concept for bisphosphonate therapy: a rationale for the development of monthly oral dosing of ibandronate. Osteoporos Int 2006; 17: 159–66
Reginster JY, Gieschke R. Clinical utility of a pharmacostatistical model for ibandronate in postmenopausal osteoporosis. Curr Drug Metab 2006; 7: 827–36
Reginster JY, Wilson KM, Dumont E, et al. Monthly oral ibandronate is well tolerated and efficacious in postmenopausal women: results from the Monthly Oral Pilot Study. J Clin Endocrinol Metab 2005; 90: 5018–24
Recker RR, Delmas PD, Halse J, et al. Effects of intravenous zoledronic acid once yearly on bone remodeling and bone structure. J Bone Miner Res 2008; 23: 6–16
Reid IR, Brown JP, Burckhardt P, et al. Intravenous zoledronic acid in postmenopausal women with low bone mineral density. N Engl J Med 2002; 346: 653–61
Cremers SC, Pillai G, Papapoulos SE. Pharmacokinetics/pharmacodynamics of bisphosphonates: use for optimisation of intermittent therapy for osteoporosis. Clin Pharmacokinet 2005; 44: 551–70
Close P, Neuprez A, Reginster JY. Developments in the pharmacotherapeutic management of osteoporosis. Expert Opin Pharmacother 2006; 7: 1603–15
Stoch SA, Wagner JA. Cathepsin K inhibitors: a novel target for osteoporosis therapy. Clin Pharmacol Ther 2008; 83: 172–6 [online]. Available from URL: http://www.nature.com/clpt [Accessed 2009 Nov 17]
Rosen CJ. Clinical practice: postmenopausal osteoporosis. N Engl J Med 2005; 353: 595–603
Riggs BL, Parfitt AM. Drugs used to treat osteoporosis: the critical need for a uniform nomenclature based on their action on bone remodeling. J Bone Miner Res 2005; 20: 177–84
Parfitt AM. Morphologic basis of bone mineral measurements: transient and steady state effects of treatment in osteoporosis. Mineral Electrolyte Metab 1980; 4: 273–87
Eastell R. Treatment of postmenopausal osteoporosis. N Engl J Med 1998; 338: 736–46
Bilezikian JP. Combination anabolic and antiresorptive therapy for osteoporosis: opening the anabolic window. Curr Osteoporos Rep 2008; 6: 24–30
Borges JL, Bilezikian JP. Update on osteoporosis therapy. Arq Bras Endocrinol Metabol 2006; 50: 755–63
Bone HG, Hosking D, Devogelaer JP, et al. Ten years’ experience with alendronate for osteoporosis in postmenopausal women. N Engl J Med 2004; 350: 1189–99
Delmas PD. The use of bisphosphonates in the treatment of osteoporosis. Curr Opin Rheumatol 2005; 17: 462–6
Bauss F, Schimmer RC. Ibandronate: the first once-monthly oral bisphosphonate for treatment of postmenopausal osteoporosis. Ther Clin Risk Manag 2006; 2: 3–18
Chapurlat RD, Delmas PD. Drug insight: bisphosphonates for postmenopausal osteoporosis. Nat Clin Pract Endocrinol Metab 2006; 2: 211–9
Chapurlat RD. Clinical pharmacology of potent new bisphosphonates for postmenopausal osteoporosis. Treat Endocrinol 2005; 4: 115–25
Colon-Emeric CS. Ten vs five years of bisphosphonate treatment for postmenopausal osteoporosis: enough of a good thing. JAMA 2006; 296: 2968–9
Cramer JA, Gold DT, Silverman SL, et al. A systematic review of persistence and compliance with bisphosphonates for osteoporosis. Osteoporos Int 2007; 18: 1023–31
McClung M. Bisphosphonates. Arq Bras Endocrinol Metabol 2006; 50: 735–44
McClung M. Use of highly potent bisphosphonates in the treatment of osteoporosis. Curr Osteoporos Rep 2003; 1: 116–22
Ravn P. Bisphosphonates for prevention of postmenopausal osteoporosis. Dan Med Bull 2002; 49: 1–18
Rodan GA, Reszka AA. Osteoporosis and bisphosphonates. J Bone Joint Surg Am 2003; 85-A Suppl. 3: 8–12
Russell RG, Watts NB, Ebetino FH, et al. Mechanisms of action of bisphosphonates: similarities and differences and their potential influence on clinical efficacy. Osteoporos Int 2008; 19: 733–59
Delmas PD. HRT in the prevention and treatment of osteoporosis. J Epidemiol Biostat 1999; 4: 155–60
Anderson GL, Limacher M, Assaf AR, et al. Effects of conjugated equine estrogen in postmenopausal women with hysterectomy: the Women’s Health Initiative randomized controlled trial. JAMA 2004; 291: 1701–12
Rossouw JE, Anderson GL, Prentice RL, et al. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results from the Women’s Health Initiative randomized controlled trial. JAMA 2002; 288: 321–33
Stepan JJ, Alenfeld F, Boivin G, et al. Mechanisms of action of antiresorptive therapies of postmenopausal osteoporosis. Endocr Regul 2003; 37: 225–38
Bjarnason NH, Byrjalsen I, Hassager C, et al. Low doses of estradiol in combination with gestodene to prevent early postmenopausal bone loss. Am J Obstet Gynecol 2000; 183: 550–60
Boivin G, Vedi S, Purdie DW, et al. Influence of estrogen therapy at conventional and high doses on the degree of mineralization of iliac bone tissue: a quantitative microradiographic analysis in postmenopausal women. Bone 2005; 36: 562–7
Bots ML, Evans GW, Riley W, et al. The effect of tibolone and continuous combined conjugated equine oestrogens plus medroxyprogesterone acetate on progression of carotid intima-media thickness: the Osteoporosis Prevention and Arterial effects of tiboLone (OPAL) study. Eur Heart J 2006; 27: 746–55
Cauley JA, Robbins J, Chen Z, et al. Effects of estrogen plus progestin on risk of fracture and bone mineral density: the Women’s Health Initiative randomized trial. JAMA 2003; 290: 1729–38
Chen Z, Bassford T, Green SB, et al. Postmenopausal hormone therapy and body composition: a substudy of the estrogen plus progestin trial of the Women’s Health Initiative. Am J Clin Nutr 2005; 82: 651–6
Chesnut III CH, Bell NH, Clark GS, et al. Hormone replacement therapy in postmenopausal women: urinary N-telopeptide of type I collagen monitors therapeutic effect and predicts response of bone mineral density. Am J Med 1997; 102: 29–37
de Valk-de Roo GW, Netelenbos JC, Peters-Muller IR, et al. Continuously combined hormone replacement therapy and bone turnover: the influence of dydrogesterone dose, smoking and initial degree of bone turnover. Maturitas 1997; 28: 153–62
Ettinger B. Rationale for use of lower estrogen doses for postmenopausal hormone therapy. Maturitas 2007; 57: 81–4
Gambacciani M, Cappagli B, Ciaponi M, et al. Ultra low-dose hormone replacement therapy and bone protection in postmenopausal women. Maturitas 2008; 59: 2–6
Gambacciani M, Ciaponi M, Cappagli B, et al. Effects of low-dose, continuous combined hormone replacement therapy on sleep in symptomatic postmenopausal women. Maturitas 2005; 50: 91–7
Hammar M, Christau S, Nathorst-Boos J, et al. A double-blind, randomised trial comparing the effects of tibolone and continuous combined hormone replacement therapy in postmenopausal women with menopausal symptoms. Br J Obstet Gynaecol 1998; 105: 904–11
Harris ST, Eriksen EF, Davidson M, et al. Effect of combined risedronate and hormone replacement therapies on bone mineral density in postmenopausal women. J Clin Endocrinol Metab 2001; 86: 1890–7
Jackson RD, Shidham S. The role of hormone therapy and calcium plus vitamin D for reduction of bone loss and risk for fractures: lessons learned from the Women’s Health Initiative. Curr Osteoporos Rep 2007; 5: 153–9
Jackson RD, Wactawski-Wende J, LaCroix AZ, et al. Effects of conjugated equine estrogen on risk of fractures and BMD in postmenopausal women with hysterectomy: results from the Women’s Health Initiative randomized trial. J Bone Miner Res 2006; 21: 817–28
Jerome CP. Hormonal therapies and osteoporosis. ILAR J 2004; 45: 170–8
Nielsen TF, Ravn P, Bagger YZ, et al. Pulsed estrogen therapy in prevention of postmenopausal osteoporosis: a 2-year randomized, double blind, placebo-controlled study. Osteoporos Int 2004; 15: 168–74
Paschalis EP, Boskey AL, Kassem M, et al. Effect of hormone replacement therapy on bone quality in early postmenopausal women. J Bone Miner Res 2003; 18: 955–9
Prestwood KM, Kenny AM, Unson C, et al. The effect of low dose micronized 17ss-estradiol on bone turnover, sex hormone levels, and side effects in older women: a randomized, double blind, placebo-controlled study. J Clin Endocrinol Metab 2000; 85: 4462–9
Raisz LG, Wiita B, Artis A, et al. Comparison of the effects of estrogen alone and estrogen plus androgen on biochemical markers of bone formation and resorption in postmenopausal women. J Clin Endocrinol Metab 1996; 81: 37–43
Bagger YZ, Tanko LB, Alexandersen P, et al. Two to three years of hormone replacement treatment in healthy women have long-term preventive effects on bone mass and osteoporotic fractures: the PERF study. Bone 2004; 34: 728–35
Delmas PD, Ensrud KE, Adachi JD, et al. Efficacy of raloxifene on vertebral fracture risk reduction in postmenopausal women with osteoporosis: four-year results from a randomized clinical trial. J Clin Endocrinol Metab 2002; 87: 3609–17
Chapurlat RD, Blackwell T, Bauer DC, et al. Changes in biochemical markers of bone turnover in women treated with raloxifene: influence of regression to the mean. Osteoporos Int 2001; 12: 1006–14
Delmas PD, Davis SR, Hensen J, et al. Effects of tibolone and raloxifene on bone mineral density in osteopenic postmenopausal women. Osteoporos Int 2008; 19: 1153–60
Ettinger B, Black DM, Mitlak BH, et al. Reduction of vertebral fracture risk in postmenopausal women with osteoporosis treated with raloxifene: results from a 3-year randomized clinical trial. Multiple Outcomes of Raloxifene Evaluation (MORE) Investigators. JAMA 1999; 282: 637–45
Hamdy RC, Chesnut III CH, Gass ML, et al. Review of treatment modalities for postmenopausal osteoporosis. South Med J 2005; 98: 1000–14
Johnell O, Kanis JA, Black DM, et al. Associations between baseline risk factors and vertebral fracture risk in the Multiple Outcomes of Raloxifene Evaluation (MORE) study. J Bone Miner Res 2004; 19: 764–72
Morii H, Ohashi Y, Taketani Y, et al. Effect of raloxifene on bone mineral density and biochemical markers of bone turnover in Japanese postmenopausal women with osteoporosis: results from a randomized placebocontrolled trial. Osteoporos Int 2003; 14: 793–800
Neele SJ, Evertz R, De Valk-De Roo RG, et al. Effect of 1 year of discontinuation of raloxifene or estrogen therapy on bone mineral density after 5 years of treatment in healthy postmenopausal women. Bone 2002; 30: 599–603
Prestwood KM, Gunness M, Muchmore DB, et al. A comparison of the effects of raloxifene and estrogen on bone in postmenopausal women. J Clin Endocrinol Metab 2000; 85: 2197–202
Seeman E, Crans GG, Diez-Perez A, et al. Anti-vertebral fracture efficacy of raloxifene: a meta-analysis. Osteoporos Int 2006; 17: 313–6
Beardsworth SA, Kearney CE, Purdie DW. Prevention of postmenopausal bone loss at lumbar spine and upper femur with tibolone: a two-year randomised controlled trial. Br J Obstet Gynaecol 1999; 106: 678–83
Berning B, Bennink HJ, Fauser BC. Tibolone and its effects on bone: a review. Climacteric 2001; 4: 120–36
Berning B, Van KC, Kuiper JW, et al. Increased loss of trabecular but not cortical bone density, 1 year after discontinuation of 2 years hormone replacement therapy with tibolone. Maturitas 1999; 31: 151–9
Berning B, Kuijk CV, Kuiper JW, et al. Effects of two doses of tibolone on trabecular and cortical bone loss in early postmenopausal women: a two-year randomized, placebo-controlled study. Bone 1996; 19: 395–9
Bjarnason NH, Bjarnason K, Haarbo J, et al. Tibolone: prevention of bone loss in late postmenopausal women. J Clin Endocrinol Metab 1996; 81: 2419–22
Bots ML, Evans GW, Riley W, et al. The Osteoporosis Prevention and Arterial effects of tiboLone (OPAL) study: design and baseline characteristics. Control Clin Trials 2003; 24: 752–75
Gallagher JC, Baylink DJ, Freeman R, et al. Prevention of bone loss with tibolone in postmenopausal women: results of two randomized, doubleblind, placebo-controlled, dose-finding studies. J Clin Endocrinol Metab 2001; 86: 4717–26
Lippuner K, Haenggi W, Birkhaeuser MH, et al. Prevention of postmenopausal bone loss using tibolone or conventional peroral or transdermal hormone replacement therapy with 17beta-estradiol and dydrogesterone. J Bone Miner Res 1997; 12: 806–12
Modelska K, Cummings S. Tibolone for postmenopausal women: systematic review of randomized trials. J Clin Endocrinol Metab 2002; 87: 16–23
Prelevic GM, Markou A, Arnold A, et al. The effect of tibolone on bone mineral density in postmenopausal women with osteopenia or osteoporosis: 8 years follow-up. Maturitas 2004; 47: 229–34
Prelevic GM, Bartram C, Wood J, et al. Comparative effects on bone mineral density of tibolone, transdermal estrogen and oral estrogen/progestogen therapy in postmenopausal women. Gynecol Endocrinol 1996; 10: 413–20
Rymer J, Robinson J, Fogelman I. Ten years of treatment with tibolone 2.5 mg daily: effects on bone loss in postmenopausal women. Climacteric 2002; 5: 390–8
Rymer J, Robinson J, Fogelman I. Effects of 8 years of treatment with tibolone 2.5 mg daily on postmenopausal bone loss. Osteoporos Int 2001; 12: 478–83
Rymer J, Chapman MG, Fogelman I. Effect of tibolone on postmenopausal bone loss. Osteoporos Int 1994; 4: 314–9
Swegle JM, Kelly MW. Tibolone: a unique version of hormone replacement therapy. Ann Pharmacother 2004; 38: 874–81
Cummings SR, Ettinger B, Delmas PD, et al. The effects of tibolone in older postmenopausal women. N Engl J Med 2008; 359: 697–708
Adami S, Passeri M, Ortolani S, et al. Effects of oral alendronate and intranasal salmon calcitonin on bone mass and biochemical markers of bone turnover in postmenopausal women with osteoporosis. Bone 1995; 17: 383–90
Adami S, Baroni MC, Broggini M, et al. Treatment of postmenopausal osteoporosis with continuous daily oral alendronate in comparison with either placebo or intranasal salmon calcitonin. Osteoporos Int 1993; 3 Suppl. 3: S21–7
Bagger YZ, Tanko LB, Alexandersen P, et al. Oral salmon calcitonin induced suppression of urinary collagen type II degradation in postmenopausal women: a new potential treatment of osteoarthritis. Bone 2005; 37: 425–30
Chesnut III CH, Majumdar S, Newitt DC, et al. Effects of salmon calcitonin on trabecular microarchitecture as determined by magnetic resonance imaging: results from the QUEST study. J Bone Miner Res 2005; 20: 1548–61
Chesnut III CH, Silverman S, Andriano K, et al. A randomized trial of nasal spray salmon calcitonin in postmenopausal women with established osteoporosis: the prevent recurrence of osteoporotic fractures study. PROOF Study Group. Am J Med 2000; 109: 267–76
Silverman SL. Calcitonin. Endocrinol Metab Clin North Am 2003; 32: 273–84
Tanko LB, Bagger YZ, Alexandersen P, et al. Safety and efficacy of a novel salmon calcitonin (sCT) technology-based oral formulation in healthy postmenopausal women: acute and 3-month effects on biomarkers of bone turnover. J Bone Miner Res 2004; 19: 1531–8
Ettinger B, San MJ, Crans G, et al. Differential effects of teriparatide on BMD after treatment with raloxifene or alendronate. J Bone Miner Res 2004; 19: 745–51
Bilezikian JP, Rubin MR. Combination/sequential therapies for anabolic and antiresorptive skeletal agents for osteoporosis. Curr Osteoporos Rep 2006; 4: 5–13
Boonen S, Marin F, Obermayer-Pietsch B, et al. Effects of previous antiresorptive therapy on the bone mineral density response to two years of teriparatide treatment in postmenopausal women with osteoporosis. J Clin Endocrinol Metab 2008; 93: 852–60
Bilezikian JP, Rubin MR, Finkelstein JS. Parathyroid hormone as an anabolic therapy for women and men. J Endocrinol Invest 2005; 28: 41–9
Bradbeer JN, Arlot ME, Meunier PJ, et al. Treatment of osteoporosis with parathyroid peptide (hPTH 1–34) and oestrogen: increase in volumetric density of iliac cancellous bone may depend on reduced trabecular spacing as well as increased thickness of packets of newly formed bone. Clin Endocrinol (Oxf) 1992; 37: 282–9
Chen P, Satterwhite JH, Licata AA, et al. Early changes in biochemical markers of bone formation predict BMD response to teriparatide in postmenopausal women with osteoporosis. J Bone Miner Res 2005; 20: 962–70
Chen P, Miller PD, Delmas PD, et al. Change in lumbar spine BMD and vertebral fracture risk reduction in teriparatide-treated postmenopausal women with osteoporosis. J Bone Miner Res 2006; 21: 1785–90
Finkelstein JS, Leder BZ, Burnett SM, et al. Effects of teriparatide, alendronate, or both on bone turnover in osteoporotic men. J Clin Endocrinol Metab 2006; 91: 2882–7
Neer RM, Arnaud CD, Zanchetta JR, et al. Effect of parathyroid hormone (1–34) on fractures and bone mineral density in postmenopausal women with osteoporosis. N Engl J Med 2001; 344: 1434–41
Tashjia Jr AH, Gagel RF. Teriparatide [human PTH(1–34)]: 2.5 years of experience on the use and safety of the drug for the treatment of osteoporosis. J Bone Miner Res 2006; 21: 354–65
Lane NE, Kelman A. A review of anabolic therapies for osteoporosis. Arthritis Res Ther 2003; 5: 214–22
Marie PJ. Strontium ranelate: a dual mode of action rebalancing bone turnover in favour of bone formation. Curr Opin Rheumatol 2006; 18 Suppl. 1: S11–5
Marie PJ. Strontium ranelate: a physiological approach for optimizing bone formation and resorption. Bone 2006; 38: S10–4
Marie PJ. Strontium as therapy for osteoporosis. Curr Opin Pharmacol 2005; 5: 633–6
Marie PJ. Strontium ranelate: a novel mode of action optimizing bone formation and resorption. Osteoporos Int 2005; 16 Suppl. 1: S7–10
Meunier PJ, Slosman DO, Delmas PD, et al. Strontium ranelate: dosedependent effects in established postmenopausal vertebral osteoporosis: a 2-year randomized placebo controlled trial. J Clin Endocrinol Metab 2002; 87: 2060–6
Meunier PJ, Roux C, Seeman E, et al. The effects of strontium ranelate on the risk of vertebral fracture in women with postmenopausal osteoporosis. N Engl J Med 2004; 350: 459–68
Reginster JY, Seeman E, De Vernejoul MC, et al. Strontium ranelate reduces the risk of nonvertebral fractures in postmenopausal women with osteoporosis: Treatment of Peripheral Osteoporosis (TROPOS) study. J Clin Endocrinol Metab 2005; 90: 2816–22
Reginster JY, Felsenberg D, Boonen S, et al. Effects of long-term strontium ranelate treatment on the risk of nonvertebral and vertebral fractures in postmenopausal osteoporosis: results of a five-year, randomized, placebocontrolled trial. Arthritis Rheum 2008; 58: 1687–95
Khosla S, Westendorf JJ, Oursler MJ. Building bone to reverse osteoporosis and repair fractures. J Clin Invest 2008; 118: 421–8
Ebeling PR, Wark JD, Yeung S, et al. Effects of calcitriol or calcium on bone mineral density, bone turnover, and fractures in men with primary osteoporosis: a two-year randomized, double blind, double placebo study. J Clin Endocrinol Metab 2001; 86: 4098–103
Jackson RD, LaCroix AZ, Gass M, et al. Calcium plus vitamin D supplementation and the risk of fractures. N Engl J Med 2006; 354: 669–83
Prestwood KM, Pannullo AM, Kenny AM, et al. The effect of a short course of calcium and vitamin D on bone turnover in older women. Osteoporos Int 1996; 6: 314–9
Scopacasa F, Need AG, Horowitz M, et al. Effects of dose and timing of calcium supplementation on bone resorption in early menopausal women. Horm Metab Res 2002; 34: 44–7
Storm D, Eslin R, Porter ES, et al. Calcium supplementation prevents seasonal bone loss and changes in biochemical markers of bone turnover in elderly New England women: a randomized placebo-controlled trial. J Clin Endocrinol Metab 1998; 83: 3817–25
Wishart JM, Scopacasa F, Horowitz M, et al. Effect of perimenopause on calcium absorption: a longitudinal study. Climacteric 2000; 3: 102–8
Dawson-Hughes B, Harris SS, Krall EA, et al. Effect of withdrawal of calcium and vitamin D supplements on bone mass in elderly men and women. Am J Clin Nutr 2000; 72: 745–50
Dawson-Hughes B, Harris SS, Krall EA, et al. Effect of calcium and vitamin D supplementation on bone density in men and women 65 years of age or older. N Engl J Med 1997; 337: 670–6
Garnero P, Munoz F, Sornay-Rendu E, et al. Associations of vitamin D status with bone mineral density, bone turnover, bone loss and fracture risk in healthy postmenopausal women: the OFELY study. Bone 2007; 40: 716–22
Heikkinen AM, Parviainen M, Niskanen L, et al. Biochemical bone markers and bone mineral density during postmenopausal hormone replacement therapy with and without vitamin D3: a prospective, controlled, randomized study. J Clin Endocrinol Metab 1997; 82: 2476–82
Jesudason D, Need AG, Horowitz M, et al. Relationship between serum 25-hydroxyvitamin D and bone resorption markers in vitamin D insufficiency. Bone 2002; 31: 626–30
Komulainen M, Tuppurainen MT, Kroger H, et al. Vitamin D and HRT: no benefit additional to that of HRT alone in prevention of bone loss in early postmenopausal women. A 2.5-year randomized placebo-controlled study. Osteoporos Int 1997; 7: 126–32
Lips P. Relative value of 25(OH)D and 1,25(OH)2D measurements. J Bone Miner Res 2007; 22: 1668–71
Murphy S, Khaw KT, Prentice A, et al. Relationships between parathyroid hormone, 25-hydroxyvitamin D, and bone mineral density in elderly men. Age Ageing 1993; 22: 198–204
Dawson-Hughes B, Chen P, Krege JH. Response to teriparatide in patients with baseline 25-hydroxyvitamin D insufficiency or sufficiency. J Clin Endocrinol Metab 2007; 92: 4630–6
Black DM, Delmas PD, Eastell R, et al. Once-yearly zoledronic acid for treatment of postmenopausal osteoporosis. N Engl J Med 2007; 356: 1809–22
Black DM, Greenspan SL, Ensrud KE, et al. The effects of parathyroid hormone and alendronate alone or in combination in postmenopausal osteoporosis. N Engl J Med 2003; 349: 1207–15
Cosman F, Nieves J, Zion M, et al. Daily and cyclic parathyroid hormone in women receiving alendronate. N Engl J Med 2005; 353: 566–75
Wasnich RD, Bagger YZ, Hosking DJ, et al. Changes in bone density and turnover after alendronate or estrogen withdrawal. Menopause 2004; 11: 622–30
Bagger YZ, Tanko LB, Alexandersen P, et al. Alendronate has a residual effect on bone mass in postmenopausal Danish women up to 7 years after treatment withdrawal. Bone 2003; 33: 301–7
Tremollieres FA, Pouilles JM, Ribot C. Withdrawal of hormone replacement therapy is associated with significant vertebral bone loss in postmenopausal women. Osteoporos Int 2001; 12: 385–90
Black DM, Schwartz AV, Ensrud KE, et al. Effects of continuing or stopping alendronate after 5 years of treatment: the Fracture Intervention Trial Long-Term Extension (FLEX): a randomized trial. JAMA 2006; 296: 2927–38
Black DM, Cummings SR, Karpf DB, et al. Randomised trial of effect of alendronate on risk of fracture in women with existing vertebral fractures. Fracture Intervention Trial Research Group. Lancet 1996; 348: 1535–41
Black DM, Thompson DE, Bauer DC, et al. Fracture risk reduction with alendronate in women with osteoporosis: the Fracture Intervention Trial. FIT Research Group. J Clin Endocrinol Metab 2000; 85: 4118–24
Ensrud KE, Barrett-Connor EL, Schwartz A, et al. Randomized trial of effect of alendronate continuation versus discontinuation in women with low BMD: results from the Fracture Intervention Trial Long-Term Extension. J Bone Miner Res 2004; 19: 1259–69
Martin RB. Toward a unifying theory of bone remodeling. Bone 2000; 26: 1–6
van der Linden JC, Verhaar JA, Pols HA, et al. A simulation model at trabecular level to predict effects of antiresorptive treatment after menopause. Calcif Tissue Int 2003; 73: 537–44
Hernandez CJ, Gupta A, Keaveny TM. A biomechanical analysis of the effects of resorption cavities on cancellous bone strength. J Bone Miner Res 2006; 21: 1248–55
Martin MJ, Buckland-Wright JC. A novel mathematical model identifies potential factors regulating bone apposition. Calcif Tissue Int 2005; 77: 250–60
Martin MJ, Buckland-Wright JC. Sensitivity analysis of a novel mathematical model identifies factors determining bone resorption rates. Bone 2004; 35: 918–28
Pioletti DP, Rakotomanana LR. Can the increase of bone mineral density following bisphosphonates treatments be explained by biomechanical considerations? Clin Biomech (Bristol, Avon) 2004; 19: 170–4
Ruffoni D, Fratzl P, Roschger P, et al. The bone mineralization density distribution as a fingerprint of the mineralization process. Bone 2007; 40: 1308–19
Hernandez CJ, Hazelwood SJ, Martin RB. The relationship between basic multicellular unit activation and origination in cancellous bone. Bone 1999; 25: 585–7
Hernandez CJ, Beaupre GS, Marcus R, et al. A theoretical analysis of the contributions of remodeling space, mineralization, and bone balance to changes in bone mineral density during alendronate treatment. Bone 2001; 29: 511–6
Hernandez CJ, Beaupre GS, Marcus R, et al. Long-term predictions of the therapeutic equivalence of daily and less than daily alendronate dosing. J Bone Miner Res 2002; 17: 1662–6
Hernandez CJ, Beaupre GS, Carter DR. A theoretical analysis of the relative influences of peak BMD, age-related bone loss and menopause on the development of osteoporosis. Osteoporos Int 2003; 14: 843–7
Beaupre GS, Orr TE, Carter DR. An approach for time-dependent bone modeling and remodeling-application: a preliminary remodeling simulation. J Orthop Res 1990; 8: 662–70
Hazelwood SJ, Bruce MR, Rashid MM, et al. A mechanistic model for internal bone remodeling exhibits different dynamic responses in disuse and overload. J Biomech 2001; 34: 299–308
Fleming DE, Chettle DR, Webber CE, et al. The O’Flaherty model of lead kinetics: an evaluation using data from a lead smelter population. Toxicol Appl Pharmacol 1999; 161: 100–9
O’Flaherty EJ. A physiologically based kinetic model for lead in children and adults. Environ Health Perspect 1998; 106 Suppl. 6: 1495–503
Moroz A, Crane MC, Smith G, et al. Phenomenological model of bone remodeling cycle containing osteocyte regulation loop. Biosystems 2006; 84: 183–90
Moroz A, Wimpenny DI. Allosteric control model of bone remodelling containing periodical modes. Biophys Chem 2007; 127: 194–212
Wimpenny DI, Moroz A. On allosteric control model of bone turnover cycle containing osteocyte regulation loop. Biosystems 2007; 90: 295–308
Komarova SV, Smith RJ, Dixon SJ, et al. Mathematical model predicts a critical role for osteoclast autocrine regulation in the control of bone remodeling. Bone 2003; 33: 206–15
Komarova SV. Mathematical model of paracrine interactions between osteoclasts and osteoblasts predicts anabolic action of parathyroid hormone on bone. Endocrinology 2005; 146: 3589–95
Komarova SV. Bone remodeling in health and disease: lessons from mathematical modeling. Ann N Y Acad Sci 2006; 1068: 557–9
Rattanakul C, Lenbury Y, Krishnamara N, et al. Modeling of bone formation and resorption mediated by parathyroid hormone: response to estrogen/PTH therapy. Biosystems 2003; 70: 55–72
Stepan JJ. Prediction of bone loss in postmenopausal women. Osteoporos Int 2000; 11 Suppl. 6: S45–54
Garnero P, Darte C, Delmas PD. A model to monitor the efficacy of alendronate treatment in women with osteoporosis using a biochemical marker of bone turnover. Bone 1999; 24: 603–9
Eastell R, Hannon RA, Garnero P, et al. Relationship of early changes in bone resorption to the reduction in fracture risk with risedronate: review of statistical analysis. J Bone Miner Res 2007; 22: 1656–60
Melsen F, Mosekilde L. Tetracycline double-labeling of iliac trabecular bone in 41 normal adults. Calcif Tissue Res 1978; 26: 99–102
Melsen F, Melsen B, Mosekilde L, et al. Histomorphometric analysis of normal bone from the iliac crest. Acta Pathol Microbiol Scand A 1978; 86: 70–81
Mosekilde L, Melsen F. Effect of antithyroid treatment on calcium-phosphorus metabolism in hyperthyroidism: II. Bone histomorphometry. Acta Endocrinol (Copenh) 1978; 87: 751–8
Mosekilde L, Melsen F. A tetracycline-based histomorphometric evaluation of bone resorption and bone turnover in hyperthyroidism and hyperparathyroidism. Acta Med Scand 1978; 204: 97–102
Deacon AC, Hulme P, Hesp R, et al. Estimation of whole body bone resorption rate: a comparison of urinary total hydroxyproline excretion with two radioisotopic tracer methods in osteoporosis. Clin Chim Acta 1987; 166: 297–306
Reeve J, Arlot ME, Chavassieux PM, et al. The assessment of bone formation and bone resorption in osteoporosis: a comparison between tetracyclinebased iliac histomorphometry and whole body 85Sr kinetics. J Bone Miner Res 1987; 2: 479–89
Eastell R, Riggs BL. Calcium homeostasis and osteoporosis. Endocrinol Metab Clin North Am 1987; 16: 829–42
Frost ML, Fogelman I, Blake GM, et al. Dissociation between global markers of bone formation and direct measurement of spinal bone formation in osteoporosis. J Bone Miner Res 2004; 19: 1797–804
Sheiner LB, Steimer JL. Pharmacokinetic/pharmacodynamic modeling in drug development. Annu Rev Pharmacol Toxicol 2000; 40: 67–95
Sheiner LB, Ludden TM. Population pharmacokinetics/dynamics. Annu Rev Pharmacol Toxicol 1992; 32: 185–209
Breimer DD, Danhof M. Relevance of the application of pharmacokineticpharmacodynamic modelling concepts in drug development: the ‘wooden shoe’ paradigm. Clin Pharmacokinet 1997; 32: 259–67
Danhof M, de Lange EC, la Pasqua OE, et al. Mechanism-based pharmacokinetic-pharmacodynamic (PK-PD) modeling in translational drug research. Trends Pharmacol Sci 2008; 29: 186–91
Holford N, Nutt JG. Disease progression, drug action and Parkinson’s disease: why time cannot be ignored. Eur J Clin Pharmacol 2008; 64: 207–16
Holford NH, Sheiner LB. Understanding the dose-effect relationship: clinical application of pharmacokinetic-pharmacodynamic models. Clin Pharmacokinet 1981;6: 429–53
Pillai G, Gieschke R, Goggin T, et al. A semimechanistic and mechanistic population PK-PD model for biomarker response to ibandronate, a new bisphosphonate for the treatment of osteoporosis. Br J Clin Pharmacol 2004; 58: 618–31
Holford N, Pillai G, Kamel S, et al. PKPD model for cathepsin K inhibition with balicatib and changes in bone turnover biomarkers, in particular NTx [abstract no. 1015]. 15th Meeting, Population Approach Group in Europe; 2006 Jun 14–16; Bruges [online]. Available from URL: http://www.page-meeting.org/default.asp?abstract=1015 [Accessed 2009 Nov 13]
Cremers S, Sparidans R, Den HJ, et al. A pharmacokinetic and pharmacodynamic model for intravenous bisphosphonate (pamidronate) in osteoporosis. Eur J Clin Pharmacol 2002; 57: 883–90
Cremers SC, Papapoulos SE, Gelderblom H, et al. Skeletal retention of bisphosphonate (pamidronate) and its relation to the rate of bone resorption in patients with breast cancer and bone metastases. J Bone Miner Res 2005; 20: 1543–7
Zierhut ML, Peterson MC, Gastonguay MR, et al. Development and evaluation of a population PK/PD model for Fc-OPG in healthy postmenopausal women [abstract no. 483]. 15th Meeting, Population Approach Group in Europe; 2004 Jun 17–18; Uppsala [online]. Available from URL: http://www.page-meeting.org/default.asp?abstract=483 [Accessed 2009 Nov 13]
Girard P, Claret L, Ebling W, et al. Evaluation of an osteoporosis biomarker model for simulation using posterior predictive check technique [abstract no. 460]. 12th Meeting, Population Approach Group in Europe; 2003 Jun 12–13; Verona [online]. Available from URL: http://www.page-meeting.org/default.asp?abstract=460 [Accessed 2009 Nov 13]
Gieschke R, Pillai G, Goggin T, et al. Population PD and clinical trial simulation: investigating oral dosing regimens for a new bisphosphonate drug for treatment of osteoporosis [abstract no. 198]. 10th Meeting, Population Approach Group in Europe; 2001 Jun 7–8; Basel [online]. Available from URL: http://www.pagemeeting.org/default.asp?abstract=198 [Accessed 2009 Nov 13]
Earp JC, Dubois DC, Molano DS, et al. Modeling corticosteroid effects in a rat model of rheumatoid arthritis I: mechanistic disease progression model for the time course of collagen-induced arthritis in Lewis rats. J Pharmacol Exp Ther 2008; 326: 532–45
Zierhut ML, Gastonguay MR, Martin SW, et al. Population PK-PD model for Fc-osteoprotegerin in healthy postmenopausal women. J Pharmacokinet Pharmacodyn 2008; 35: 379–99
Garnett C, Holford N. Bone mineral density progression to dropout and time-to-fracture: application to postmenopausal women taking hormone replacement therapy [abstract]. 5th International Symposium on Measurement and Kinetics of In Vivo Drug Effects; 2006 Apr 26–29; Leiden
Holford NHG, Bååthe S, Karlsson M. Auckland bones and summer sun [abstract no. 231]. 10th Meeting, Population Approach Group in Europe; 2001 Jun 7–8; Basel [online]. Available from URL: http://www.page-meeting.org/default.asp?abstract=231 [Accessed 2009 Nov 13]
Defranoux NA, Stokes CL, Young DL, et al. In silico modeling and simulation of bone biology: a proposal. J Bone Miner Res 2005; 20: 1079–84
NONMEM® users’ guides. Ellicott City (MD): Icon Development Solutions, 1989
Chan PL, Holford NH. Drug treatment effects on disease progression. Annu Rev Pharmacol Toxicol 2001; 41: 625–59
Bhattaram VA, Bonapace C, Chilukuri DM, et al. Impact of pharmacometric reviews on new drug approval and labeling decisions: a survey of 31 new drug applications submitted between 2005 and 2006. Clin Pharmacol Ther 2007; 81: 213–21
Lalonde RL, Kowalski KG, Hutmacher MM, et al. Model-based drug development. Clin Pharmacol Ther 2007; 82: 21–32
Miller R, Ewy W, Corrigan BW, et al. How modeling and simulation have enhanced decision making in new drug development. J Pharmacokinet Pharmacodyn 2005; 32: 185–97
Gieschke R, Steimer JL. Pharmacometrics: modelling and simulation tools to improve decision making in clinical drug development. Eur J Drug Metab Pharmacokinet 2000; 25: 49–58
Steimer JL, Holford NHG, Kaila N, et al. Population PK model of balicatib, a cathepsin K inhibitor, and a PKPD model for changes in rapid and slow bone biomarkers [abstract]. Pharmacokinetics UK; 2006 Nov 15–17; Sheffield [online]. Available from URL: http://www.pkuk.org.uk/ContentImages/PKUK2006ABSTRACTS.pdf [Accessed 2009 Nov 13]
Peterson MC, Riggs MM. A physiologically based mathematical model of integrated calcium homeostasis and bone remodeling. Bone. Epub 2009 Sep 2
Acknowledgements
This research was performed within the framework of project no. D2-104 of the Dutch Top Institute Pharma (Leiden, the Netherlands; www.tipharma.com). The authors have no conflicts of interest that are directly relevant to the contents of this review.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Post, T.M., Cremers, S.C.L.M., Kerbusch, T. et al. Bone Physiology, Disease and Treatment. Clin Pharmacokinet 49, 89–118 (2010). https://doi.org/10.2165/11318150-000000000-00000
Published:
Issue Date:
DOI: https://doi.org/10.2165/11318150-000000000-00000