
Abstract
The main functions of the osteoblast lineage (pre-osteoblasts, osteoblasts and osteocytes) are the production of bone matrix (osteoid), its mineralisation and the support of osteoclast formation. EphrinB2 and its contact-dependent receptor, EphB4, are stably expressed through all these stages of the lineage, and their expression of ephrinB2 is stimulated by parathyroid hormone (PTH), an agent that stimulates bone formation. Recent work has shown that the ephrinB2/EphB4 interaction is required for late stages of osteoblast differentiation and PTH action. Furthermore, specific deletion of ephrinB2 within the osteoblast lineage delays the process of bone mineralisation resulting in impaired bone strength and its response to anabolic PTH in both trabecular and periosteal bone. EphrinB2 and EphB4 are also expressed in growth plate chondrocytes, cells that also support osteoclast formation in development and fracture healing. This review will discuss the roles of forward and reverse signalling through EphB4 and ephrinB2, respectively, in bone growth and remodelling.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Skeletal structure depends on two key processes. In bone modelling, bone formation and resorption occur on different surfaces, resulting in changes in the size and shape of the skeleton, including, for example, bone formation on the periosteal (outer) surface of the cortical bone [1]. Secondly, in bone remodelling, bone formation follows prior resorption on the same surface. Bone remodelling occurs asynchronously at many sites throughout the skeleton and throughout life to replace old or damaged bone with new bone matrix and thereby maintain the structural integrity of the skeleton. The sequence of events by which bone formation follows bone resorption is termed “coupling”, and for bone mass to be maintained, balance between these two activities is necessary. The process of coupling, and its optimal balance, involves communication between the bone resorbing cells (osteoclasts) and those that form bone (osteoblasts) [2] and within the osteoblast lineage itself [3]. Recent studies suggest a cell-contact-dependent receptor tyrosine kinase ephrinB2, and its interaction with the receptor EphB4, mediates communication within the osteoblast lineage both during bone remodelling and periosteal growth (modelling).
Three Functions of the Osteoblast Lineage
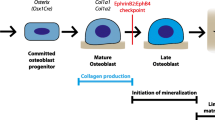
The osteoblast lineage includes osteoblast precursors, bone matrix-producing osteoblasts and matrix-embedded osteocytes (Fig. 1). This lineage carries out three major functions to determine skeletal structure: bone matrix (osteoid) production, regulation of osteoid mineralisation and support of osteoclast formation. While they are known to contribute to other biological activities, such as maintenance of the haematopoietic stem cell niche [4], phosphate homeostasis [5] and B cell homeostasis [6], the focus of this review will be on those functions that directly influence skeletal structure.

EphrinB2 and EphB4 are expressed within the osteoblast and chondrocyte lineages: from pluripotent stem cells through to mature osteoblasts, osteocytes and chondrocytes. Osteoblastic ephrinB2 is upregulated by PTH treatment and its interaction with EphB4, and subsequent reverse signalling, is required as a checkpoint for late stages of osteoblast/osteocyte differentiation on both trabecular and periosteal surfaces. Without this ephrinB2/EphB4 interaction, osteoblasts undergo early apoptosis, and bone formation is impaired. In chondrocytes, ephrinB2/EphB2 signalling promotes chondrocyte commitment, while ephrinB2/EphB4 signalling supports osteoclast formation and late-stage chondrocyte differentiation. EphrinB2 and EphB4 expression in chondrocytes is stimulated by IGF-I
The osteoblast lineage derives from multipotent mesenchymal progenitors, which can also differentiate into chondrocytes or adipocytes [7]. The expression of early osteoblast markers, such as runt-related transcription factor 2 (Runx2) and osterix (Osx), allows mesenchymal stem cells to become committed osteo-chondro-progenitors with restricted differentiation capacity [8, 9]. Once osteoblasts mature to the stage of actively forming new bone matrix (osteoid), they express high levels of alkaline phosphatase (Alpl), type 1 collagen (Col1a1) and parathyroid hormone 1 receptor (Pthr1), among other markers [10]. Matrix-producing osteoblasts exist as groups of cells on the bone surface, and they require cell-cell contact to produce matrix [11–13]. During later stages of osteoblast differentiation, matrix-producing osteoblasts express osteocalcin (Bglap) [14] and osteopontin (Spp1) [15], which inhibit the process of mineralisation.
After depositing osteoid matrix, osteoblasts may undergo one of three fates: (1) become bone lining cells, (2) undergo apoptosis or (3) become embedded in the matrix and differentiate into osteocytes. Differentiated osteocytes form an extensive intercellular network throughout the bone matrix and regulate both bone formation and resorption. Cell-cell contact is also a feature of this network: multiple dendritic projections from each cell form a total of ~3.7 trillion connections throughout the adult skeleton [16]. In addition to controlling osteoblast activity on the bone surface by the release of local factors such as sclerostin (Sost) [17], and oncostatin M (Osm) [18], osteocytes also regulate bone matrix mineralisation by expressing factors such as dentin matrix protein 1 (Dmp1) [19] and matrix extracellular phosphoglycoprotein (Mepe) [20].
Bone structure is influenced not only by matrix production and its mineralisation but also by the process of osteoclastic bone resorption. During bone remodelling, osteoclast generation depends on production of macrophage colony-stimulating factor (M-CSF) and receptor activator of nuclear factor kappa-B ligand (RANKL) by cells of the osteoblast lineage [21], although whether the key source of RANKL is the osteocyte or the preosteoblast remains controversial [22–24]. During bone development and growth, hypertrophic chondrocytes are also a source of RANKL [23]. They support the formation of osteoclasts that resorb the calcified cartilage matrix, leaving a template on which trabecular bone formation occurs.
EphrinB2 in Osteoblasts and Osteocytes
Parathyroid hormone (PTH) acts via its receptor (PTHR1) in osteoblasts both to stimulate bone formation [25, 26] and to stimulate their production of RANKL, thereby also increasing support of osteoclast formation [27]. Intermittent administration of PTH is the only currently available therapeutic agent that can increase bone mass [25]. It achieves this by promoting differentiation of committed osteoblast precursors [28, 29], inhibiting osteoblast and osteocyte apoptosis [30] and inhibiting production of the osteocytic bone formation inhibitor, sclerostin [31]. This anabolic effect of PTH reproduces the action of PTH-related protein (PTHrP), a locally produced paracrine stimulus of bone formation [32]. PTHrP not only acts through the same receptor but also has autocrine and nuclear activity [33].
In a microarray analysis of the effects of PTH and PTHrP on cultured osteoblasts, ephrinB2 was rapidly upregulated [34]. This effect was reproduced in PTH-treated rats in vivo [34]. EphrinB2 expression by osteoblasts and osteocytes was confirmed in rat bone [34] and human bone [35] by immunohistochemistry and was particularly prominent in osteoblasts on the surface of mature, lamellar bone, undergoing the process of remodelling.
EphrinB2 is a member of the largest family of receptor tyrosine kinases [36]. This family has the unique ability to generate simultaneous bidirectional signals: forward signalling through Eph receptors and reverse signalling through the ephrin ligand [37]. For signalling to occur, direct cell-cell contact must be made since both receptor and ligand are membrane-bound [36], although soluble forms of some family members exist [38–40]. The Eph/ephrin family can be divided into two groups, A and B. EphrinA ligands are membrane-anchored proteins, while ephrinB ligands are transmembrane proteins. These ephrin ligands bind to EphA and EphB receptors, with some promiscuity. Although many members of this family are expressed in osteoblasts in vitro and in vivo [34, 41, 42], only ephrinB2 was stimulated by PTH and PTHrP [34]. Given the extensive connections within the osteoblast lineage, the contact-dependent communication system of ephrins and the specific stimulation of one family member by PTH/PTHrP suggested a possible role in the process of bone formation.
The EphrinB2/EphB4 Interaction is a Checkpoint for Osteoblast Differentiation
Osteoblasts and osteocytes in both human and mouse also express EphB4 [35, 43•] The importance of the particular interaction of ephrinB2 with one of its receptors, EphB4, was identified in studies using inhibitors of this interaction: a peptide antagonist, TNYLFSPNGPIARAW (TNYL-RAW) [34] or the recombinant extracellular domain of EphB4 (sEphB4) [43•, 44]. Because they block the interaction, both directions of signalling are blocked [45, 46]: this was confirmed in osteoblasts, where both reagents inhibited both ephrinB2 and EphB4 phosphorylation [43•]. Addition of either reagent to cultured osteoblasts or a murine stromal cell line inhibited osteoblast differentiation, as indicated by reduced mRNA levels of late-stage osteoblast markers, such as Dmp1, Mepe and Sost [34, 43•] without any change in early osteoblast markers such as Runx2, Col1a1 and Alpl [43•]. Inhibition of the interaction of ephrinB2 with its other receptors EphB2 and EphA4 did not show the same effect [43•, 47••]. In addition, TNYL-RAW addition to human mesenchymal stem cells inhibited their ability to form mineral in vitro, a finding consistent with impaired osteoblast differentiation [35]. This suggested that the specific interaction between ephrinB2 and EphB4 within the osteoblast lineage is a checkpoint through which the differentiating osteoblast must pass for continued differentiation in vitro (Fig. 1). This interaction did not require the presence of any other cell type indicating that signalling within the osteoblast lineage is necessary for late stages of osteoblast differentiation.
When such inhibition of the ephrinB2/EphB4 interaction was tested in vivo, using sEphB4, late stages of osteoblast differentiation were again impaired [43•]. The evidence of this was twofold. Firstly, there was the observation of a change in osteoblast function: sEphB4 treatment in mice increased the number of osteoblasts and the level of osteoid production, but the rate of bone mineralisation was not increased. Secondly, mRNA extracted from bones of mice treated with sEphB4 showed high levels of early osteoblast markers with no modification in late stage osteoblast markers [43•]. This suggested an accumulation of matrix-producing osteoblasts with reduced ability to fully differentiate. These data showed the ephrinB2/EphB4 interaction is not required for osteoblasts to produce osteoid matrix and confirmed its requirement for osteoblasts to pass a checkpoint into the later stages of differentiation that promote osteoid mineralisation.
Inhibition of the ephrinB2/EphB4 interaction in vivo also impaired the anabolic action of PTH, but not only by impairing late stages of osteoblast differentiation [43•]. EphrinB2/EphB4 inhibition in the presence of PTH (but not in its absence) also increased osteoclast formation in vivo. This was reproduced in co-culture studies of osteoblasts with osteoclast precursors, suggesting the early stages of osteoblast differentiation are more supportive of osteoclast formation. In addition, in vitro studies showed that sEphB4 or TNYL-RAW treatment of osteoblasts rapidly increased their support of osteoclastogenesis by promoting RANKL transcription in cells of the osteoblast linage and rapidly stimulated mRNA levels of other pro-osteoclastic factors, such as interleukin-6 (Il6) and oncostatin M receptor (Osmr) [43•]. Therefore, the interaction of ephrinB2 and EphB4 might inhibit the production of a range of osteoclast inhibitors in response to PTH. Surprisingly, this contrasted with other works using a similar co-culture system showing that TNYL-RAW treatment suppressed osteoclastogenesis in a RANKL-independent manner when insulin-like growth factor 1 (IGF-I) was used as a stimulus of osteoclast formation in vitro [48•].
The Role of EphrinB2 in Osteoclast Formation
EphrinB2 is expressed not only by osteoblast lineage cells but also in osteoclasts and their precursors [41]. However, ephrinB2 signalling does not play a cell-lineage autonomous role in osteoclasts, since these cells do not express any Eph receptors that bind ephrinB2 [41]. This was confirmed by a lack of effect of ephrinB2/EphB4 inhibition on osteoclast formation in vitro in the absence of osteoblasts [43•]. However, when exogenous EphB4-Fc was used to stimulate ephrinB2 signalling in osteoclast precursors, their ability to form osteoclasts in vitro was impaired [41]. This suggested that interaction of osteoclast precursors with EphB4-expressing cells, such as osteoblasts, could suppress osteoclast formation by activating ephrinB2 signalling in osteoclast precursors [41]. The increased osteoclast formation we observed in response to sEphB4 in vivo and in co-cultures could be explained by this alternative model, except for the finding that sEphB4 treatment retained its ability to stimulate osteoclast formation in co-cultures of ephrinB2-expressing osteoblasts with ephrinB2-deficient osteoclasts [47••]. This confirmed that suppression of ephrinB2 reverse signalling within the osteoclast lineage did not cause the increased osteoclast formation induced by sEphB4. Rather, the osteoclast formation was induced by increased production of RANKL by osteoblasts in which ephrinB2/EphB4 signalling was suppressed. Therefore, ephrinB2/EphB4 signalling in osteoblasts either limits the expression of osteoblastic RANKL or maintains osteoblasts at an immature stage in which they produce more RANKL.
Defective osteoclastogenesis in mice with RANKL deficiency at specific stages of osteoblast and osteocyte differentiation have led to the suggestion that osteocytes are an important source of RANKL expression [23, 24]. Although early studies reported conflicting evidence of whether RANKL mRNA levels are higher in osteocytes than osteoblasts [24, 49, 50], recent work using more specific purification indicates that RANKL mRNA levels are higher in osteoblasts than in osteocytes [22]. Thus, the interaction between ephrinB2 and EphB4 was required for the continued differentiation of osteoblasts; without it, osteoblasts are held at an immature state when their support of osteoclastogenesis is high and their ability to produce factors that promote bone mineralisation is low.
EphrinB2 Signalling Within the Osteoblast Lineage Promotes Osteoblast Differentiation by Limiting Apoptosis
Pharmacological inhibition of the ephrinB2/EphB4 interaction impaired late stages of osteoblast differentiation both in vivo and in cultured osteoblasts [43•]. However, two questions remained unanswered: (1) which signalling direction (through ephrinB2 or EphB4) is most important and (2) whether all effects observed resulted from inhibiting an osteoblast-lineage specific interaction? While effects were consistent between the two systems, the in vivo effects may also have resulted from inhibition of ephrinB2 and EphB4 signalling in other cells such as embryonic stem cells [51], endothelial cells [52] T cells [53] and neurons [54], all of which are known to influence bone remodelling. The specific role of ephrinB2 within the osteoblast lineage was further investigated using Osx1-Cre-directed deletion of ephrinB2 [47••].
In the absence of ephrinB2 in adult female Osx1-Cre.ephrinB2f/f mice, osteoblast numbers were greater than in controls, but bone formation rate was not increased [47••]. This phenotype is similar to the effects of sEphB4 treatment in vivo [43•]. Furthermore, while osteoid thickness was greater in Osx1-Cre.ephrinB2f/f mice, a reduction in mineral apposition rate in these mice indicated that, rather than osteoid production being increased, it was the progression of bone mineralisation that was delayed [47••], a similar observation to the clinical condition of osteomalacia [55].
This phenotype was not restricted to a change in trabecular bone formation. These mice also exhibited thinner cortical bone, smaller femoral width and reduced polar moment of inertia compared to controls. This and the osteomalacia-like phenotype resulted in reduced bone stiffness, allowing a greater level of bone deformation before fracture by 3-point bending tests and reduced loading and unloading slopes by reference point indentation; in simplest terms, they had soft, rubbery bones [47••].
mRNA from Osx1-Cre.ephrinB2f/f bones revealed higher expression levels of early osteoblast markers and lower levels of late osteoblast/osteocyte markers [47••], again consistent with the impaired late stage osteoblast differentiation observed with sEphB4 treatment [43•]. One mechanism identified that maybe responsible for the impairment in late-stage osteoblast differentiation was a higher level of osteoblast and osteocyte apoptosis in Osx1-Cre.ephrinB2f/f bones [47••]. This was identified by TUNEL staining and electron microscopy in situ and higher levels of caspase 8-mediated apoptosis in cultured ephrinB2-deficient osteoblasts [47••]. This supported the model of an ephrinB2/EphB4 checkpoint in osteoblast differentiation indicated by the pharmacological inhibition studies. Without expression of anti-apoptotic ephrinB2, osteoblasts undergo apoptosis rather than making the transition to the fully mature stages expressing Dmp1, Mepe and Sost that promote osteoid mineralisation.
Does EphB4 Forward or EphrinB2 Reverse Signalling Promote Osteoblast Differentiation?
Due to the bidirectional signalling induced within the osteoblast lineage by the ephrinB2/EphB4 interaction, identifying the signalling direction that promotes osteoblast differentiation has been challenging. For example, when Osx1-Cre.ephrinB2f/f osteoblasts were analysed by Western blot and immunofluorescence, although the EphB4 phosphorylation response (forward signalling) to exogenous ephrinB2 remained intact, their basal levels of EphB4 phosphorylation were low, presumably due to the lack of ephrinB2 reverse signalling in neighbouring cells [47••]. This implied that the phenotype might relate to impairments of both EphB4 and ephrinB2 signalling.
That EphB4 signalling within the osteoblast lineage might promote osteoblast differentiation was suggested when EphB4 phosphorylation was stimulated in cultured osteoblasts by treatment with clustered ephrinB2-Fc [41]. This treatment increased expression of early osteoblast markers such as Runx2, Osx, Alpl, Col1a1 and Bglap, suggesting that EphB4 forward signalling within osteoblasts enhanced osteoblast differentiation. In addition, trabecular bone volume was increased in transgenic mice overexpressing EphB4 in the osteoblast lineage [41]. Compact bone and marrow isolated from these mice also exhibit greater numbers of osteoprogenitor cells [56•] and, thus, increased support of haematopoiesis [57•]. Although it was suggested that this phenotype occurred due to increased EphB4 forward signalling, it should be noted that EphB4 overexpression in osteoblasts would also stimulate ephrinB2 signalling.
To determine which direction of signalling is most important for osteoblast differentiation, we compared the effects of ephrinB2 knockdown to knockdown of EphB4. Surprisingly, although the ephrinB2 knockdown reproduced the effects of sEphB4 treatment on osteoblast differentiation, EphB4 knockdown had the opposite effect in cultured osteoblasts: mineralized nodule formation and early markers of osteoblast differentiation were increased [47••]. The contrasting effects of EphB4 knockdown and ephrinB2 deletion in osteoblasts supports the conclusion that reverse signalling through ephrinB2 mediates the ephrinB2/EphB4 checkpoint through which osteoblasts pass to reach late stages of differentiation.
EphrinB2 as a Mediator of PTH and IGF-I Anabolic Actions
Since PTH stimulates ephrinB2 expression, the response of Osx1-Cre.ephrinB2f/f mice to anabolic PTH treatment was also investigated. The PTH-induced increase in osteoblast number, osteoid thickness and mineral apposition rate in both trabecular and periosteal bone was impaired, but not fully blocked, in Osx1-Cre.ephrinB2f/f mice [47••]. Consistent with a limited response to PTH, the ephrinB2-deficient mice also showed a deficiency in the usual PTH-induced changes in gene expression. Early osteoblast markers (Runx2, Alpl and Col1a1) were not increased, and Sost expression was not reduced. However, the RANKL gene response to PTH was retained [47••]. This indicated that ephrinB2 signalling within the osteoblast is required for the anabolic response of PTH to stimulate osteoblast differentiation and reduce osteocytic sclerostin expression, but is not required for the RANKL response to PTH. Again, this may relate to retention of early osteoblast-lineage cells that have higher levels of RANKL expression, but have reached the stage of PTH receptor expression.
It has also been suggested that ephrinB2/EphB4 signalling may mediate IGF-I-induced bone formation since global genetic deletion of IGF-I, and osteoblast-targeted insulin-like growth factor receptor (IGF-IR) deletion in mice resulted in low levels of both ephrinB2 and EphB4 [48•]. Cell culture studies reported that siRNA deletion of EphB4 and TNYL-RAW treatment blocked the IGF-I-induced increase in mRNA levels of osteoblast markers, Runx2, Alpl and Bglap [58]. It is possible that the effects of IGF-I on ephrinB2 expression may be required for PTH anabolic action, since the PTH-induced increase in ephrinB2 was not observed in IGF-I-deficient bone marrow stromal cells [48•], and PTH anabolic action is reduced in IGF-I-deficient mice [59]. The interdependency of these pathways has not yet been shown nor has the requirement for ephrinB2/EphB4 signalling in the increased bone formation resulting from IGF-I treatment in vivo.
What is the Role of EphrinB2 Signalling in the Osteoclast Lineage?
The possible involvement of ephrinB2 signalling within the osteoclast in bone remodelling has been a question that is difficult to answer. Since osteoclasts express only ephrinB2, but not EphB4, the signalling of ephrinB2 in osteoclasts depends on an interaction with a different cell type that expresses ephrinB2. The earliest model, based on results from in vitro co-culture studies, proposed that the main interaction of ephrinB2-expressing osteoclasts that suppressed their differentiation was an interaction with EphB4-expressing osteoblasts [41]. This proposed that this direct interaction provided a mechanism for coupling balance, by which the activity of osteoblasts is matched to the prior activity of osteoclasts on the same surface. However, since there is a time delay of some weeks between the processes of bone resorption and formation on bone remodelling surfaces, termed the “reversal phase” [60], a direct interaction between mature osteoclasts and osteoblasts on the bone surface, is rare. Alternative proposals have been suggested, including the possibility that the interaction occurs between osteoclast precursors and osteoblast precursors or between osteoclast precursors and some other ephrinB2-expressing cells such as endothelial cells [52, 61]. Finally, recent work has identified that the cells that line the bone surface during the reversal phase are osteoblast precursors [62]; if these express ephrinB2 and interact directly with EphB4-expressing osteoclasts, these could provide a mechanism, at the cellular basis, by which this might work. However, co-culture studies using osteoclasts deficient in ephrinB2 showed that the stimulatory effects of sEphB4 on osteoclast differentiation did not result from modifying this cell-specific pathway [47••], questioning the role of osteoclastic ephrinB2 in osteoblast-stimulated osteoclastogenesis. Since haematopoietic specific knockout of ephrinB2 did not result in a detectable bone phenotype [41, 47••], the situations in which ephrinB2 signalling within the osteoclast lineage inhibits osteoclast differentiation remain obscure and may be restricted to an in vitro phenomenon.
A very recent study of mice with an osteoclast-driven deletion of the NF-κB inhibitor CHMP5 has suggested the phenotype of exuberant “Pagetic” periosteal remodelling in these mice results from an interaction of ephrinB2 in osteoclasts with EphB4 in osteoblasts [63]. In this work, high ephrinB2 mRNA levels were noted in cultured osteoclasts from these mice, likely a simple reflection of greater osteoclast numbers (also indicated by high mRNA levels of osteoclast markers). Addition of EphB4-Fc in osteoclast-osteoblast co-cultures mildly reduced Alizarin Red staining. This was interpreted as evidence that ephrinB2/EphB4 signalling between osteoclasts and osteoblasts supported osteoblast differentiation, based on the incorrect assumption that EphB4-Fc acted as an inhibitor. The levels of ephrinB2 and EphB4 phosphorylation in cells from these mice were not assessed nor the effects of EphB4-Fc in the co-culture system. This highlights the complexity of studies of ephrinB2/EphB4 interactions and the need for careful examination of the effects of pharmacological stimuli and inhibitors on both directions of signalling.
EphrinB2/EphB4 Signalling During Endochondral Ossification
In addition to regulating osteoblast differentiation in periosteal growth and bone remodelling, ephrinB2/EphB4 signalling has also been suggested as a regulator of endochondral ossification. This process occurs during bone development and longitudinal growth at the metaphyseal growth plate (Fig. 1) and describes the way in which a cartilage model is resorbed by osteoclasts and replaced with bone by osteoblasts.
Chondrocytes and osteoblasts share common mesenchymal stem cell progenitors, and like osteoblasts, chondrocytes express both ephrinB2 and EphB4 during the proliferating and hypertrophic stages at the growth plate [48•], as well as during fracture healing [64], in articular cartilage [65] and in the ATDC5 chondrocyte cell line [64]. This suggests a role of ephrinB2 and EphB4 in endochondral ossification, although chondrocytes are not cells that have been previously noted to depend on cell-cell contact for their activity.
In vitro studies using human multipotent mesenchymal stem cells and a small peptide of the interaction of ephrinB2 with EphB2, but not EphB4 significantly inhibited their ability to produce glycosaminoglycans, a major component of the cartilage matrix [35]. More recently, however, work with the committed chondrocytic ATDC5 cell line showed that TNYL-RAW treatment blunted the normal progression of chondrocyte differentiation, usually indicated by increased type II collagen followed by type X collagen mRNA levels [48•]. This suggests that ephrinB2/EphB4 signalling, and possibly ephrinB2/EphB2 signalling, stimulates the differentiation of committed chondrocytes, and ephrinB2/EphB2 signalling may promote chondrocytic commitment.
The ephrinB2/EphB4 interaction has also been reported to mediate IGF-I/IGF-IR signalling in the growth plate chondrocytes [48•]. IGF-I promotes chondrocyte proliferation during longitudinal bone growth [66]. Immunohistochemistry of wild-type bones showed ephrinB2 and EphB4 protein expression in the prehypertrophic chondrocytes of the growth plate, and levels were reduced in mice with germline IGF-I deletion. RNA extracted from these growth plates also showed reduced ephrinB2, EphB4 and RANKL mRNA levels. This suggested that IGF-I/IGF-IR signalling is either required for chondrocytes to reach the stage of ephrinB2/EphB4 expression or that IGF-I signalling directly stimulates ephrinB2 and EphB4 in growth plate chondrocytes, a regulatory pathway that may also exist in osteoblasts. A direct regulatory pathway was supported by the finding that chondrocytic ATDC5 cells treated with IGF-I significantly increased ephrinB2 and EphB4 mRNA levels [48•].
Since chondrocytes also support osteoclast differentiation, it might be expected that ephrinB2/EphB4 inhibition in chondrocytes would promote RANKL, as observed in osteoblasts [43•]. However, TNYL-RAW treatment of co-cultures of ATDC-5 cells with osteoclast precursors reduced the formation of osteoclasts when stimulated with IGF-I and did not stimulate their production of RANKL [48•]. Although different to the effects of sEphB4 treatment in osteoblasts, the result was consistent with the impaired ability of osteoblasts from Osx1Cre.EphrinB2f/f mice to support osteoclast differentiation when stimulated with 1,25-dihydroxyvitamin-D3 or oncostatin M [47••]. There are two possibilities to explain these effects. One is that, since expression of RANKL increases as chondrocytes differentiate [67], and TNYL-RAW treatment suppresses chondrocyte differentiation, these late stages of chondrocyte differentiation may not be reached. Alternatively, the authors suggested that the ephrinB2/EphB4 interaction might mediate RANKL-independent osteoclastogenesis in the co-culture system used. In vivo studies are required to resolve this question and determine how this system might influence endochondral ossification both during the process of bone development and in fracture healing.
The role of the ephrinB2/EphB4 interaction in endochondral ossification has also been investigated in the context of fracture healing in transgenic mice overexpressing EphB4 in osteoblasts [56•]. Fractured femora from EphB4 transgenic mice displayed a significantly larger extent of cartilaginous tissue matrix in intermediate and distal regions compared to controls. This delay in endochondral remodelling may result from impaired resorption of the cartilage matrix due to reduced osteoclast formation [56•], which in turn could result either from enhanced ephrinB2 signalling in the osteoclast lineage [41] or greater EphB4-mediated suppression of RANKL production by osteoblasts [47••]. The authors also suggested that this may result from increased chondrocyte proliferation induced by EphB4, which they observed in cultured chondrocytes, but since EphB4 overexpression in the in vivo model was directed to the osteoblast, this conclusion seems less likely. Nevertheless, these two studies provide intriguing evidence for a role of ephrinB2/EphB4 signalling in endochondral ossification, both in growth and in fracture healing.
Conclusion
The current body of knowledge suggests that ephrinB2 and EphB4 expressed on the cell membrane of osteoblast lineage cells interact with each other to promote osteoblast differentiation and mineralisation (Fig. 1). It appears that ephrinB2 reverse signalling within the osteoblast lineage is most important for osteoblast survival and the anabolic action of PTH during bone remodelling and periosteal growth. Forward signalling through EphB4 may also play a role, but this is difficult to discern from the currently available data. EphrinB2 reverse signalling within osteoclasts, while inhibitory in vitro does not yet have a clear role in vivo, although a number of possible EphB4-expressing cells that may interact with osteoclastic ephrinB2 are being identified. Finally, a similar EphrinB2/EphB4 interaction in chondrocytes may also play a role in regulating longitudinal bone growth or the process of endochondral ossification in fracture healing.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Dempster DW, Raisz LG. Bone physiology: bone cells, modeling, and remodeling. Nutrition and bone health. Springer; 2015. p. 37–56.
Sims NA, Martin TJ. Coupling the activities of bone formation and resorption: a multitude of signals within the basic multicellular unit. Bonekey Rep. 2014;3:481. doi:10.1038/bonekey.2013.215.
Tonna S, Sims NA. Talking among ourselves: paracrine control of bone formation within the osteoblast lineage. Calcif Tissue Int. 2014;94(1):35–45. doi:10.1007/s00223-013-9738-2.
Askmyr M, Sims NA, Martin TJ, Purton LE. What is the true nature of the osteoblastic hematopoietic stem cell niche? Trends Endocrinol Metab. 2009;20(6):303–9. doi:10.1016/j.tem.2009.03.004.
Fukumoto S, Martin TJ. Bone as an endocrine organ. Trends Endocrinol Metab. 2009;20(5):230–6. doi:10.1016/j.tem.2009.02.001.
Wu JY, Purton LE, Rodda SJ, Chen M, Weinstein LS, McMahon AP, et al. Osteoblastic regulation of B lymphopoiesis is mediated by Gs{alpha}-dependent signaling pathways. Proc Natl Acad Sci U S A. 2008;105(44):16976–81. doi:10.1073/pnas.0802898105.
Bianco P, Robey PG, Simmons PJ. Mesenchymal stem cells: revisiting history, concepts, and assays. Cell Stem Cell. 2008;2(4):313–9. doi:10.1016/j.stem.2008.03.002.
Ducy P, Starbuck M, Priemel M, Shen J, Pinero G, Geoffroy V, et al. A Cbfa1-dependent genetic pathway controls bone formation beyond embryonic development. Genes Dev. 1999;13(8):1025–36.
Nakashima K, Zhou X, Kunkel G, Zhang Z, Deng JM, Behringer RR, et al. The novel zinc finger-containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell. 2002;108(1):17–29.
Aubin JE, Liu F, Malaval L, Gupta AK. Osteoblast and chondroblast differentiation. Bone. 1995;17(2 Suppl):77S–83.
Ecarot-Charrier B, Glorieux FH, van der Rest M, Pereira G. Osteoblasts isolated from mouse calvaria initiate matrix mineralization in culture. J Cell Biol. 1983;96(3):639–43.
Abe Y, Akamine A, Aida Y, Maeda K. Differentiation and mineralization in osteogenic precursor cells derived from fetal rat mandibular bone. Calcif Tissue Int. 1993;52(5):365–71.
Gerber I, ap Gwynn I. Influence of cell isolation, cell culture density, and cell nutrition on differentiation of rat calvarial osteoblast-like cells in vitro. Eur Cell Mater. 2001;2:10–20.
Sims NA, White CP, Sunn KL, Thomas GP, Drummond ML, Morrison NA, et al. Human and murine osteocalcin gene expression: conserved tissue restricted expression and divergent responses to 1,25-dihydroxyvitamin D3 in vivo. Mol Endocrinol. 1997;11(11):1695–708. doi:10.1210/mend.11.11.0008.
Aubin JE. Regulation of osteoblast formation and function. Rev Endocr Metab Disord. 2001;2(1):81–94.
Buenzli PR, Sims NA. Quantifying the osteocyte network in the human skeleton. Bone. 2015;75:144–50. doi:10.1016/j.bone.2015.02.016.
van Bezooijen RL, Roelen BA, Visser A, van der Wee-Pals L, de Wilt E, Karperien M, et al. Sclerostin is an osteocyte-expressed negative regulator of bone formation, but not a classical BMP antagonist. J Exp Med. 2004;199(6):805–14. doi:10.1084/jem.20031454.
Walker EC, McGregor NE, Poulton IJ, Solano M, Pompolo S, Fernandes TJ, et al. Oncostatin M promotes bone formation independently of resorption when signaling through leukemia inhibitory factor receptor in mice. J Clin Invest. 2010;120(2):582–92. doi:10.1172/JCI40568.
Toyosawa S, Shintani S, Fujiwara T, Ooshima T, Sato A, Ijuhin N, et al. Dentin matrix protein 1 is predominantly expressed in chicken and rat osteocytes but not in osteoblasts. J Bone Miner Res. 2001;16(11):2017–26. doi:10.1359/jbmr.2001.16.11.2017.
Gowen LC, Petersen DN, Mansolf AL, Qi H, Stock JL, Tkalcevic GT, et al. Targeted disruption of the osteoblast/osteocyte factor 45 gene (OF45) results in increased bone formation and bone mass. J Biol Chem. 2003;278(3):1998–2007. doi:10.1074/jbc.M203250200.
Teitelbaum SL, Ross FP. Genetic regulation of osteoclast development and function. Nat Rev Genet. 2003;4(8):638–49. doi:10.1038/nrg1122.
Chia LY, Walsh NC, Martin TJ, Sims NA. Isolation and gene expression of haematopoietic-cell-free preparations of highly purified murine osteocytes. Bone. 2015;72:34–42. doi:10.1016/j.bone.2014.11.005.
Xiong J, Onal M, Jilka RL, Weinstein RS, Manolagas SC, O'Brien CA. Matrix-embedded cells control osteoclast formation. Nat Med. 2011;17(10):1235–41. doi:10.1038/nm.2448.
Nakashima T, Hayashi M, Fukunaga T, Kurata K, Oh-Hora M, Feng JQ, et al. Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat Med. 2011;17(10):1231–4. doi:10.1038/nm.2452.
Neer RM, Arnaud CD, Zanchetta JR, Prince R, Gaich GA, Reginster JY, et al. Effect of parathyroid hormone (1–34) on fractures and bone mineral density in postmenopausal women with osteoporosis. N Engl J Med. 2001;344(19):1434–41. doi:10.1056/NEJM200105103441904.
Lindsay R, Zhou H, Cosman F, Nieves J, Dempster DW, Hodsman AB. Effects of a one-month treatment with PTH(1–34) on bone formation on cancellous, endocortical, and periosteal surfaces of the human ilium. J Bone Miner Res. 2007;22(4):495–502. doi:10.1359/jbmr.070104.
Martin TJ, Sims NA. RANKL/OPG; critical role in bone physiology. Rev Endocr Metab Disord. 2015. doi:10.1007/s11154-014-9308-6.
Dobnig H, Turner RT. Evidence that intermittent treatment with parathyroid hormone increases bone formation in adult rats by activation of bone lining cells. Endocrinology. 1995;136(8):3632–8. doi:10.1210/endo.136.8.7628403.
Kim SW, Pajevic PD, Selig M, Barry KJ, Yang JY, Shin CS, et al. Intermittent parathyroid hormone administration converts quiescent lining cells to active osteoblasts. J Bone Miner Res. 2012;27(10):2075–84. doi:10.1002/jbmr.1665.
Jilka RL, Weinstein RS, Bellido T, Roberson P, Parfitt AM, Manolagas SC. Increased bone formation by prevention of osteoblast apoptosis with parathyroid hormone. J Clin Invest. 1999;104(4):439–46. doi:10.1172/JCI6610.
Kramer I, Loots GG, Studer A, Keller H, Kneissel M. Parathyroid hormone (PTH)-induced bone gain is blunted in SOST overexpressing and deficient mice. J Bone Miner Res. 2010;25(2):178–89. doi:10.1359/jbmr.090730.
Miao D, He B, Jiang Y, Kobayashi T, Soroceanu MA, Zhao J, et al. Osteoblast-derived PTHrP is a potent endogenous bone anabolic agent that modifies the therapeutic efficacy of administered PTH 1–34. J Clin Invest. 2005;115(9):2402–11. doi:10.1172/JCI24918.
Danks J, Freeman A, Martin T. Historical perspective and evolutionary origins of parathyroid hormone-related protein. Clin Rev Bone Miner Metab. 2014;12:104–18. doi:10.1007/s12018-014-9163-8.
Allan EH, Hausler KD, Wei T, Gooi JH, Quinn JM, Crimeen-Irwin B, et al. EphrinB2 regulation by PTH and PTHrP revealed by molecular profiling in differentiating osteoblasts. J Bone Miner Res. 2008;23(8):1170–81. doi:10.1359/jbmr.080324.
Arthur A, Zannettino A, Panagopoulos R, Koblar SA, Sims NA, Stylianou C, et al. EphB/ephrin-B interactions mediate human MSC attachment, migration and osteochondral differentiation. Bone. 2011;48(3):533–42. doi:10.1016/j.bone.2010.10.180.
Pasquale EB. Eph receptors and ephrins in cancer: bidirectional signalling and beyond. Nat Rev Cancer. 2010;10(3):165–80. doi:10.1038/nrc2806.
Murai KK, Pasquale EB. ‘Eph’ective signaling: forward, reverse and crosstalk. J Cell Sci. 2003;116(Pt 14):2823–32. doi:10.1242/jcs.00625.
Segura I, Essmann CL, Weinges S, Acker-Palmer A. Grb4 and GIT1 transduce ephrinB reverse signals modulating spine morphogenesis and synapse formation. Nat Neurosci. 2007;10(3):301–10. doi:10.1038/nn1858.
Holmberg J, Genander M, Halford MM, Anneren C, Sondell M, Chumley MJ, et al. EphB receptors coordinate migration and proliferation in the intestinal stem cell niche. Cell. 2006;125(6):1151–63. doi:10.1016/j.cell.2006.04.030.
Munro KM, Dixon KJ, Gresle MM, Jonas A, Kemper D, Doherty W, et al. EphA4 receptor tyrosine kinase is a modulator of onset and disease severity of experimental autoimmune encephalomyelitis (EAE). Plos One. 2013;8(2), e55948. doi:10.1371/journal.pone.0055948.
Zhao C, Irie N, Takada Y, Shimoda K, Miyamoto T, Nishiwaki T, et al. Bidirectional ephrinB2-EphB4 signaling controls bone homeostasis. Cell Metab. 2006;4(2):111–21. doi:10.1016/j.cmet.2006.05.012.
Irie N, Takada Y, Watanabe Y, Matsuzaki Y, Naruse C, Asano M, et al. Bidirectional signaling through ephrinA2-EphA2 enhances osteoclastogenesis and suppresses osteoblastogenesis. J Biol Chem. 2009;284(21):14637–44. doi:10.1074/jbc.M807598200.
Takyar FM, Tonna S, Ho PW, Crimeen-Irwin B, Baker EK, Martin TJ, et al. EphrinB2/EphB4 inhibition in the osteoblast lineage modifies the anabolic response to parathyroid hormone. J Bone Miner Res. 2013;28(4):912–25. doi:10.1002/jbmr.1820. This work shows that the interaction of EphrinB2 with the receptor EphB4 stimulates partial differentiation of osteoblasts, but their ability to promote bone mineralisation is defective. This was the first evidence of an ephrinB2/EphB4 checkpoint in osteoblast differentiation.
Martin TJ, Allan EH, Ho PW, Gooi JH, Quinn JM, Gillespie MT, et al. Communication between EphrinB2 and EphB4 within the osteoblast lineage. Adv Exp Med Biol. 2010;658:51–60. doi:10.1007/978-1-4419-1050-9_6.
Kertesz N, Krasnoperov V, Reddy R, Leshanski L, Kumar SR, Zozulya S, et al. The soluble extracellular domain of EphB4 (sEphB4) antagonizes EphB4-EphrinB2 interaction, modulates angiogenesis, and inhibits tumor growth. Blood. 2006;107(6):2330–8. doi:10.1182/blood-2005-04-1655.
Koolpe M, Burgess R, Dail M, Pasquale EB. EphB receptor-binding peptides identified by phage display enable design of an antagonist with ephrin-like affinity. J Biol Chem. 2005;280(17):17301–11. doi:10.1074/jbc.M500363200.
Tonna S, Takyar FM, Vrahnas C, Crimeen-Irwin B, Ho PW, Poulton IJ, et al. EphrinB2 signaling in osteoblasts promotes bone mineralization by preventing apoptosis. FASEB J. 2014;28(10):4482–96. doi:10.1096/fj.14-254300. This confirmed the requirement of transition through the ephrinB2/EphB4 checkpoint for osteoblast differentiation, and showed that in the absence of ephrinB2, osteoblasts undergo apoptosis, leading to a bone phenotype of reduced bone strength and delayed mineralisation.
Wang Y, Menendez A, Fong C, ElAlieh HZ, Chang W, Bikle DD. Ephrin B2/EphB4 mediates the actions of IGF-I signaling in regulating endochondral bone formation. J Bone Miner Res. 2014;29(8):1900–13. doi:10.1002/jbmr.2196. This work points to potential cross regulation of ephrinB2/EphB4 by IGF1 and suggests a role for this interaction in chondrocytic support of osteoclast formation.
Kramer I, Halleux C, Keller H, Pegurri M, Gooi JH, Weber PB, et al. Osteocyte Wnt/beta-catenin signaling is required for normal bone homeostasis. Mol Cell Biol. 2010;30(12):3071–85. doi:10.1128/MCB.01428-09.
Fumoto T, Takeshita S, Ito M, Ikeda K. Physiological functions of osteoblast lineage and T cell-derived RANKL in bone homeostasis. J Bone Miner Res. 2014;29(4):830–42. doi:10.1002/jbmr.2096.
Wang Z, Cohen K, Shao Y, Mole P, Dombkowski D, Scadden DT. Ephrin receptor, EphB4, regulates ES cell differentiation of primitive mammalian hemangioblasts, blood, cardiomyocytes, and blood vessels. Blood. 2004;103(1):100–9. doi:10.1182/blood-2003-04-1063.
Gerety SS, Wang HU, Chen ZF, Anderson DJ. Symmetrical mutant phenotypes of the receptor EphB4 and its specific transmembrane ligand ephrin-B2 in cardiovascular development. Mol Cell. 1999;4(3):403–14.
Sharfe N, Nikolic M, Cimpeon L, Van De Kratts A, Freywald A, Roifman CM. EphA and ephrin-A proteins regulate integrin-mediated T lymphocyte interactions. Mol Immunol. 2008;45(5):1208–20. doi:10.1016/j.molimm.2007.09.019.
Yamaguchi Y, Pasquale EB. Eph receptors in the adult brain. Curr Opin Neurobiol. 2004;14(3):288–96. doi:10.1016/j.conb.2004.04.003.
Melsen F, Mosekilde L. Trabecular bone mineralization lag time determined by tetracycline double-labeling in normal and certain pathological conditions. Acta Pathol Microbiol Scand A Pathol. 1980;88(2):83–8.
Arthur A, Panagopoulos RA, Cooper L, Menicanin D, Parkinson IH, Codrington JD, et al. EphB4 enhances the process of endochondral ossification and inhibits remodeling during bone fracture repair. J Bone Miner Res. 2013;28(4):926–35. doi:10.1002/jbmr.1821. This work suggests that EphrinB2/EphB4 signalling within osteoblasts can promote early stages of callus formation and bone formation in fracture repair.
Nguyen TM, Arthur A, Panagopoulos R, Paton S, Hayball JD, Zannettino AC, et al. EphB4 expressing stromal cells exhibit an enhanced capacity for hematopoietic stem cell maintenance. Stem Cells. 2015. doi:10.1002/stem.2069. The first indication that EphrinB2/EphB4 signalling in stromal cells contributes to their support of the haematopoietic stem cell niche.
Wang Y, Nishida S, Boudignon BM, Burghardt A, Elalieh HZ, Hamilton MM, et al. IGF-I receptor is required for the anabolic actions of parathyroid hormone on bone. J Bone Miner Res. 2007;22(9):1329–37. doi:10.1359/jbmr.070517.
Bikle DD, Sakata T, Leary C, Elalieh H, Ginzinger D, Rosen CJ, et al. Insulin-like growth factor I is required for the anabolic actions of parathyroid hormone on mouse bone. J Bone Miner Res. 2002;17(9):1570–8. doi:10.1359/jbmr.2002.17.9.1570.
Sims NA, Martin TJ. Coupling signals between the osteoclast and osteoblast: how are messages transmitted between these temporary visitors to the bone surface? Front Endocrinol. 2015;6:41. doi:10.3389/fendo.2015.00041.
Wang Y, Nakayama M, Pitulescu ME, Schmidt TS, Bochenek ML, Sakakibara A, et al. Ephrin-B2 controls VEGF-induced angiogenesis and lymphangiogenesis. Nature. 2010;465(7297):483–6. doi:10.1038/nature09002.
Andersen TL, Sondergaard TE, Skorzynska KE, Dagnaes-Hansen F, Plesner TL, Hauge EM, et al. A physical mechanism for coupling bone resorption and formation in adult human bone. Am J Pathol. 2009;174(1):239–47. doi:10.2353/ajpath.2009.080627.
Greenblatt MB, Park KH, Oh H, Kim JM, Shin DY, Lee JM, et al. CHMP5 controls bone turnover rates by dampening NF-kappaB activity in osteoclasts. J Exp Med. 2015;212(8):1283–301. doi:10.1084/jem.20150407.
Ito M, Tokunaga K, Kitahara H, Ito T, Kondoh N, Endo N. ephrinB2 and EphB4 are expressed on the chondrocyte during fracture healing. Acta Med Biol. 2006;54(1):21–34.
Kwan Tat S, Pelletier JP, Amiable N, Boileau C, Lavigne M, Martel-Pelletier J. Treatment with ephrin B2 positively impacts the abnormal metabolism of human osteoarthritic chondrocytes. Arthritis Res Ther. 2009;11:R119. doi:10.1186/ar2782.
Sims NA, Clement-Lacroix P, Da Ponte F, Bouali Y, Binart N, Moriggl R, et al. Bone homeostasis in growth hormone receptor-null mice is restored by IGF-I but independent of Stat5. J Clin Invest. 2000;106(9):1095–103. doi:10.1172/JCI10753.
Kartsogiannis V, Zhou H, Horwood NJ, Thomas RJ, Hards DK, Quinn JM, et al. Localization of RANKL (receptor activator of NF kappa B ligand) mRNA and protein in skeletal and extraskeletal tissues. Bone. 1999;25(5):525–34.
Compliance with Ethics Guidelines
Conflict of Interest
Christina Vrahnas and Natalie A. Sims declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is part of the Topical Collection on Molecular Biology of Skeletal Development
Rights and permissions
About this article

Cite this article
Vrahnas, C., Sims, N.A. EphrinB2 Signalling in Osteoblast Differentiation, Bone Formation and Endochondral Ossification. Curr Mol Bio Rep 1, 148–156 (2015). https://doi.org/10.1007/s40610-015-0024-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40610-015-0024-0