
Abstract
A novel aminopeptidase B (APB-AN) was identified from Aspergillus niger CGMCC 3.1454 for the first time and was cloned and expressed in Pichia pastoris. The mature enzyme of approximately 100 kDa was purified for characterization. The optimum pH and temperature of the recombinant APB-AN were determined to be 7.0 and 40 °C, respectively. The enzyme was stable below 40 °C and at pH values from 5.0 to 8.0. The Km and Vmax values were determined to be 0.61 mmol/L and 11.45 mmol/L/min, respectively, using Arg-pNA as the substrate. APB-AN was inhibited by Cu2+ and Fe2+ and activated by Co2+ and Na+. Most metal chelators (Ca2+, Mg2+ and Mn2+) and aminopeptidase inhibitors (bestatin and puromycin) suppressed its activity. APB-AN was found to be active towards 13 kinds of amino acid p-nitroanilide (pNA) substrates:Arg-pNA, Lys-pNA, Tyr- pNA, Trp-pNA, Phe-pNA, His-pNA, Ala-pNA, Met-pNA, Leu-pNA, Glu-pNA, Val-pNA, Pro-pNA and Ile-pNA, and the most preferred N-terminal amino acids were arginine and lysine. APB-AN also hydrolyzed 4 natural proteins: casein, bovine serum albumin, soy protein isolate and water-soluble wheat protein. It is expected that APB-AN has potential food processing applications.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Aminopeptidases (APs), a group of exopeptidases that release N-terminal amino acid residues from peptides (Sanderink et al. 1988), are classified into a variety of categories based on their preference for substrates, sensitivity to inhibitors and catalytic mechanism. Among aminopeptidases, aminopeptidase B (AP-B, EC 3.4.11.6), also named arginine aminopeptidase or arginyl aminopeptidase (Attri et al. 2021; Zhang et al. 2013; Bolumar et al. 2003), is one of the main enzymes used in food processing, such as modifying the flavor of aquatic products and meat (Zhang et al. 2013; Toldra et al. 2000; Nishimura et al. 2006).
In general, aminopeptidases, including AP-Bs, are intracellular enzymes (Rao et al. 1998). It is difficult to maintain the activities of AP-Bs in the process of disrupting cells to extract them, and natural enzyme content is always low in animals and plants, so aminopeptidase production by microbial fermentation has become a feasible method. Many aminopeptidases derived from different microorganisms have been cloned and expressed (Song et al. 2020; Yang et al. 2016; Xi et al. 2015). Among them, four aminopeptidases from Aspergillus niger have been purified and characterized, including lysine aminopeptidase (ApsA) (Basten et al. 2001), dipeptidyl peptidase IV (DapB) (Jalving et al. 2005), prolyl aminopeptidase (PapA) (Basten et al. 2005) and phenylalanine aminopeptidase (ApsC) (Basten et al. 2003).
A. niger, a member of the black aspergilli, is widely used in biotechnology for the production of industrial enzymes. Although the genome sequencing and analysis of A. niger strains had been completed and released (Pel et al. 2007; Andersen et al. 2011), there are still quite a few hypothetical proteins in the database that are functionally unknown. P. pastoris is a powerful and versatile heterologous expression system, and its low background levels of proteases and other secreted proteins are advantageous for the production of relatively pure enzyme products. Recently, a number of prolyl or leucyl aminopeptidases have been functionally expressed in P. pichia (Yang et al. 2016; Xi et al. 2015; Tang et al. 2016). In the present study, a gene (named apb-AN) encoding AP-B from A. niger was cloned and expressed in Pichia pastoris. The enzymatic properties of recombinant APB-AN were then systematically investigated.
Materials and methods
Strains, plasmids and growth conditions
The A. niger CGMCC 3.1454 strain was obtained from the China General Microbiological Culture Collection Center (CGMCC). Escherichia coli JM109 and P. pastoris GS115 (Invitrogen) served as the hosts for plasmid amplification and gene expression, respectively. The shuttle vector pPIC9K was used for gene cloning. A. niger was grown in PDA medium and incubated in a 250 mL flask at 32 °C and 240 rpm for 24 h, and the mycelium was recovered for total RNA isolation. E. coli JM109 was cultivated in Luria–Bertani (LB) medium at 37 °C for 16 h to clone the recombinant plasmids. P. pastoris GS115 and its recombinants were cultured in YPD medium, minimal dextrose medium (MD), buffered minimal glycerol-complex medium (BMGY), and buffered minimal methanol-complex medium (BMMY) according to the Multi-Copy Pichia Expression Kit (Invitrogen) instructions.
Cloning of the A. niger apb-AN gene
Total RNA from A. niger was extracted using the RNAqueous-Micro Total RNA Isolation Kit (Invitrogen) according to the manufacturer’s protocol. cDNA was synthesized by the SuperScript III First-Strand Synthesis System for RT-PCR (Invitrogen). The sequences of the primers were as follows: forward primer, 5′-GCATCTAAGGACAGAGATATCCTCC-3′; reverse primer, 5′-TGCTCTAGATTAAGCGTAGCCATGGGCCT-3′ (the Bln I (5′) restriction site is in italics). The PCR conditions were as follows: 95 °C for 5 min; 30 cycles of 95 °C for 30 s, 58 °C for 30 s and 72 °C for 2.5 min; and 72 °C for 10 min.
Bioinformatics analysis of the apb-AN gene
The theoretical molecular mass of the protein was calculated by the ProtParam tool (http://us.expasy.org/tools/protparam.html). Conserved domain analysis was carried out with the Conserved Domain Architecture Retrieval Tool (CDART). The signal peptide was predicted using SignalP 4.1 Server (http://www.cbs.dtu.dk/services/SignalP). The protease family was identified by referencing to the peptidase database of MEROPS (http://merops.sanger.ac.uk). Alignments of the deduced amino acid sequence of APB-AN with the sequences of other proteins were performed by the NCBI protein BLAST program (http://www.ncbi.nlm.nih.gov).
Construction of recombinant P. pastoris
The mature apb-AN gene was obtained by PCR, as described above. The PCR product was then digested with Xba I and ligated with vector pPIC9K digested with SnaB I and Bln I, generating the recombinant plasmid pPIC9K-apb. After the correctness of the recombinant plasmid pPIC9K-apb was confirmed by restriction analysis and DNA sequencing (Sangon Biotech Co., Ltd., Shanghai, China), the plasmid was linearized with Stu I and then transformed into P. pastoris GS115 by electroporation. His+ transformants were screened on MD agar plates at 30 °C for 3 days, and then grown on YPD agar plates with geneticin (G418, Sigma) at a concentration of 2.0 mg/mL.
Expression of recombinant APB-AN in P. pastoris GS115
A single colony of the transformants exhibiting resistance to 2.0 mg/mL G418 was cultured at 30 °C in 25 mL of BMGY medium with shaking at 250 rpm until the OD600 reached 4–6. The cells were then transferred to 50 mL of BMMY medium, with a final OD600 value of 1.0. For induction, pure methanol was added once every 24 h to reach a final concentration of 0.5% (v/v) during the 5-day induction period. The induction of cells harboring the empty pPIC9K vector as a negative control was carried out in parallel. All operations were carried out according to the Multi-Copy Pichia Expression Kit (Invitrogen) instructions.
Purification of recombinant APB-AN
Cell-free supernatant was collected by centrifugation at 8000×g for 20 min at 4 °C, and precipitated by ammonium sulfate between 30 and 85% saturation followed by recentrifugation. The precipitate was then suspended in 20 mmol/L phosphate–citrate buffer (pH 7.0), dialyzed against the same buffer, and concentrated by PEG8000. The concentrated supernatant was loaded onto a Sephacryl S-200 column (GE Healthcare, Waukesha, WI, USA) equilibrated with 20 mmol/L phosphate–citrate buffer (pH 7.0) and the active fractions were collected and concentrated using an ultrafiltration membrane (Pall, cutoff value 30 kDa). All of the above procedures were performed at 4 °C.
Enzyme activity and protein concentration assay
The enzyme assay described by Xi et al (2015) was carried out in 50 mmol/L phosphate–citrate buffer, at a pH 7.0, containing 2 mmol/L Arg-pNA substrate (obtained from GL Biochem, Shanghai, China) at 40 °C for 10 min. The produced p-nitroaniline was identified according to the absorbance at 405 nm in a microplate reader (TECAN, Männedorf, The Netherlands). One unit was defined as the amount of enzyme that released 1 μmol p-nitroaniline per minute at 40 °C.
The protein concentration was assayed by the Bradford method using bovine serum albumin (BSA, Sigma) as the standard (Bradford 1976).
Effects of pH, temperature, metal ions and protease inhibitors on enzyme activity
The optimal pH of APB-AN was determined at 40 °C using Arg-pNA as substrate in the following buffers (50 mM): phosphate–citrate buffer (pH 3.0–8.0), phosphate buffer (pH 8.0–9.0), and glycine/NaOH (pH 9.0–10.0). The pH stability of the enzyme was evaluated by measuring the residual enzymatic activity at 40 °C and pH 7.0 (50 mM phosphate–citrate buffer) for 10 min after incubating the enzyme at 40 °C for 60 min in different pH buffers (pH 4.0, 5.0, 6.0, 7.0, 8.0 and 9.0). The optimal temperature of APB-AN was analyzed at temperatures ranging from 20 to 50 °C in 50 mM phosphate–citrate buffer (pH 7.0). To investigate thermal stability, APB-AN was incubated for 60 min at different temperatures (20 °C, 30 °C, 40 °C and 50 °C) in 50 mmol/L phosphate–citrate buffer (pH 7.0) and immediately cooled on ice before the remaining activities were measured. To determine the effects of metal ions and protease inhibitors on APB-AN activity, the enzyme was preincubated with 1 mmol/L divalent cations, or 100 mmol/L Na+, or 0.05 mmol/L Bestatin, E-64, pepstatin, PMSF and puromycin (all purchased from Sigma), or 1 mmol/L EDTA (Sigma). The enzyme activity in the absence of both metal ions and protease inhibitors served as the control. All the above experiments were conducted in triplicate and the resulting data are expressed as the mean ± standard deviation (SD).
Determination of kinetic parameters
Enzyme assays were performed in 50 mmol/L phosphate–citrate buffer, pH 7.0, at 40 °C, using Arg-pNA as a substrate. Substrate concentrations were in the range of 0.2–2.0 mmol/L. Three independent experiments were performed. The kinetic parameters Km and Vmax were then calculated by Eadie–Hofstee plots (Yang et al. 2011).
Substrate specificity towards synthetic substrates and hydrolysis of natural proteins
The activity of the purified APB-AN in the presence of thirteen amino acid-pNA substrates (GL Biochem, Shanghai, China) of at 2 mmol/L was assayed to determine the substrate specificity towards synthetic substrates.
To investigate the selectivity of APB-AN towards natural proteins, 2% (w/v) casein (Sigma), BSA (Sigma), soy protein isolate (SPI, Yuanye Biotech, Shanghai, China) and water-soluble wheat protein (WSWP, Yuanye Biotech, Shanghai, China) were separately used as substrates. The enzyme activity was determined by measuring the amino acids released from the natural proteins. The ninhydrin method described by Morita et al (2009) was adopted to measure the enzyme activity. One unit of activity was defined as the amount of enzyme that produced 1 μg of free amino acid per min under the assay conditions.
Results
Molecular cloning and analysis of the APB-AN gene
The APB-AN gene was amplified by PCR using the reverse transcription product as the template and cloned into the pPIC9K vector. Sequence analysis of the target fragment demonstrated the presence of an ORF of 2646 bp. This ORF was predicted to encode a polypeptide of 881 amino acids with a molecular mass of approximately 98.1 kDa. The cDNA sequence encoding this enzyme was found to be the same as XM_025611793 assigned in the A. niger CBS 115572 genome. The analysis of the predicted protein sequence suggested that no signal peptide was present. Two conserved domains the peptidase M1 aminopeptidase N family (M1_APN-Q_like, cd09601) domain at amino acid positions 16–475 and the ERAP1-like C-terminal (ERAP1_C, pfam11838) domain at amino acids 544–858 were found in the enzyme by using the CDART program within the NCBI website. Furthermore, as predicted by the InterProScan tool, this enzyme was found to belong to the peptidase M1 family which contains mainly aminopeptidases (Rawlings et al. 1993).
Multiple sequence alignment showed that the deduced amino acid sequence of APB-AN only had 14.08, 11.98 and 13,12% identity with identified AP-Bs derived from rat liver (Fukasawa et al. 1996), human lymphocyte (Belhacene et al. 1993) and goat brain (Bogra et al. 2009), respectively, indicating that the present APB-AN is a new AP-B. The four peptidases shared a consensus zinc-binding sequence HEXXHX18E, which is the canonical signature of M1 family metallopeptidases (Fig. 1) (Rawlings et al. 1993).

Multiple sequence alignment of the deduced amino acid sequence of APB-AN with other identified aminopeptidase Bs. RAT APMP (from rat liver); HUMAN AMPB (from human lymphocyte); GOAT AMPB (from goat brain). Amino acids are numbered with respect to the first amino acid of the sequence. Gaps are introduced to optimize the alignment. “Red box”: HEXXHX18E motif. “*”: conservative amino acids; “:”: conservative substitution; “.”: semi-conservative substitution
Heterologous expression of APB-AN in P. pastoris
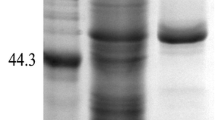
The target fragment was integrated into the plasmid pPIC9K and transformed into P. pastoris GS115. The transformants were grown on MD agar plates and selected on YPD agar plates with G418 (2.0 mg/mL). The insertion of the APB-AN gene in recombinant P. pastoris was confirmed by PCR. Then, the recombinant transformants were induced in 50 mL of BMMY medium at 30 °C in 250 mL shaker flasks. After 120 h of fermentation, the yield and total enzyme activity of APB-AN reached a maximum of 1.2 mg/mL and 184.7 U/mL, respectively, while the control (P. pastoris harboring the empty pPIC9K vector) showed no activity under the same conditions. Unlike the control transformant harboring empty pPIC9K, the transformant harboring the APB-AN gene produced a visible protein band with a molecular mass of approximately 100 kDa on the SDS-PAGE gel (Fig. 2, lane 2), consistent with the theoretical molecular mass of the enzyme (98.1 kDa).

SDS-PAGE analysis of the expression and purification of recombinant APB-AN. Lane 1, purified recombinant APB-AN; lane 2, culture supernatant of APB-AN; lane M, protein molecular weight marker
Purification of APB-AN
The recombinant APB-AN was purified to homogeneity by ammonium sulfate precipitation and gel filtration chromatography of the culture supernatant (Table 1). The purified recombinant enzyme showed a clear single band after Coomassie blue staining an SDS-PAGE gel (Fig. 2, lane 1). The final recovery and purification factors of the enzyme were 23.6% and 2.2, respectively (Table 1).
Enzymatic properties of APB-AN
As shown in Fig. 3a, the optimum pH of APB-AN was 7.0. The activity rapidly increased with an increase in pH from 4.0 to 6.0 and decreased with an increase in pH from 8.0 to 10.0. APB-AN exhibited more than 80% of its maximal activity between pH 5.5 and 8.0. The stability curve of APB-AN at different pH values is shown in Fig. 3b. APB-AN displayed optimal stability at pH 7.0 and retained more than 80% of the maximum activity between pH 5.0 and 8.0 after incubation at 40 °C for 60 min.

Effects of pH and temperature on the activity of the purified APB-AN using Arg-pNA as substrate. a Effects of pH on the activity of APB-AN; b Effects of pH on the stability of APB-AN; c Effects of temperature on the activity of APB-AN; d Effects of temperature on the stability of APB-AN
As shown in Fig. 3c, the activity of APB-AN increased at temperatures ranging from 20 to 40 °C and decreased rapidly at temperatures between 40 and 50 °C. The optimum temperature was 40 °C, and the activity at the optimum temperature was fourfold higher than that at 20 °C. Figure 3d shows that the APB-AN retained more than 70% of its activity after incubation at 20 °C and 30 °C for 60 min; however, its activity declined rapidly at temperatures above 40 °C for 60 min, and little activity (< 4%) was detected after incubation at 50 °C for 60 min.
As shown in Table 2, the effect of different metal ions on APB-AN activity was remarkable. Ca2+, Mg2+ and Mn2+ (1 mmol/L) significantly inhibited the recombinant enzyme. Cu2+ and Fe2+ markedly inhibited protease activity in the presence of 1 mmol/L Cu2+ and Fe2+, only 7.4% and 16.5% of enzyme activity was retained, respectively. The enzyme was strongly activated by 1 mmol/L Co2+ (127.3%) and slightly activated by high concentrations of Na+ (108.7%). Zn2+ was the only cation that did not affect APB-AN. In addition, protease activity was severely inhibited by the metallopeptidase inhibitor 1,10-phenanthroline and EDTA, whereas, an aspartic protease inhibitor (pepstatin) and a cysteine protease inhibitor (E-64) only slightly suppressed the activity. A serine protease inhibitor (PMSF) has almost no effect on enzyme activity. As typical and effective inhibitors of aminopeptidases, bestatin and puromycin both severely inhibited APB-AN.
To determine the kinetic parameters of recombinant APB-AN, different concentrations of Arg-pNA ranging from 0.2 to 1.4 mmol/L were used according to standard methods. From the Lineweaver–Burk plot, Km and Vmax were estimated to be 0.61 mmol/L and 11.45 mmol/L/min, respectively.
Substrate specificity and natural protein hydrolysis
To investigate the substrate specificity of the APB-AN, several synthetic amino acid-pNA substrates and natural protein substrates were examined, and the results are shown in Table 3 and Fig. 4. Among the thirteen amino acid-pNA substrates tested, the recombinant APB-AN rapidly hydrolyzed Arg-pNA (100%) and Lys-pNA (74.3%), followed by Tyr-pNA (22.1%), Trp-pNA (17.6%), Phe-pNA (14.9%), His-pNA (11.2%), Ala-pNA (9.7%), Met-pNA (7.4%), Leu-pNA (6.5%), Glu-pNA (4.3%), Val-pNA (2.7%), Pro-pNA (1.4%) and Ile-pNA (1.1%) (Table 3). According to substrate specificity, the enzyme is regarded as an AP-B (Zhang et al. 2013). The recombinant protease also exerted considerable hydrolytic activity on casein, BSA, SPI, and WSWP and exhibited the highest activity (6.4 U/mL) against casein (Fig. 4).

Proteolytic hydrolysis of different substrates by purified APB-AN. Each substrate (2%, w/v) was dissolved or suspended in 100 mM phosphate–citrate buffer, pH 7.0, mixed with an equal volume (250 μL each) of enzyme solution, and incubated at 40 °C for 20 min. The activity on casein was regarded as 100%. Bars are means ± SD of 3 replicate samples
Discussion
This report described the cloning and expression of APB-AN from A. niger in P. pichia and subsequently the purification, characterization, and sequence analysis of the recombinant enzyme. This study is the first time an AP-B from A. niger has been expressed and identified. The enzyme was first purified from rat liver and is known to be distributed in some animal tissues (Foulon et al. 1999). In contrast, the presence of this exopeptidase in microorganisms is not well documented, only one in the yeast D. hansenii (Bolumar et al. 2003) has been described.
APB-AN was a monomer with a molecular mass of approximately 98.1 kDa according to SDS-PAGE, which is similar to that the molecular mass of AP-Bs from L. vannamei (100 kDa) (Zhang et al. 2013), D. hanseii (101 kDa) (Bolumar et al. 2003) and rat liver (95.5 kDa) (Hopsu et al. 1966); this molecular weight is higher than that of AP-Bs from goat brain (80 kDa) (Bogra et al. 2009) and porcine muscle (76 kDa) (Flores et al. 1993). It seems that the molecular mass of AP-Bs vary depending on origin.
APB-AN was revealed to have an optimum pH of 7.0 and an optimum temperature of 40 °C using Arg-pNA as the substrate. The optimum temperature of APB-AN was lower than that of AP-B from goat brain (45 °C) (Bogra et al. 2009) and was slightly higher than that of AP-Bs from A. parasiticus (37 °C) (Sharma et al. 1989) and D. hansenii (37 °C) (Bolumar et al. 2003). Moreover, the optimum temperature of APB-AN was much higher than that of an AP-B from L. vannamei (30 °C) (Zhang et al. 2013). This temperature-dependent characteristic of AP-Bs may be ascribed to the accommodation temperature of A.niger and the difference among species. The optimum pH of APB-AN was the same as that of AP-Bs from A. parasiticus (Sharma et al. 1989) and D. hansenii (Bolumar et al. 2003), and most of the purified AP-Bs had an optimal pH between 6.5 and 8.0 (Ohishi et al. 2010; Mercado-Flores et al. 2004; Bogra et al. 2009). However, the enzyme possessed obvious activity in a broad pH range (pH 3.0–10.0) in contrast to previously reported AP-Bs (Zhang et al. 2013; Bolumar et al. 2003).
Our present results indicated that APB-AN activity was severely inhibited by the metallopeptidase inhibitors 1,10-phenanthroline and EDTA or the aminopeptidase inhibitors bestatin and puromycin. However, a serine protease inhibitor (PMSF), an aspartic protease inhibitor (pepstatin), and a cysteine protease inhibitor (E-64) only slightly suppressed the activity. These results strongly suggested that the present enzyme was a metalloaminopeptidase. Metalloenzymes contain metal cofactors that are essential for enzyme activity. APB-AN was activated by Co2+ and inhibited by Cu2+ and Fe2+. Zn2+ was the only cation that did not affect APB-AN, suggesting that Zn2+ was a constitutive compound of A. niger APB-AN, which was similar to the AP-B of D. hansenii (Bolumar et al. 2003). This hypothesis was supported by the data showing that the structure of AP-B has a Zn2+-binding site (HEXXHX18E) (Foulon et al. 1999). High concentrations of Na+ slightly activated the enzyme, which was in agreement with previous findings that an appropriate concentration of NaCl can promote aminopeptidase activity (Bolumar et al. 2003; Uraji et al. 2007) and that NaCl might induce conformational changes in aminopeptidase to promote easy access to substrate (Uraji et al. 2007). Co2+ may promote the activity of APB-AN; substituting Co2+ for Zn2+ may improve the catalysis of the enzyme (Holland et al. 1995). APB-AN was severely inhibited by competitive and specific inhibitors of aminopeptidases, bestatin and puromycin and the same effect was observed in other AP-Bs from L. vannamei (Zhang et al. 2013) and D. hansenii (Bolumar et al. 2003).
Previous work has identified two kinds of AP-Bs: broad-substrate AP-Bs and Arg-specific AP-Bs, each capable of hydrolyzing N-terminal arginine residues (Goldstein et al. 2002). APB-AN, identified in this study, had the highest activity against Arg-pNA and showed activity towards all thirteen amino acid-pNA substrates tested, which is consistent with broad-substrate AP-Bs. However, APB-AN has remarkably broader substrate specificity than do other broad-substrate AP-Bs. Most broad-substrate AP-Bs cleave N-terminal amino acid residues, but these enzymes are restricted to Arg and Lys or sometimes Leu and Met (Bolumar et al. 2003; Zhang et al. 2013). This phenomenon could be explained by amino acid changes in the AP-B catalytic core leading to cleavage sites specificity (Fukasawa et al. 2006).
Animal and plant proteins are crucial ingredients in food, and the hydrolysis of these proteins is very meaningful for some types of food processing as it allows for the release of free amino acids and develops flavor (Nishimura et al. 1990). Considering that 100 mmol/L NaCl activated APB-AN, NaCl may promote APB-AN activity during the processing of numerous fermented foods that contain high levels of salt, such as sausage, cheese and soy sauce (Yang et al. 2016; Zhao et al. 2011).
In the present research, APB-AN was expressed in P. pastoris for the first time. After culturing for 120 h, protein expression reached a maximum of 1.2 mg/mL, which was higher than the reported expression of most heterologous proteins in P. pastoris at the flask fermentation level. It has potential applications for production and industrial usage by codon optimization and high cell density cultivation.
Conclusion
The present paper described the heterologous expression, purification and characterization of a novel AP-B from A. niger CGMCC 3.1454. The optimal temperature and pH of the recombinant APB-AN were 40 °C and 7.0, respectively. The enzyme was found to be active towards a number of amino acid p-nitroanilides (pNA) and natural protein substrates. Two unique characteristics of APB-AN were confirmed in contrast to other purified AP-Bs: broader substrate specificity and more larger pH range. In addition, the tolerance of APB-AN to high concentrations of NaCl (100 mmol/L) makes it an attractive candidate for food processing, such as sausage and cheese making and soy sauce fermentation.
References
Andersen MR, Salazar MP, Schaap PJ et al (2011) Comparative genomics of citric-acid-producing Aspergillus niger ATCC 1015 versus enzyme-producing CBS 513.88. Genome Res 21:885–897. https://doi.org/10.1101/gr.112169.110
Attri P, Jodha D, Bansal P et al (2021) Membrane bound aminopeptidase B of a potential probiotic Pediococcus acidilactici NCDC 252: purification, physicochemical and kinetic characterization. Int J Pept Res Ther. https://doi.org/10.1007/s10989-021-10197-w
Basten DE, Visser J, Schaap PJ (2001) Lysine aminopeptidase of Aspergillus niger. Microbiology 147:2045–2050. https://doi.org/10.1006/mpat.2001.0448
Basten DE, Dekker PJ, Schaap PJ (2003) Aminopeptidase C of Aspergillus niger is a novel phenylalanine aminopeptidase. Appl Environ Microbiol 69:1246–1250. https://doi.org/10.1128/AEM.69.2.1246-1250.2003
Basten DEJW, Moers APHA, Ooyen AJJV et al (2005) Characterisation of Aspergillus niger prolyl aminopeptidase. Mol Genet Genomics 272:673–679. https://doi.org/10.1007/s00438-004-1094-5
Belhacene N, Mari B, Rossi B et al (1993) Characterization and purification of T lymphocyte aminopeptidase B: a putative marker of T cell activation. Eur J Immunol 23:1948–1955. https://doi.org/10.1002/eji.1830230833
Bogra P, Singh J, Singh H (2009) Purification and characterization of aminopeptidase B from goat brain. Process Biochem 44:776–780. https://doi.org/10.1016/j.procbio.2009.03.001
Bolumar T, Sanz Y, Aristoy MC et al (2003) Purification and properties of an arginyl aminopeptidase from Debaryomyces hansenii. Int J Food Microbiol 86:141–151. https://doi.org/10.1016/S0168-1605(03)00069-2
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254. https://doi.org/10.1016/0003-2697(76)90527-3
Flores M, Aristoy MC, Toldrá F (1993) HPLC purification and characterization of porcine muscle aminopeptidase B. Biochimie 75:861–867. https://doi.org/10.1016/0300-9084(93)90040-Y
Foulon T, Cadel S, Cohen P (1999) Aminopeptidase B (EC 3.4.11.6). Int J Biochem Cell B 31:747–750. https://doi.org/10.1016/S1357-2725(99)00021-7
Fukasawa KM, Fukasawa K, Kanai M et al (1996) Molecular cloning and expression of rat liver aminopeptidase B. J Biol Chem 271:30731–30735. https://doi.org/10.1074/jbc.271.48.30731
Fukasawa KM, Hirose J, Hata T et al (2006) Aspartic acid 405 contributes to the substrate specificity of aminopeptidase B. Biochemistry 45:11425–11431. https://doi.org/10.1021/bi0604577
Goldstein JM, Nelson D, Kordula T et al (2002) Extracellular arginine aminopeptidase from Streptococcus gordonii FSS2. Infect Immun 70:836–843. https://doi.org/10.1128/IAI.70.2.836-843.2002
Holland DR, Hausrath AC, Juers D et al (1995) Structural analysis of zinc substitutions in the active site of thermolysin. Protein Sci 4:1955–1965. https://doi.org/10.1002/pro.5560041001
Hopsu VK, Mäkinen KK, Glenner GG (1966) Purification of a mammalian peptidase selective for N-terminal arginine and lysine residues: aminopeptidase B. Arch Biochem Biophys 114:557–566. https://doi.org/10.1016/0003-9861(66)90380-8
Jalving R, Godefrooij J, Veen WJT et al (2005) Characterization of the Aspergillus niger dapB gene, which encodes a novel fungal type IV dipeptidyl aminopeptidase. Mol Genet Genomics 273:319–325. https://doi.org/10.1007/s00438-005-1134-9
Mercado-Flores Y, Noriega-Reyes Y, Ramírez-Zavala B et al (2004) Purification and characterization of aminopeptidase (pumAPE) from Ustilago maydis. Fems Microbiol Lett 234:247–253. https://doi.org/10.1016/j.femsle.2004.03.035
Morita H, Okamoto A, Yamagata Y et al (2009) Heterologous expression and characterization of CpI, OcpA, and novel serine-type carboxypeptidase OcpB from Aspergillus oryzae. Appl Microbiol Biot 85:335–346. https://doi.org/10.1007/s00253-009-2087-4
Nishimura T, Okitani A, Rhyu MR, Kato H (1990) Survey of neutral aminopeptidases in bovine, porcine, and chicken skeletal muscles. Agric Biol Chem 54:2769–2775. https://doi.org/10.1080/00021369.1990.10870427
Nishimura T, Okitani A, Kato H (2006) Identification of neutral aminopeptidases responsible for peptidolysis in postmortem rabbit skeletal muscle. Agric Biol Chem 52:2183–2190. https://doi.org/10.1080/00021369.1988.10869008
Ohishi K, Yamamoto T, Tomofuji T et al (2010) Isolation and characterization of aminopeptidase from Capnocytophaga granulosa ATCC 51502. Mol Oral Microbiol 20:67–72. https://doi.org/10.1111/j.1399-302X.2005.00183.x
Pel HJ, De Winde JH, Archer DB et al (2007) Genome sequencing and analysis of the versatile cell factory Aspergillus niger CBS 513.88. Nat Biotechnol 25:221–231. https://doi.org/10.1038/nbt1282
Rao MB, Tanksale AM, Ghatge MS et al (1998) Molecular and biotechnological aspects of microbial proteases. FEMS Microbiol Rev 23:411–456. https://doi.org/10.1128/MMBR.62.3.597-635.1998
Rawlings ND, Barrett AJ (1993) Evolutionary families of peptidases. Biochem J 290(Pt 1):205
Sanderink GJ, Artur Y, Siest G (1988) Human aminopeptidases: a review of the literature. Clin Chem Lab Med 26:795–807. https://doi.org/10.1515/cclm.1988.26.12.795
Sharma A, Padwaldesai SR, Ninjoor V (1989) Intracellular hydrolases of Aspergillus parasiticus and Aspergillus flavus. Biochem Biophys Res Co 159:464–471. https://doi.org/10.1016/0006-291X(89)90015-6
Song P, Cheng L, Tian K et al (2020) A novel aminopeptidase with potential debittering properties in casein and soybean protein hydrolysates. Food Sci Biotechnol 29:1491–1499. https://doi.org/10.1007/s10068-020-00813-8
Tang W, Li Z, Li C et al (2016) High-level expression and characterization of the Bacillus subtilis subsp. subtilis str. BSP1 YwaD aminopeptidase in Pichia pastoris. Protein Exp Purif 122:23–30. https://doi.org/10.1016/j.pep.2016.02.009
Toldra F, Aristoy MC, Flores M (2000) Contribution of muscle aminopeptidases to flavor development in dry-cured ham. Food Res Int 33:181–185. https://doi.org/10.1016/S0963-9969(00)00032-6
Uraji M, Arima J, Uesugi Y et al (2007) Effect of salt on the activity of Streptomyces prolyl aminopeptidase. Biochim Biophys Acta 1774:1462–1469. https://doi.org/10.1016/j.bbapap.2007.08.022
Xi H, Tian Y, Zhou N et al (2015) Characterization of an N-glycosylated Bacillus subtilis leucine aminopeptidase expressed in Pichia pastoris. J Basic Microb 55:236–246. https://doi.org/10.1002/jobm.201400368
Yang H, Long L, Li J et al (2011) Heterologous expression, biochemical characterization, and overproduction of alkaline α-amylase from Bacillus alcalophilus in Bacillus subtilis. Microb Cell Fact 10:77. https://doi.org/10.1186/1475-2859-10-77
Yang H, Zhu Q, Zhou N et al (2016) Optimized expression of prolyl aminopeptidase in Pichia pastoris and its characteristics after glycosylation. World J Microb Biot 32:176. https://doi.org/10.1007/s11274-016-2135-z
Zhang L, Cai QF, Wu GP et al (2013) Arginine aminopeptidase from white shrimp (Litopenaeus vannamei) muscle: purification and characterization. Eur Food Res Technol 236:759–769. https://doi.org/10.1007/s00217-013-1941-x
Zhao W, Zheng J, Zhou HB (2011) A thermotolerant and cold-active mannan endo-1,4-β-mannosidase from Aspergillus niger CBS 513.88: constitutive overexpression and high-density fermentation in Pichia pastoris. Bioresour Technol 102:7538–7547. https://doi.org/10.1016/j.biortech.2011.04.070
Acknowledgements
This work was funded by National key research and development program of China (2017YFD0502005).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that there is no conflict of interest.
Rights and permissions
About this article

Cite this article
Song, P., Feng, W. Functional expression and characterization of a novel aminopeptidase B from Aspergillus niger in Pichia pastoris. 3 Biotech 11, 366 (2021). https://doi.org/10.1007/s13205-021-02915-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s13205-021-02915-4