
Abstract
We have cloned a gene (papA) that encodes a prolyl aminopeptidase from Aspergillus niger. Homologous genes are present in the genomes of the Eurotiales A. nidulans, A. fumigatus and Talaromyces emersonii, but the gene is not present in the genome of the yeast Saccharomyces cerevisiae. Cell extracts of strains overexpressing the gene under the control of its own promoter showed a fourfold to sixfold increase in prolyl aminopeptidase activity, but no change in phenylalanine or leucine aminopeptidase activity. The overexpressed enzyme was subsequently purified and characterised. The enzyme specifically removes N-terminal proline and hydroxyproline residues from peptides. It is the first enzyme of its kind from a eukaryotic organism that has been characterised.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
All living organisms maintain a particular rate of protein turnover by continuous degradation and synthesis of proteins. Outside the cell, proteins are degraded into oligopeptides by extracellular broad-specificity proteases. Further degradation of these oligopeptides into amino acids can be achieved by the interaction of extracellular or intracellular oligopeptidases, dipeptidases and tripeptidases and finally by carboxypeptidases and aminopeptidases. The turnover of proteins by proteases provides a ready pool of amino acids as precursors for the synthesis of new proteins (Bennet and Klich 1992).
Proteases normally do not hydrolyse bonds adjacent to proline residues. Instead a specialised group of enzymes has evolved that hydrolyses these bonds. Their activity depends on both the isomeric state of the proline residue and its position in the peptide chain (Vanhoof et al. 1995; Cunningham and O’Connor 1997). Proline aminopeptidases (Pap, prolyl iminopeptidase, EC 3.4.11.5) are serine proteases (Barret et al. 1998) that catalyse the cleavage of N-terminal proline residues from peptides, and so far these enzymes have only been characterised from bacteria. Based on their molecular mass and substrate specificity, these enzymes can be divided into two classes. The first class consists of monomeric enzymes with a molecular mass of approximately 30 kDa. Enzymes of this class appear to have a strict specificity for N-terminal proline residues (Kitazono et al. 1994b). Such monomeric enzymes have been characterised from Bacillus coagulans (Kitazono et al. 1992), Lactobacillus delbrueckii (Atlan et al. 1994; Gilbert et al. 1994), Neisseria gonorrhoeae (Albertson and Koomey 1993), Serratia marcescens (Yoshimoto et al. 1999; Ito et al. 2000) and Xanthomonas campestris (Alonso and Garcia 1996; Medrano et al. 1998). The second class of enzymes comprise large multimeric proteins that are not only capable of hydrolysing terminal prolines but have also been reported to be capable of hydrolysing N-terminal hydroxyproline residues. Enzymes of this second class have only been cloned and characterised from the prokaryotes Aeromonas sobria (Kitazono et al. 1994b), Hafnia alvei (Kitazono et al. 1996) and Propionibacterium shermanii (Leenhouts et al. 1998).
Our aim is to fully understand the protease spectrum of Aspergillus niger (Van den Hombergh et al. 1997). Products synthesised by A. niger usually have the GRAS (Generally Regarded As Safe) status and a prolyl aminopeptidase from such an organism thus could also be interesting for the food industry. No prolyl aminopeptidase function appears to be present in the genome of Saccharomyces cerevisiae (http://mips.biochem.mpg.de). Surprisingly, however, we found an expressed sequence tag (EST) that showed homology to the multimeric class of enzymes in the ascomycete A. nidulans. This EST was used to clone the corresponding gene from A. niger gene. This paper describes the characterisation of this first eukaryotic prolyl aminopeptidase, isolated from A. niger.
Materials and methods
Bacterial strains, DNA and RNA techniques and sequence analysis
The Escherichia coli strains LE392 and DH5α (Promega, Madison, WI, USA) were used for phage amplification and for plasmid transformation and propagation, respectively. Standard DNA and RNA manipulations were carried out essentially as described by Sambrook et al. (1989). Protein and nucleotide sequence analyses were performed using the program DNAstar (Lasergene, Madison, WI, USA). Public databases were searched with the BLAST search tools (Altschul et al. 1997). Multiple alignments were done with CLUSTAL X (Jeanmougin et al. 1998). A dendrogram of the multiple alignment was displayed with the program Treeview (Page 1996).
For Northern analysis strains were grown in minimal medium (Pontecorvo et al. 1953) supplemented with 1% glucose as carbon source and 0.5% yeast extract for 17 h. Cultures (50 ml) were set up in 250-ml Erlenmeyer flasks, which were incubated at 30°C in an shaker at 250 rpm. Total RNA was isolated from mycelium samples with the Trizol reagent (Life Technologies, Rockville, MD, USA).
Cloning and overexpression of papA
A papA fragment was amplified by PCR from genomic DNA of A. nidulansWG096 using the primers Forward (5′-TCTTCGAGGTTCCCCTC-3′) and Reverse (5′-CTATGTATGTCGCTGCG-3′). Routinely PCR was carried out for 30 cycles of 1 min at 94°C, 1 min at 48°C, and 1 min at 72°C, preceded by an incubation of 4 min at 94°C and followed by incubation for 5 min at 72°C. The DNA fragment obtained was cloned in the vector pGEM-T (Promega) and its identity was confirmed by direct sequencing. Genomic sequences of the A. niger papA gene were obtained by screening an λEMBL4 genomic library of the A. niger Strain N400 by standard methods using the A. nidulans PCR product as a probe and non-stringent hybridisation conditions. From a hybridising plaque a 3.7-kb EcoRI fragment was recovered and cloned in pUC19, resulting in the plasmid pIM4105, which contains the papA gene with its 5′ and 3′ flanking regions.
The sequence data for the A. niger papA gene and the papA fragment from A. nidulans have been deposited in the DDBJ/EMBL/GenBank databases under the Accession Nos. AJ315565 and AJ315566.
The plasmids pIM4105 and pGW635 (Goosen et al. 1989), which contains the A. niger pyrA gene encoding orotidine-5′-phosphate decarboxylase, were used to co-transform A. niger NW219 (cspA1, leuA1, nicA1, pyrA6; Kusters van Someren et al. 1991). Transformants were first selected for uridine prototrophy and subsequently screened for enhanced prolyl aminopeptidase activity in cell extracts (CE). For this, strains were grown in shake-flask cultures as described for Northern analysis. Ground mycelium was extracted with 20 mM Tris–HCl pH 7.5 and clarified by centrifugation (10,000 g for 15 min at 4°C). Aminopeptidase activity in these cell extracts was assayed as described below.
Aminopeptidase enzyme assay
Aminopeptidase activity was determined using both artificial substrates and peptides. In the first case activity was measured as described by Atlan et al. (1994) using different amino acids coupled to para-nitroanilide (pNA) as substrates. Standard reactions contained 2 mM P-pNA as substrate in 20 mM TRIS-HCl (pH 7.5) and were performed at 30°C. Aminopeptidase activity was also determined as described by Frey and Röhm (1979) using amino acids coupled to β-naphthylamide (β-NA) as substrate. These reactions contained 1 mM β-NA in 20 mM Tris-HCl (pH 7.5) and were carried out at 30°C. L-pNA, P-pNA, R-pNA, F-pNA, A-pNA, M-pNA and K-pNA were obtained from Sigma (St. Louis, MO, USA). V-pNA, G-pNA, I-pNA, E-pNA, P-βNA, hydroxy-P-βNA were obtained from Bachem (Bubendorf, Switzerland).
When peptide substrates were used, proline releasing activity was determined using the discontinuous ninhydrin assay described by Troll and Lindsley (1955). An aliquot of enzyme was mixed with 1 mM peptide in 20 mM Tris–HCl (pH 7.5) and incubated at 30°C. At various times thereafter samples were taken and the reaction was terminated by the addition of 1 M sodium acetate (pH 2.8). Next, 25 μl of 10% ninhydrin in ethanol was added, and the mixtures were incubated for 10 min at 80°C. Colour development was measured spectrophotometrically at 450 nm and the amount of proline liberated was calculated from a proline calibration curve generated in a similar way. The reaction rate was determined from the slope of the line through the linear part of the activity plot. The peptides AP, PA, PLG(NH2), PPGFSPFR, and PLSRTLSVAAKK were obtained from Sigma.
One unit of enzyme activity is defined as the amount of enzyme that produces 1 μmol of pNA, βNA or proline per min.
Protein concentrations were determined with bicinchoninic acid as described by the manufacturer (Sigma).
KM and Kcat values were determined by fitting initial rate measurements to the Michaelis Menten equation and are expressed as the means (±SD) of three independent determinations.
Purification of A niger proline aminopeptidase A
Strain Pap-1 was pre-grown in 300 ml of MM (Pontecorvo et al. 1953) supplemented with 2% (w/v) glucose, 0.2% (w/v) meat peptone, 0.1% (w/v) yeast extract, 0.1% (w/v) peptone 140, and 0.03% (w/v) yeast ribonucleic acids. After 6.5 h this culture was poured into 5 l of MM supplemented with 2% (w/v) glucose, 0.2% (w/v) meat peptone, 0.1% (w/v) yeast extract, and 0.1% (w/v) peptone 140, and growth was allowed continue for 17 h.
The mycelium was harvested, ground and resuspended in 20 mM TRIS-HCl (pH 7.5) and stirred for 15 min at 4°C. Cellular debris was removed by centrifugation. Then (NH4)2SO4 was added to 40% saturation. After centrifugation (15 min, 10,000 g), (NH4)2SO4was added to the supernatant to a concentration equivalent to 70% saturation. After centrifugation the pellet was dissolved in 20 mM Tris–HCl (pH 7.5) and dialysed against the same buffer containing 1.5 M (NH4)2SO4.
The protein was purified using four chromatographic steps. The dialysed fraction was loaded onto a 20-ml Phenyl Sepharose high performance column (Pharmacia Biotech, Uppsala, Sweden), pre-equilibrated with the same buffer, and bound protein was eluted using a 200-ml linear gradient from 1.5 to 0 M (NH4)2SO4 in 20 mM Tris–HCl (pH 7.5). The active fractions were pooled, dialysed against 20 mM bis–Tris (pH 6.5) and loaded onto a 15.5-ml Source 30 Q column (Pharmacia Biotech), pre-equilibrated with the same buffer. A 155-ml linear gradient from 0 to 0.5 M NaCl in 20 mM bis–Tris (pH 6.5) was then applied. The active fractions were pooled, diluted tenfold in 20 mM piperazine (pH 6.0) and loaded onto a 1-ml Resource Q column (Pharmacia Biotech) pre-equilibrated with the same buffer. Bound protein was eluted using a 50-ml linear gradient from 0 to 0.5 M NaCl in the loading buffer. The active fractions were pooled and loaded onto a 318-ml Superdex 200 column (Pharmacia Biotech) equilibrated in 20 mM Tris–HCl (pH 7.5) + 0.1 M NaCl, and eluted with the same buffer.
Biochemical characterisation of prolyl aminopeptidase A
The optimal pH for enzymatic activity was determined using McIlvaine buffers ranging from pH 5 to 8, 200 mM HEPES buffered in the range from pH 7.2 to 8 and 50 mM Tris–glycine buffers from pH 8 to 10. P-pNA was used as the substrate.
For determination of its thermal stability, the purified enzyme was pre-incubated at 0, 30, 37, 50, and 60°C for 30 min, followed by the standard enzyme reaction. A sample of the enzyme that had been preincubated at 0°C was used as the reference.
The pH stability of PapA was determined by preincubation of the purified enzyme in McIlvaine buffers ranging from pH 2.2 to 8 at 30°C, for 30 min, followed by the standard enzyme reaction. A sample preincubated at pH 7 was used as a reference.
The effects of the protease inhibitors bestatin, 1,10-phenanthroline, EDTA, EGTA, tosyl phenylalanyl chloromethyl ketone (TPCK), tosyl lysyl chloromethyl ketone (TLCK), leupeptin, iodoacetamide, and 4-chloromercuribenzoic acid (PCMB) on the enzymatic activity were measured in 200 mM HEPES buffer at pH 7.5. The purified enzyme was incubated with the respective compound for 30 min at 30°C, and its activity was then measured using the standard enzyme assay.
The molecular mass of the native enzyme was determined using HPLC (Dionex, CA, USA). Three gel-filtration columns were used in cascade, with 10 mM sodium phosphate as eluent: BIO-Gel SEC 40 XL, BIO-Gel SEC 30 XL and BIO-Gel SEC 20 XL (Biorad, CA, USA). As reference proteins ovalbumin (45 kDa), hexokinase (100 kDa), alcohol dehydrogenase (145 kDa), aldolase (160 kDa), catalase (240 kDa), and ferritin (450 kDa) were used. Retention times were plotted against molecular mass and compared with the retention time of the native enzyme.
Results and discussion
Cloning of the papA gene
By database searching at http://aspergillus-genomics.org using textstrings with the keyword aminopeptidase in combination with BLAST searches of selected expressed sequence tags (ESTs) against the non-redundant protein database at NCBI (http://www.ncbi.nlm.nih.gov/), we identified two A. nidulans ESTs, (Accession Nos. AA785412 and AA78541). These ESTs could encode different parts of a putative fungal prolyl aminopeptidase (PapA) similar to the multimeric prolyl aminopeptidases of prokaryotes. We designed two primers based on these EST sequences and amplified a 1534-bp PCR product from A. nidulans genomic DNA. The PCR product specified a large fragment of the ORF of a putative A. nidulans papA gene and the 5′ and 3′ ends of the sequence overlapped with the sequences of the two selected ESTs (Fig. 1). This indicated that the EST sequences originated from the same gene. When all available sequences were joined, more than 95% of the ORF of the putative gene could be reconstructed. A comparison with the sequenced A. nidulans genome revealed that the reconstructed A. nidulans gene is a single-copy gene and the ORF is identical to predicted protein AN2092 (Aspergillus Sequencing Project, Center for Genome Research, http://www.broad.mit.edu). Under conditions of low stringency a 3.7-kb EcoRI fragment of A. niger genomic DNA hybridised with the PCR product from A. nidulans. The fragment was recovered from an A. niger genomic library and cloned, yielding plasmid pIM4105. The cloned fragment contained the complete ORF of the gene and approximately 1600 bp of upstream and 300 bp of downstream sequence (Fig. 1). The ORF encodes a protein of 491 amino acids interrupted by seven introns. Using RT-PCR we able to obtain overlapping cDNA fragments, and sequencing confirmed that the predicted introns were positioned correctly (Fig. 1). Southern analysis using conditions of low stringency indicated that the papA gene is also a single-copy gene in A. niger (results not shown). Northern analysis revealed that the gene is constitutively expressed (results not shown).

A comparison of the A. niger and A. nidulans papA genes. The ORFs are indicated by the filled boxes, the arrow indicates the direction of transcription. The positions of the introns in the A. niger papA gene (Accession No. AJ315565) and the partial A. nidulans papA gene (Accession No. AJ315566) are indicated by the open boxes. The A. nidulans ESTs (Accession Nos. AA785412 and AA78541) are shaded in grey
Analysis of the papAORF sequence
The inferred amino acid sequence of PapA was analysed for known subcellular sorting signals and none were found, suggesting that, like ApsA (Basten et al. 2001) and ApsC (Basten et al. 2003), the enzyme is located in the cytoplasm. In addition, we were unable to detect proline aminopeptidase activity in the culture fluid. Proline aminopeptidases from bacteria have also been reported to be intracellular aminopeptidases (Albertson and Koomey 1993; Atlan et al. 1994; Kitazono et al. 1994b; Alonso and Garcia 1996; Yoshimoto et al. 1999).
The PapA protein sequence was compared to previously characterised bacterial prolyl aminopeptidases and putative homologs from other fungi.
The PapA protein displayed highest identity with the multimeric prokaryotic prolyl aminopeptidases, exhibiting 39 and 37% identity with the enzymes from A. sobria and H. alvei, respectively. Only very limited similarity to the monomeric B. coagulans prolyl aminopeptidase (14%) was detected. The inferred A. niger papA protein sequence is 80 and 79% identical to the corresponding polypeptides from A. fumigatus and A. nidulans, respectively. A dendrogram depicting the sequence relationships of these proteins with the A. niger papA was constructed (Fig. 2) The dendogram shows that the fungal enzymes share sequence homology with the multimeric bacterial enzymes but form a separate branch. There is no ortholog of papA in the sequenced genome of S. cerevisiae (http://mips.biochem.mpg.de). The GXSXG motif, which contains the reactive serine in serine proteases (Polgar 1992; Kanatani et al. 1993; Mitta et al. 1998) is located at amino acid residues 163–167: the GQSFG is 100% conserved between the three Aspergillus Species and is also present in the Pap enzyme from Taleromyces emersonii. This suggests that Ser 165 is the active-site serine. The motif has also been found in the enzymes from H. alvei (Kitazono et al. 1996), A. sobria (Kitazono et al. 1994b) and P. shermanii (Leenhouts et al. 1998). By site-directed mutagenesis, the serine in the GXSXG motif was proven to be responsible for the catalytic activity of the enzymes from A. sobria and B. coagulans (Kitazono et al. 1994a). This motif is also found in the monomeric class of enzymes; however, its precise sequence differs from that in the multimeric class of enzymes: in most cases, GGSWG is found, but GHSWG and GQSWG also occur.

Dendrogram showing the relationships between prolyl aminopeptidases. The A. niger PapA was compared with the predicted prolyl aminopeptidases AN2092 from A. nidulans (Accession No. EAA64924), from A. fumigatus (translated from genomic sequences available at http://www.tigr.org), T. emersonii (AAL40648), N. crassa (XP327701), M. grisea (EAA50657), F. graminearum (XP387012), and the prolyl aminopeptidases from A. sobria (P46547), X. campestris (BAA11623), B. coagulans (P46541), L. delbrueckii (P46544), H. alvei (JC4623),N. gonorrhoeae (P42786), S. marcescens (O32449), P shermanii (CAA04698) and C. albicans (EAK96989)
Characterisation of papA-overexpressing strains
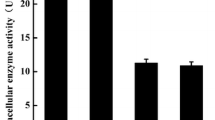
We transformed A. niger with plasmid pIM4105, and at least five transformants with elevated expression of the papA gene were identified by Northern analysis. Highest mRNA levels were observed in the transformant Pap-1. Southern analysis of transformants Pap-1 and 2 indicated that these transformants have integrated approximately 30 (Pap-1) and 10 (Pap-2) copies of the gene in their respective genomes (results not shown). We determined the proline, phenylalanine and leucine aminopeptidase activities in cell-free extracts of Pap-1 and Pap-2 and of a wild-type strain, using pNA substrates. Pap-1 and Pap-2 show a sixfold and fourfold increase in prolyl aminopeptidase activity, respectively, relative to the wild type. No change in hydrolysis of the phenylalanine and leucine pNA substrates was measured. In agreement with the elevated prolyl aminopeptidase activity, the highest papA copy number and the highest mRNA levels were found in transformant Pap-1.
Purification and biochemical characterisation of the A niger prolyl aminopeptidase
Five purification steps were needed to purify PapA (1.4×103-fold) from transformant Pap-1, resulting in an enzyme preparation with a specific activity of 2.9×102 U mg, and a final yield of 16% (Table 1). The low yield in the first step is apparently due to inefficient precipitation by ammonium sulphate.
An SDS-PAGE analysis of the purified PapA (Fig. 3) shows a single band with an apparent molecular mass of 50 kDa, in agreement with the molecular mass of 50.0 kDa calculated from the ORF. The molecular mass of the native enzyme, as determined by gel filtration, is 316 kDa, suggesting that PapA is a hexameric enzyme. The enzymes from Aeromonas and Hafnia both occur as tetramers (Kitazono et al. 1994b, 1996).

Purification of prolyl aminopeptidase A from A. niger. Coomassie-stained SDS polyacrylamide gel (10%) of 20 μl of the active fractions 38–44 eluted from the Superdex 200 column. Fraction numbers are indicated above the lanes. M protein molecular weight marker
The substrate specificity was determined using an extensive set of pNA and βNA substrates and several peptide substrates (dimers, trimers and oligomers). PapA was unable to hydrolyse artificial substrates other than P-pNA and hydroxy-P-βNA. Remarkably, PapA efficiently hydrolyses P-pNA but not P-βNA. Cell extracts from A. niger were also tested for activity with P-βNA but no (other) activity could be detected. However, the hydroxy-P-βNA substrate can be hydrolysed by PapA, as well as by the enzymes from Aeromonas and Hafnia (Kitazono et al. 1994b, 1996). The monomeric Lactobacillus and Bacillus enzymes were reported not to be active on hydroxy-P-βNA (Kitazono et al. 1994b). The KM of PapA for P-pNA is 0.037±0.002 mM, kcat is 4.1±0.06 μkat/mg.
The activity on peptide substrates was tested using peptides of increasing length and compared to the rates of hydrolysis obtained with the Aeromonas and Bacillus enzymes (Kitazono et al. 1994b) (Table 2). Upon complete hydrolysis 2 mM proline is released from 1 mM PPG(8), so PapA is apparently able to efficiently hydrolyse both proline residues from the peptide substrate PPGFSPFR; therefore the overall hydrolysis rate for both proline residues is reported in Table 2. In contrast to the Aeromonas enzyme, PapA from A. niger hydrolyses the PLG substrate more rapidly than the longer peptide substrate PLSR(12). Hydrolysis rates for dipeptides have not been not reported for the Bacillus and Aeromonas enzymes. The pH optimum for P-pNA was also determined. The pH optimum is between 7 and 8, highest activity is found at pH 7.5 (Fig. 4); these values are similar to those previously found for other intracellular aminopeptidases of A. niger (Basten et al. 2001, 2003) and to the pH optimum for the Hafnia enzyme (pH 7.0–7.5), but the enzyme from A. sobria has a different pH optimum (8.5) (Kitazono et al. 1994b, 1996). Furthermore, the enzyme is stable in the range from pH 5 to pH 8 (Fig. 4). Highest stability was found at pH 6 to 7. The enzyme is stable up to a temperature of 37°C (Fig. 4), and thus differs in this respect from the Aeromonas and Hafnia enzymes, which are reported to be stable up to temperatures of 57 and 55°C, respectively (Kitazono et al. 1994b, 1996).

Biochemical characterisation of PapA. a pH optimum of PapA. The following buffers were used: McIlvaine (diamonds), Hepes (triangles) and Tris–glycine (circles). b pH stability of PapA. Residual activity was calculated relative to a sample kept at pH 7.0 at 30°C. c Thermal stability of PapA. Residual activity after a 30-min incubation at the indicated temperatures is expressed relative to a sample kept at 0°C. Results are the mean of three independent experiments
Several potential protease inhibitors were also tested (Table 3). EDTA did not influence enzyme activity, implying that the enzyme is not a metalloprotease. The serine protease inhibitors TLCK and TPCK were able to inhibit the enzyme activity, and 50% inhibition was achieved with 4 mM TLCK. However, PapA was strongly inhibited by PCMB. This inhibition may be caused by the modification of a cysteine residue located near the active site (Ito et al. 2000). Similar inhibitory effects of PCMB were also reported for the prolyl aminopeptidases from A. sobria and B. coagulans, although these enzymes are known to be serine proteases (Kitazono et al. 1994a).
In this report we have described the biochemical characteristics of the first prolyl aminopeptidase to be identified in a eukaryotic organism. The results clearly indicate that this fungal enzyme is a prolyl aminopeptidase that is able to hydrolyse both artifical and peptide substrates. This aminopeptidase, homologues of which are encoded in the genomes of a number of Eurotiales but not in the yeast S. cerevisiae, shares homology with, but is not similar to, the previously reported multimeric class of bacterial prolyl aminopeptidases. Both the bacterial enzymes and the fungal enzyme can hydrolyse N-terminal proline and hydroxyproline residues. However, the artificial P-βNA substrate is not hydrolysed by the A. niger enzyme. Furthermore, the active fungal enzyme appears to be a hexamer. In contrast to the bacterial enzymes, hydrolysis by the fungal enzyme shows an increase in rate with decreasing substrate length.
References
Albertson NH, Koomey M (1993) Molecular cloning and characterization of a proline iminopeptidase gene from Neisseria gonorrhoeae. Mol Microbiol 9:1203–1211
Alonso J, Garcia JL (1996) Proline iminopeptidase gene from Xanthomonas campestris pv citri. Microbiology 142:2951–2957
Altschul SF, Madden TL, Schaffer A, Zhang j, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402
Atlan D, Gilbert C, Blanc B, Portalier R (1994) Cloning, sequencing and characterization of the pepIP gene encoding a proline iminopeptidase from Lactobacillus delbrueckii subsp bulgaricus CNRZ 397. Microbiology 140:527–535
Barret AJ, Rawlings ND, Woessner JF (1998) Handbook of proteolytic enzymes. Academic Press, San Diego
Basten DEJW, Visser J, Schaap PJ (2001) Lysine aminopeptidase of Aspergillus niger. Microbiology 147:2045–2050
Basten DEJW, Dekker PJT, Schaap PJ (2003) Aminopeptidase C of Aspergillus niger is a novel phenylalanine aminopeptidase. Appl Environ Microbiol 69:1246–1250
Bennet JW, Klich MA (1992) Aspergillus: biology and industrial applications. Butterworth-Heinemann, Boston
Cunningham DF, O’Connor B (1997) Proline specific peptidases. Biochim Biophys Acta 1343:160–186
Frey J, Röhm K-H (1979) External and internal forms of yeast aminopeptidase II. Eur J Biochem 97:169–173
Gilbert C, Atlan D, Blanc B, Portalier R (1994) Proline iminopeptidase from Lactobacillus delbrueckii subsp. bulgaricus CNRZ 397: purification and characterization. Microbiology 140:537–542
Goosen T, van Engelenburg F, Debets F, Swart K, Bos K, Van den Broek H (1989) Tryptophan auxotrophic mutants in Aspergillus niger: inactivation of the trpC gene by cotransformation mutagenesis. Mol Gen Genet 219:282–288
Ito K, Inoue T, Kabashima T, Kanada M, Huang HS, Ma XH, Azmi N, Azab E, Yoshimoto T (2000) Substrate recognition mechanism of prolyl aminopeptidase from Serratia marcescens. J Biochem 128:673–678
Jeanmougin F, Thompson JD, Gouy M, Higgins DG, Gibson TJ (1998) Multiple sequence alignment with Clustal X. Trends Biochem Sci 23:403–405
Kanatani A, Yoshimoto T, Kitazono A, Kokubo T, Tsuru D (1993) Prolyl endopeptidase from Aeromonas hydrophila: cloning, sequencing and expression of the enzyme gene and characterization of the expressed enzyme. J Biochem 113:790–796
Kitazono A, Yoshimoto T, Tsuru D (1992) Cloning, sequencing and high expression of the proline iminopeptidase gene from Bacillus coagulans. J Bacteriol 174:7919–7925
Kitazono A, Ito K, Yoshimoto T (1994a) Prolyl aminopeptidase is not a sulfhydryl enzyme: identification of the active serine residue by site-directed mutagenesis. J Biochem 116:943–945
Kitazono A, Kitano A, Tsuru D, Yoshimoto T (1994b) Isolation and characterization of the prolyl aminopeptidase gene (pap) from Aeromonas sobria: comparison with the Bacillus coagulans enzyme. J Biochem 116:818–825
Kitazono A, Kitano A, Kabashima T, Ito K, Yoshimoto T (1996) Prolyl aminopeptidase is also present in Enterobacteriaceae: cloning and sequencing of the Hafnia alvei enzyme-gene and characterization of the expressed enzyme. J Biochem 119:468–474
Kusters van Someren MA, Harmsen JAM, Kester HCM, Visser J (1991) Structure of the Aspergillus pelA gene and its expression in Aspergillus niger and Aspergillus nidulans. Curr Genet 20:293–299
Leenhouts K, Bolhuis A, Boot J, Deutz I, Toonen M, Venema G, Kok J, Ledeboer A (1998) Cloning, expression and chromosomal stabilization of the Propionibacterium shermanii proline iminopeptidase gene (pip) for food-grade application in Lactococcus lactis. Appl Environ Microbiol 64:4736–4742
Medrano FJ, Alonso J, Garcia JL, Romero A, Bode W, Gomis-Ruth FX (1998) Structure of proline iminopeptidase from Xanthomonas campestris pv citri: a prototype for the prolyl oligopeptidase family. EMBO J 17:1–9
Mitta M, Miyagi M, Kato I, Tsunasawa S (1998) Identification of the catalytic triad residues of porcine liver acylamino acid-releasing enzyme. J Biochem 123:924–931
Page RDM (1996) TREEVIEW: an application to display phylogenetic trees on personal computers. Comput Appl Biosci 12:357–358
Polgar L (1992) Structural relationship between lipases and peptidases of the prolyl oligopeptidase family. FEBS Lett 311:281–284
Pontecorvo G, Roper JA, Hemmons LJ, MacDonald KD, Bufton AWJ (1953) The genetics of Aspergillus nidulans. Adv Genet 5:141–238
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: a laboratory manual 2nd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York
Troll W, Lindsley J (1955) A photometric method for the determination of proline. J Biol Chem 215:655–660
Van den Hombergh JPT, van de Vondervoort PJI, Fraissinet-Tachet L, Visser J (1997) Aspergillus as a host for heterologous protein production: the problem of proteases. Trends Biotechnol 15:256–263
Vanhoof G, Goossens F, Meester ID, Hendriks D, Scharpe S (1995) Proline motifs in peptides and their biological processing. FASEB J 9:736–744
Yoshimoto T, Kabashima T, Uchikawa K, Inoue T, Tanaka N, Nakamura KT, Tsuru M, Ito K (1999) Crystal structure of prolyl aminopeptidase from Serratia marcescens. J Biochem 126:559–565
Acknowledgements
The authors thank H. Kester for assistance with the biochemical work. Daniëlle Basten was supported by DSM. P. Schaap was partially supported by a grant (WBI 4100) from the Dutch Technology Foundation (STW). All the work was carried out in compliance with Dutch and local laws governing genetic experimentation.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by C.P. Hollenberg
Rights and permissions
About this article
Cite this article
Basten, D.E.J.W., Moers, A.P.H.A., Ooyen, A.J.J.v. et al. Characterisation of Aspergillus niger prolyl aminopeptidase. Mol Genet Genomics 272, 673–679 (2005). https://doi.org/10.1007/s00438-004-1094-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00438-004-1094-5