
Abstract
Two new aspartic proteases, PepAb and PepAc (encoded by pepAb and pepAc), were heterologously expressed and biochemically characterized from Aspergillus niger F0215. They possessed a typical structure of pepsin-type aspartic protease with the conserved active residues D (84, 115), Y (131, 168) and D (281, 326), while their identity in amino acid sequences was only 19.0%. PepAb had maximum activity at pH 2.5 and 50 °C and PepAc at 3.0 and 50 °C. The specific activities of PepAb and PepAc toward casein were 1368.1 and 2081.4 U/mg, respectively. Their activities were significantly promoted by Cu2+ and Mn2+ and completely inhibited by pepstatin. PepAb exhibited higher catalytic efficiency (kcat/Km) toward soy protein isolates than casein, while PepAc showed higher catalytic efficiency toward casein. The hydrolysis capacities of PepAb and PepAc on soy protein isolates were slightly lower than that of previously identified A. niger aspartic protease, PepA (aspergillopepsin I), while the resultant peptide profiles were remarkably different for all three proteases.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Aspartic proteases (EC 3.4.23), commonly known as acid proteases, is a catalytic type of protease enzyme that uses an activated water molecule bound to one or more aspartate residues for catalysis of its peptide substrates (Rawlings et al. 2018; Coates et al. 2006). In general, it contains two highly conserved aspartates in the catalytic sites and is optimally active at acidic pHs (Coates et al. 2006). The interest on aspartic proteases is continually growing due to their significant application potential in food, fermentation, animal feed, beverage and pharmaceutical industries (Theron and Divol 2014; Nout and Rombouts 1990; Marangon et al. 2012; Pinelo et al. 2010). For example, aspartic proteases have been used in manufacturing of cheese, soy sauce and other fermented foods, as well as in the baking industry for the modification of wheat proteins in bread dough systems (Theron and Divol 2014; Nout and Rombouts 1990). They have also been applied in meat and fish protein hydrolysis under acidic conditions (Nout and Rombouts 1990) and utilized as digestive aids for treating certain enzyme deficiency syndromes (Theron and Divol 2014). Recently, aspartic proteases have been used in the degradation of proteins that cause white wine haze (Marangon et al. 2012), fruit juice turbidity reduction (Pinelo et al. 2010), and in oligopeptides preparation from different sources of protein (Rizzello et al. 2016; Chalamaiah et al. 2018; Maestri et al. 2016). However, limited aspartic proteases are available commercially and most of them are a mixture of peptidases which may result in undesirable side reaction and/or unintended protein degradation (Theron and Divol 2014; Marangon et al. 2012).
The first aspartic protease from Aspergillus niger was isolated in the culture filtrate of A. niger var. macrosporus, named aspergillopepsin I (EC 3.4.23.18), which is a typical pepsin-type aspartic protease, active between pH 2 and 4 and inhibited by pepstatin (Koaze et al. 1964; Rao et al. 1998; Inoue et al. 1996). The sequencing and analysis of A. niger CBS 513.88 genome has revealed more aspartic protease-like open reading frames (ORFs) (Pel et al. 2007). Since these new aspartic proteases have not been reported previously, the characterization of these may provide protease with novel or expanded biotechnological or food processing application. This study, analyzed three putative aspergillopepsin-like aspartic proteases, elucidating their biochemical properties and potential application in casein and soybean protein-related processes.
Materials and methods
Strains and cultural conditions
A. niger F0215 (Niu et al. 2017) was grown in potato dextrose agar (PDA) medium (2% potato, 2% dextrose and 1% agar) with 1% casein (Sigma, USA) at 32 °C and 240 rpm. After 24 h incubation, the mycelia were harvested for total RNA isolation. Escherichia coli JM 109 was grown in Luria–Bertani (LB) medium (10% tryptone, 5% yeast extract and 10% NaCl) at 37 °C and used for cloning experiment. Pichia pastoris GS115 was used for extracellular expression of the recombinant proteins and recombinants were cultivated according to the Instructions Manual from the supplier (Invitrogen, USA).
DNA manipulation, molecular cloning and expression
DNA manipulation and the transformation of E. coli were carried out according to standard procedures (Sambrook et al. 1989). Total RNA was extracted from mycelia of A. niger using RNAqueous™-Micro Total RNA Isolation Kit (Invitrogen, USA). The cDNA was synthesized by SuperScript III First-Strand Synthesis System for RT-PCR (Invitrogen, USA). Three sets of oligonucleotide primers (Table 1) were designed and the predicted genes (PepA, PepAb and PepAc) were amplified by Pyrobest DNA polymerase (Takara, China) with cDNA of A. niger F0215 as template. The PCR products were digested with XbaI and ligated into the pPIC9K vector (Invitrogen, USA) linearized with BlnI and SnaBI. The recombinant plasmids were confirmed by restriction digestion and nucleotide sequencing. GenBank accession numbers of pepA, pepAb and pepAc were XM_001401056, XM_001388448 and XM_001395370, respectively.
The verified recombinant plasmids carrying the three protease genes were linearized with restriction enzymes (SalI, SacI and NcoI, respectively) and transformed into P. pastoris GS115 using a Micropulser Electroporator (Bio-Rad, USA) with condition 1.5 kV, 200 Ω, and 25 mF. The transformants were selected on minimal dextrose (MD) plates (1.34% yeast nitrogen base without amino acids, 2% dextrose, 0.00004% biotin, and 1.5% agar) and further selected on yeast extract peptone dextrose (YPD) plates (1% yeast extract, 2% peptone, 2% dextrose and 1.5% agar) containing Geneticin (2 mg/L). The positive colonies were confirmed by colony PCR and aspartic protease activity assay. Production of the recombinant proteases from P. pastoris transformants was performed according to the Multi-Copy Pichia Expression Kit Instructions (Invitrogen, USA). The enzyme induction was carried out in 50 mL cultures (OD600 of 1.0) at 30 °C and 200 rpm for 5 days with 0.5% (v/v) methanol supplemented once every 24 h.
Sequence analysis and modeling
The conserved domain analysis was conducted using CDART programs (https://www.ncbi.nlm.nih.gov/Structure/lexington/lexington.cgi). Protein sequence analysis and classification was carried out with InterPro (https://www.ebi.ac.uk/interpro/). Protease family assorting was identified by reference to the peptidase database of Merops (https://www.merops.sanger.ac.uk). The theoretical molecular mass of the protein was calculated using the ProtParam tool (https://www.us.expasy.org/tools/protparam.html). Signal peptide was predicted using SignalP 4.1 Server (https://www.cbs.dtu.dk/services/SignalP). The three-dimensional models of aspartic proteases were generated by the online Swiss-Model server and viewed with Swiss-Pdb Viewer program (https://www.swissmodel.expasy.org).
Protease activity assay
The aspartic protease activity was measured by the Folin–phenol method using casein as substrate and the reaction was carried out at pH 3.0 and 50 °C for 10 min (Oda and Murao 1974). The reaction mixture contained equal volume of enzyme solution and 1% (w/v) casein dissolved in 50 mM lactic acid/sodium lactate buffer. One unit of enzyme activity was defined as the enzyme quantity which liberates 1 μg of tyrosine per ml of the reaction mixture per minute under the conditions.
Purification of recombinant proteases
The recombinant enzymes were collected by ammonium sulfate gradient precipitation followed by centrifugation at 10,000×g for 15 min. The precipitated enzymes were resuspended in 10 mM lactic acid/sodium lactate buffer (pH 3.0) and dialyzed (Dialysis bag, MWCO 8000–14,000 Da, Spectrum Labs Inc.) overnight at 4 °C in the same buffer and then lyophilized. The lyophilized products were re-dissolved in 10 mM lactic acid/sodium lactate buffer (pH 3.0) and loaded onto a Sephacryl S-200 column (GE Healthcare, Waukesha, WI, USA) equilibrated with the same buffer, and the active fractions were collected. Protein concentration was determined using the Bradford assay (1976) with bovine serum albumin Fractionation V (Sigma) as reference standard.
Effects of temperature, pH, metal ions and inhibitors on the activity of recombinant aspartic proteases
To determine the optimum temperature, aspartic protease activity was measured at temperatures range from 30 to 65 °C. The thermostability was determined by incubating the enzyme samples at different temperatures (30–70°C) for 60 min and the residual activities were analyzed. The optimal pH was detected by performing enzyme assays at 50 °C with 50 mM lactic acid–sodium lactate buffer with different pH ranges (pH 1.5–4.0). To determinate the pH stability of the enzymes, the enzymes were pre-incubated in different pH buffers at 25 °C for 60 min and the residual activities were subsequently assayed according to the standard enzyme assay.
The effect of metal ions and inhibitors (Pepstatin, PMSF and E-64; Sigma, USA) was investigated by incubating the enzymes with individual metal ions (5 mM) or inhibitors (5 mM) for 60 min, followed by the measurement of the residual activities under optimal conditions for each protease.
Determination of kinetic parameters
The enzyme assays were performed using optimal conditions with different concentrations (0.5–50 mg/mL) of casein or soy protein isolates as substrates. The kinetic parameters Km and Vmax were calculated using the Lineweaver–Burk plot (1934) and the kcat was the value of Vmax divided by E (enzyme concentration).
Protein hydrolysis with the enzymes and the hydrolytes analysis by HPLC
Protein hydrolysis with the recombinant enzymes was carried out using 1% soy protein isolates (Yuanye Biotechnology Co. Ltd, China) as substrate. The reactions were performed in 10 mL volumes with 10 units of the enzyme at optimal conditions for 3 h. The degree of hydrolysis (DH) was calculated from ratio of α-amino nitrogen and total nitrogen according to Nilsang et al. (2005). The resulting peptides and their molecular weights (MW) were determined by HPLC (Agilent 1200 Series HPLC System), equipped with a TSKgel G2000SWxl column (300 mm × 7.8 mm, Tosoh, Japan). Acetonitrile: water: trifluoroacetic acid solution (45:55:0.1, v/v) was used for eluting at a flow rate of 0.5 mL/min at 30 °C. The initial substrate and reaction products were detected by UV detector at a wavelength of 280 nm. Cytochrome (12,500 Da), aprotinin (6500 Da), bacitracin (1450 Da), Gly-Gly-Tyr-Arg (451 Da) and Gly-Gly-Gly (189 Da) (National Institute of Metrology, China) were used as MW standards.
Results and discussion
Cloning and sequence analysis of aspergillopepsin-like proteases from A. niger
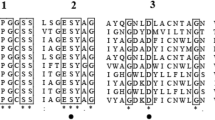
Aspergillopepsin I encoded by pepA in A. niger has been identified and well characterized (Koaze et al. 1964; Rao et al. 1998; Inoue et al. 1996; Ichishima 2012). By using sequence analysis and prediction tools, two putative aspergillopepsin-like proteases, PepAb and PepAc (encoded by pepAb and pepAc, respectively), were predicated from genomic sequence of A. niger. The genes (pepA, pepAb and pepAc) were successfully amplified by PCR and ligated into the pPIC9K vector. The coding nucleotide sequences of pepAb and pepAc from A. niger F0215 were the same as that identified from the A. niger CBS 513.88 genome. PepAb and PepAc were all found to belong to aspartic peptidase A1 family according to Rawlings et al. (2018). An aspergillopepsin-like domain and a eukaryotic aspartyl protease domain were identified in PepAb, whereas PepAc had only the eukaryotic aspartyl protease domain. PepAb and PepAc contained highly conserved catalytic motifs Asp-Thr-Gly-Ser in the N-terminal domain and Asp-Ser/Thr-Gly-Thr in the C-terminal domain, which are also observed in PepA and other pepsin-type aspartic protease (Fig. 1) (Inoue et al. 1996; Fujimoto et al. 2004; Wlodawer et al. 2004). The active site residues D (D101, D84 and D115 for PepA, PepAb and PepAc, respectively), Y (143, 131, 168) and D (283, 281, 326) and inhibitor (pepstatin) binding amino acid residues D (D101, D84 and D115 for PepA, PepAb and PepAc, respectively), G (103, 86, 117), Y (143, 131, 168), G (144, 132, 169), D (145, 133, 170), D (283, 281, 326), G (285, 283, 328), T (286, 284, 329) and T (287, 285, 330) were conserved in PepA, PepAb and PepAc (Fig. 1) (Wlodawer et al. 2004).

Multiple sequence alignment of the deduced amino acid sequences of PepA, PepAb and PepAc using ClustalW. Signal peptides for PepA and PepAc are indicated by gray shading blocks. Catalytic motif was boxed. Inhibitor binding sites were indicated by green shading blocks. The active site flap is underlined. Active site residues are marked with a solid black spot
The predicted models of PepAb and PepAc created by SWISS-MODEL were similar to that of PepA (Fig. 2a). Each of them had two independent domains mainly composed of β-barrel domain structures. The first domain contains only a catalytic Asp residue, while the second contains a catalytic Asp residue and a conserved Tyr residue that is reported to affect specificity (Hong and Tang 2004; Dunn 2002). The catalytic sites are located between the two lobes of the molecules (Fig. 2a) and the spatial structure of catalytic residues are shown by overlapping the three aspartic proteases (Fig. 2b). Surprisingly, although their amino acid sequences are significantly different, the overlapping structures are evenly matched. PepAb and PepAc showed only 35.90% and 22.17% sequence identity to PepA, respectively, while PepAb showed only 19.0% to PepAc (Fig. 1).

Predicted 3D structures of PepA, PepAb and PepAc which were created by SWISS-MODEL. Box a presents the 3D protein models of the A. niger F0215 aspartic proteases PepA, PepAb and PepAc. Box b presents the overlapped active residues of three 3D models. Green residues are from PepA, yellow residues from PepAb and blue residues are from PepAc. The amino acid residues in active sites are shown in three-letter codes
Expression and biochemical characteristics of the recombinant aspartic proteases
P. pastoris recombinants expressing PepA, PepAb and PepAc were successfully constructed. The recombinant enzymes were prepared by shake flask fermentation with methanol induction and then purified (Table 2, Fig. 3). The theoretical MWs of PepA, PepAb and PepAc were 39.3, 46.7 and 46.0 kDa, respectively, consistent with the apparent MWs revealed by SDS-PAGE (Fig. 3). The specific activities of PepAb and PepAc were 1368.1 U/mg and 2081.4 U/mg, slightly lower than that of PepA which was 2289.6 U/mg (Table 2).

SDS-PAGE analysis of purified proteases. Lane M: Pierce™ unstained protein molecular weight marker; lane 1: PepA; lane 2: PepAb; lane 3: PepAc. The proteases were purified by fraction precipitation by ammonium sulfate and Sephacryl S-200 column chromatography
Analysis of biochemical properties of recombinant proteases was conducted and the results are summarized in Fig. 4, Tables 3 and 4. PepAb and PepA showed similar temperature profiles with a temperature optimum of 50 °C and a broad temperature range of activity (Fig. 4a). These enzymes also retained more than 60% activity between 35 and 55 °C, while PepAc was 5 °C higher, ranging from 40 to 60 °C (Fig. 4a). PepAc and PepAb retained more than 90% and 70% of the maximum activity, respectively, after incubation for 60 min at 50 °C (Fig. 4b). The optimum pH of PepAb and PepAc was 2.5 and 3.0, respectively (Fig. 4c), and both enzymes retained more than 70% of maximum activity after incubation for 60 min at their respective pH optima (Fig. 4d). Interestingly, compared with previously reported aspartic proteases, PepAb and PepAc revealed lower temperature and pH optima (Vishwanatha et al. 2009; Souza et al. 2017; Lakshman et al. 2010; Hsiao et al. 2014). Their low temperature and pH optima can be used for potential application such as food processing, as these characters will contribute to decrease of energy consumption and restraining microbial spoilage (Santiago et al. 2016; Shiby and Mishra 2013).

Effect of temperature and pH on enzyme activity with casein as a substrate. All tests were carried out using 10 U of enzymes. a Temperature profiles and b thermal stability as determined by the activity remaining after incubation for 60 min, c pH profiles and d pH stability as determined by the activity remaining after incubation for 60 min. The data presented with mean ± standard deviation of triplicate determinations
The effect of metal ions on PepAb and PepAc was similar to that on PepA, which is significantly enhanced by Cu2+ and Mn2+, but inhibited by Fe3+ (Table 3). These observations were different from previous reports with aspartic protease from Rizopus oryzae inhibited by both Cu2+ and Mn2+ (Hsiao et al. 2014), whereas Fe3+ activated aspartic proteases from Monascus pilosus (Lakshman et al. 2010) and Aspergillus foetidus (Souza et al. 2017). Ewert et al. (2018) suggested that metal ions bond with some amino acids in the aspartic proteases which cause conformational changes and consequently affect enzyme activity. Therefore, effects of metal ions on PepA, PepAb and PepAc may also due to the same mechanism. Pepstatin is a well-known inhibitor of aspartic proteases (Fujimoto et al. 2004) and PepAb, PepAc and PepA were completely inhibited by 5 mM pepstatin (Table 3). PMSF (inhibitor for serine protease) and E-64 (inhibitor for cysteine protease) did not cause detectable effect on the enzyme activity (Table 3). The inhibition by pepstatin and the invalidity of PMSF confirmed that PepAb and PepAc belong to aspartic proteases.
Casein and soy protein isolates were used as substrates for determining the enzyme kinetic parameters and the results are summarized in Table 4. The PepAb showed a higher affinity (Km 2.5 mg/mL) to casein, whereas PepAc had higher affinity to soy protein isolates (Km 1.9 mg/mL). PepAc showed significantly higher catalytic efficiency (kcat/Km) on both casein and soy protein isolates than PepA and PepAb. However, both PepAb and PepAc exhibited higher affinity than the PepA to casein and soy protein substrates. The results indicated that PepA, PepAb and PepAc could play a collaborative role in enzymatic hydrolysis of protein in industrial application.
Hydrolysis of soy protein isolates with PepAb and PepAc and peptide analysis
To further elucidate the hydrolytic characteristics of PepAb and PepAc on a natural protein substrate, soy protein isolates were hydrolyzed with PepAb, PepAc or PepA under optimal conditions for 3 h until a constant DH was obtained for each reaction, which was 7.6, 6.9 and 8.7% for PepAb, PepAc and PepA, respectively. Furthermore, peptide profiles for PepAb and PepAc were remarkably different from that of PepA (Fig. 5). PepAb and PepAc acting on soy protein isolates released more oligopeptides with the MWs between 189 and 1450 Da (about 2–11 amino acid residues), while PepA tended to release oligopeptides with relatively higher MWs (Fig. 5). The results strongly suggested that the cleavage specificity of PepAb and PepAc toward soy protein isolates are different from that of PepA, which was consistent with that each aspartic protease has a unique cleavage specificity (Wlodawer et al. 2004; Souza et al. 2017; Lakshman et al. 2010; Hsiao et al. 2014). Pepsin hydrolysis of soy protein isolates has been studied for modification of functional properties (Tsumura et al. 2004; Cui et al. 2013; Yang et al. 2016; Liu et al. 2018). The glycinin fraction in native soybean protein isolates was selectively hydrolyzed by pepsin in the pH 1.5–2.5 range, while the β-conglycinin fraction was not susceptible to pepsin digestion (Tsumura et al. 2004). With another pepsin, strong degradation effect was found on glycinin, along with a little effect on the α-subunit of β-conglycinin (Cui et al. 2013). Varied effects of pepsin digestion were also reported on soybean protein isolates from different soybean varieties (Yang et al. 2016). Diverse properties of the resultant hydrolysates were obtained based on the specificity of pepsins to soy protein isolates (Tsumura et al. 2005; Cui et al. 2013; Yang et al. 2016). Compared with PepA, PepAb and PepAc exhibited different hydrolytic characteristics on soybean protein isolates, their detailed specificity to certain protein substrates and potential applications in protein modification and preparation of functional oligopeptides require further investigation.

HPLC analysis of peptide profiles of soy protein isolates and its hydrolysates digested with PepA, PepAb and PepAc, respectively. ①: untreated soy protein isolates; ②: soy protein isolates digested with PepA; ③: soy protein isolates digested with PepAb; ④: soy protein isolates digested with PepAc; ⑤: protein molecular weight standards. Protein hydrolysis with PepA, PepAb or PepAc was carried out using 1% soy protein isolates as substrate. The reactions were performed in 10 mL volumes with 10 U of enzyme at optimal conditions for 3 h
Conclusion
Two new A. niger aspartic proteases were heterologously expressed and characterized with significant biochemical and catalytic differences from previously reported A. niger aspartic protease, aspergillopepsin I. They possess functional properties suitable for application in preparation of protein hydrolysate products for food industry individually or in combination.
References
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254. https://doi.org/10.1016/0003-2697(76)90527-3
Chalamaiah M, Yu W, Wu J (2018) Immunomodulatory and anticancer protein hydrolysates (peptides) from food proteins: a review. Food Chem 245:205–222. https://doi.org/10.1016/j.foodchem.2017.10.087
Coates L, Erskine PT, Mall S et al (2006) X-ray, neutron and NMR studies of the catalytic mechanism of aspartic proteinases. Eur Biophys J 35:559–566. https://doi.org/10.1007/s00249-006-0065-7
Cui C, Zhao M, Yuan B et al (2013) Effect of pH and pepsin limited hydrolysis on the structure and functional properties of soybean protein hydrolysates. J Food Sci 78:C1871–C1877. https://doi.org/10.1111/1750-3841.12309
Dunn BM (2002) Structure and mechanism of the pepsin-like family of aspartic peptidases. Chem Rev 102:4431–4458. https://doi.org/10.1021/cr010167q
Ewert J, Glück C, Strasdeit H et al (2018) Influence of the metal ion on the enzyme activity and kinetics of PepA from Lactobacillus delbrueckii. Enzyme Microb Technol 110:69–78. https://doi.org/10.1016/j.enzmictec.2017.10.002
Fujimoto Z, Fujii Y, Kaneko S et al (2004) Crystal structure of aspartic proteinase from Irpex lacteus in complex with inhibitor pepstatin. J Mol Biol 341:1227–1235. https://doi.org/10.1016/j.jmb.2004.06.049
Hong L, Tang J (2004) Flap position of free memapsin 2 (β-secretase), a model for flap opening in aspartic protease catalysis. Biochemistry 43:4689–4695. https://doi.org/10.1021/bi0498252
Hsiao NW, Chen Y, Kuan YC et al (2014) Purification and characterization of an aspartic protease from the Rhizopus oryzae protease extract, Peptidase R. Electron J Biotechn 17:89–94. https://doi.org/10.1016/j.ejbt.2014.02.002
Ichishima E (2012) Chapter 27-Aspergillopepsin I. Rawlings ND, Salvesen GS Handbook of proteolytic enzymes, 3rd edn. Elsevier, Amsterdam, pp 135–141
Inoue H, Hayashi T, Huang XP et al (1996) Heterologous expression and site-directed mutagenesis studies on the activation mechanism and the roles of the basic residues in the prosegment of aspergillopepsinogen I. Eur J Biochem 237(3):719–725. https://doi.org/10.1111/j.1432-1033.1996.0719p.x
Koaze Y, Goi H, Ezawa K et al (1964) Fungal proteolytic enzymes. Part I. Isolation of two kinds of acid-proteases excreted by Aspergillus niger var. macrosporus. Agric Biol Chem 28:216–223. https://doi.org/10.1080/00021369.1964.10858233
Lakshman PLN, Toyokawa Y, Toyama H et al (2010) Purification and characterisation of two extracellular acid proteinases from Monascus pilosus. Food Chem 121:1216–1224. https://doi.org/10.1016/j.foodchem.2010.02.007
Lineweaver H, Burk D (1934) The determination of enzyme dissociation constants. J Am Chem Soc 56:658–666. https://doi.org/10.1021/ja01318a036
Liu H, Zhang R, Li L et al (2018) The high expression of Aspergillus pseudoglaucus protease in Escherichia coli for hydrolysis of soy protein and milk protein. Prep Biochem Biotechnol 48:725–733. https://doi.org/10.1080/10826068.2018.1508035
Maestri E, Marmiroli M, Marmiroli N (2016) Bioactive peptides in plant-derived foodstuffs. J Proteom 147:140–155. https://doi.org/10.1016/j.jprot.2016.03.048
Marangon M, Van Sluyter SC, Robinson EMC et al (2012) Degradation of white wine haze proteins by Aspergillopepsin I and II during juice flash pasteurization. Food Chem 135:1157–1165. https://doi.org/10.1016/j.foodchem.2012.05.042
Nilsang S, Lertsiri S, Suphantharika M et al (2005) Optimization of enzymatic hydrolysis of fish soluble concentrate by commercial proteases. J Food Eng 70:571–578. https://doi.org/10.1016/j.jfoodeng.2004.10.011
Niu D, Tian X, Mchunu NP et al (2017) Biochemical characterization of three Aspergillus niger β-galactosidases. Electron J Biotechnol 27:37–43. https://doi.org/10.1016/j.ejbt.2017.03.001
Nout MJR, Rombouts FM (1990) Recent developments in tempe research. J Appl Bacteriol 69:609–633. https://doi.org/10.1111/j.1365-2672.1990.tb01555.x
Oda K, Murao S (1974) Purification and some enzymatic properties of acid protease A and B of Scytalidium lignicolum ATCC 24568. Agric Biol Chem 38:2435–2444. https://doi.org/10.1080/00021369.1974.10861522
Pel HJ, de Winde JH, Archer DB et al (2007) Genome sequencing and analysis of the versatile cell factory Aspergillus niger CBS 513.88. Nat Biotechnol 25:221–231. https://doi.org/10.1038/nbt1282
Pinelo M, Zeuner B, Meyer AS (2010) Juice clarification by protease and pectinase treatments indicates new roles of pectin and protein in cherry juice turbidity. Food Bioprod Process 88:259–265. https://doi.org/10.1016/j.fbp.2009.03.005
Rao MB, Tanksale AM, Ghatge MS et al (1998) Molecular and biotechnological aspects of microbial proteases. Microbiol Mol Biol Rev 62:597–635. https://doi.org/10.1128/MMBR.62.3.597-635.1998
Rawlings ND, Barrett AJ, Thomas PD et al (2018) The MEROPS database of proteolytic enzymes, their substrates and inhibitors in 2017 and a comparison with peptidases in the PANTHER database. Nucleic Acids Res 46:D624–D632. https://doi.org/10.1093/nar/gkx1134
Rizzello CG, Tagliazucchi D, Babini E et al (2016) Bioactive peptides from vegetable food matrices: research trends and novel biotechnologies for synthesis and recovery. J Funct Foods 27:549–569. https://doi.org/10.1016/j.jff.2016.09.023
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: a laboratory manual, 2nd edn. Cold Spring Harbor Laboratory Press, New York
Santiago M, Ramirez-Sarmiento CA, Zamora RA et al (2016) Discovery, molecular mechanisms, and industrial applications of cold-active enzymes. Front Microbiol 7:1408. https://doi.org/10.3389/fmicb.2016.01408
Shiby VK, Mishra HN (2013) Fermented milks and milk products as functional foods—a review. Crit Rev Food Sci Nutr 53(5):482–496. https://doi.org/10.1080/10408398.2010.547398
Souza PM, Werneck G, Aliakbarian B et al (2017) Production, purification and characterization of an aspartic protease from Aspergillus foetidus. Food Chem Toxicol 109:1103–1110. https://doi.org/10.1016/j.fct.2017.03.055
Theron LW, Divol B (2014) Microbial aspartic proteases: current and potential applications in industry. Appl Microbiol Biotechnol 98:8853–8868. https://doi.org/10.1007/s00253-014-6035-6
Tsumura K, Saito T, Kugimiya W et al (2004) Selective proteolysis of the glycinin and β-conglycinin fractions in a soy protein isolate by pepsin and papain with controlled pH and temperature. J Food Sci 69:363–367. https://doi.org/10.1111/j.1365-2621.2004.tb10698.x
Tsumura K, Saito T, Tsuge K et al (2005) Functional properties of soy protein hydrolysates obtained by selective proteolysis. Lwt-Food Sci Technol 38:255–261. https://doi.org/10.1016/j.lwt.2004.06.007
Vishwanatha K, Rao AA, Singh SA (2009) Characterisation of acid protease expressed from Aspergillus oryzae MTCC 5341. Food Chem 114:402–407. https://doi.org/10.1016/j.foodchem.2008.09.070
Wlodawer A, Gustchina A, James MNG (2004) Catalytic pathway of aspartic peptidases. In: Barrett AJ, Rawlings ND, Woessner JF (eds) Handbook of proteolytic enzymes. Elsevier, London, pp 12–19
Yang Y, Wang Z, Wang R et al (2016) Secondary structure and subunit composition of soy protein in vitro digested by pepsin and its relation with digestibility. Biomed Res Int 2016:1–11. https://doi.org/10.1155/2016/5498639
Acknowledgements
This work was kindly supported by the Intergovernmental International Scientific and Technological Innovation Cooperation program, MOST, China (Grant no.: 2018YFE0100400), the Raising Program of Innovation Team for Tianjin Universities, Tianjin, China (Grant no.: TD12-5002) to ZXWANG and the National Natural Science Foundation of Fujian Province, China (Grant no.: 2016J01157) to DDNIU
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Rights and permissions
About this article

Cite this article
Song, P., Cheng, L., Tian, K. et al. Biochemical characterization of two new Aspergillus niger aspartic proteases. 3 Biotech 10, 303 (2020). https://doi.org/10.1007/s13205-020-02292-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s13205-020-02292-4