
Abstract
Hypertrophic cardiomyopathy (HCM) is a cardiac genetic disease characterized by ventricular enlargement, diastolic dysfunction, and increased risk for sudden cardiac death. Sarcomeric genetic defects are the predominant known cause of HCM. In particular, mutations in the myosin-binding protein C gene (MYBPC3) are associated with ~ 40% of all HCM cases in which a genetic basis has been established. A decade ago, our group reported a 25–base pair deletion in intron 32 of MYBPC3 (MYBPC3Δ25bp) that is uniquely prevalent in South Asians and is associated with autosomal dominant cardiomyopathy. Although our studies suggest that this deletion results in left ventricular dysfunction, cardiomyopathies, and heart failure, the precise mechanism by which this variant predisposes to heart disease remains unclear. Increasingly appreciated, however, is the contribution of secondary risk factors, additional mutations, and lifestyle choices in augmenting or modifying the HCM phenotype in MYBPC3Δ25bp carriers. Therefore, the goal of this review article is to summarize the current research dedicated to understanding the molecular pathophysiology of HCM in South Asians with the MYBPC3Δ25bp variant. An emphasis is to review the latest techniques currently applied to explore the MYBPC3Δ25bp pathogenesis and to provide a foundation for developing new diagnostic strategies and advances in therapeutics.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Hypertrophic cardiomyopathy (HCM), one of the leading forms of cardiovascular disease (CVD) in the world, is a genetic disorder that affects ~ 20 million people globally, including 750,000 Americans. HCM is characterized by ventricular expansion, resulting in diastolic dysfunction and an increased risk of sudden death (Gersh et al. 2011). Sarcomeric genetic defects are the predominant cause of HCM (Bonne et al. 1995; Carrier et al. 1998; Gersh et al. 2011; Harris et al. 2011; Spirito et al. 1997; Watkins et al. 1995; Watkins et al. 1993). Several reports have established a direct link between sarcomeric mutations and cardiomyopathies, including HCM and dilated cardiomyopathy (DCM) (Hitomi et al. 2010; Niimura et al. 2002; Tabish et al. 2019; Tanjore et al. 2008; Van Driest et al. 2005).
HCM is termed a disease of the sarcomere as mutations in sarcomeric genes, such as MYH7, MYBPC3, MYL3, TPM1, TNNI3, and TNNT2, are the principal causes of pathology (Geisterfer-Lowrance et al. 1990; Seidman and Seidman 2011; Thierfelder et al. 1994). However, 5–10% of HCM cases are caused by mutations in genes involving metabolic pathways (MacRae et al. 1995; Murphy et al. 2005). Importantly, mutations in both MYH7 and MYBPC3 genes contribute to more than 80% of HCM cases (Viswanathan et al. 2017), of which MYBPC3 is the leading gene known to cause HCM and DCM (Bonne et al. 1995; Previs et al. 2012; Viswanathan et al. 2017). The MYBPC3 gene encodes a thick-filament cardiac muscle protein, a cardiac paralog of myosin-binding protein C (cMyBP-C), which is involved in regulating myosin function, rate of force generation, and cardiac contractility. Also, MYBPC3 mutations present a high risk for heart failure (HF) (Adalsteinsdottir et al. 2014; Barefield et al. 2014; Dhandapany et al. 2009; Helms et al. 2014; Michels et al. 2009; Page et al. 2012). Previously, our group discovered a polymorphic variant in MYBPC3, a 25–base pair deletion (MYBPC3Δ25bp) at intron 32 (Fig. 1). One of the molecular consequences of MYBPC3Δ25bp is the replacement of 65 wild-type amino acids by 58 novel amino acids in the carboxyl region of cMyBP-C (Dhandapany et al. 2009; Kuster et al. 2015; Kuster and Sadayappan 2014; Waldmuller et al. 2003). The variant is associated with HCM, DCM, and HF (Dhandapany et al. 2009; Simonson et al. 2010; Srivastava et al. 2011).

Genotype of MYBPC3Δ25bp in intron 32 of the MYBPC3 gene. (a) Two options of 25-bp deletion are indicated with the same outcome. (b) The location of the splice branch point and polypyrimidine track at the junction of intron 32/exon 33 splicing (modified from Sadayappan et al. 2020)
Discovery of the MYBPC3Δ25bp mutation and its association with cardiomyopathies was first noted at the International Society for Heart Research in Winnipeg, Canada, on July 6–11, 2001 (Sakthivel et al. 2001). In our first study, MYBPC3Δ25bp was discovered in two unrelated Indian families in association with the development of HCM (Waldmuller et al. 2003). Since MYBPC3Δ25bp is familial with late-onset in aging and a mild form of HCM in carriers, the existence of unequivocal disease penetrance has been concluded. The study further determined that MYBPC3Δ25bp could be a canonical monogenic risk with low expressivity and penetrance. However, whole-genome DNA sequencing was not available at that time, and it was unclear whether the deletion was, in fact, both necessary and sufficient to cause HCM, or, indeed, whether other known or unidentified genetic or risk factors were at play (Waldmuller et al. 2003).
Following these initial investigations, a series of population-based surveys revealed a striking prevalence in the South Asian (SA) population (Table 1), estimated to be present in 6% and translating to ~ 100 million people worldwide (Dhandapany et al. 2009; Viswanathan et al. 2018) (Fig. 2). For example, Dhandapany and colleagues suggested MYBPC3Δ25bp prevalence around 2–6% in Southeast Asians. They further suggested that the higher incidence of cardiomyopathies in SAs, compared with other ethnicities, could be, at least partly, attributed to this polymorphism (Dhandapany et al. 2009; Dodani 2008; Gupta and Brister 2006; Jones et al. 2014; Omariba 2015; Uppaluri 2002). More recently, Kumar et al. (2016) studied the frequency of sarcomeric gene polymorphisms with left ventricular dysfunction (LVD) in patients with coronary artery disease (CAD). Contrary to other sarcomeric gene mutations, such as titin, troponin T type 2, and myospryn, they showed the MYBPC3Δ25bp polymorphism to be associated with an elevated risk of left ventricular pathologic remodeling and cardiac dysfunction post-myocardial infarction (Kumar et al. 2016; Srivastava et al. 2011). The association of MYBPC3 mutations with HCM and sudden death, the high prevalence of this mutation in the SA community, and preclinical studies revealing pathologic consequences of the MYBPC3 mutation all support ongoing effort to understand the molecular mechanisms underlying MYBPC3 mutation–mediated HCM. The objective of our review is to detail the association of MYBPC3Δ25bp and HCM/HF in SAs, describe the pathophysiology of HCM/HF in the setting of MYBPC3 mutations, and explore newly proposed diagnostic and therapeutic strategies. Overall, we aim to provide a constructive answer to the frequently posed question by SAs: “I recently found out through genetic testing (23andme) that I have one copy of the MYBPC3 mutation [25bp deletion]… should I be taking any precautions?”

Prevalence of MYBPC3Δ25bp worldwide. The frequency of MYBPC3Δ25bp distribution in various countries is shown as a percentage. Details are provided in Table 1 with the total number of samples screened by the country concerning the existing literature
Increased risk of cardiovascular disease in South Asians
At 1.8 billion people, SAs comprise approximately 20% of the world’s population. An estimated 3.5 million SAs live in the USA, constituting approximately 1% of the American population (Tang et al. 2012). Despite being only 20% of the world’s population, SAs represent about 60% of CVD cases worldwide (Kraker et al. 2016), suggesting a unique predisposition worthy of increased investigation. The World Health Organization (WHO) reports that CVD takes the lives of 17.9 million people every year, accounting for 31% of all deaths globally. Strikingly, out of those 17.9 million people, 13.6%, or 2.43 million, are of SA origin (Finegold et al. 2013; Volgman et al. 2018), including Sri Lanka, Nepal, Bangladesh, Bhutan, India, and Pakistan. Interestingly, the SA population has a higher risk of CVD than any other ethnic group (Gupta and Brister 2006). While researchers have advanced many causes to explain the high incidence of CVD among SAs, the literature has been circumspect and inconclusive, and, to date, no reports have pinpointed with precision the etiology of increased risk for CVD among SAs. MetS (metabolic syndrome) is a group of disease conditions of metabolism (Pan et al. 2008) associated with an increased risk for heart disease (Ram and Farmer 2012). The conditions associated with this designation are all risk factors for CVD (Kaur 2014), and they include hypertension, elevated triglyceride/HDL ratio, hyperinsulinemia, insulin resistance, abdominal adiposity, and dyslipidemia (Huang 2009; Pan et al. 2008; Ram and Farmer 2012). Around 20–25% of SAs are known to currently have MetS (Eapen et al. 2009), indicating that MetS could play an important pathogenic role in the susceptibility of SAs to CVD. A key component of MetS is insulin resistance (Eapen et al. 2009). When compared with Caucasians, SAs have notably been found to be at more risk for insulin resistance (Simmons et al. 1991), which is a key component of MetS (Eapen et al. 2009). Similarly, the high frequency of MYBPC3Δ25bp mutation among SAs (~ 6%) (Dhandapany et al. 2009; Simonson et al. 2010) (Viswanathan et al. 2018) is presumed to be a significant contributor to the high incidence of CVD, along with these secondary risk factors (Srivastava et al. 2011). The challenge is to determine if and how comorbidities, such as diabetes and hypertension, contribute to the ultimate phenotype and natural history of MYBPC3Δ25bp in a specific individual.
cMyBP-C is a regulator of contractility
MYBPC3 encodes cMyBP-C, a key structural protein of the heart muscle that interacts with myosin (Flavigny et al. 2003; Flavigny et al. 1999; Gruen and Gautel 1999; Shaffer et al. 2009), titin (Freiburg and Gautel 1996), and actin (Shaffer et al. 2009; Squire et al. 2003) to support sarcomeric integrity (Fig. 3). cMyBP-C was first discovered in the 1970s as a contaminant in a myosin preparation (Bennett et al. 1986; Craig and Offer 1976; Starr and Offer 1971). The gene encoding the cardiac isoform was later identified and characterized (Carrier et al. 1997; Carrier et al. 1993; Freiburg and Gautel 1996; Oakley et al. 2004). MYBPC3 is a single-copy, 24-kb gene, consisting of 35 codons encoding a 1274–amino acid and 140-kDa protein. Mutations involving the MYBPC3 gene cause significant cardiac disease at all ages (Barefield and Sadayappan 2010; Chung et al. 2003; Maron 1996; Van Driest et al. 2004). Experimental evidence, mainly resulting from in vitro protein-protein interaction studies, suggests that cMyBP-C may serve two functions: one as a molecular “ruler,” coordinating the spacing between thick and thin filaments, and one as a regulator of actomyosin interaction by association with the myosin II neck region (S2) and F-actin. cMyBP-C encodes multiple repeats of fibronectin type III–like and immunoglobulin (Ig)-like domains but differs from skeletal isoforms in that it contains an N-terminal C0 domain, a proline/alanine-rich linker between C0 and C1 and an ~ 100 amino acid segment between C1 and C2 Ig domains (the M-domain). The M-domain contains multiple serine residues that are conserved between mice and humans and are reversibly phosphorylated in response to systolic pressure and adrenergic stimulation (Kulikovskaya et al. 2003b), thereby modulating myofilament affinity (Kooij et al. 2013; Mun et al. 2011). Genetic profiles have been used to study how changes in cMyBP-C regulation at the molecular and cellular levels affect the sarcomere structure. However, the correlation between sarcomeric defects and organ-scale defects of myocyte organization remains an open area for investigation. The phosphorylation of cMyBP-C plays an important role in the regulation of cardiac mechanics (Rosas et al. 2015; Rosas et al. 2019; Tong et al. 2008; Tong et al. 2015). Sadayappan and colleagues have demonstrated that total cMyBP-C phosphorylation affects cardiac contractility, sarcomere organization, and the response to ischemia-reperfusion (I/R) injury (Sadayappan et al. 2009; Sadayappan et al. 2005; Sadayappan et al. 2006). In vitro experiments suggest that genetic mutations in cMyBP-C could alter interactions within the sarcomere, such as the α-tropomyosin and light meromyosin region of myosin (James and Robbins 2011; Okagaki et al. 1993), the S2 region of myosin (Gruen and Gautel 1999), F-actin (Colson et al. 2012; Kensler et al. 2011; Shaffer et al. 2009), and titin (Al-Khayat et al. 2013). Moreover, the M-domain, containing serine residues differentially phosphorylated by cAMP-dependent protein kinase (Gautel et al. 1995; Hartzell and Titus 1982) and endogenous calcium/calmodulin-dependent kinase (Hartzell and Glass 1984), may modulate contractility (Stelzer et al. 2006) through tethering of the S2 region of myosin (Weisberg and Winegrad 1996) and by modulation of actin-binding at the N-terminus (Kulikovskaya et al. 2003a; Razumova et al. 2006). Nuclear magnetic resonance studies suggest that the first 140 amino acids of the M-domain contain regulatory phosphoserines flanked by N-terminal charged residues. Phosphorylation of the M-domain leads to a transient helical structure, whereas a more stable trihelical structure comprises the C-terminal portion of the M-domain (Howarth et al. 2012).

cMyBP-C: structure, localization, and function illustrating the location of both MYBPC3Δ25bp and MYBPC3D389V variants. (a) MYBPC3 gene comprising 35 exons and 35 introns. (b) cMyBP-C codes for 1273 amino acids of cMyBP-C protein containing several domains. (c) cMyBP-C is located on 7–9 stripes of 43 nm spacing in each half of the A-band (cross-bridge bearing zone, C-region) of the sarcomere exclusively in cardiac myocytes. Both MYBPC3Δ25bp and MYBPC3D389V variants are indicated in the MYBPC3 gene and cMyBP-C protein. cMyBP-C is an important structural component of the sarcomere which plays a regulatory role in cardiac muscle function via interacting with actin, myosin, and titin. cMyBP-C, cardiac myosin-binding protein C; MYBPC3, cardiac myosin-binding protein C3 gene
As revealed in previously published in vitro results (Kulikovskaya et al. 2003a), the N-terminal region relative to the M-domain is required for the interaction between M-domain and F-actin. Together, these data support the hypothesis that mutations in the gene encoding cMyBP-C affect the signaling platform regulating actin and myosin and cause an altered contraction in the heart (Charron et al. 1998; Kooij et al. 2013; Niimura et al. 1998). In addition, we previously demonstrated in rats with induced myocardial infarction that the normal left ventricular architectural pattern is replaced by a largely disordered region, upon which a mesh-like network of orthogonally oriented myofibers is superimposed, extending from within the infarct to the septal and basal border zones. Addressing the underlying mechanism of this architectural disorder, the Sadayappan laboratory demonstrated that an N-terminal 40-kDa fragment of cMyBP-C during I-R injury is generated from calpain-dependent cleavage at 272-TSLAGAGRRTS, near a conserved protein kinase A phosphorylation motif, containing the cardiac-specific C0 domain (99 amino acids), C1 domain, and the first 17 residues of the M-domain (C0-C1f) (Govindan et al. 2012; Sadayappan and de Tombe 2012). Since C0-C1f lacks the phosphorylation sites for dynamic interplay involving actin and myosin S2, the presence of the cleaved product during I/R may be responsible for impaired sarcomere structure and function, leading to altered macroscopic myoarchitecture and impaired mechanics (Barefield et al. 2019; Hoffman et al. 2018; Taylor et al. 2016). Moreover, it is well known that I/R promotes a reduction in cMyBP-C phosphorylation, notably via a loss in triphosphorylated species (Sadayappan et al. 2006), and that cMyBP-C phosphorylation confers resistance to proteolysis (Barefield et al. 2019) while protecting against I/R injury (Sanada et al. 2004). Extending these concepts regarding the central role of cMyBP-C in affecting sarcomere structure, it is reasonable to postulate that the tissue architectural response to I/R would similarly be regulated by cMyBP-C and its degree of phosphorylation. Therefore, it is obvious that mutations in genes encoding for cMyBP-C would likely affect sarcomere structure, regulation, and function.
Potential mechanism of cardiomyopathies owing to MYBPC3Δ25bp
Currently, more than 350 disease-causing MYBPC3 mutations have been identified, and most are associated with HCM (Carrier et al. 2015; Dhandapany et al. 2009; Morimoto 2008; Stenson et al. 2014). Importantly, several founder mutations in different populations/countries were identified and reported, including Japan (Kubo et al. 2005), Finland (Jaaskelainen et al. 2013), Iceland (Adalsteinsdottir et al. 2014), France (Teirlinck et al. 2012), Spain (Oliva-Sandoval et al. 2010), the USA (Saltzman et al. 2010), the Netherlands (Alders et al. 2003) and MYBPC3Δ25bp from South Asia (Dhandapany et al. 2009). It is likely that such an effect existed in SA countries some 40,000 years ago (Dhandapany et al. 2009). Furthermore, the chance of developing HCM among MYBPC3Δ25bp carriers is increased by 5.3-fold odds ratios among independent subjects with HF (Dhandapany et al. 2009). This calls for research focused on discovering the pathophysiological mechanism of MYBPC3Δ25bp relative to cardiomyopathies. Interestingly, 70% of total MYBPC3 mutations are premature stop codons or frameshifts at the carboxyl region of cMyBP-C, leading to truncations lacking key myosin-binding residues. Truncations in domains C7 to C10 of the carboxyl region of cMyBP-C prevent its incorporation into the sarcomere (Barefield and Sadayappan 2010; Harris et al. 2011; Kuster and Sadayappan 2014). Other underlying mechanisms may include (1) “splicing defects,” an insufficient amount, or “haploinsufficiency,” of MYBPC3 product (Barefield et al. 2015; Marston et al. 2009); (2) a direct pathogenic effect of a mutant on typical myofilament function, i.e., “poison polypeptide effect” (Kuster et al. 2019); and (3) accumulation of misfolded proteins, i.e., “proteotoxicity” (Kuster et al. 2019) (Fig. 4). The possible mechanisms for MYBPC3Δ25bp pathogenesis remain uncertain, but evidence suggests the following three possibilities: (i) exon skipping and inclusion of novel amino acids in the coded protein, (ii) haploinsufficiency, or (iii) combination with other mutations on the same allele (Fig. 4). The following sections will discuss each of these mechanisms aiming to understand how MYBPC3Δ25bp could be pathogenic and cause contractile dysfunction.

Possible mechanisms for MYBPC3Δ25bp pathogenesis. Representation of various possible mechanisms of MYBPC3Δ25bp in cardiomyocytes with the strong probability of another unidentified phenomenon causing left ventricular hypertrophy (LVH), left ventricular dysfunction (LVD), HCM, and HF. Eight related mechanisms range from abnormal gene transcription to defective translated protein
Altered splicing and exon 33 skipping owing to MYBPC3Δ25bp could be pathogenic
The deletion in intron 32 was potentially expected to cause full skipping of the downstream exon 33, resulting in a reading frameshift, subsequently skipping the stop codon in exon 34. Translation then continued through exon 34 and a part of the 3′-UTR, finally stopping in 3′-UTR (Figs. 3 and 5) (Dhandapany et al. 2009; Waldmuller et al. 2003). In the mouse model, MYBPC3Δ25bp leads to the replacement of the last C-terminal 65 amino acids with 50 unique amino acids (cMyBP-C∆C10mut) (Kuster et al. 2015). In this way, MYBPC3Δ25bp creates a newly altered C10 domain sequence and translates cMyBP-C∆C10mut protein lacking exon 33 (Dhandapany et al. 2009; Waldmuller et al. 2003). Reports show that cMyBP-C∆C10mut cannot link cMyBP-C to the myosin LMM region, resulting in contractile dysfunction (Hossain et al. 2019; Kuster et al. 2015). Kumar et al. have studied models for cMyBP-C that depict the front and back helical views of cMyBP-C and cMyBP-C∆C10mut protein. Interestingly, cMyBP-C∆C10mut introduces β-pleated sheets and α-helix that might decrease the binding capacity of cMyBP-C to the myosin LMM region (Kumar et al. 2016). Since carboxyl domains of cMyBP-C directly bind to myosin, titin, and connectin to form normal sarcomere structure, conformational changes caused by cMyBP-C∆C10mut will affect these interactions, resulting in severe sarcomere disorganization and leading to structural and functional changes in cardiac muscle (Hossain et al. 2019; Kumar et al. 2016).

MYBPC3Δ25bp and MYBPC3Δ25bp/D389V variants and associated pathogenesis. (a) Schematic illustration showing transcription of non-carrier MYBPC3 allele coding normal cMyBP-C protein. (b) Intron 32 carrying MYBPC3Δ25bp mutation drives splicing defect causing exon 33 skipping and coding mutated C10 domain (cMyBP-CΔC10mut). (c) Similarly, an additional point mutation, i.e., D389V, along with MYBPC3Δ25bp (together represented as MYBPC3Δ25bp/D389V), is also presented. Such mutations drive abnormal MYBPC3 gene transcription and translation leading to diverse processes/pathways that have adverse effects on cardiomyocyte health that may result in cardiomyopathy phenotype
In earlier studies, the molecular consequence of MYBPC3Δ25bp was determined using neonatal cardiomyocytes in vitro and demonstrated that MYBPC3Δ25bp could skip exon 33 in the transcription (Waldmuller et al. 2003). In a follow-up study, the presence of exon 33 skipping in mRNA was confirmed using a biopsy from an HCM patient with MYBPC3Δ25bp (Dhandapany et al. 2009). Exon 33 skipping leads to a loss of 62 native cMyBP-C amino acids in the C10 domain (cMyBP-C∆C10mut). The C10 domain of cMyBP-C directly interacts with the myosin LMM region, which is critical for cMyBP-C anchoring, localization, and stabilization of the cardiac sarcomere. Furthermore, previous studies have established that modification of the C10 domain of cMyBP-C will result in the removal of mutant cMyBP-C since it is not incorporated in the sarcomere (McConnell et al. 1999). In addition, carboxyl domains C7 to C10 of cMyBP-C interact with titin, promoting cMyBP-C integration into the C-zone of the A-band. Kuster et al. (2019) have analyzed the effects of cMyBP-CΔC10mut overexpression in isolated cultured cardiomyocytes in vitro (Kuster et al. 2015) and in transgenic mouse hearts in vivo. Results showed that cMyBP-CΔC10mut expression is sufficient to cause the HCM phenotype. Altogether, these data suggest that the expression of cMyBP-CΔC10mut in cardiomyocytes results in the development of contractile dysfunction and HCM. Alternatively, it could also be hypothesized that the expression of cMyBP-C∆C10mut mRNA in cardiomyocytes would result in susceptibility to rapid degradation. This effect was previously shown in a mouse model where exon 31 was mutated such that the mutant RNA does not exist and if it does exist, it does not translate (McConnell et al. 1999). In that specific mouse model, a homozygous mutation of MYBPC3 in the C-terminus is guaranteed to cause cMyBP-C null, resulting in the HCM phenotype and possible sudden death at an early age (Zahka et al. 2008). On this basis, we propose that MYBPC3∆25bp could be pathogenic. However, we still do not know how MYBPC3∆25bp promotes HCM. Recent studies suggest that MYBPC3∆25bp may not be pathogenic in the context of HCM (Harper et al. 2020; Sadayappan et al. 2020; Viswanathan et al. 2018). However, we need a systematic study to determine whether MYBPC3∆25bp is or is not pathogenic (Hossain et al. 2019). One possibility is that the presence of MetS may aggravate the effect of MYBPC3∆25bp activation and cause HF (Sadayappan et al. 2020). Future research should consider questions such as (i) the factors that promote MYBPC3∆25bp to skip exon 33, (ii) exon 33 skipping under all circumstances or only in the presence of MetS, (iii) any genetic cofactors required to cause exon 33 skipping, and (iv) gender and age association with exon 33 skipping and the relationship with left ventricular dysfunction (Sadayappan et al. 2020).
Haploinsufficiency
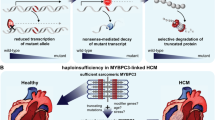
According to the National Institutes of Health, haploinsufficiency is a condition in which “one allele of a gene is inactivated or deleted, and the remaining second functional and normal allele of the gene is not adequate to produce a sufficient amount protein to preserve normal function.” In the case of one affected MYBPC3 allele in a truncated mutation, it appears that the truncated protein is never found in the heart (Rottbauer et al. 1997; van Dijk et al. 2009). Likewise, in patients with the MYBPC3Δ25bp, mutant cMyBP-C was not detected in the biopsies (Dhandapany et al. 2009). It is plausible that haploinsufficiency with the MYBPC3Δ25bp allele could reduce the total normal cMyBP-C protein level by the failure of the normal allele to fully compensate (Dhandapany et al. 2009). Based on a recent report, the relatively inadequate compensation induces a primary increase in calcium sensitivity which would explain the features observed in HCM patients, including an increased percentage of fractional shortening and ejection fraction, increased calcium transients, presence of myocardial hypertrophy and evidence of relaxation impairments, and increased cardiac remodeling causing myofibrillar disarray and/or life-threatening arrhythmias (Hossain et al. 2019).
The mechanism by which MYBPC3Δ25bp causes increased myofilament calcium sensitivity is unknown. Nonetheless, mouse models and HCM patient studies suggest that either poison polypeptide or haploinsufficiency of MYBPC3 would have such an effect (Fraysse et al. 2012; Kuster et al. 2019; Najafi et al. 2016; Sequeira et al. 2013). On the myofilament level, several different mechanisms are proposed to cause HCM, followed by HF and sudden cardiac death (SCD). For example, increased Ca2+ handling, inefficient ATP utilization, and accelerated mechanical force in combination could play a role in the development of HCM (Ashrafian et al. 2011; Huke and Knollmann 2010). In addition, mitochondrial energetic abnormalities could also cause increased reactive oxygen species and oxidative stress (Ashrafian et al. 2011; Brouwer et al. 2011), leading to pathologic cardiac remodeling associated with cardiomyopathies like HCM. In support of this mechanism, a recent study shows that cardiac troponin C, the calcium-binding protein in the myofilament, showed increased sensitivity to calcium, meaningless calcium generates more force, which would lead to prolonged muscle tension (Davis et al. 2016). Our group has determined another potential effect of cMyBP-C haploinsufficiency, namely, a reduction in the level of super-relaxed state myosin in which myosin heavy chain is in a low energy state and regulated by cMyBP-C (McNamara et al. 2017; McNamara et al. 2016). This can cause increased energy consumption and, hence, contribute to HCM pathophysiology. In support of this possibility, a recent study of mouse cardiomyocytes with cMyBP-C haploinsufficiency showed that treatment with the myosin inhibitor mavacamten improved super-relaxed myosin to contractile function (Toepfer et al. 2019b), suggesting a clear relationship between cMyBP-C haploinsufficiency and contractile dysfunction during HCM.
Poison polypeptide effect
Another possible pathological mechanism in HCM is the “poison polypeptide” effect, in which the mutant proteins, either missense or truncated, cause dysfunction through integration into the sarcomere (Knoll 2012). In the case of cMyBP-CΔC10mut, improper assembly of the cardiac sarcomere can occur, resulting in the development of contractile dysfunction and HCM (Kuster et al. 2019). Using transgenic mice expressing the cardiac-specific cMyBP-CΔC10mut protein, Kuster et al. demonstrated that cMyBP-CΔC10mut protein does not incorporate into its native sarcomeric C-zone. Instead, it preferentially localizes to the cytosol and Z-line. Increased toxic mutant and truncated proteins, if the poisoning process is operative, result in altered contractile function, thereby altering calcium handling and inducing pro-hypertrophic signaling in the cardiomyocytes that express cMyBP-CΔC10mut proteins. Both in vitro (Kuster et al. 2015) and in vivo (Kuster et al. 2019) experiments have concluded that cMyBP-CΔC10mut could be expressed in the cardiomyocytes, which would have turned into “poison polypeptide” effects on cardiomyocyte contractility. When cMyBP-CΔC10mut is expressed and localized improperly, it could alter the speed of contraction by regulating actomyosin interactions as the amino terminus of cMyBP-CΔC10mut is unaltered (Razzaque et al. 2013; Witayavanitkul et al. 2014). However, this hypothesis remains speculative by the failure of several studies to detect the mutant cMyBP-C proteins in the biopsies of patients with HCM (Jacques et al. 2008; Knoll 2012; van Dijk et al. 2009).
Proteotoxicity: inflammation and impaired ubiquitin-proteasome system
Several reports have suggested that MYBPC3 mutations could produce toxic effects and that truncated proteins induce inflammatory proteotoxicity and HCM (Bahrudin et al. 2011; Gupta et al. 2014; Schlossarek et al. 2014; Wang and Robbins 2014). More recently, Lipps et al. showed that cMyBP-C is degraded and released into myocardial tissue as a predominantly N-terminal 40-kDa fragment (C0C1f) with pro-inflammatory characteristics in the systemic circulation (Lipps et al. 2016). Alternatively, cMyBP-CΔC10mut could involve impairment of the ubiquitin-proteasome system (UPS) (Bahrudin et al. 2008; Sarikas et al. 2005). Eukaryotic cells use the lysosomal pathway (autophagy) (Bhuiyan et al. 2013; Maloyan and Robbins 2010; Pattison and Robbins 2011; Wang and Robbins 2014) and the UPS to remove unwanted proteins from the cytosol (Bahrudin et al. 2011; Carrier et al. 2010; Predmore et al. 2010; Sarikas et al. 2005; Schlossarek et al. 2014). In theory, the elevated levels of mutant protein could overwhelm and impair the UPS (Bahrudin et al. 2008; Schlossarek et al. 2012). Consistent with this notion, HCM patients with sarcomeric mutations have been observed with reduced proteasome activity compared with patients without the sarcomeric mutation (Predmore et al. 2010). Moreover, improper functioning of UPS may contribute to the disease process via (1) accumulation of malfunctioning proteins like cMyBP-C and (2) aggregated formation of unfolded proteins. Because cardiac UPS system efficiency is reduced with aging (Bulteau et al. 2002), we propose that disease penetrance in MYBPC3Δ25bp carriers is determined by the level of UPS impairment, which could be age-dependent (Dhandapany et al. 2009). It is also possible that MYBPC3 mutations destabilize the UPS system by accumulating more in the cytosol, further contributing to cardiomyocyte dysfunction (Sarikas et al. 2005). Finally, considering the well-described decline in process and pathways of the UPS with increasing oxidative stress and secondary risk factors, such as age (Bulteau et al. 2002; Okada et al. 1999), mutant cMyBP-C proteins may store in the cardiomyocyte, affect cellular homeostasis, activate autophagy, and cause LV dysfunction (Kumar et al. 2016). This is supported by recent studies in cMyBP-CΔC10mut transgenic mouse heart showing differential expression of several genes associated with proteasome function (Kuster et al. 2019), suggesting the direct association among MYBPC3Δ25bp, UPS impairment, and pathologic cardiac remodeling, leading to cardiomyopathy.
The endoplasmic reticulum (ER) and unfolded protein response (UPR) pathway regulate cellular homeostasis by mediating protein synthesis, folding, and maturation (Glembotski 2008; Liu and Dudley Jr 2014). The ER lumen contains three parallel signaling transmembrane sensor proteins, namely PRKR-like ER kinase (PERK), activating transcription factor 6 (ATF6), and inositol-requiring protein 1α (IRE1α), all of which detect signs of ER stress in the cytosol and selectively recognize the accumulation of unfolded and misfolded proteins (Groenendyk et al. 2010; Minamino and Kitakaze 2010; Minamino et al. 2010). In the absence of ER stress, glucose-regulated protein 78 (GRP78, or BiP) binds and inhibits these three pathways. However, when misfolded proteins accumulate, GRP78 translocates from the ER lumen by dissociating from PERK, IRE1α, and ATF6, thereby activating those pathways. Accumulation of unfolded and misfolded proteins in the cell results in ER stress and activates PERK by autophosphorylation. Activated PERK, in turn, phosphorylates eukaryotic initiation factor 2α (eIF2α), which reduces cellular translation by ATF4 and CHOP activation (Halliday et al. 2014; Kitakaze and Tsukamoto 2010; Thorp 2012). PERK activation promotes apoptosis by inhibiting translational machinery, inducing the pro-apoptotic proteins Bax and Bim, and preventing the function of antiapoptotic protein Bcl2. Conversely, the activation of IRE1α and ATF6 induces cell survival by upregulating ER chaperones. Furthermore, the accumulation of high-molecular-weight cMyBP-CΔC10 proteins in the cytoplasm might alter the proteolytic capacity of the UPS and limit the ability of proteasomes to degrade other substrates. If shown, the results could elucidate novel therapeutic targets to augment the UPR pathway, as well as proteasome activity, and maintain sarcomere integrity, thereby attenuating the numerous types of heart disease linked to proteotoxicity. Therefore, it is possible that preventing activation of the PERK-mediated pathway in cMyBP-CΔC10mut expression would prevent or rescue the HCM phenotype (Groenendyk et al. 2010).
Nonsense-mediated decay
Nonsense-mediated decay (NMD) is a cellular process in the cytosol that detects mRNA with nonsense mutations and prevents their translations to mutant proteins. Specifically, NMD degrades mRNAs with premature termination codons (PTCs). NMD performs a dual role as it not only degrades PTC-containing transcripts but also regulates the expression of normal transcripts. HCM-associated MYBPC3 mutations are predominantly premature stop codons. As such, the NMD pathway is the preferred pathway to remove them from the cellular process (Schlossarek et al. 2011; Vignier et al. 2009). Interestingly, using human-induced pluripotent stem cell–derived cardiomyocytes (hiPSC-CMs), Seeger et al. demonstrated that MYBPC3 PTC mutations involve abnormal calcium signaling and molecular dysregulation in the absence of significant haploinsufficiency of cMyBP-C protein (Seeger et al. 2019). Also, modulation of the cMyBP-C degradation pathway through therapeutic approaches may protect normal levels of cMyBP-C in HCM patients (Helms et al. 2020). Also, acute inhibition of NMD leads to a restoration of the HCM phenotype in hiPSC-CMs in vitro. Demonstration of such a direct relationship between NMD system activity and HCM development thus challenges haploinsufficiency or poison peptide mechanism as likely causes of MYBPC3 PTC-mediated HCM (Seeger et al. 2019). We believe that the NMD could remove and prevent mutant MYBPC3Δ25bp mRNA exon 33 skipping or alternative splicing to translate into cMyBP-CΔC10mut protein, leading to haploinsufficiency and causing contractile dysfunction, left ventricular hypertrophy, and HF.
Impact of MYBPC3Δ25bp with compound mutations
Not all carriers of the genetic variant MYBPC3Δ25bp show a cardiomyopathic phenotype (Dhandapany et al. 2009; Viswanathan et al. 2018). Cardiomyopathy is more likely and more pronounced when MYBPC3Δ25bp exists with other sarcomeric gene mutations (Bashyam et al. 2012; Tanjore et al. 2008; Waldmuller et al. 2003). For example, patients carrying both MYBPC3Δ25bp and another mutant in MYH7-E927del present with a severe HCM disease (Waldmuller et al. 2003). Likewise, when MYBPC3Δ25bp coexists with TNNT2 mutants, HCM appears to be more likely (Kumar et al. 2016). Although heterozygous MYBPC3Δ25bp carriers predominantly present with incomplete penetrance and develop HCM symptoms at late-onset (Dhandapany et al. 2009), a compound effect of MYBPC3Δ25bp with other sarcomeric gene mutations (termed “two-hit phenomenon”) can cause severe HCM and early sudden death (Marian 2001; Morita et al. 2008; Srivastava et al. 2011) and CAD (Kumar et al. 2016). Homozygous carriers of this MYBPC3Δ25bp variant, which is an example, in principle, of “two hits” can develop severe HCM phenotype and early clinical symptoms (Dhandapany et al. 2009).
Recent studies by our group show an association of cardiomyopathy/HCM with MYBPC3Δ25bp and MYBPC3D389V mutations in SA descendants (Viswanathan et al. 2018) (Fig. 5). SAs living in the USA have a ~ 6% prevalence of the MYBPC3Δ25bp variant (Viswanathan et al. 2018); ~ 9.6% of these individuals also harbor a unique missense mutation (aspartic acid (D) to valine (V) at codon 389 in cMyBP-C) represented as MYBPC3D389V. This variant is located at exon 12 which codes for the C2 domain of cMyBP-C. The C2 domain is a critical region of cMyBP-C that directly interacts with the S2 region of myosin in a phosphorylation-dependent manner and thereby regulates actomyosin and speed of contractions (Ratti et al. 2011). Interestingly, MYBPC3D389V coexists with the MYBPC3Δ25bp variant in the same allele. The combined impact of both mutations predicts changes in actin-myosin interaction, abnormal Ca2+ transients, and cardiac dysfunction (heart hyperdynamic state) predisposing to HCM/HF (Viswanathan et al. 2018). Because MYBPC3D389V is linked to HCM, it is proposed that MYBPC3D389V is translated and incorporated into the sarcomere to exert HCM. For instance, if a carrier of the MYBPC3Δ25bp variant also bears a mutation in MYH7, this grouping often results in sudden cardiac death (Dhandapany et al. 2009). Systematic studies are needed to define whether MYBPC3Δ25bp alone is sufficient to cause LV dysfunction, LVH, and HCM or whether a definite association with another mutation or a secondary risk factor is needed (Van Driest et al. 2004).
. Compound mutations with MYBPC3Δ25bp may also cause cardiac arrhythmias, leading to the likelihood of organ damage, organ failure, and SCD (Al-Khatib et al. 2018; Hunt et al. 2001). More than 4 million Americans suffer from recurrent arrhythmias, and this statistic is expected to increase as the population ages. Almost 40% of all patients seen in cardiology clinics, including one in four seen for the first time, present with cardiac arrhythmia or conduction defect (Vazquez Ruiz de Castroviejo et al. 2005). Current medical therapies for treating cardiac arrhythmias suppress neurohormonal activation first and then treat cardiac fluid volume overload and function. It has been proposed that antiarrhythmic drugs might slow the progression of HCM. Nevertheless, the prognosis for patients, including those receiving optimal treatments, remains poor (Bristow et al. 1996; Coats 2002; Colucci et al. 1996)These circumstances call for the development of novel therapies for cardiac arrhythmia (Bristow 2000; From 1998; Sabbah and Stanley 2002; Tang and Francis 2003). Cardiac arrhythmias such as tachycardia, ventricular and atrial fibrillation, and premature contractions) are the major causes of SCD in HF (Swaminathan et al. 2012). Arrhythmias also lead to the worsening of HF by decreasing the efficiency of the whole organ and disrupting the link between heart rate and cardiac output. Unfortunately, certain treatments for HF, such as inotropic drugs that change the force of the heart’s contractions, are contraindicated owing to increased SCD events (Krell et al. 1986), while conventional antiarrhythmic treatments, like Ca2+ channel blockers, cannot be used in HF owing to the worsening function of the failing myocardium with use of this class of drugs (Echt et al. 1991). Therefore, notwithstanding the association between cardiac arrhythmias and HF, it is difficult to strategize and develop novel therapies that can be used to treat both diseases without any side effects. In this process, the important first step is to identify novel therapeutic targets that can reduce HCM-induced cardiac arrhythmias and HF. Future studies should involve the elucidation of molecular mechanisms that cause the abnormal Ca2+ handling in HCM and SCD. We propose that the confluence of modifiers, including calcium handling proteins, such as phospholamban, sarco/endoplasmic reticulum Ca2+-ATPase, ryanodine receptor (RyR), or histidine-rich Ca2+-binding protein (HRC) with MYBPC3Δ25bp variant, could contribute to the development and manifestation of a severe cardiomyopathy phenotype and SCD, indicating that a more complex genetic architecture is involved in the disease (Wu et al. 2015).
Epigenetic changes and MYBPC3Δ25bp
Epigenetics means “on top of” or “in addition to” the traditional genetic features involved in gene regulation. Epigenetics involves DNA methylation, histone modifications, and RNA-mediated silencing. DNA methylation, such as methylcytosine and hydroxymethylcytosine, occurs in cytosine nucleotides at CpG sites specifically linked to guanine by a phosphate. Utilizing an MYBPC3 mutant mouse model of cardiomyopathy, Tabish et al. reported significant epigenetic changes within the introns of several cardiac genes associated with pathological cardiac remodeling and heart contractile dysfunction (Tabish et al. 2019). Based on these data, epigenetic changes in the intronic region of MYBPC3 may induce altered splicing causing exon skipping and the rise of MYBPC3Δ25bp variant. Similarly, some patients within the same family cohort carrying an MYBPC3Δ25bp variant show HCM phenotype at the age of 30 years, while other carriers have healthy hearts, even in old age (Dhandapany et al. 2009). Others and we have hypothesized that the difference in phenotype could be explained by the epigenetic impact of comorbidities, such as promoting exon skipping or increasing alternative splicing. In addition, it has been suggested that HCM is directly associated with hypertension (Karam et al. 1989), a condition well known to exert epigenetic effects (Arif et al. 2019). Thus, the MYBPC3Δ25bp variant–related occurrence of HCM phenotype in patients at different ages and MetS may be classified by differences in epigenetic maps that activate and promote exon 33 skipping and increase alternative splicing. Such a notion may also determine the mechanism behind the differences in severity and onset of HCM at different ages in MYBPC3Δ25bp carriers.
Diagnosis and innovative therapeutic approaches
As mentioned earlier, one of the key conundrums of research in MYBPC3Δ25bp is incomplete penetrance and variable expressivity (Dhandapany et al. 2009; Waldmuller et al. 2003). The challenge is, in part, addressed by improving screening methods to detect asymptomatic cardiac disease. Diagnosis of HCM using contemporary technologies, such as magnetic resonance imaging, PCR, and biomarkers, is still in its early stages.
Next-generation sequencing–based strategies
Recent advances in DNA sequencing, such as next-generation sequencing (NGS) screening for genetic variants, are robust and high-throughput (Gomez et al. 2014), resulting in the identification of many new variants, as well as establishing new HCM databases. Such resources would help physicians make timely diagnostic decisions and start treatment (McNally and Puckelwartz 2015). The future of precision medicine will rely on genetic testing, genetic counseling, and therapeutic decisions based on collaborative efforts. Whole-genome DNA sequencing allows for the identification of multiple mutations within the same individual. Given the phenotypic implications of the association of MYBPC3Δ25bp with other gene mutations, such as MYH7, TNNT2, or TNNI3, this constitutes a major advance. The importance is emphasized by the understanding that mutations in both MYBPC3 and MYH7 consist about of 80% of the identified HCM-associated mutations, while another 10% are derived from TNNI3 and TNNT2 genes (McNally et al. 2015; Morimoto 2008). Therefore, at least 90% of HCM cases caused by mutations in these four major sarcomeric genes can be diagnosed via the design of an NGS panel. In fact, recent technological developments enabled researchers to use NGS as a molecular diagnostic tool for HCM (Chen et al. 2019) and other cardiomyopathies (Daoud et al. 2019; Forleo et al. 2017). In 2016, Barefield and coworkers performed a pilot study to develop a variant-specific diagnostic assay and quickly screen for the presence of the MYBPC3Δ25bp variant using either a small volume of blood or saliva from a cohort of SAs (Barefield et al. 2016). The RNaseH-mediated qPCR assay was used to diagnose the MYBPC3Δ25bp gene-positive with specific RNaseH-blocked nucleotide primers. Such assay provides faster prescreening of SAs who are at a high risk for cardiomyopathies with MYBPC3Δ25bp for earlier clinical care. Likewise, other non-sarcomeric gene mutations, such as ACE and extracardiac disease, can also be associated with the onset of HCM (Kraker et al. 2016); nevertheless, this is beyond the scope of this review.
Gene-editing technologies
Gene-editing technologies can be used to cure genetically caused HCM (Strong and Musunuru 2017). Clustered, regularly interspaced, short palindromic repeats (CRISPR) and CRISPR-associated 9 (Cas9) can provide specific site-directed gene editing. These technologies can also correct individual and selective gene mutations. Once such a system is validated, it could be an ideal means to treat patients with a homozygous MYBPC3Δ25bp variant. Recent studies show that the MYBPC3 mutation can be corrected using the CRISPR/Cas9 in human pre-implanted embryos and that such non-carrier embryos, without evidence of off-target mutations, could be candidates for in vitro fertilization (Ma et al. 2017). However, in addition to possible ethical issues, it should be noted that the CRISPR/Cas9 system has some major technical limitations, e.g., ineffectiveness for repairing homozygous mutations or the possibility of off-targeting editing which affects other genes and thus induces secondary phenotypes.
Stem cell technologies: hiPSCs, spheroids, organoids, and engineered heart
Limitations in the availability of human cells and tissues present considerable obstacles to the study of human mutations. Furthermore, cardiomyocytes, a post-mitotic cell type, are almost impossible to maintain under culture conditions, leading to the inability to study such cells in vitro. A solution is a use of human-induced pluripotent stem cell (HiPSC) technology, a method that enables the use of renewable patient-specific biological material (Shi et al. 2017). The hiPSC system provides a unique opportunity to study genetic disease in vitro. These hiPSC-derived cardiomyocytes (hiPSC-CMs) are an ideal in vitro tool for testing drug cardiotoxicity, screening, and validation. HiPSCs can be differentiated into 2D cardiomyocytes (Kamdar et al. 2015), providing researchers the opportunity to study the importance of sarcomeric gene mutation and associated mechanisms of cardiomyopathy. For example, hiPSCs-CMs have been successfully utilized to study genetic diseases, including Pompe disease (Huang et al. 2011) and LEOPARD syndrome (Carvajal-Vergara et al. 2010). Similarly, the impact of MYBPC3 gene mutations has been studied in hiPSC-CMs (Helms et al. 2020; Ross et al. 2016; Seeger et al. 2019; Tanaka et al. 2014). However, HiPS-CMs have several limitations including incomplete reprogramming, an immature adult phenotype with embryonic genetic and epigenetic markers, variabilities, lack of t-tubules, and normal contractility (Knollmann 2013). Therefore, alternative and improved in vitro models are warranted. HiPS-CMs, cultured in a three-dimensional (3D) matrix architecture, termed as “spheroids” (Campbell et al. 2019) or “organoids” (Hoang et al. 2018), is another advancement in the field of cardiac tissue research. More importantly, these small 3D units under in vitro culture can be utilized as disease models (Ho et al. 2018; Mattapally et al. 2018). Another recent breakthrough in the field of cardiac research is the whole organ bioprinting using 3D tissue printing (Noor et al. 2019). Such pioneering research could allow cardiac studies to advance to a level that facilitates the investigation of cardiomyocyte and non-cardiomyocyte crosstalk and physiological function in a complex cardiac tissue–like environment. Recent developments in the use of hiPSCs differentiated into cardiomyocytes have allowed the fabrication of 3D heart muscle and cardiac organoids. Modern cell biology techniques have led to the generation of iPSC-derived cardiac organoids, resulting in a valuable approach toward assessing disease mechanisms and phenotype in vitro. Indeed, for recapitulating genetic diseases in Petri dishes, such as HCM, cardiac organoids may be essential. In particular, in vitro cardiac organogenesis using hiPSCs may lead to new opportunities for high-throughput modeling to study disease phenotype, determine disease mechanism, and perform preclinical studies to screen for therapeutic reagents. Such cardiac organoids may provide a tool with which to study organ development, cardiac function, and cardiac injury. HiPSCs-derived cardiac organoids may be considered a potential method to validate and assess disease phenotype and preclinical studies in a quick turnaround time and closely resemble human disease conditions, compared with hiPSC-CMs and mouse models (Marti-Figueroa and Ashton 2017; Silva et al. 2019). Organogenesis from hiPSCs may also provide new opportunities to study disease phenotype, define disease mechanisms, and help in screening for therapeutic reagents (Li et al. 2014). HiPSCs are now used to generate cardiomyocytes and engineer tissue, including 3D heart muscle and cardiac organoids (Mills et al. 2019; Mills et al. 2017). These cardiac organoids can act as a mechanical tool to study cardiac development, cardiac function, and cardiac injury (Moretti et al. 2013; Richards et al. 2017).
Gene therapies and pharmacological inhibitors to treat HCM
The percentage of HCM patients with MYBPC3 mutations is estimated to be ~ 40%, making it the most common HCM-associated gene. Importantly, 70% of all MYBPC3 mutations are predicted to result in incomplete, or truncated, proteins, that alter sarcomere structure and function and impact cardiac mechanics (Barefield et al. 2015; Harris et al. 2011; Kuster et al. 2015; Sadayappan and de Tombe 2014). Recent drug development has focused on direct manipulation of the contractile apparatus by affecting myosin activators (Cleland et al. 2006; Solaro 2009; Teerlink 2009), modification of single histidine buttons in cardiac troponin I (cTnI) (Day et al. 2006; Palpant et al. 2009), the introduction of therapeutic genes via the expression of full-length cMyBP-C (Mamidi et al. 2014; Mearini et al. 2014; Merkulov et al. 2012), or the removal of mutant myosin transcripts (Jiang et al. 2013). Mearini et al. used AAV9-mediated expression of full-length cMyBP-C to improve haploinsufficiency and affect polypeptide expression. However, this study was limited by the fact that AAV9-mediated cMyBP-C was administered in 1-day-old mice to prevent HCM, rather than testing in adult mice once the HCM phenotype had been established (Mearini et al. 2014), or partially rescued. Alternatively, in vitro systems, such as hiPSC-derived cardiomyocytes (Prondzynski et al. 2017) and engineered heart tissues (Dutsch et al. 2019), have been used to test the efficacy of gene therapy designed to improve MYBPC3 mutant–mediated contractile dysfunction; however, such systems require further in vivo validation using preclinical models (Prondzynski et al. 2019).
Direct targeting of contractile proteins, effectively bypassing receptor signaling, could lead to more promising therapeutic results (Day et al. 2006; James et al. 2005; Sadayappan et al. 2009; Solaro 2009; Teerlink 2009; Tissier et al. 2008). Unfortunately, most pharmacologic options available today at best only modestly improve symptoms associated with HF. However, Ho et al. have demonstrated that diltiazem, a calcium channel blocker used to treat hypertension, also prevents the development of HCM in patients with MYBPC3 mutations (Ho et al. 2015). A recently studied alternative to normalizing actomyosin interactions by manipulating signaling cascades is direct inactivation or activation of myosin (Mamidi et al. 2018). Such drugs might be prescribed to family members at risk at an earlier time before they develop a functional reduction of cardiac output. This approach has largely been directed toward improving cardiac function in inherited disease featuring diastolic dysfunction, namely restrictive cardiomyopathy (Davis et al. 2007) and HCM (Green et al. 2016; Kawas et al. 2017). Mavacamten, a myosin inhibitor, was established to treat HCM with left ventricular hypertrophy, disorganized sarcomere, fibrosis, and diastolic dysfunction. Early research has shown that chronic administration of mavacamten reduces hypercontractility in LVH, as well as disarray and fibrosis in the myocardium, and normalizes gene expression to rescue HCM in a mouse model (Green et al. 2016). In addition, Mamidi et al. demonstrated that Myk461-induced force reduction is regulated via cMyBP-C expression levels in the sarcomere (Mamidi et al. 2018). Mavacamten is a very promising drug to treat HCM and was recently approved for use in clinical trials (Phase 3 pivotal EXPLORER-HCM clinical trial, NCT03470545) for HCM (Heitner et al. 2019). Recently, mavacamten was also used to improve the hiPSC-CM function carrying a heterozygous MYBPC3 mutation in vitro (Toepfer et al. 2019a). Based on this information, molecular inhibitors seem to have the potential to target the HCM phenotype, yet more focused studies to design relevant future therapies are needed. Thus, direct manipulation of the contractile apparatus is now a focus on drug development and could have significant therapeutic advantages by bypassing receptor-ligand signaling pathways (Cleland et al. 2006).
Concluding remarks
In SAs, MYBPC3Δ25bp is specifically inherited with an increased, but poorly understood, risk of HCM, HF, and sudden death. The high prevalence of MYBPC3Δ25bp and associated risk of cardiomyopathy have prompted urgency for understanding pathophysiology as a means to improve outcomes. This review highlights some of the mechanisms that may explain the association of MYBPC3Δ25bp with HCM, HF, and sudden death. Also, we summarize some of the current tools and techniques that may be helpful for the development of precision medicine to address the clinical problem of HCM.
References
Adalsteinsdottir B et al (2014) Nationwide study on hypertrophic cardiomyopathy in Iceland: evidence of a MYBPC3 founder mutation. Circulation 130:1158–1167. https://doi.org/10.1161/CIRCULATIONAHA.114.011207
Alders M et al (2003) The 2373insG mutation in the MYBPC3 gene is a founder mutation, which accounts for nearly one-fourth of the HCM cases in the Netherlands. Eur Heart J 24:1848–1853
Al-Khatib SM et al (2018) 2017 AHA/ACC/HRS guideline for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: executive summary: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Rhythm Society. J Am Coll Cardiol 72:1677–1749. https://doi.org/10.1016/j.jacc.2017.10.053
Al-Khayat HA, Kensler RW, Squire JM, Marston SB, Morris EP (2013) Atomic model of the human cardiac muscle myosin filament. Proc Natl Acad Sci U S A 110:318–323. https://doi.org/10.1073/pnas.1212708110
Arif M, Sadayappan S, Becker RC, Martin LJ, Urbina EM (2019) Epigenetic modification: a regulatory mechanism in essential hypertension. Hypertens Res 42:1099–1113. https://doi.org/10.1038/s41440-019-0248-0
Ashrafian H, McKenna WJ, Watkins H (2011) Disease pathways and novel therapeutic targets in hypertrophic cardiomyopathy. Circ Res 109:86–96. https://doi.org/10.1161/CIRCRESAHA.111.242974
Bahrudin U et al (2008) Ubiquitin-proteasome system impairment caused by a missense cardiac myosin-binding protein C mutation and associated with cardiac dysfunction in hypertrophic cardiomyopathy. J Mol Biol 384:896–907. https://doi.org/10.1016/j.jmb.2008.09.070
Bahrudin U et al (2011) Impairment of ubiquitin-proteasome system by E334K cMyBPC modifies channel proteins, leading to electrophysiological dysfunction. J Mol Biol 413:857–878. https://doi.org/10.1016/j.jmb.2011.09.006
Barefield D, Sadayappan S (2010) Phosphorylation and function of cardiac myosin binding protein-C in health and disease. J Mol Cell Cardiol 48:866–875. https://doi.org/10.1016/j.yjmcc.2009.11.014
Barefield D, Kumar M, de Tombe PP, Sadayappan S (2014) Contractile dysfunction in a mouse model expressing a heterozygous MYBPC3 mutation associated with hypertrophic cardiomyopathy. Am J Physiol Heart Circ Physiol 306:H807–H815. https://doi.org/10.1152/ajpheart.00913.2013
Barefield D, Kumar M, Gorham J, Seidman JG, Seidman CE, de Tombe PP, Sadayappan S (2015) Haploinsufficiency of MYBPC3 exacerbates the development of hypertrophic cardiomyopathy in heterozygous mice. J Mol Cell Cardiol 79:234–243. https://doi.org/10.1016/j.yjmcc.2014.11.018
Barefield DY, Lynch TL, Jagadeesan A, Sanagala T, Sadayappan S (2016) High-throughput diagnostic assay for a highly prevalent cardiomyopathy-associated mybpc3 variant. J Mol Biomark Diagn 7. https://doi.org/10.4172/2155-9929.1000303
Barefield DY et al (2019) Ablation of the calpain-targeted site in cardiac myosin binding protein-C is cardioprotective during ischemia-reperfusion injury. J Mol Cell Cardiol 129:236–246. https://doi.org/10.1016/j.yjmcc.2019.03.006
Bashyam MD et al (2012) A low prevalence of MYH7/MYBPC3 mutations among familial hypertrophic cardiomyopathy patients in India. Mol Cell Biochem 360:373–382. https://doi.org/10.1007/s11010-011-1077-x
Bennett P, Craig R, Starr R, Offer G (1986) The ultrastructural location of C-protein, X-protein and H-protein in rabbit muscle. J Muscle Res Cell Motil 7:550–567. https://doi.org/10.1007/BF01753571
Bhuiyan MS et al (2013) Enhanced autophagy ameliorates cardiac proteinopathy. J Clin Invest 123:5284–5297. https://doi.org/10.1172/JCI70877
Bonne G et al (1995) Cardiac myosin binding protein-C gene splice acceptor site mutation is associated with familial hypertrophic cardiomyopathy. Nat Genet 11:438–440
Bristow M (2000) Etomoxir: a new approach to treatment of chronic heart failure. Lancet 356:1621–1622. https://doi.org/10.1016/S0140-6736(00)03149-4
Bristow MR et al (1996) Carvedilol produces dose-related improvements in left ventricular function and survival in subjects with chronic heart failure. MOCHA Investigators Circ 94:2807–2816
Brouwer WP, van Dijk SJ, Stienen GJ, van Rossum AC, van der Velden J, Germans T (2011) The development of familial hypertrophic cardiomyopathy: from mutation to bedside. Eur J Clin Investig 41:568–578. https://doi.org/10.1111/j.1365-2362.2010.02439.x
Bulteau AL, Szweda LI, Friguet B (2002) Age-dependent declines in proteasome activity in the heart. Arch Biochem Biophys 397:298–304. https://doi.org/10.1006/abbi.2001.2663
Campbell M, Chabria M, Figtree GA, Polonchuk L, Gentile C (2019) Stem cell-derived cardiac spheroids as 3D in vitro models of the human heart microenvironment methods. Mol Biol 2002:51–59. https://doi.org/10.1007/7651_2018_187
Carrier L et al (1993) Mapping of a novel gene for familial hypertrophic cardiomyopathy to chromosome 11. Nat Genet 4:311–313. https://doi.org/10.1038/ng0793-311
Carrier L et al (1997) Organization and sequence of human cardiac myosin binding protein C gene (MYBPC3) and identification of mutations predicted to produce truncated proteins in familial hypertrophic cardiomyopathy. Circ Res 80:427–434
Carrier L, Bonne G, Schwartz K (1998) Cardiac myosin-binding protein C and hypertrophic cardiomyopathy. Trends Cardiovasc Med 8:151–157. https://doi.org/10.1016/S1050-1738(97)00144-8
Carrier L, Schlossarek S, Willis MS, Eschenhagen T (2010) The ubiquitin-proteasome system and nonsense-mediated mRNA decay in hypertrophic cardiomyopathy. Cardiovasc Res 85:330–338. https://doi.org/10.1093/cvr/cvp247
Carrier L, Mearini G, Stathopoulou K, Cuello F (2015) Cardiac myosin-binding protein C (MYBPC3) in cardiac pathophysiology. Gene 573:188–197. https://doi.org/10.1016/j.gene.2015.09.008
Carvajal-Vergara X et al (2010) Patient-specific induced pluripotent stem-cell-derived models of LEOPARD syndrome. Nature 465:808–812. https://doi.org/10.1038/nature09005
Charron P et al (1998) Genotype-phenotype correlations in familial hypertrophic cardiomyopathy. A comparison between mutations in the cardiac protein-C and the beta-myosin heavy chain genes. Eur Heart J 19:139–145. https://doi.org/10.1053/euhj.1997.0575
Chen X, Jiang J, Zhu W, Wu Y, Su M (2019) Next-generation sequencing (NGS) as a molecular diagnostic tool for hypertrophic cardiomyopathy in a Chinese boy due to novel compound heterozygous mutations in the MYBPC3 gene: a case report. Medicine (Baltimore) 98:e14676. https://doi.org/10.1097/MD.0000000000014676
Chowdry AB, Mandegar MA, Benton GM, Naughton BT, Conklin BR (2012) Population Sampling and in vitro Modeling of a 25bp Deletion in MYBPC3 Associated With Hypertrophic Cardiomyopathy. https://blog.23andme.com/wp-content/uploads/2012/11/HCM-ASHG-TTAM.pdf
Chung MW, Tsoutsman T, Semsarian C (2003) Hypertrophic cardiomyopathy: from gene defect to clinical disease. Cell Res 13:9–20. https://doi.org/10.1038/sj.cr.7290146
Cleland JG, Coletta AP, Clark AL (2006) Clinical trials update from the Heart Failure Society of America meeting: FIX-CHF-4, selective cardiac myosin activator and OPT-CHF. Eur J Heart Fail 8:764–766. https://doi.org/10.1016/j.ejheart.2006.10.001
Coats AJ (2002) Angiotensin type-1 receptor blockers in heart failure. Prog Cardiovasc Dis 44:231–242
Colson BA, Rybakova IN, Prochniewicz E, Moss RL, Thomas DD (2012) Cardiac myosin binding protein-C restricts intrafilament torsional dynamics of actin in a phosphorylation-dependent manner. Proc Natl Acad Sci U S A 109:20437–20442. https://doi.org/10.1073/pnas.1213027109
Colucci WS et al (1996) Carvedilol inhibits clinical progression in patients with mild symptoms of heart failure US Carvedilol Heart Failure Study Group. Circulation 94:2800–2806
Craig R, Offer G (1976) The location of C-protein in rabbit skeletal muscle. Proc R Soc Lond B Biol Sci 192:451–461. https://doi.org/10.1098/rspb.1976.0023
Daoud H et al (2019) Genetic diagnostic testing for inherited cardiomyopathies: considerations for offering multi-gene tests in a health care setting. J Mol Diagn 21:437–448. https://doi.org/10.1016/j.jmoldx.2019.01.004
Davis J, Wen H, Edwards T, Metzger JM (2007) Thin filament disinhibition by restrictive cardiomyopathy mutant R193H troponin I induces Ca2+-independent mechanical tone and acute myocyte remodeling. Circ Res 100:1494–1502
Davis J et al (2016) A tension-based model distinguishes hypertrophic versus dilated cardiomyopathy. Cell 165:1147–1159. https://doi.org/10.1016/j.cell.2016.04.002
Day SM et al (2006) Histidine button engineered into cardiac troponin I protects the ischemic and failing heart. Nat Med 12:181–189. https://doi.org/10.1038/nm1346
Dhandapany PS et al (2009) A common MYBPC3 (cardiac myosin binding protein C) variant associated with cardiomyopathies in South Asia. Nat Genet 41:187–191. https://doi.org/10.1038/ng.309
Dodani S (2008) Excess coronary artery disease risk in South Asian immigrants: can dysfunctional high-density lipoprotein explain increased risk? Vasc Health Risk Manag 4:953–961
Dutsch A et al (2019) Phosphomimetic cardiac myosin-binding protein C partially rescues a cardiomyopathy phenotype in murine engineered heart tissue. Sci Rep 9:18152. https://doi.org/10.1038/s41598-019-54665-2
Eapen D, Kalra GL, Merchant N, Arora A, Khan BV (2009) Metabolic syndrome and cardiovascular disease in South Asians. Vasc Health Risk Manag 5:731–743
Echt DS et al (1991) Mortality and morbidity in patients receiving encainide, flecainide, or placebo The Cardiac Arrhythmia Suppression Trial. N Engl J Med 324:781–788. https://doi.org/10.1056/NEJM199103213241201
Finegold JA, Asaria P, Francis DP (2013) Mortality from ischaemic heart disease by country, region, and age: statistics from World Health Organisation and United Nations. Int J Cardiol 168:934–945. https://doi.org/10.1016/j.ijcard.2012.10.046
Flavigny J et al (1999) COOH-terminal truncated cardiac myosin-binding protein C mutants resulting from familial hypertrophic cardiomyopathy mutations exhibit altered expression and/or incorporation in fetal rat cardiomyocytes. J Mol Biol 294:443–456. https://doi.org/10.1006/jmbi.1999.3276
Flavigny J, Robert P, Camelin JC, Schwartz K, Carrier L, Berrebi-Bertrand I (2003) Biomolecular interactions between human recombinant beta-MyHC and cMyBP-Cs implicated in familial hypertrophic cardiomyopathy. Cardiovasc Res 60:388–396
Forleo C et al (2017) Targeted next-generation sequencing detects novel gene-phenotype associations and expands the mutational spectrum in cardiomyopathies. PLoS One 12:e0181842. https://doi.org/10.1371/journal.pone.0181842
Fraysse B et al (2012) Increased myofilament Ca2+ sensitivity and diastolic dysfunction as early consequences of Mybpc3 mutation in heterozygous knock-in mice. J Mol Cell Cardiol 52:1299–1307. https://doi.org/10.1016/j.yjmcc.2012.03.009
Freiburg A, Gautel M (1996) A molecular map of the interactions between titin and myosin-binding protein C. Implications for sarcomeric assembly in familial hypertrophic cardiomyopathy. Eur J Biochem 235:317–323. https://doi.org/10.1111/j.1432-1033.1996.00317.x
From AH (1998) Should manipulation of myocardial substrate utilization patterns be a component of the congestive heart failure therapeutic paradigm? J Card Fail 4:127–129
Gautel M, Zuffardi O, Freiburg A, Labeit S (1995) Phosphorylation switches specific for the cardiac isoform of myosin binding protein-C: a modulator of cardiac contraction? EMBO J 14:1952–1960
Geisterfer-Lowrance AA, Kass S, Tanigawa G, Vosberg HP, McKenna W, Seidman CE, Seidman JG (1990) A molecular basis for familial hypertrophic cardiomyopathy: a beta cardiac myosin heavy chain gene missense mutation. Cell 62:999–1006. https://doi.org/10.1016/0092-8674(90)90274-i
Gersh BJ et al (2011) 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: executive summary: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation 124:2761–2796. https://doi.org/10.1161/CIR.0b013e318223e230
Glembotski CC (2008) The role of the unfolded protein response in the heart. J Mol Cell Cardiol 44:453–459. https://doi.org/10.1016/j.yjmcc.2007.10.017
Gomez J et al (2014) Mutation analysis of the main hypertrophic cardiomyopathy genes using multiplex amplification and semiconductor next-generation sequencing. Circ J 78:2963–2971. https://doi.org/10.1253/circj.cj-14-0628
Govindan S et al (2012) Cardiac myosin binding protein-C is a potential diagnostic biomarker for myocardial infarction. J Mol Cell Cardiol 52:154–164. https://doi.org/10.1016/j.yjmcc.2011.09.011
Green EM et al (2016) A small-molecule inhibitor of sarcomere contractility suppresses hypertrophic cardiomyopathy in mice. Science 351:617–621. https://doi.org/10.1126/science.aad3456
Groenendyk J, Sreenivasaiah PK, Kaimdo H, Agellon LB, Michalak M (2010) Biology of endoplasmic reticulum stress in the heart. Circ Res 107:1185–1197. https://doi.org/10.1161/CIRCRESAHA.110.227033
Gruen M, Gautel M (1999) Mutations in beta-myosin S2 that cause familial hypertrophic cardiomyopathy (FHC) abolish the interaction with the regulatory domain of myosin-binding protein-C. J Mol Biol 286:933–949. https://doi.org/10.1006/jmbi.1998.2522
Gupta M, Brister S (2006) Is South Asian ethnicity an independent cardiovascular risk factor? Can J Cardiol 22:193–197
Gupta MK, Gulick J, Liu R, Wang X, Molkentin JD, Robbins J (2014) Sumo E2 enzyme UBC9 is required for efficient protein quality control in cardiomyocytes. Circ Res 115:721–729. https://doi.org/10.1161/CIRCRESAHA.115.304760
Halliday M, Radford H, Mallucci GR (2014) Prions: generation and spread versus neurotoxicity. J Biol Chem 289:19862–19868. https://doi.org/10.1074/jbc.R114.568477
Harper AR et al (2020) A re-evaluation of the South Asian MYBPC3(Delta25) intronic deletion in hypertrophic cardiomyopathy. Circ Genom Precis Med. https://doi.org/10.1161/CIRCGEN.119.002783
Harris SP, Lyons RG, Bezold KL (2011) In the thick of it: HCM-causing mutations in myosin binding proteins of the thick filament. Circ Res 108:751–764. https://doi.org/10.1161/CIRCRESAHA.110.231670
Hartzell HC, Glass DB (1984) Phosphorylation of purified cardiac muscle C-protein by purified cAMP-dependent and endogenous Ca2+-calmodulin-dependent protein kinases. J Biol Chem 259:15587–15596
Hartzell HC, Titus L (1982) Effects of cholinergic and adrenergic agonists on phosphorylation of a 165,000-Dalton myofibrillar protein in intact cardiac muscle. J Biol Chem 257:2111–2120
Heitner SB et al (2019) Mavacamten treatment for obstructive hypertrophic cardiomyopathy: a clinical trial. Ann Intern Med. https://doi.org/10.7326/M18-3016
Helms AS et al (2014) Sarcomere mutation-specific expression patterns in human hypertrophic cardiomyopathy. Circ Cardiovasc Genet 7:434–443. https://doi.org/10.1161/CIRCGENETICS.113.000448
Helms AS et al (2020) Effects of MYBPC3 loss-of-function mutations preceding hypertrophic cardiomyopathy. JCI Insight 5. https://doi.org/10.1172/jci.insight.133782
Hitomi N et al (2010) A frameshift deletion mutation in the cardiac myosin-binding protein C gene associated with dilated phase of hypertrophic cardiomyopathy and dilated cardiomyopathy. J Cardiol 56:189–196. https://doi.org/10.1016/j.jjcc.2010.04.003
Ho CY et al (2015) Diltiazem treatment for pre-clinical hypertrophic cardiomyopathy sarcomere mutation carriers: a pilot randomized trial to modify disease expression. JACC Heart Fail 3:180–188. https://doi.org/10.1016/j.jchf.2014.08.003
Ho BX, Pek NMQ, Soh BS (2018) Disease modeling using 3D organoids derived from human induced pluripotent stem cells. Int J Mol Sci 19. https://doi.org/10.3390/ijms19040936
Hoang P, Wang J, Conklin BR, Healy KE, Ma Z (2018) Generation of spatial-patterned early-developing cardiac organoids using human pluripotent stem cells. Nat Protoc 13:723–737. https://doi.org/10.1038/nprot.2018.006
Hoffman MP, Taylor EN, Aninwene GE 2nd, Sadayappan S, Gilbert RJ (2018) Assessing the multiscale architecture of muscular tissue with Q-space magnetic resonance imaging: review. Microsc Res Tech 81:162–170. https://doi.org/10.1002/jemt.22777
Hossain MB, Elbeck Z, Siga H, Knoll R (2019) Myosin binding protein C (MYBPC) and hypertrophic cardiomyopathy: role of altered C10 domain. Cardiovasc Res. https://doi.org/10.1093/cvr/cvz167
Howarth JW, Ramisetti S, Nolan K, Sadayappan S, Rosevear PR (2012) Structural insight into unique cardiac myosin-binding protein-C motif: a partially folded domain. J Biol Chem 287:8254–8262. https://doi.org/10.1074/jbc.M111.309591
Huang PL (2009) A comprehensive definition for metabolic syndrome. Dis Model Mech 2:231–237. https://doi.org/10.1242/dmm.001180
Huang HP et al (2011) Human Pompe disease-induced pluripotent stem cells for pathogenesis modeling, drug testing and disease marker identification. Hum Mol Genet 20:4851–4864. https://doi.org/10.1093/hmg/ddr424
Huke S, Knollmann BC (2010) Increased myofilament Ca2+-sensitivity and arrhythmia susceptibility. J Mol Cell Cardiol 48:824–833. https://doi.org/10.1016/j.yjmcc.2010.01.011
Hunt SA et al. (2001) ACC/AHA guidelines for the evaluation and management of chronic heart failure in the adult: executive summary A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Committee to Revise the 1995 Guidelines for the Evaluation and Management of Heart Failure): Developed in Collaboration With the International Society for Heart and Lung Transplantation; Endorsed by the Heart Failure Society of America Circulation 104:2996–3007
Jaaskelainen P et al (2013) Two founder mutations in the alpha-tropomyosin and the cardiac myosin-binding protein C genes are common causes of hypertrophic cardiomyopathy in the Finnish population. Ann Med 45:85–90. https://doi.org/10.3109/07853890.2012.671534
Jacques A, Hoskins AC, Kentish JC, Marston SB (2008) From genotype to phenotype: a longitudinal study of a patient with hypertrophic cardiomyopathy due to a mutation in the MYBPC3 gene. J Muscle Res Cell Motil 29:239–246. https://doi.org/10.1007/s10974-009-9174-0
James J, Robbins J (2011) Signaling and myosin-binding protein C. J Biol Chem 286:9913–9919. https://doi.org/10.1074/jbc.R110.171801
James J et al (2005) Forced expression of alpha-myosin heavy chain in the rabbit ventricle results in cardioprotection under cardiomyopathic conditions. Circulation 111:2339–2346. https://doi.org/10.1161/01.CIR.0000164233.09448.B1
Jiang J, Wakimoto H, Seidman JG, Seidman CE (2013) Allele-specific silencing of mutant Myh6 transcripts in mice suppresses hypertrophic cardiomyopathy. Science 342:111–114. https://doi.org/10.1126/science.1236921
Jones DA et al (2014) Mortality in South Asians and Caucasians after percutaneous coronary intervention in the United Kingdom: an observational cohort study of 279,256 patients from the BCIS (British Cardiovascular Intervention Society) National Database. JACC Cardiovasc Interv 7:362–371. https://doi.org/10.1016/j.jcin.2013.11.013
Kamdar F, Klaassen Kamdar A, Koyano-Nakagawa N, Garry MG, Garry DJ (2015) Cardiomyopathy in a dish: using human inducible pluripotent stem cells to model inherited cardiomyopathies. J Card Fail 21:761–770. https://doi.org/10.1016/j.cardfail.2015.04.010
Karam R, Lever HM, Healy BP (1989) Hypertensive hypertrophic cardiomyopathy or hypertrophic cardiomyopathy with hypertension? A study of 78 patients. J Am Coll Cardiol 13:580–584. https://doi.org/10.1016/0735-1097(89)90596-2
Kaur J (2014) A comprehensive review on metabolic syndrome. Cardiol Res Pract 2014:943162. https://doi.org/10.1155/2014/943162
Kawas RF, Anderson RL, Ingle SRB, Song Y, Sran AS, Rodriguez HM (2017) A small-molecule modulator of cardiac myosin acts on multiple stages of the myosin chemomechanical cycle. J Biol Chem 292:16571–16577. https://doi.org/10.1074/jbc.M117.776815
Kensler RW, Shaffer JF, Harris SP (2011) Binding of the N-terminal fragment C0-C2 of cardiac MyBP-C to cardiac F-actin. J Struct Biol 174:44–51. https://doi.org/10.1016/j.jsb.2010.12.003
Kitakaze M, Tsukamoto O (2010) What is the role of ER stress in the heart? Introduction and series overview. Circ Res 107:15–18. https://doi.org/10.1161/CIRCRESAHA.110.222919
Knoll R (2012) Myosin binding protein C: implications for signal-transduction. J Muscle Res Cell Motil 33:31–42. https://doi.org/10.1007/s10974-011-9281-6
Knollmann BC (2013) Induced pluripotent stem cell-derived cardiomyocytes: boutique science or valuable arrhythmia model? Circ Res 112:969–976; discussion 976. https://doi.org/10.1161/CIRCRESAHA.112.300567
Kooij V, Holewinski RJ, Murphy AM, Van Eyk JE (2013) Characterization of the cardiac myosin binding protein-C phosphoproteome in healthy and failing human hearts. J Mol Cell Cardiol 60:116–120. https://doi.org/10.1016/j.yjmcc.2013.04.012
Kraker J, Viswanathan SK, Knoll R, Sadayappan S (2016) Recent advances in the molecular genetics of familial hypertrophic cardiomyopathy in South Asian descendants. Front Physiol 7:499. https://doi.org/10.3389/fphys.2016.00499
Krell MJ et al (1986) Intermittent, ambulatory dobutamine infusions in patients with severe congestive heart failure. Am Heart J 112:787–791
Kubo T et al (2005) Lifelong left ventricular remodeling of hypertrophic cardiomyopathy caused by a founder frameshift deletion mutation in the cardiac myosin-binding protein C gene among Japanese. J Am Coll Cardiol 46:1737–1743. https://doi.org/10.1016/j.jacc.2005.05.087
Kulikovskaya I, McClellan G, Flavigny J, Carrier L, Winegrad S (2003a) Effect of MyBP-C binding to actin on contractility in heart muscle. J Gen Physiol 122:761–774. https://doi.org/10.1085/jgp.200308941
Kulikovskaya I, McClellan G, Levine R, Winegrad S (2003b) Effect of extraction of myosin binding protein C on contractility of rat heart. Am J Physiol Heart Circ Physiol 285:H857–H865. https://doi.org/10.1152/ajpheart.00841.2002
Kumar S et al (2016) Role of common sarcomeric gene polymorphisms in genetic susceptibility to left ventricular dysfunction. J Genet 95:263–272
Kuster DW, Sadayappan S (2014) MYBPC3’s alternate ending: consequences and therapeutic implications of a highly prevalent 25 bp deletion mutation. Pflugers Arch 466:207–213. https://doi.org/10.1007/s00424-013-1417-7
Kuster DW, Govindan S, Springer TI, Martin JL, Finley NL, Sadayappan S (2015) A hypertrophic cardiomyopathy-associated MYBPC3 mutation common in populations of South Asian descent causes contractile dysfunction. J Biol Chem 290:5855–5867. https://doi.org/10.1074/jbc.M114.607911
Kuster DWD et al (2019) Altered C10 domain in cardiac myosin binding protein-C results in hypertrophic cardiomyopathy. Cardiovasc Res. https://doi.org/10.1093/cvr/cvz111
Li Y, Xu C, Ma T (2014) In vitro organogenesis from pluripotent stem cells. Organogenesis 10:159–163. https://doi.org/10.4161/org.28918
Lipps C et al (2016) N-terminal fragment of cardiac myosin binding protein-C triggers pro-inflammatory responses in vitro. J Mol Cell Cardiol 99:47–56. https://doi.org/10.1016/j.yjmcc.2016.09.003
Liu M, Dudley SC Jr (2014) Targeting the unfolded protein response in heart diseases. Expert Opin Ther Targets 18:719–723. https://doi.org/10.1517/14728222.2014.918605
Ma H et al (2017) Correction of a pathogenic gene mutation in human embryos. Nature 548:413–419. https://doi.org/10.1038/nature23305
MacRae CA et al (1995) Familial hypertrophic cardiomyopathy with Wolff-Parkinson-White syndrome maps to a locus on chromosome 7q3. J Clin Invest 96:1216–1220. https://doi.org/10.1172/JCI118154
Maloyan A, Robbins J (2010) Autophagy in desmin-related cardiomyopathy: thoughts at the halfway point. Autophagy 6:665–666. https://doi.org/10.1161/CIRCRESAHA.109.212639
Mamidi R, Li J, Gresham KS, Stelzer JE (2014) Cardiac myosin binding protein-C: a novel sarcomeric target for gene therapy. Pflugers Arch 466:225–230. https://doi.org/10.1007/s00424-013-1412-z
Mamidi R, Li J, Doh CY, Verma S, Stelzer JE (2018) Impact of the myosin modulator mavacamten on force generation and cross-bridge behavior in a murine model of hypercontractility. J Am Heart Assoc 7:e009627. https://doi.org/10.1161/JAHA.118.009627
Marian AJ (2001) On genetic and phenotypic variability of hypertrophic cardiomyopathy: nature versus nurture. J Am Coll Cardiol 38:331–334. https://doi.org/10.1016/s0735-1097(01)01389-4
Maron BJ (1996) Triggers for sudden cardiac death in the athlete. Cardiol Clin 14:195–210. https://doi.org/10.1016/s0733-8651(05)70273-3
Marston S et al (2009) Evidence from human myectomy samples that MYBPC3 mutations cause hypertrophic cardiomyopathy through haploinsufficiency. Circ Res 105:219–222. https://doi.org/10.1161/CIRCRESAHA.109.202440
Marti-Figueroa CR, Ashton RS (2017) The case for applying tissue engineering methodologies to instruct human organoid morphogenesis. Acta Biomater 54:35–44. https://doi.org/10.1016/j.actbio.2017.03.023
Mattapally S et al (2018) Spheroids of cardiomyocytes derived from human-induced pluripotent stem cells improve recovery from myocardial injury in mice. Am J Physiol Heart Circ Physiol 315:H327–H339. https://doi.org/10.1152/ajpheart.00688.2017
McConnell BK et al (1999) Dilated cardiomyopathy in homozygous myosin-binding protein-C mutant mice. J Clin Invest 104:1235–1244. https://doi.org/10.1172/JCI7377
McNally EM, Puckelwartz MJ (2015) Genetic variation in cardiomyopathy and cardiovascular disorders. Circ J 79:1409–1415. https://doi.org/10.1253/circj.CJ-15-0536
McNally EM, Barefield DY, Puckelwartz MJ (2015) The genetic landscape of cardiomyopathy and its role in heart failure. Cell Metab 21:174–182. https://doi.org/10.1016/j.cmet.2015.01.013
McNamara JW et al (2016) Ablation of cardiac myosin binding protein-C disrupts the super-relaxed state of myosin in murine cardiomyocytes. J Mol Cell Cardiol 94:65–71. https://doi.org/10.1016/j.yjmcc.2016.03.009
McNamara JW et al (2017) MYBPC3 mutations are associated with a reduced super-relaxed state in patients with hypertrophic cardiomyopathy. PLoS One 12:e0180064. https://doi.org/10.1371/journal.pone.0180064
Mearini G et al (2014) Mybpc3 gene therapy for neonatal cardiomyopathy enables long-term disease prevention in mice. Nat Commun 5:5515. https://doi.org/10.1038/ncomms6515
Merkulov S, Chen X, Chandler MP, Stelzer JE (2012) In vivo cardiac myosin binding protein-C gene transfer rescues myofilament contractile dysfunction in cardiac myosin binding protein-C null mice. Circ Heart Fail 5:635–644. https://doi.org/10.1161/CIRCHEARTFAILURE.112.968941
Michels M et al (2009) Disease penetrance and risk stratification for sudden cardiac death in asymptomatic hypertrophic cardiomyopathy mutation carriers. Eur Heart J. https://doi.org/10.1093/eurheartj/ehp306
Mills RJ et al (2017) Functional screening in human cardiac organoids reveals a metabolic mechanism for cardiomyocyte cell cycle arrest. Proc Natl Acad Sci U S A 114:E8372–E8381. https://doi.org/10.1073/pnas.1707316114
Mills RJ et al (2019) Drug screening in human PSC-cardiac organoids identifies pro-proliferative compounds acting via the mevalonate pathway. Cell Stem Cell 24:895–907 e896. https://doi.org/10.1016/j.stem.2019.03.009
Minamino T, Kitakaze M (2010) ER stress in cardiovascular disease. J Mol Cell Cardiol 48:1105–1110. https://doi.org/10.1016/j.yjmcc.2009.10.026
Minamino T, Komuro I, Kitakaze M (2010) Endoplasmic reticulum stress as a therapeutic target in cardiovascular disease. Circ Res 107:1071–1082. https://doi.org/10.1161/CIRCRESAHA.110.227819
Moretti A, Laugwitz KL, Dorn T, Sinnecker D, Mummery C (2013) Pluripotent stem cell models of human heart disease. Cold Spring Harb Perspect Med:3. https://doi.org/10.1101/cshperspect.a014027
Morimoto S (2008) Sarcomeric proteins and inherited cardiomyopathies. Cardiovasc Res 77:659–666 doi:https://doi.org/10.1093/cvr/cvm084
Morita H et al (2008) Shared genetic causes of cardiac hypertrophy in children and adults. N Engl J Med 358:1899–1908. https://doi.org/10.1056/NEJMoa075463
Mun JY, Gulick J, Robbins J, Woodhead J, Lehman W, Craig R (2011) Electron microscopy and 3D reconstruction of F-actin decorated with cardiac myosin-binding protein C (cMyBP-C). J Mol Biol 410:214–225. https://doi.org/10.1016/j.jmb.2011.05.010
Murphy RT et al (2005) Adenosine monophosphate-activated protein kinase disease mimicks hypertrophic cardiomyopathy and Wolff-Parkinson-White syndrome: natural history. J Am Coll Cardiol 45:922–930. https://doi.org/10.1016/j.jacc.2004.11.053
Najafi A et al (2016) Selective phosphorylation of PKA targets after beta-adrenergic receptor stimulation impairs myofilament function in Mybpc3-targeted HCM mouse model. Cardiovasc Res 110:200–214. https://doi.org/10.1093/cvr/cvw026
Niimura H et al (1998) Mutations in the gene for cardiac myosin-binding protein C and late-onset familial hypertrophic cardiomyopathy. N Engl J Med 338:1248–1257. https://doi.org/10.1056/NEJM199804303381802
Niimura H, Patton KK, McKenna WJ, Soults J, Maron BJ, Seidman JG, Seidman CE (2002) Sarcomere protein gene mutations in hypertrophic cardiomyopathy of the elderly. Circulation 105:446–451
Noor N, Shapira A, Edri R, Gal I, Wertheim L, Dvir T (2019) 3D printing of personalized thick and perfusable cardiac patches and hearts. Adv Sci (Weinh) 6:1900344. https://doi.org/10.1002/advs.201900344
Oakley CE, Hambly BD, Curmi PM, Brown LJ (2004) Myosin binding protein C: structural abnormalities in familial hypertrophic cardiomyopathy. Cell Res 14:95–110. https://doi.org/10.1038/sj.cr.7290208
Okada K, Wangpoengtrakul C, Osawa T, Toyokuni S, Tanaka K, Uchida K (1999) 4-Hydroxy-2-nonenal-mediated impairment of intracellular proteolysis during oxidative stress. Identification of proteasomes as target molecules. J Biol Chem 274:23787–23793. https://doi.org/10.1074/jbc.274.34.23787
Okagaki T, Weber FE, Fischman DA, Vaughan KT, Mikawa T, Reinach FC (1993) The major myosin-binding domain of skeletal muscle MyBP-C (C protein) resides in the COOH-terminal, immunoglobulin C2 motif. J Cell Biol 123:619–626. https://doi.org/10.1083/jcb.123.3.619
Oliva-Sandoval MJ et al (2010) Insights into genotype-phenotype correlation in hypertrophic cardiomyopathy. Findings from 18 Spanish families with a single mutation in MYBPC3. Heart 96:1980–1984. https://doi.org/10.1136/hrt.2010.200402
Omariba DW (2015) Immigration, ethnicity, and avoidable mortality in Canada, 1991-2006. Ethn Health:1–28. https://doi.org/10.1080/13557858.2014.995155
Page SP et al (2012) Cardiac myosin binding protein-C mutations in families with hypertrophic cardiomyopathy: disease expression in relation to age, gender, and long term outcome. Circ Cardiovasc Genet 5:156–166. https://doi.org/10.1161/CIRCGENETICS.111.960831
Palpant NJ, D'Alecy LG, Metzger JM (2009) Single histidine button in cardiac troponin I sustains heart performance in response to severe hypercapnic respiratory acidosis in vivo. FASEB J 23:1529–1540. https://doi.org/10.1096/fj.08-121996
Pan WH, Yeh WT, Weng LC (2008) Epidemiology of metabolic syndrome in Asia Asia. Pac J Clin Nutr 17(Suppl 1):37–42
Pattison JS, Robbins J (2011) Autophagy and proteotoxicity in cardiomyocytes. Autophagy 7:1259–1260. https://doi.org/10.4161/auto.7.10.16882
Predmore JM et al (2010) Ubiquitin proteasome dysfunction in human hypertrophic and dilated cardiomyopathies. Circulation 121:997–1004. https://doi.org/10.1161/CIRCULATIONAHA.109.904557
Previs MJ, Beck Previs S, Gulick J, Robbins J, Warshaw DM (2012) Molecular mechanics of cardiac myosin-binding protein C in native thick filaments. Science 337:1215–1218. https://doi.org/10.1126/science.1223602
Prondzynski M et al (2017) Evaluation of MYBPC3 trans-splicing and gene replacement as therapeutic options in human iPSC-derived cardiomyocytes. Mol Ther Nucleic Acids 7:475–486. https://doi.org/10.1016/j.omtn.2017.05.008
Prondzynski M, Mearini G, Carrier L (2019) Gene therapy strategies in the treatment of hypertrophic cardiomyopathy. Pflugers Arch 471:807–815. https://doi.org/10.1007/s00424-018-2173-5
Ram CV, Farmer JA (2012) Metabolic syndrome in South Asians. J Clin Hypertens (Greenwich) 14:561–565. https://doi.org/10.1111/j.1751-7176.2012.00652.x
Ratti J, Rostkova E, Gautel M, Pfuhl M (2011) Structure and interactions of myosin-binding protein C domain C0: cardiac-specific regulation of myosin at its neck? J Biol Chem 286:12650–12658. https://doi.org/10.1074/jbc.M110.156646
Razumova MV, Shaffer JF, Tu AY, Flint GV, Regnier M, Harris SP (2006) Effects of the N-terminal domains of myosin binding protein-C in an in vitro motility assay: evidence for long-lived cross-bridges. J Biol Chem 281:35846–35854. https://doi.org/10.1074/jbc.M606949200
Razzaque MA, Gupta M, Osinska H, Gulick J, Blaxall BC, Robbins J (2013) An endogenously produced fragment of cardiac myosin-binding protein C is pathogenic and can lead to heart failure. Circ Res 113:553–561. https://doi.org/10.1161/CIRCRESAHA.113.301225
Richards DJ et al (2017) Inspiration from heart development: biomimetic development of functional human cardiac organoids. Biomaterials 142:112–123. https://doi.org/10.1016/j.biomaterials.2017.07.021
Rosas PC et al (2015) Phosphorylation of cardiac myosin-binding protein-C is a critical mediator of diastolic function. Circ Heart Fail 8:582–594. https://doi.org/10.1161/CIRCHEARTFAILURE.114.001550
Rosas PC, Warren CM, Creed HA, Trzeciakowski JP, Solaro RJ, Tong CW (2019) Cardiac myosin binding protein-C phosphorylation mitigates age-related cardiac dysfunction: hope for better aging? JACC Basic Transl Sci 4:817–830. https://doi.org/10.1016/j.jacbts.2019.06.003
Ross SB, Fraser ST, Semsarian C (2016) Induced pluripotent stem cells in the inherited cardiomyopathies: from disease mechanisms to novel therapies. Trends Cardiovasc Med 26:663–672. https://doi.org/10.1016/j.tcm.2016.05.001
Rottbauer W et al (1997) Novel splice donor site mutation in the cardiac myosin-binding protein-C gene in familial hypertrophic cardiomyopathy. Characterization of cardiac transcript and protein. J Clin Invest 100:475–482. https://doi.org/10.1172/JCI119555
Sabbah HH, Stanley WC (2002) Partial fatty acid oxidation inhibitors: a potentially new class of drugs for heart failure. Eur J Heart Fail 4:3–6
Sadayappan S, de Tombe PP (2012) Cardiac myosin binding protein-C: redefining its structure and function. Biophys Rev 4:93–106. https://doi.org/10.1007/s12551-012-0067-x
Sadayappan S, de Tombe PP (2014) Cardiac myosin binding protein-C as a central target of cardiac sarcomere signaling: a special mini review series. Pflugers Arch 466:195–200. https://doi.org/10.1007/s00424-013-1396-8
Sadayappan S et al (2005) Cardiac myosin-binding protein-C phosphorylation and cardiac function. Circ Res 97:1156–1163. https://doi.org/10.1161/01.RES.0000190605.79013.4d
Sadayappan S et al (2006) Cardiac myosin binding protein C phosphorylation is cardioprotective. Proc Natl Acad Sci U S A 103:16918–16923. https://doi.org/10.1073/pnas.0607069103
Sadayappan S, Gulick J, Klevitsky R, Lorenz JN, Sargent M, Molkentin JD, Robbins J (2009) Cardiac myosin binding protein-C phosphorylation in a {beta}-myosin heavy chain background. Circulation 119:1253–1262. https://doi.org/10.1161/CIRCULATIONAHA.108.798983
Sadayappan S, Puckelwartz MJ, McNally EM (2020) South Asian-specific MYBPC3(Delta25bp) intronic deletion and its role in cardiomyopathies and heart failure. Circ Genom Precis Med 13:e002986. https://doi.org/10.1161/CIRCGEN.120.002986
Sakthivel S et al (2001) Novel mutations in MYH7 and MYBPC3 of an Indian family causing hypertrophic cardiomyopathy. J Mol Cell Cardiol 33:A105 (Abstract)
Saltzman AJ et al (2010) Short communication: the cardiac myosin binding protein C Arg502Trp mutation: a common cause of hypertrophic cardiomyopathy. Circ Res 106:1549–1552. https://doi.org/10.1161/CIRCRESAHA.109.216291
Sanada S et al (2004) Protein kinase A as another mediator of ischemic preconditioning independent of protein kinase C. Circulation 110:51–57. https://doi.org/10.1161/01.CIR.0000133390.12306.C7
Sarikas A et al (2005) Impairment of the ubiquitin-proteasome system by truncated cardiac myosin binding protein C mutants. Cardiovasc Res 66:33–44. https://doi.org/10.1016/j.cardiores.2005.01.004
Schlossarek S, Mearini G, Carrier L (2011) Cardiac myosin-binding protein C in hypertrophic cardiomyopathy: mechanisms and therapeutic opportunities. J Mol Cell Cardiol 50:613–620. https://doi.org/10.1016/j.yjmcc.2011.01.014
Schlossarek S, Englmann DR, Sultan KR, Sauer M, Eschenhagen T, Carrier L (2012) Defective proteolytic systems in Mybpc3-targeted mice with cardiac hypertrophy. Basic Res Cardiol 107:235. https://doi.org/10.1007/s00395-011-0235-3
Schlossarek S, Singh SR, Geertz B, Schulz H, Reischmann S, Hubner N, Carrier L (2014) Proteasome inhibition slightly improves cardiac function in mice with hypertrophic cardiomyopathy. Front Physiol 5:484. https://doi.org/10.3389/fphys.2014.00484
Seeger T et al (2019) A premature termination codon mutation in MYBPC3 causes hypertrophic cardiomyopathy via chronic activation of nonsense-mediated decay. Circulation 139:799–811. https://doi.org/10.1161/CIRCULATIONAHA.118.034624
Seidman CE, Seidman JG (2011) Identifying sarcomere gene mutations in hypertrophic cardiomyopathy: a personal history. Circ Res 108:743–750. https://doi.org/10.1161/CIRCRESAHA.110.223834
Sequeira V et al (2013) Perturbed length-dependent activation in human hypertrophic cardiomyopathy with missense sarcomeric gene mutations. Circ Res 112:1491–1505. https://doi.org/10.1161/CIRCRESAHA.111.300436
Shaffer JF, Kensler RW, Harris SP (2009) The myosin-binding protein C motif binds to F-actin in a phosphorylation-sensitive manner. J Biol Chem 284:12318–12327. https://doi.org/10.1074/jbc.M808850200
Shi Y, Inoue H, Wu JC, Yamanaka S (2017) Induced pluripotent stem cell technology: a decade of progress. Nat Rev Drug Discov 16:115–130. https://doi.org/10.1038/nrd.2016.245
Silva TP, Cotovio JP, Bekman E, Carmo-Fonseca M, Cabral JMS, Fernandes TG (2019) Design principles for pluripotent stem cell-derived organoid engineering. Stem Cells Int 2019:4508470. https://doi.org/10.1155/2019/4508470
Simmons D, Williams DR, Powell MJ (1991) The Coventry Diabetes Study: prevalence of diabetes and impaired glucose tolerance in Europids and Asians. Q J Med 81:1021–1030
Simonson TS et al (2010) Limited distribution of a cardiomyopathy-associated variant in India. Ann Hum Genet 74:184–188. https://doi.org/10.1111/j.1469-1809.2010.00561.x
Solaro RJ (2009) CK-1827452, a sarcomere-directed cardiac myosin activator for acute and chronic heart disease. IDrugs 12:243–251
Spirito P, Seidman CE, McKenna WJ, Maron BJ (1997) The management of hypertrophic cardiomyopathy. N Engl J Med 336:775–785. https://doi.org/10.1056/NEJM199703133361107
Squire JM, Luther PK, Knupp C (2003) Structural evidence for the interaction of C-protein (MyBP-C) with actin and sequence identification of a possible actin-binding domain. J Mol Biol 331:713–724. https://doi.org/10.1016/s0022-2836(03)00781-2
Srivastava A, Garg N, Mittal T, Khanna R, Gupta S, Seth PK, Mittal B (2011) Association of 25 bp deletion in MYBPC3 gene with left ventricle dysfunction in coronary artery disease patients. PLoS One 6:e24123. https://doi.org/10.1371/journal.pone.0024123
Starr R, Offer G (1971) Polypeptide chains of intermediate molecular weight in myosin preparations. FEBS Lett 15:40–44. https://doi.org/10.1016/0014-5793(71)80075-3
Stelzer JE, Fitzsimons DP, Moss RL (2006) Ablation of myosin-binding protein-C accelerates force development in mouse myocardium. Biophys J 90:4119–4127. https://doi.org/10.1529/biophysj.105.078147
Stenson PD, Mort M, Ball EV, Shaw K, Phillips A, Cooper DN (2014) The Human Gene Mutation Database: building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum Genet 133:1–9. https://doi.org/10.1007/s00439-013-1358-4
Strong A, Musunuru K (2017) Genome editing in cardiovascular diseases. Nat Rev Cardiol 14:11–20. https://doi.org/10.1038/nrcardio.2016.139
Swaminathan PD, Purohit A, Hund TJ, Anderson ME (2012) Calmodulin-dependent protein kinase II: linking heart failure and arrhythmias. Circ Res 110:1661–1677. https://doi.org/10.1161/CIRCRESAHA.111.243956
Tabish AM, Arif M, Song T, Elbeck Z, Becker RC, Knoll R, Sadayappan S (2019) Association of intronic DNA methylation and hydroxymethylation alterations in the epigenetic etiology of dilated cardiomyopathy. Am J Physiol Heart Circ Physiol 317:H168-H180 https://doi.org/10.1152/ajpheart.00758.2018
Tanaka A et al (2014) Endothelin-1 induces myofibrillar disarray and contractile vector variability in hypertrophic cardiomyopathy-induced pluripotent stem cell-derived cardiomyocytes. J Am Heart Assoc 3:e001263. https://doi.org/10.1161/JAHA.114.001263
Tang WH, Francis GS (2003) Novel pharmacological treatments for heart failure. Expert Opin Investig Drugs 12:1791–1801. https://doi.org/10.1517/13543784.12.11.1791
Tang JW, Mason M, Kushner RF, Tirodkar MA, Khurana N, Kandula NR (2012) South Asian American perspectives on overweight, obesity, and the relationship between weight and health. Prev Chronic Dis 9:E107. https://doi.org/10.5888/pcd9.110284
Tanjore RR, Rangaraju A, Kerkar PG, Calambur N, Nallari P (2008) MYBPC3 gene variations in hypertrophic cardiomyopathy patients in India. Can J Cardiol 24:127–130. https://doi.org/10.1016/s0828-282x(08)70568-3
Taylor EN et al (2016) Alterations in multi-scale cardiac architecture in association with phosphorylation of myosin binding protein-C. J Am Heart Assoc 5:e002836. https://doi.org/10.1161/JAHA.115.002836
Teerlink JR (2009) A novel approach to improve cardiac performance: cardiac myosin activators. Heart Fail Rev 14:289–298. https://doi.org/10.1007/s10741-009-9135-0
Teirlinck CH et al (2012) A human MYBPC3 mutation appearing about 10 centuries ago results in a hypertrophic cardiomyopathy with delayed onset, moderate evolution but with a risk of sudden death. BMC Med Genet 13:105. https://doi.org/10.1186/1471-2350-13-105
Thierfelder L et al (1994) Alpha-tropomyosin and cardiac troponin T mutations cause familial hypertrophic cardiomyopathy: a disease of the sarcomere. Cell 77:701–712. https://doi.org/10.1016/0092-8674(94)90054-x
Thorp EB (2012) The myocardial unfolded protein response during ischemic cardiovascular disease. Biochem Res Int 2012:583170. https://doi.org/10.1155/2012/583170
Tissier R, Berdeaux A, Ghaleh B, Couvreur N, Krieg T, Cohen MV, Downey JM (2008) Making the heart resistant to infarction: how can we further decrease infarct size? Front Biosci 13:284–301
Toepfer CN et al (2019a) SarcTrack Circ Res 124:1172–1183. https://doi.org/10.1161/CIRCRESAHA.118.314505
Toepfer CN et al (2019b) Hypertrophic cardiomyopathy mutations in MYBPC3 dysregulate myosin. Sci Transl Med 11. https://doi.org/10.1126/scitranslmed.aat1199
Tong CW, Stelzer JE, Greaser ML, Powers PA, Moss RL (2008) Acceleration of crossbridge kinetics by protein kinase A phosphorylation of cardiac myosin binding protein C modulates cardiac function. Circ Res 103:974–982. https://doi.org/10.1161/CIRCRESAHA.108.177683
Tong CW et al (2015) Phosphoregulation of cardiac inotropy via myosin binding protein-C during increased pacing frequency or beta1-adrenergic stimulation. Circ Heart Fail 8:595–504. https://doi.org/10.1161/CIRCHEARTFAILURE.114.001585
Uppaluri CR (2002) Heart disease and its related risk factors in Asian Indians. Ethn Dis 12:45–53
van Dijk SJ et al (2009) Cardiac myosin-binding protein C mutations and hypertrophic cardiomyopathy: haploinsufficiency, deranged phosphorylation, and cardiomyocyte dysfunction. Circulation 119:1473–1483. https://doi.org/10.1161/CIRCULATIONAHA.108.838672
Van Driest SL, Vasile VC, Ommen SR, Will ML, Tajik AJ, Gersh BJ, Ackerman MJ (2004) Myosin binding protein C mutations and compound heterozygosity in hypertrophic cardiomyopathy. J Am Coll Cardiol 44:1903–1910. https://doi.org/10.1016/j.jacc.2004.07.045
Van Driest SL, Ommen SR, Tajik AJ, Gersh BJ, Ackerman MJ (2005) Sarcomeric genotyping in hypertrophic cardiomyopathy. Mayo Clin Proc 80:463–469. https://doi.org/10.1016/S0025-6196(11)63196-0
Vazquez Ruiz de Castroviejo E et al (2005) Analysis of the frequency of cardiac arrhythmias and conduction disturbances from a health-care perspective. Rev Esp Cardiol 58:657–665
Vignier N et al (2009) Nonsense-mediated mRNA decay and ubiquitin-proteasome system regulate cardiac myosin-binding protein C mutant levels in cardiomyopathic mice. Circ Res 105:239–248. https://doi.org/10.1161/CIRCRESAHA.109.201251
Viswanathan SK, Sanders HK, McNamara JW, Jagadeesan A, Jahangir A, Tajik AJ, Sadayappan S (2017) Hypertrophic cardiomyopathy clinical phenotype is independent of gene mutation and mutation dosage. PLoS One 12:e0187948. https://doi.org/10.1371/journal.pone.0187948
Viswanathan SK et al (2018) Association of cardiomyopathy with MYBPC3 D389V and MYBPC3Delta25bpIntronic deletion in South Asian descendants. JAMA Cardiol 3:481–488. https://doi.org/10.1001/jamacardio.2018.0618
Volgman AS et al (2018) Atherosclerotic cardiovascular disease in South Asians in the United States: epidemiology, risk factors, and treatments: a scientific statement from the American Heart Association. Circulation 138:e1–e34. https://doi.org/10.1161/CIR.0000000000000580
Waldmuller S et al (2003) Novel deletions in MYH7 and MYBPC3 identified in Indian families with familial hypertrophic cardiomyopathy. J Mol Cell Cardiol 35:623–636
Wang X, Robbins J (2014) Proteasomal and lysosomal protein degradation and heart disease. J Mol Cell Cardiol 71:16–24. https://doi.org/10.1016/j.yjmcc.2013.11.006
Watkins H et al (1993) A disease locus for familial hypertrophic cardiomyopathy maps to chromosome 1q3. Nat Genet 3:333–337. https://doi.org/10.1038/ng0493-333
Watkins H et al (1995) Mutations in the cardiac myosin binding protein-C gene on chromosome 11 cause familial hypertrophic cardiomyopathy. Nat Genet 11:434–437. https://doi.org/10.1038/ng1295-434
Weisberg A, Winegrad S (1996) Alteration of myosin cross bridges by phosphorylation of myosin-binding protein C in cardiac muscle. Proc Natl Acad Sci U S A 93:8999–9003. https://doi.org/10.1073/pnas.93.17.8999
Witayavanitkul N et al (2014) Myocardial infarction-induced N-terminal fragment of cardiac myosin-binding protein C (cMyBP-C) impairs myofilament function in human myocardium. J Biol Chem 289:8818–8827. https://doi.org/10.1074/jbc.M113.541128
Wu W et al (2015) Novel phenotype-genotype correlations of restrictive cardiomyopathy with myosin-binding protein C (MYBPC3) gene mutations tested by next-generation sequencing. J Am Heart Assoc 4:e001879. https://doi.org/10.1161/JAHA.115.001879
Zahka K et al (2008) Homozygous mutation of MYBPC3 associated with severe infantile hypertrophic cardiomyopathy at high frequency among the Amish. Heart 94:1326–1330. https://doi.org/10.1136/hrt.2007.127241
Funding
Dr. Sadayappan has received support from the National Institutes of Health grants (R01 HL130356, R01 HL105826, R01 AR067279, and RO1/R56 HL139680), the American Heart Association, Cardiovascular Genome-Phenome Study (15CVGPSD27020012), Catalyst (17CCRG33671128), Institutional Undergraduate Student (19UFEL34380251) and Transformation (19TPA34830084) awards, and the PLN Foundation (PLN crazy idea) awards, as well as from AstraZeneca, MyoKardia, Merck, and Amgen. Dr. Dhandapany is funded by an American Heart Association-Scientist Development Grant (15SDG23250005) and Wellcome Trust-Intermediate Fellowship (IA/I/16/1/502367). Drs. Kranias and Sadayappan are funded by the Leducq Foundation (Transatlantic Network 18CVD01, PLN-CURE). Dr. Becker has received support from the National Institutes of Health grants R01-HL065222 and U54-HL112307 and the American Heart Association 14FRN24110000. Dr. Kumar (Predoctoral Fellowship, 17PRE33630192), Ms. Desai (Predoctoral Fellowship, 20PRE35120272) and Dr. Song (Postdoctoral Fellowship, 19POST34380448) were supported by the American Heart Association fellowship training grants.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Dr. Sadayappan provides consulting and collaborative research studies to the Leducq Foundation (CURE-PLAN), Red Saree Inc., Greater Cincinnati Tamil Sangam, AstraZeneca, MyoKardia, Merck, and Amgen, but such work is unrelated to the content of this manuscript. Dr. Becker serves on scientific advisory boards for following: Janssen and DSMB Committees for Ionis Pharmaceuticals, Akcea Therapeutics, and Novartis. Dr. Singh has been a post-doctoral fellow of Amgen, starting from June, 2019 and performs research at the University of Cincinnati. No other disclosures are reported.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Arif, M., Nabavizadeh, P., Song, T. et al. Genetic, clinical, molecular, and pathogenic aspects of the South Asian–specific polymorphic MYBPC3Δ25bp variant. Biophys Rev 12, 1065–1084 (2020). https://doi.org/10.1007/s12551-020-00725-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12551-020-00725-1