
Abstract
Several lines of evidence suggest that alterations of the ubiquitin-proteasome system (UPS) and autophagy-lysosome pathway (ALP) may be involved in cardiac diseases. Little is known, however, in hypertrophic cardiomyopathy (HCM). This study studied these pathways in two mouse models of HCM that mainly differ by the presence or absence of truncated mutant proteins. Analyses were performed in homozygous Mybpc3-targeted knock-in (KI) mice, carrying a HCM mutation and exhibiting low levels of mutant cardiac myosin-binding protein C (cMyBP-C), and in Mybpc3-targeted knock-out (KO) mice expressing no cMyBP-C, thus serving as a model of pure cMyBP-C insufficiency. In the early postnatal development of cardiac hypertrophy, both models showed higher levels of ubiquitinated proteins and greater proteasomal activities. To specifically monitor the degradation capacity of the UPS with age, mice were crossed with transgenic mice that overexpress UbG76V-GFP. UbG76V-GFP protein levels were fourfold higher in 1-year-old KI, but not KO mice, suggesting a specific UPS impairment in mice expressing truncated cMyBP-C. Whereas protein levels of key ALP markers were higher, suggesting ALP activation in both mutant mice, their mRNA levels did not differ between the groups, underlying rather defective ALP-mediated degradation. Analysis of key proteins regulated in heart failure did not reveal specific alterations in KI and KO mice. Our data suggest (1) UPS activation in early postnatal development of cardiac hypertrophy, (2) specific UPS impairment in old KI mice carrying a HCM mutation, and (3) defective ALP as a common mechanism in genetically engineered mice with cardiac hypertrophy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Eukaryotic cells possess two major proteolytic systems, the ubiquitin-proteasome system (UPS) and the autophagy-lysosome pathway (ALP) [26, 61]. The UPS is a highly selective degradation pathway of short-lived cytosolic, nuclear and myofibrillar proteins [61]. A main function of the UPS is to prevent accumulation of damaged, misfolded and mutant proteins, but it is also involved in several biological processes including cell proliferation, adaptation to stress and cell death [61]. The UPS is an ATP-dependent system that requires polyubiquitination of the target protein prior to its degradation by the 26S proteasome (for review, see [34]). Polyubiquitination involves the concerted action of three different ubiquitin enzymes: E1 (ubiquitin-activating), E2 (ubiquitin-conjugating) and E3 (ubiquitin ligase). The 26S proteasome is a large, multicatalytic protein complex composed of the 19S regulatory particle, which recognizes, deubiquitinates and unfolds the target protein, and the 20S core, which subsequently degrades the target protein through three major proteolytic activities (chymotrypsin-like, trypsin-like and caspase-like).
The ALP is a bulk protein degradation process that engulfs long-lived proteins as well as cytoplasmic organelles into autophagosomes, which subsequently fuse with lysosomes to form autophagolysosomes, in which lysosomal proteases degrade autophagosomal content [26]. ALP has been shown to play a pro-survival role in a variety of physiological and pathophysiological processes such as cell growth, tissue homeostasis and intracellular clearance of mutated and misfolded proteins as well as damaged organelles [26]. On the other hand, excessive activity of ALP leads to self-digestion, degradation of essential intracellular components and finally cell death [35]. Although autophagy has been considered to be non-selective and independent of the UPS, growing lines of evidence indicate the presence of selective autophagy, including mitophagy, pexophagy, and the p62/sequestosome 1 (p62/SQSTM1)-dependent degradation of ubiquitin-positive aggregates (for reviews, see [23, 49]).
The activity of the UPS and ALP has been found to be altered in several human and experimental cardiac diseases (for reviews, see [12, 34, 59]). In failing human hearts, prominent accumulation of ubiquitinated proteins has been reported [4, 25, 39, 51, 56]. Anti-proteasomal auto-immunity was found to be markedly increased in patients with dilated cardiomyopathy [54, 55]. Experimental animal models of hypertrophy induced by transverse aortic constriction (TAC) showed either an increase [13] or a decrease [51] in proteasomal activity, increased transcript levels of UPS regulators [13, 39, 40], as well as induced autophagic activity [50]. Involvement of the UPS in genetic cardiac diseases has been first suspected in myocardial tissue of patients with hypertrophic cardiomyopathy (HCM; for review see [8]). HCM is characterized by left ventricular hypertrophy (LVH), which is frequently asymmetric, involving the interventricular septum. It is caused by more than 450 mutations in at least 13 genes encoding proteins of the cardiac sarcomere (for recent reviews, see [8, 46]). One of the two most frequently mutated genes [41], MYBPC3 encoding cardiac myosin-binding protein C (cMyBP-C), mainly exhibits frameshift mutations, which were expected to produce C-terminal truncated cMyBP-C [6]. However, despite the presence of nonsense mRNAs, truncated cMyBP-Cs were consistently undetectable in myocardial tissue of patients with frameshift MYBPC3 mutations [31, 36, 43, 52]. Similarly, protein levels of truncated cMyBP-Cs were very low after gene transfer in cells or in transgenic mice [16, 44, 57], and blockade of the UPS by proteasome inhibitors markedly increased mutant protein levels [1, 44]. Importantly, marked degradation of truncated cMyBP-C was associated with the formation of ubiquitin-positive aggregates and impairment of the UPS as evidenced with a fluorescent reporter system [44]. The initial signal that promotes targeting of mutant cMyBP-C to the UPS is not yet elucidated. It could be due to low steady-state levels of cMyBP-C phosphorylation [52], which is known to be regulated by cAMP-dependent protein kinase [17], Ca2+/Calmodulin kinase II [32], protein kinase C ε [24], protein kinase D [3] and/or by p90 ribosomal S6 kinase [10]. More recently, proteasomal activities were found to be lower in myocardial tissue from HCM patients [39]. Involvement of ALP in HCM has not been described. However, we previously showed that the lysosome inhibitor bafilomycin A1 slightly increased the level of truncated cMyBP-C after gene transfer in cardiomyocytes [44], suggesting potential involvement of ALP in the degradation of mutant sarcomeric proteins. Taken together, these data led us to hypothesize that HCM mutations that result in the production of abnormal sarcomeric proteins can impair the activity of the major proteolytic systems in the heart and that this may play a pathophysiological role in HCM on its own.
In order to gain more insights into the pattern and specificity of UPS and ALP alterations during the evolution of HCM, and to test our hypothesis in an in vivo context we investigated these protein quality controls at different postnatal windows in two mouse models of HCM. Homozygous Mybpc3-targeted knock-in (KI) mice exhibit LVH with reduced fractional shortening and express low levels of mutant cMyBP-C proteins (10% of normal). The reduction in cMyBP-C results from the activation of both the nonsense-mediated mRNA decay and the UPS [53]. KI mice were compared to homozygous Mybpc3-targeted knock-out (KO) mice, which also exhibit LVH with reduced fractional shortening, but do not express any cMyBP-C [7], thus serving as a pure model of cMyBP-C insufficiency.
Methods
The investigation conforms to the guide for the care and use of laboratory animals published by the NIH (Publication No. 85-23, revised 1985).
Animal models
Creation and initial characterizations of Mybpc3-targeted KI- and KO-mice were previously reported [7, 53]. Both mouse lines were created and maintained on the Black Swiss genetic background and wild-type (WT) mice were used as controls. The UbG76V-GFP/1 mice ubiquitously express a green fluorescent protein (GFP)-fused proteasome substrate [30], and were on the C57BL/6 J background. Crossed KI/, KO/ and WT/UbG76V-GFP mice were therefore on a mixed genetic background.
Analysis of mRNAs
Total RNA was extracted from mouse ventricles using the SV Total RNA Isolation Kit (Promega) according to the manufacturer’s instructions. RNA concentration, purity and quality were determined using the NanoDrop® ND-1000 spectrophotometer. Reverse transcription was performed using oligo-dT primers with the RevertAid™ First Strand cDNA Synthesis Kit (Fermentas) from 100 ng RNA. The quantitative determination of the UbG76V-GFP, beclin-1, microtubule-associated protein 1 light chain 3 (LC3), p62/sequestosome 1 (p62/SQSTM1) and GαS mRNAs was performed by qPCR using the Maxima™ SYBR Green/ROX qPCR Master Mix (Fermentas), and primers specific for every mouse sequence (see supplemental Table). GαS was used as an endogenous control to normalize the quantification of the target mRNAs for difference in the amount of total RNA added to each reaction. Experiments were performed on the ABI PRISM® 7900HT Sequence Detection System (Applied Biosystems). The mRNA amount was estimated according to the comparative Ct method with the 2-∆∆Ct formula.
Cytosolic and crude protein extracts from ventricular tissue
About 50 mg of mouse ventricular tissue was frozen–thawed 3 times in 250 μl of H2O containing a protease-inhibitor cocktail (complete mini™, Roche Diagnostics), and homogenized using the Tissue Lyser (2 × 30 s at 20 Hz). Soluble material (cytosolic fraction) was recovered by centrifugation at 13,200 rpm for 30 min at 4°C. Crude protein extract was obtained from about 50 mg of ventricular tissue homogenized in 3% SDS, 30 mM Tris-base, pH 8.8, 5 mM EDTA, 30 mM NaF, 10% glycerol and 1 mM DTT and centrifuged at 13,200 rpm for 10 min. The supernatants were collected and their concentrations were determined using the BioRad protein assay reagent (BioRad).
Western blot analysis
Proteins were loaded on acrylamide/bisacrylamide (29:1) gels and electrotransferred to nitrocellulose membranes with a 0.45 μm-pore size. For LC3 analysis, proteins were electrotransferred to polyvinylidene fluoride membranes. Membranes were stained overnight with the monoclonal antibodies directed against ubiquitin (FK2, Biomol, 1:50,000), phospholamban (total PLB, A1, Badrilla, 1:5,000) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH, HyTest, 1:2,000) and the polyclonal antibodies directed against ubiquitin (Santa-Cruz Biotechnology, 1:750) GFP (Santa-Cruz Biotechnology, 1:2,000), p62/Sequestosome 1 (p62/SQSTM1; Sigma, 1:1,000), beclin-1 (Cell Signaling Technology, 1:1,000), microtubule-associated protein-1 light chain 3 (LC3; Novus Biologicals, 1:500), phosphorylated phospholamban (Ser16-PLB and Thr17-PLB, Badrilla, 1:5,000), phosphorylated cardiac troponin I (Ser23/24-cTnI, Cell Signaling Technology, 1:1,000), sarcoplasmic reticulum (SR) Ca2+-ATPase (SERCA2; Santa-Cruz Biotechnology, 1:500) and calsequestrin (CSQ; Dianova, 1:2,500). The secondary antibodies were coupled to HRP (Dianova). Signals were revealed with SuperSignal® West Dura extended duration substrate (Pierce) and acquired with the Chemie Genius2 Bio Imaging System. Quantification of the signal was done using the Gene Tools software.
Determination of proteasomal activities
The chymotrypsin-like, trypsin-like and caspase-like activities of the proteasome were assessed in ventricular cytosolic lysates using the synthetic peptide substrates Suc-LLVY-AMC, Bz-VGR-AMC and Z-LLE-ßNA, respectively, as described previously [33, 53]. For determination of the activities, 30 μg of protein were incubated in the dark for 1 h at 37°C in an incubation buffer (225 mM Tris–HCl, pH 8.2, 45 mM KCl, 7.5 mM Mg(CH3COO)2-4H2O, 7.5 mM MgCl2-6H2O, 1.1 mM DTT) containing an ATP-regenerating system (6 mM ATP, 5 mM phosphocreatine, 0.2 U phosphocreatinekinase) and the specific fluorogenic substrates Suc-LLVY-AMC (60 μM, Merck Biosciences), Bz-VGR-AMC (20 μM, Biomol) and Z-LLE-βNA (200 μM, Biomol) for measuring the chymotrypsin-like, the trypsin-like and the caspase-like activity, respectively. The fluorescence of the released AMC or βNA reporter was measured using the TECAN Safire2 microplate reader at an excitation wavelength of 380 and 350 nm, respectively, and an emission wavelength of 460 and 450 nm, repectively. Each sample was measured in triplicates. The mean of the blank (incubation buffer and H2O) was subtracted from the mean of each sample triplicate.
Statistical analysis
Data are presented as mean ± SEM. Statistical analyses were performed using the unpaired Student’s t test or two-way ANOVA as indicated in the legends. All analyses were realized using the GraphPad Software Inc. A value of P < 0.05 was considered statistically significant.
Results
Cardiac hypertrophy in Mybpc3-KI and -KO mice
We first evaluated the heart-to-body weight ratio (HW/BW) from birth to 13 weeks of age. Whereas it did not differ between KO and WT mice at birth, it was higher in KO than WT mice after 2 weeks of age and during all subsequent postnatal windows (Fig. 1). Interestingly, the HW/BW was already 22% higher in KI than in WT at birth and remained higher at all investigated ages (Fig. 1). This was not associated with a difference in BW between the groups.

Degree of cardiac hypertrophy during the early postnatal development in KO, KI and WT mice. Heart-to-body weight ratio (HW/BW) was determined at different postnatal windows in KO (black squares), KI (black triangles) and WT (empty symbols) mice. Data are expressed as mean ± SEM (n = 8 animals per group and age) with ***P < 0.001, two-way ANOVA
Activation of the ubiquitin-proteasome system during early postnatal development in KI and KO mice
To investigate whether the UPS is altered during the evolution of hypertrophy, we determined the steady-state levels of ubiquitinated proteins by Western blot. Ventricular protein extracts from eight gender-matched mice were first pooled to have an overview of the evolution in the early postnatal development on one blot (Fig. 2a). The steady-state levels of ubiquitinated proteins were elevated at birth, dropped thereafter and remained low with postnatal development in WT mice (Fig. 2a). Similarly, in both KI and KO mice, the amount of ubiquitinated proteins was also high at birth and dropped in the first 2–4 weeks of age, but then increased again with time (Fig. 2a). We then precisely quantified the levels of ubiquitinated proteins in KI and KO and compared them to those in WT mice at birth and at 2, 4, 6, 9 and 13 weeks of age (Fig. 2b, c). In both KO and KI, the steady-state level of ubiquitinated proteins was higher than in WT mice.

Steady-state levels of ubiquitinated proteins during the early postnatal development in KO, KI and WT mice. Analyses were performed in ventricular crude protein extracts. a Expression pattern of ubiquitinated proteins in the first 3 months. Ventricular proteins of eight gender-matched mice were pooled for each time point (from neonates (NN) to 13 weeks of age) in KO, KI and WT mice. Western blot stained with an anti-ubiquitin antibody (from Biomol for KO and WT littermates and from Santa-Cruz for KI and WT littermates). b Representative Western blot of ubiquitinated proteins and corresponding Ponceau in 13-week-old KO and WT mice. c Representative time courses of the steady-state levels of ubiquitinated proteins normalized to Ponceau in neonatal to 13-week-old KO (black squares) and KI (black triangles) mice related to WT (empty symbols). Data are expressed as mean ± SEM with ***P < 0.001, two-way ANOVA. The number of animals per timepoint was n = 6
To evaluate whether the higher steady-state levels of ubiquitinated proteins result from proteasome impairment, proteasomal activities (chymotrypsin-like, trypsin-like and caspase-like activity) were determined in ventricular cytosolic protein extracts at each predefined postnatal time point. At birth, the three proteasomal activities did not differ between KO and WT mice, whereas they were slightly higher in KI than in WT mice (Fig. 3a, b, c). In both KO and KI mice, the three proteasomal activities were higher throughout the development and showed a progressive relative increase over WT between 9 and 13 weeks of age. The increase in proteasomal activities appeared overall similar in KI and KO and positively correlated with the degree of cardiac hypertrophy as determined by HW/BW ratio (Fig. 4). No such correlation was observed in WT (Fig. 4). These data suggest activation rather than the inhibition of the UPS during the early postnatal development of cardiac hypertrophy in both KI and KO mice.

Proteasomal activities during the early postnatal development in KO, KI and WT mice. Analyses were performed in ventricular cytosolic protein extracts. Representative time courses of a chymotrypsin-like activity, b trypsin-like activity and c caspase-like activity of the proteasome in KO (black squares), KI (black triangles) and WT (empty symbols) mice. Data are related to WT and expressed as mean ± SEM (n = 8 animals per group and age) with *P < 0.05, **P < 0.01 ***P < 0.001, two-way ANOVA

Correlation between the degree of cardiac hypertrophy and the activities of the proteasome in KO, KI and WT mice. Relationship between the heart-to-body weight ratio (HW/BW) and a the chymotrypsin-like activity or b caspase-like activity. Analyses of Spearman correlation factor (r) were performed in KO (green), KI (blue) and WT (black) mice, with P < 0.05 and P < 0.001 versus WT
Impairment of the ubiquitin-proteasome system with age only in KI mice
Neither the steady-state levels of ubiquitinated proteins nor the in vitro-determined proteasomal activities answer the question of whether the UPS is meeting its demand or not. To answer this question, KI, KO and WT mice were crossed with the UbG76V-GFP mice, which provide the opportunity to monitor the global function of the UPS in vivo [30]. The UbG76V-GFP mice ubiquitously express a fluorescent substrate of the UPS, which is normally degraded by this system. When the UPS is pharmacologically inhibited or impaired/saturated, the UbG76V-GFP substrate is accumulated [11]. We hypothesized that chronic usage of the UPS to degrade mutant cMyBP-Cs leads to UPS saturation with age in KI, but not in KO mice. The HW/BW was 54 and 32% higher in 57 ± 5-week-old KI/UbG76V-GFP and KO/UbG76V-GFP than in WT/UbG76V-GFP mice, respectively (Fig. 5a). Similarly, the HW/tibia length was 64 and 30% higher in KI/UbG76V-GFP and KO/UbG76V-GFP than in WT/UbG76V-GFP mice (Fig. 5b). This indicates marked cardiac hypertrophy in KI, and, to a lesser extent, in KO mice. The protein level of UbG76V-GFP was determined by Western blot using an antibody directed against GFP (Fig. 5c). Whereas UbG76V-GFP level did not differ between KO and WT, it was fourfold higher in KI than in WT and KO mice (Fig. 5c, d). In contrast, UbG76V-GFP mRNA level did not differ between the groups (Fig. 5e), suggesting a posttranslational mechanism for the observed increase in protein level. Furthermore, whereas the amount of ubiquitinated proteins was higher in mutant than in WT mice (Fig. 5g), the chymotrypsin-like activity was >25% lower in KI than in WT, but normal in KO mice (Fig. 5F). These data suggest UPS saturation/impairment in old KI mice.

Evaluation of the global function of the ubiquitin-proteasome system in 1-year-old KO/UbG76V-GFP, KI/UbG76V-GFP and WT/UbG76V-GFP mice. Analyses were performed in hearts isolated from ~ 57-week-old KO (light gray), KI (dark gray) and WT (white) mice. a Heart-to-body weight ratio (HW/BW). b Heart weight-to-tibia length ratio (HW/TL). c Representative Western blots of ventricular crude protein extracts stained with anti-GFP and anti-GAPDH antibodies. d Levels of UbG76V-GFP protein normalized to GAPDH. e UbG76V-GFP mRNA levels normalized to Gαs determined by RT-qPCR in ventricular total RNA extracts. f Chymotryspin-like activity of the proteasome determined in ventricular cytosolic protein extracts. g Steady-state levels of ubiquitinated proteins normalized to Ponceau determined by Western blot of ventricular crude protein extracts using an anti-ubiquitin antibody. Data are related to WT and expressed as the mean ± SEM with *P < 0.05, **P < 0.01 and ***P < 0.001 versus WT, and with # P < 0.05 and ### P < 0.001 versus KO, Student’s t test. The number of animals is indicated in the bars
Defective autophagy-lysosome pathway in KI and KO mice
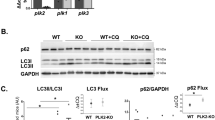
In cell lines, inhibition of the proteasome has been shown to activate ALP in order to clear polyubiquitinated proteins [14, 50]. We therefore hypothesized that UPS impairment results in activation of ALP in KI, but not in KO mice. ALP was evaluated at different steps in 1-year-old KI/UbG76V-GFP, KO/UbG76V-GFP and WT/UbG76V-GFP mice (Fig. 6). It has been previously shown that higher protein levels of beclin-1, microtubule-associated protein 1 light chain 3 (LC3), and p62/sequestosome 1 (p62/SQSTM1) underline heightened autophagic activity [22, 23, 60]. We first investigated beclin-1, which is required in membrane nucleation, an early step of autophagy. Beclin-1 protein levels were 74 and 32% higher in KO and KI than in WT mice, respectively (Fig. 6a, b), suggesting autophagy induction. However, beclin-1 mRNA levels did not differ between the groups (Fig. 6f), supporting reduced beclin-1 degradation rather than activated synthesis. Then we investigated the conversion of soluble LC3-I to lipidated LC3-II, which is involved in the formation of autophagosomes [26]. The level of LC3-II was 66% higher in KI and 52% higher, although not significant in KO than in WT mice (Fig. 6a, c). However, and in contrast to our expectations, the level of LC3-I did vary and was also higher, resulting in almost twofold higher level of total LC3 in both KO and KI than in WT mice (Fig. 6d). The absence of regulation at the mRNA levels (Fig. 6g), together with higher levels of LC3 protein suggests a reduced lysosomal-mediated LC3 degradation. We finally investigated p62/SQSTM1, which acts as a shuttle protein that binds ubiquitinated proteins and LC3 and directs ALP-mediated degradation of ubiquitinated proteins [38]. The p62/SQSTM1 protein level was more than twofold higher in both KI and KO than in WT mice (Fig. 6a,e), whereas p62/SQSTM1 mRNA level did not differ between the groups (Fig. 6h). Furthermore, a marked and significant positive correlation was found between the levels of ubiquitinated proteins and p62/SQSTM1 in KI and KO mice (Spearman correlation factor r = 0.81, p < 0.001; Fig. 7). These data suggest ALP impairment rather than activation in both KI and KO mice.

Evaluation of the autophagy/lysosome pathway in 1-year-old KO/UbG76V-GFP, KI/UbG76V-GFP and WT/UbG76V-GFP mice. Analyses were performed in ventricular protein or RNA extracts isolated from ~57-week-old KO (light gray), KI (dark gray) and WT (white) mice. a Representative Western blots stained with antibodies directed against the indicated proteins; b Protein levels of beclin-1 normalized to Ponceau; c Protein levels of LC3-II normalized to CSQ; d Protein levels of total LC3 (LC3-I + LC3-II) normalized to CSQ; e Protein levels of p62/SQSTM1 normalized to Ponceau, f Beclin-1 mRNA levels normalized to Gαs; g LC3 mRNA levels normalized to Gαs; h p62/SQSTM1 mRNA levels normalized to Gαs. Data are related to WT. Bars represent the mean ± SEM with *P < 0.05, **P < 0.01 and ***P < 0.001 versus WT, and with # P < 0.05 versus KO, Student’s t test. The number of animals is indicated in the bars

Correlation between the steady-state levels of ubiquitinated proteins and the levels of p62/SQSTM1 in KO plus KI mice
No molecular signature of heart failure in KI mice
Since KI mice specifically exhibit UPS impairment with age, we suspected a specific phenotype in these mice, and therefore evaluated whether this could be associated with a specific molecular signature of heart failure [15]. Protein levels of phosphorylated cTnI (Ser23/24-cTnI), SERCA2, as well as total and phosphorylated phospholamban (total PLB, Ser16-PLB and Thr17-PLB) were determined in all groups. The levels of Ser23/24-cTnI, SERCA2 and total PLB did not differ between KI than WT mice (Fig. 8). On the other hand, the levels of SERCA2/PLB ratio and Thr17-PLB were higher in KI (Fig. 8). Overall, KO did not differ to WT mice, except for a slightly lower Ser16-PLB level (Fig. 8). These data suggest no specific signature of heart failure in KI and KO mice.

Evaluation of the levels of heart failure markers in 1-year-old KO/UbG76V-GFP, KI/UbG76V-GFP and WT/UbG76V-GFP mice. Analyses were performed in ventricular protein extracts isolated from ~57-week-old KO (light gray), KI (dark gray) and WT (white) mice. a Representative Western blots stained with the indicated antibodies; b Levels of the indicated proteins (normalized to ponceau or calsequestrin, CSQ). Data are related to WT and expressed as the mean ± SEM with *P < 0.05 versus WT and with # P < 0.05 versus KO, Student’s t test. The number of animals is indicated in the bars
Discussion
In the present study we investigated the major proteolytic systems UPS and ALP at different postnatal windows in homozygous Mybpc3-targeted KI and KO mice. Both models develop LVH and cardiac dysfunction [7, 53]. Whereas KI mice exhibit low levels of mutant cMyBP-C proteins due, at least in part, to their degradation by the UPS [53], KO mice do not express any cMyBP-C [7], thus serving as a model of pure cMyBP-C insufficiency. We hypothesized that chronic degradation of mutant cMyBP-C proteins may specifically cause saturation of the UPS and compensatory ALP activation, which might play an own role in the evolution of the disease in KI mice. All analyses were performed in neonates to 1-year-old mice. The major findings of the present study are: (1) accumulation of ubiquitinated proteins and greater proteasomal activities, suggesting UPS activation during the early postnatal development of cardiac hypertrophy in both KI and KO mice, (2) accumulation of the UPS substrate UbG76V-GFP protein, but not mRNA, suggesting UPS impairment in 1-year-old KI, but not KO mice, (3) higher levels of proteins, but not mRNA of beclin-1, LC3-I, LC3-II and p62/SQSTM1, suggesting defective ALP in 1-year-old KI and KO mice, and (4) no overall molecular signature of heart failure in 1-year-old KI and KO mice. Our findings support the view of (i) a specific impairment of the UPS in a KI mouse model of HCM, and (ii) ALP impairment as a common mechanism in genetically engineered mice with cardiac hypertrophy.
Over the last two decades, the UPS has been increasingly recognized as a major system in several biological processes including cell proliferation, adaption to stress and cell death [5, 20, 21, 27, 37, 42]. More recently, activation or impairment of the UPS has been reported in cardiac disease (for recent reviews, see [34, 45, 48]). Particularly, accumulation of ubiquitinated proteins has been reported in failing human hearts [4, 25, 51, 56]. This argues in principle for an impairment of the UPS, which in turn is expected to be associated with lower proteasomal activities as previously observed in human and experimental model of heart failure [39, 51], and in human HCM [39]. Interestingly, in the early postnatal development, KI and KO mice exhibited elevated steady-state levels of ubiquitinated proteins, but higher proteasomal activities, which were positively correlated to the degree of LVH in both mutant mice. These data are in agreement with previous observations of greater proteasomal activities in murine, canine and feline models of TAC-induced cardiac hypertrophy [2, 13], and in transgenic mice with cardiac hypertrophy [9, 19]. Therefore, we propose that the UPS is rather activated than inhibited in the early postnatal development of cardiac hypertrophy in KI and KO mice. Both accumulation of ubiquitinated proteins and activation of the proteasome could result from accelerated protein turnover in the development of cardiac hypertrophy. Alternatively, accelerated degradation may be an adaptive mechanism to reduce the level of hypertrophic, hypertrophy-promoting and proapoptotic factors and therefore to prevent further cardiac hypertrophy. Indeed, a number of key factors that promote cardiac hypertrophy such as β-catenin or calcineurin are targeted for degradation by the UPS [18, 28].
The examination of KI and KO mouse lines crossed with the UbG76V-GFP transgenic mice gave the unique opportunity to evaluate the global function of the UPS in vivo [30]. In contrast to the determination of steady-state levels of ubiquitinated proteins, which monitors the balance between ubiquitination and deubiquitination, the level of UbG76V-GFP protein specifically underlines the degradation rate since the UbG76V-GFP protein is a specific substrate of the UPS, which is accumulated when proteasome function is inhibited [30]. The UbG76V-GFP protein levels, but not the mRNA levels were markedly higher in 1-year-old KI than in KO or WT mice. This suggests reduced proteasomal-mediated degradation, which was supported by a lower chymotrypsin-like activity in KI than in KO mice. These in vivo data support our previous findings of UPS impairment after gene transfer of truncated cMyBP-C in cardiac myocytes [44]. Furthermore, our data are in agreement with recent findings of proteasomal dysfunction in human HCM, and particularly in patients with sarcomeric gene mutations [39], as well as in heterozygous KI mice after adrenergic stress [47]. We propose that chronic degradation of mutant cMyBP-Cs leads to saturation of the UPS in aged KI mice. Although no specific molecular signature of heart failure was detected in KI mice, our data underlined specific differences between KI and KO mice, which, we propose, are related to the presence/absence of mutant cMyBP-C. Further analyses will evaluate whether inhibition/activation of proteasome function will worsen or ameliorate the phenotype in old KI mice, as recently shown in another cardiac proteinopathy [29].
It has been generally assumed that the UPS and ALP act separately. However, several studies suggest that they may function in concert to regulate the turnover of proteins (reviewed in [58, 59]). Particularly the induction of autophagy by inadequate UPS proteolytic function would be critical to alleviate proteotoxicity. In line with this, it was shown that pharmacological inhibition of the proteasome by MG132 induced autophagy in rat cardiac myocytes and other mammalian cell types [14, 50]. Therefore, we hypothesized that UPS dysfunction may induce autophagy in KI mice. The findings of higher protein levels of three major markers of ALP, beclin-1, LC3-II, and p62/SQTSM1 at first suggest activation of autophagy and formation of autophagosomes [49]. However, the absence of transcriptional activation of these proteins supports rather the view of defective degradation via ALP in KI, and also even to a greater extent in KO mice. The marked positive correlation between levels of ubiquitinated protein and p62/SQSTM1 in both mutant mice, support previous findings of selective autophagy, in which p62/SQSTM1 binds, via LC3, ubiquitinated proteins and sequester them into autophagosomes [23, 38]. Our findings suggest a blockade of the fusion between autophagosome and lysosome, which prevents the formation of the autophagolysosome and therefore degradation of the autophagosome content.
In conclusion, our data show overall alterations of the UPS and ALP in genetically engineered cardiomyopathic mice. Most of these alterations appear to be part of the general pathology related to the massive cardiac hypertrophy present in both mouse strains and are in accordance with published data in other animal models of cardiac hypertrophy. Importantly, our data support the view that (1) chronic degradation of mutant cMyBP-Cs via the UPS results in UPS impairment only in mutant mice carrying a human HCM mutation, and (2) defective ALP-mediated degradation is a common mechanism in genetically engineered mice with cardiac hypertrophy.
References
Bahrudin U, Morisaki H, Morisaki T, Ninomiya H, Higaki K, Nanba E, Igawa O, Takashima S, Mizuta E, Miake J, Yamamoto Y, Shirayoshi Y, Kitakaze M, Carrier L, Hisatome I (2008) Ubiquitin-proteasome system impairment caused by a missense cardiac myosin-binding protein C mutation and associated with cardiac dysfunction in hypertrophic cardiomyopathy. J Mol Biol 384:896–907. doi:10.1016/j.jmb.2008.09.070
Balasubramanian S, Mani S, Shiraishi H, Johnston RK, Yamane K, Willey CD, Cooper IV G, Tuxworth WJ, Kuppuswamy D (2006) Enhanced ubiquitination of cytoskeletal proteins in pressure overloaded myocardium is accompanied by changes in specific E3 ligases. J Mol Cell Cardiol 41:669–679 doi:10.1016/j.yjmcc.2006.04.022
Bardswell SC, Cuello F, Rowland AJ, Sadayappan S, Robbins J, Gautel M, Walker JW, Kentish JC, Avkiran M (2010) Distinct sarcomeric substrates are responsible for protein kinase D-mediated regulation of cardiac myofilament Ca2+ sensitivity and cross-bridge cycling. J Biol Chem 285:5674–5682. doi:10.1074/jbc.M109.066456
Birks EJ, Latif N, Enesa K, Folkvang T, le Luong A, Sarathchandra P, Khan M, Ovaa H, Terracciano CM, Barton PJ, Yacoub MH, Evans PC (2008) Elevated p53 expression is associated with dysregulation of the ubiquitin-proteasome system in dilated cardiomyopathy. Cardiovasc Res 79:472–480. doi:10.1093/cvr/cvn083
Breitschopf K, Zeiher AM, Dimmeler S (2000) Ubiquitin-mediated degradation of the proapoptotic active form of bid: a functional consequence on apoptosis induction. J Biol Chem 275:21648–21652. doi:10.1074/jbc.M001083200
Carrier L, Bonne G, Bahrend E, Yu B, Richard P, Niel F, Hainque B, Cruaud C, Gary F, Labeit S, Bouhour JB, Dubourg O, Desnos M, Hagege AA, Trent RJ, Komajda M, Fiszman M, Schwartz K (1997) Organization and sequence of human cardiac myosin binding protein C gene (MYBPC3) and identification of mutations predicted to produce truncated proteins in familial hypertrophic cardiomyopathy. Circ Res 80:427–434
Carrier L, Knoell R, Vignier N, Keller DI, Bausero P, Prudhon B, Isnard R, Ambroisine ML, Fiszman M, Ross J Jr, Schwartz K, Chien KR (2004) Asymmetric septal hypertrophy in heterozygous cMyBP-C null mice. Cardiovasc Res 63:293–304. doi:10.1016/j.cardiores.2004.04.009
Carrier L, Schlossarek S, Willis MS, Eschenhagen T (2010) The ubiquitin-proteasome system and nonsense-mediated mRNA decay in hypertrophic cardiomyopathy. Cardiovasc Res 85:330–338. doi:10.1093/cvr/cvp247
Chen Q, Liu JB, Horak KM, Zheng H, Kumarapeli AR, Li J, Li F, Gerdes AM, Wawrousek EF, Wang X (2005) Intrasarcoplasmic amyloidosis impairs proteolytic function of proteasomes in cardiomyocytes by compromising substrate uptake. Circ Res 97:1018–1026. doi:10.1161/01.RES.0000189262.92896.0b
Cuello F, Bardswell SC, Haworth RS, Ehler E, Sadayappan S, Kentish JC, Avkiran M (2011) Novel role for p90 ribosomal S6 kinase in the regulation of cardiac myofilament phosphorylation. J Biol Chem 286:5300–5310. doi:10.1074/jbc.M110.202713
Dantuma NP, Lindsten K, Glas R, Jellne M, Masucci MG (2000) Short-lived green fluorescent proteins for quantifying ubiquitin/proteasome-dependent proteolysis in living cells. Nat Biotechnol 18:538–543. doi:10.1038/75406
De Meyer GR, Martinet W (2009) Autophagy in the cardiovascular system. Biochim Biophys Acta 1793:1485–1495. doi:10.1016/j.bbamcr.2008.12.011
Depre C, Wang Q, Yan L, Hedhli N, Peter P, Chen L, Hong C, Hittinger L, Ghaleh B, Sadoshima J, Vatner DE, Vatner SF, Madura K (2006) Activation of the cardiac proteasome during pressure overload promotes ventricular hypertrophy. Circulation 114:1821–1828. doi:10.1161/CIRCULATIONAHA.106.637827
Ding WX, Ni HM, Gao W, Yoshimori T, Stolz DB, Ron D, Yin XM (2007) Linking of autophagy to ubiquitin-proteasome system is important for the regulation of endoplasmic reticulum stress and cell viability. Am J Pathol 171:513–524. doi:10.2353/ajpath.2007.070188
El-Armouche A, Pohlmann L, Schlossarek S, Starbatty J, Yeh YH, Nattel S, Dobrev D, Eschenhagen T, Carrier L (2007) Decreased phosphorylation levels of cardiac myosin-binding protein-C in human and experimental heart failure. J Mol Cell Cardiol 43:223–229. doi:10.1016/j.yjmcc.2007.05.003
Flavigny J, Souchet M, Sebillon P, Berrebi-Bertrand I, Hainque B, Mallet A, Bril A, Schwartz K, Carrier L (1999) COOH-terminal truncated cardiac myosin-binding protein C mutants resulting from familial hypertrophic cardiomyopathy mutations exhibit altered expression and/or incorporation in fetal rat cardiomyocytes. J Mol Biol 294:443–456. doi:10.1006/jmbi.1999.3276S0022-2836(99)93276-X
Gautel M, Zuffardi O, Freiburg A, Labeit S (1995) Phosphorylation switches specific for the cardiac isoform of myosin binding protein-C: a modulator of cardiac contraction? EMBO J 14:1952–1960
Glickman MH, Ciechanover A (2002) The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev 82:373–428. doi:10.1152/physrev.00027.2001
Hedhli N, Wang L, Wang Q, Rashed E, Tian Y, Sui X, Madura K, Depre C (2008) Proteasome activation during cardiac hypertrophy by the chaperone H11 Kinase/Hsp22. Cardiovasc Res 77:497–505. doi:10.1093/cvr/cvm054
Helin K (1998) Regulation of cell proliferation by the E2F transcription factors. Curr Opin Genet Dev 8:28–35. doi:10.1016/S0959-437X(98)80058-0
King RW, Deshaies RJ, Peters JM, Kirschner MW (1996) How proteolysis drives the cell cycle. Science 274:1652–1659
Kirkin V, Dikic I (2007) Role of ubiquitin- and Ubl-binding proteins in cell signaling. Curr Opin Cell Biol 19:199–205. doi:10.1016/j.ceb.2007.02.002
Kirkin V, McEwan DG, Novak I, Dikic I (2009) A role for ubiquitin in selective autophagy. Mol Cell 34:259–269. doi:10.1016/j.molcel.2009.04.026
Kooij V, Boontje N, Zaremba R, Jaquet K, dos Remedios C, Stienen GJ, van der Velden J (2010) Protein kinase C alpha and epsilon phosphorylation of troponin and myosin binding protein C reduce Ca2+ sensitivity in human myocardium. Basic Res Cardiol 105:289–300. doi:10.1007/s00395-009-0053-z
Kostin S, Pool L, Elsasser A, Hein S, Drexler HC, Arnon E, Hayakawa Y, Zimmermann R, Bauer E, Klovekorn WP, Schaper J (2003) Myocytes die by multiple mechanisms in failing human hearts. Circ Res 92:715–724. doi:10.1161/01.RES.0000067471.95890.5C
Levine B, Kroemer G (2008) Autophagy in the pathogenesis of disease. Cell 132:27–42. doi:10.1016/j.cell.2007.12.018
Li B, Dou QP (2000) Bax degradation by the ubiquitin/proteasome-dependent pathway: involvement in tumor survival and progression. Proc Natl Acad Sci USA 97:3850–3855. doi:10.1073/pnas.070047997
Li HH, Kedar V, Zhang C, McDonough H, Arya R, Wang DZ, Patterson C (2004) Atrogin-1/muscle atrophy F-box inhibits calcineurin-dependent cardiac hypertrophy by participating in an SCF ubiquitin ligase complex. J Clin Invest 114:1058–1071
Li J, Horak KM, Su H, Sanbe A, Robbins J, Wang X (2011) Enhancement of proteasomal function protects against cardiac proteinopathy and ischemia/reperfusion injury in mice. J Clin Invest 121:3689–3700. doi:10.1172/JCI45709
Lindsten K, Menendez-Benito V, Masucci MG, Dantuma NP (2003) A transgenic mouse model of the ubiquitin/proteasome system. Nat Biotechnol 21:897–902
Marston S, Copeland O, Jacques A, Livesey K, Tsang V, McKenna WJ, Jalilzadeh S, Carballo S, Redwood C, Watkins H (2009) Evidence from human myectomy samples that MYBPC3 mutations cause hypertrophic cardiomyopathy through haploinsufficiency. Circ Res 105:219–222. doi:10.1161/CIRCRESAHA.109.202440
McClellan G, Kulikovskaya I, Winegrad S (2001) Changes in cardiac contractility related to calcium-mediated changes in phosphorylation of myosin-binding protein C. Biophys J 81:1083–1092
Mearini G, Gedicke C, Schlossarek S, Witt CC, Kramer E, Cao P, Gomes MD, Lecker SH, Labeit S, Willis MS, Eschenhagen T, Carrier L (2010) Atrogin-1 and MuRF1 regulate cardiac MyBP-C levels via different mechanisms. Cardiovasc Res 85:357–366. doi:10.1093/cvr/cvp348
Mearini G, Schlossarek S, Willis MS, Carrier L (2008) The ubiquitin-proteasome system in cardiac dysfunction. Biochim Biophys Acta 1782:749–763. doi:10.1016/j.bbadis.2008.06.009
Mizushima N, Levine B, Cuervo AM, Klionsky DJ (2008) Autophagy fights disease through cellular self-digestion. Nature 451:1069–1075. doi:10.1038/nature06639
Moolman JA, Reith S, Uhl K, Bailey S, Gautel M, Jeschke B, Fischer C, Ochs J, McKenna WJ, Klues H, Vosberg HP (2000) A newly created splice donor site in exon 25 of the MyBP-C gene is responsible for inherited hypertrophic cardiomyopathy with incomplete disease penetrance. Circulation 101:1396–1402
Palombella VJ, Rando OJ, Goldberg AL, Maniatis T (1994) The ubiquitin-proteasome pathway is required for processing the NF-kappa B1 precursor protein and the activation of NF-kappa B. Cell 78:773–785 pii:S0092-8674(94)90482-0
Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Overvatn A, Bjorkoy G, Johansen T (2007) p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 282:24131–24145. doi:10.1074/jbc.M702824200
Predmore JM, Wang P, Davis F, Bartolone S, Westfall MV, Dyke DB, Pagani F, Powell SR, Day SM (2010) Ubiquitin proteasome dysfunction in human hypertrophic and dilated cardiomyopathies. Circulation 121:997–1004. doi:10.1161/CIRCULATIONAHA.109.904557
Razeghi P, Baskin KK, Sharma S, Young ME, Stepkowski S, Essop MF, Taegtmeyer H (2006) Atrophy, hypertrophy, and hypoxemia induce transcriptional regulators of the ubiquitin proteasome system in the rat heart. Biochem Biophys Res Commun 342:361–364. doi:10.1016/j.bbrc.2006.01.163
Richard P, Charron P, Carrier L, Ledeuil C, Cheav T, Pichereau C, Benaiche A, Isnard R, Dubourg O, Burban M, Gueffet JP, Millaire A, Desnos M, Schwartz K, Hainque B, Komajda M (2003) Hypertrophic Cardiomyopathy: Distribution of disease genes, spectrum of mutations and implications for molecular diagnosis strategy. Circulation 107:2227–2232. doi:10.1161/01.CIR.0000066323.15244.54
Rock KL, Gramm C, Rothstein L, Clark K, Stein R, Dick L, Hwang D, Goldberg AL (1994) Inhibitors of the proteasome block the degradation of most cell proteins and the generation of peptides presented on MHC class I molecules. Cell 78:761–771 pii:S0092-8674(94)90462-6
Rottbauer W, Gautel M, Zehelein J, Labeit S, Franz WM, Fischer C, Vollrath B, Mall G, Dietz R, Kubler W, Katus HA (1997) Novel splice donor site mutation in the cardiac myosin-binding protein-C gene in familial hypertrophic cardiomyopathy: characterization of cardiac transcript and protein. J Clin Invest 100:475–482. doi:10.1172/JCI119555
Sarikas A, Carrier L, Schenke C, Doll D, Flavigny J, Lindenberg KS, Eschenhagen T, Zolk O (2005) Impairment of the ubiquitin-proteasome system by truncated cardiac myosin binding protein C mutants. Cardiovasc Res 66:33–44. doi:10.1016/j.cardiores.2005.12.021
Schlossarek S, Carrier L (2011) The ubiquitin-proteasome system in cardiomyopathies. Curr Opin Cardiol 26:190–195. doi:10.1097/HCO.0b013e32834598fe
Schlossarek S, Mearini G, Carrier L (2011) Cardiac myosin-binding protein C in hypertrophic cardiomyopathy: mechanisms and therapeutic opportunities. J Mol Cell Cardiol 50:613–620. doi:10.1016/j.yjmcc.2011.01.014
Schlossarek S, Schuermann F, Mearini G, Geertz B, Eschenhagen T, Carrier L (2012) Adrenergic stress reveals septal hypertrophy and proteasome impairment in heterozygous Mybpc3-targeted knock-in mice. J Muscle Res Cell Motil doi:10.1007/s10974-011-9273-6 (in press)
Su H, Wang X (2010) The ubiquitin-proteasome system in cardiac proteinopathy: a quality control perspective. Cardiovasc Res 85:253–262. doi:10.1093/cvr/cvp287
Tanida I (2011) Autophagosome formation and molecular mechanism of autophagy. Antioxid Redox Signal 14:2201–2214. doi:10.1089/ars.2010.3482
Tannous P, Zhu H, Nemchenko A, Berry JM, Johnstone JL, Shelton JM, Miller FJ Jr, Rothermel BA, Hill JA (2008) Intracellular protein aggregation is a proximal trigger of cardiomyocyte autophagy. Circulation 117:3070–3078. doi:10.1161/CIRCULATIONAHA.107.763870
Tsukamoto O, Minamino T, Okada K, Shintani Y, Takashima S, Kato H, Liao Y, Okazaki H, Asai M, Hirata A, Fujita M, Asano Y, Yamazaki S, Asanuma H, Hori M, Kitakaze M (2006) Depression of proteasome activities during the progression of cardiac dysfunction in pressure-overloaded heart of mice. Biochem Biophys Res Commun 340:1125–1133 doi:10.1016/j.bbrc.2005.12.120
van Dijk SJ, Dooijes D, Dos Remedios C, Michels M, Lamers JM, Winegrad S, Schlossarek S, Carrier L, Ten Cate FJ, Stienen GJ, van der Velden J (2009) Cardiac myosin-binding protein C mutations and hypertrophic cardiomyopathy haploinsufficiency, deranged phosphorylation, and cardiomyocyte dysfunction. Circulation 119:1473–1483. doi:10.1161/CIRCULATIONAHA.108.838672
Vignier N, Schlossarek S, Fraysse B, Mearini G, Kramer E, Pointu H, Mougenot N, Guiard J, Reimer R, Hohenberg H, Schwartz K, Vernet M, Eschenhagen T, Carrier L (2009) Nonsense-mediated mRNA decay and ubiquitin-proteasome system regulate cardiac myosin-binding protein C mutant levels in cardiomyopathic mice. Circ Res 105:239–248. doi:10.1161/CIRCRESAHA.109.201251
Voigt A, Bartel K, Egerer K, Trimpert C, Feist E, Gericke C, Kandolf R, Klingel K, Kuckelkorn U, Stangl K, Felix SB, Baumann G, Kloetzel PM, Staudt A (2010) Humoral anti-proteasomal autoimmunity in dilated cardiomyopathy. Basic Res Cardiol 105:9–18. doi:10.1007/s00395-009-0061-z
Voigt A, Trimpert C, Bartel K, Egerer K, Kuckelkorn U, Feist E, Gericke C, Klingel K, Kandolf R, Felix SB, Baumann G, Kloetzel PM, Stangl K, Staudt A (2010) Lack of evidence for a pathogenic role of proteasome-directed autoimmunity in dilated cardiomyopathy. Basic Res Cardiol 105:557–567. doi:10.1007/s00395-010-0096-1
Weekes J, Morrison K, Mullen A, Wait R, Barton P, Dunn MJ (2003) Hyperubiquitination of proteins in dilated cardiomyopathy. Proteomics 3:208–216. doi:10.1002/(SICI)1522-2683(19990101)20:4/5<898:AID-ELPS898>3.0.CO;2-B
Yang Q, Sanbe A, Osinska H, Hewett TE, Klevitsky R, Robbins J (1999) In vivo modeling of myosin binding protein C familial hypertrophic cardiomyopathy. Circ Res 85:841–847
Zheng Q, Li J, Wang X (2009) Interplay between the ubiquitin-proteasome system and autophagy in proteinopathies. Int J Physiol Pathophysiol Pharmacol 1:127–142
Zheng Q, Wang X (2010) Autophagy and the ubiquitin-proteasome system in cardiac dysfunction. Panminerva Med 52:9–25 pii:R41102476
Zhu H, Tannous P, Johnstone JL, Kong Y, Shelton JM, Richardson JA, Le V, Levine B, Rothermel BA, Hill JA (2007) Cardiac autophagy is a maladaptive response to hemodynamic stress. J Clin Invest 117:1782–1793. doi:10.1172/JCI27523
Zolk O, Schenke C, Sarikas A (2006) The ubiquitin-proteasome system: focus on the heart. Cardiovasc Res 70:410–421. doi:10.1016/j.cardiores.2005.12.021
Acknowledgments
We thank Nico Dantuma (Stockholm, Sweden) for providing the UbG76V-GFP transgenic mice, as well as Elisabeth Krämer and Birgit Geertz (Hamburg) for technical help. This work was supported by the sixth and seventh Framework Programs of the European Union (Marie Curie EXT-014051; Health-F2-2009-241577-Big-Heart project), the Deutsche Forschungsgemeinschaft (FOR-604-CA 618/1-1 and 1-2), and the Leducq Foundation (Research grant Nr. 11, CVD 04).
Conflict of interest
None.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Schlossarek, S., Englmann, D.R., Sultan, K.R. et al. Defective proteolytic systems in Mybpc3-targeted mice with cardiac hypertrophy. Basic Res Cardiol 107, 235 (2012). https://doi.org/10.1007/s00395-011-0235-3
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00395-011-0235-3