
Abstract
Eurycope producta Sars, 1868 and Eurycope inermis Hansen, 1916 are two widely distributed and highly abundant isopod species complexes within Icelandic waters, a region known for its highly variable environment. The two species complexes have bathymetric depth ranges from 103 to 2029 m (E. producta) and from 302 to 2113 m (E. inermis). Molecular evidence was used for species delimitation within these species complexes by analyzing nuclear (18S rDNA, H3) and mitochondrial (16S rDNA, COI) sequence data. Tree-based methods (BI and ML) and four species delimitation methods (ABGD, GMYC, NDT, PTP) were applied, in order to disentangle the two species complexes. A total of eight and four species clades could be identified within samples of the E. producta and E. inermis complexes and respectively included the closely related species E. dahli Svavarsson, 1987; E. hanseni Ohlin, 1901; and E. cornuta Sars, 1864. The morphological findings coincide with the observed molecular species clades. The elucidated species clades were geographically and bathymetrically much more restricted than previously assumed. Eight species clades featured depth spans of less than 400 m and only four species clades featured depth spans of 1000 to 1500 m. Only two species clades (E. producta sensu stricto and E. inermis sensu stricto) were found on both sides of the Greenland-Scotland Ridge. Further, species distribution maps were generated using random forest, to predict potential distributional patterns for the resolved species clades of the two species complexes. We present the first attempt of combining morphological, molecular, and species distribution models in marine isopods thus far.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Species are regarded as the fundamental unit of biodiversity (Claridge et al. 1997) and, thus, are of major importance not only for taxonomists and evolutionary biologists, but also for ecologists and conservationists (Harrison 1998; Kunz 2001). Species delimitation was unavoidably dominated by morphological data evaluation for centuries (Fujita et al. 2012). New integrative taxonomic approaches of species delimitation that include morphological, genetic, behavioral, and/or ecological data can make species delimitation more robust (Sites and Marshall 2004; Dayrat 2005; Leaché et al. 2009; Padial et al. 2010).
Molecular analyses of the population structure and diversity of deep-sea benthic invertebrates have become more common within the last two decades and suggest that recently morphologically determined widespread species are likely to represent cryptic species (e.g., France and Kocher 1996; Etter et al. 1999; Raupach and Wägele 2006; Raupach et al. 2007; Brix et al. 2011) or species complexes (e.g., Brökeland and Raupach 2008; Havermans et al. 2013). Molecular species identification has been well supported by the classical gene for DNA barcoding, the mitochondrial cytochrome oxidase subunit I (COI; Hebert et al. 2003). However, COI can be difficult to amplify in asellote isopods (e.g., Raupach et al. 2007; Brökeland and Raupach 2008; Riehl and Kaiser 2012; Riehl et al. 2017). Thus, the mitochondrial ribosomal RNA large subunit (16S) has been used for asellote isopods as a replacement barcode marker in various studies (e.g., Raupach and Wägele 2006; Riehl and Kaiser 2012; Kaiser et al. 2017; Riehl et al. 2017; Bober et al. 2018; Brix et al. 2018). Further, the inclusion of a nuclear gene has been shown to prevent the challenges of incomplete lineage sorting and introgression (Rubinoff and Holland 2005; Galtier et al. 2009).
Crustaceans (Arthropoda) are ubiquitous in the marine benthos and appear to be very diverse, considering the number of species and their large range of observed morphologies (Hessler 1981). Asellote isopods in particular are considered to be the most numerous crustacean taxon encountered within the deep-sea macrobenthos (Sanders et al. 1965; Sanders and Hessler 1969; Brandt et al. 2007). Munnopsidae Lilljeborg, 1864 is one of the most diverse and abundant isopod families in the deep sea (Sanders and Hessler 1969; Wilson and Hessler 1987) and features a known depth range from 4 m (Svavarsson et al. 1993) to 9345 m (Birstein 1971). The family contains 42 genera and currently more than 320 species (Wilson and Schotte 2017). Munnopsids lack (like all other peracarid crustaceans) planktonic larvae; instead, their development takes place in the brood pouch (marsupium) of females. Most munnopsids are, in contrast to other asellote isopods, able to swim, or at least able to be active in the near-bottom water layer. Gene flow depends only on the active and/or passive (e.g., by currents) dispersal of adults (Wilson 1989; Brandt 1992; Marshall and Diebel 1995).
Munnopsid isopods are a common component of the fauna within the highly variable environment at the transition between the northernmost North Atlantic and the Nordic Seas (Svavarsson et al. 1993; Schnurr et al. 2014). The subfamily Eurycopinae Hansen, 1916 is the most diverse group within munnopsid isopods (Svavarsson 1987). The genus Eurycope Sars, 1864 is especially speciose and known to be complex in comparison to the other genera within the subfamily (Wilson 1983a; Kussakin 2003). Molecular phylogenetic analysis showed the paraphyly of the genus (Osborn 2009), and multiple authors have discussed the diversity of Eurycope, as well as the presence of species complexes within the clade (Wolff 1962; Wilson and Hessler 1981; Wilson 1989; Malyutina and Brandt 2006). This problematic genus is in need of revision, in no small part because of its ubiquitous presence in a topographically and hydrologically complex region.
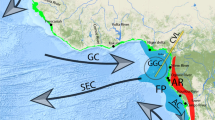
The oceanic conditions around Iceland are shaped by the Greenland-Scotland Ridge (GSR), a topographic feature that separates the deep-sea basins of the northernmost North Atlantic and the Greenland, Iceland, and Norwegian Seas (the Nordic Seas). The ridge system features a mean depth of around 500 m with three deep sills, each on a different portion of the ridge. The maximum depth of the GSR (840 m) is located in the Faroe Bank Channel between the Faroe Islands and Scotland. The maximum depth in the Denmark Strait between Greenland and Iceland is 620 m, whereas the maximum depth of the Iceland-Faroe Ridge (IFR) is 480 m (Hansen and Østerhus 2000). The near-bottom water masses exhibit major temperature differences ranging from -1 up to 12–14 °C (Jochumsen et al. 2016). Direct exchanges of deep water masses between the deep basins of the northern North Atlantic and the Nordic Seas across the GSR are not possible, and thus, only limited exchanges of intermediate layers take place through the deep channels (Hansen and Østerhus 2000). Water transport across the ridge at depth is of major importance to global thermohaline circulation and thus for the regional climate and oceanic regions north of this submarine barrier (Hansen and Østerhus 2000). Hence, species distributional patterns and distributional limits within this highly variable environment are especially interesting. Previous studies on benthic invertebrates within the area observed distributional limits in connection to the GSR and abiotic factors associated with the ridge (e.g., Svavarsson et al. 1990, 1993; Svavarsson 1997; Dijkstra et al. 2009; Brix and Svavarsson 2010; Dauvin et al. 2012).
Combining morphological, genetic, and ecological approaches in order to determine mechanisms that shape the geographic distribution of species has become more common, especially in terrestrial environments (e.g., Johnson and Cicero 2002; McCallum et al. 2014). However, sampling in the vast oceanic environment relies on more localized data, and the major limitations of sampling make it difficult to collect sufficient data for species distribution modeling. Although species distribution models (SDMs), which use spatial environmental variables, can lead to a better understanding of species distribution patterns even within the less accessible marine environment (Elith and Graham 2009), only a few SDMs of benthic marine invertebrates have been constructed so far (e.g., Meißner et al. 2008; Elith and Graham 2009; Meißner et al. 2014). However, a combination of morphological, genetic, and ecological approaches has not been applied to marine benthic isopods thus far.
In this study, we sampled and analyzed specimens of Eurycope producta Sars, 1868 and Eurycope inermis Hansen, 1916 around Iceland, which were suspected to represent species complexes (Wilson 1982; Svavarsson 1987). We hypothesize that (1) multiple species clades within both taxa can be identified using multiple genetic loci, (2) genetically distinct clades within each species complex can be identified by morphological key characters, (3) the resolved species clades are separated from each other by natural geological or hydrological barriers, and (4) species distribution maps for the resolved species clades within both species complexes can predict more complete species distribution patterns.
Material and methods
Sampling and sequencing
Specimens of the E. producta complex and of the E. inermis complex were examined morphologically and genetically. The datasets of both species complexes included closely related sister species, which are morphologically similar and also present within the sampled research area. Those known species were included in the analyses particularly in regard to the need of a morphological revision of the genus Eurycope, which will be part of a future study. Thus, species that are already known to science, but look similar to the E. producta and E. inermis complexes, were also included in the dataset. The analyzed E. producta complex dataset contained 83 specimens (including specimens of E. dahli Svavarsson, 1987) and the E. inermis complex dataset contained 102 specimens (including specimens of E. hanseni Ohlin, 1901 and E. cornuta Sars, 1864). Hence, hereafter they will be referred to as E. producta and E. inermis species complexes and base our confirmation of named species and identification of new species on our genetic and morphological analyses.
All specimens were sampled around Iceland with three different types of epibenthic sleds (EBS; Rothlisberg and Pearcy 1977; Brenke 2005; Brandt et al. 2013) during the IceAGE1 and IceAGE2 (Icelandic marine Animals: Genetics and Ecology) expeditions in 2011 and 2013, respectively (Fig. 1). Bulk samples were immediately fixed on deck in chilled 96% nondenatured ethanol and kept cool throughout the sorting process according to Riehl et al. (2014). Subsamples from the EBS stations were sorted on board and at the DZMB (German Centre for Marine Biodiversity Research, Hamburg). One to three posterior pereopods (legs, depending on the size of the individual) of E. producta and E. inermis specimens were dissected and separately stored for tissue digestion and DNA amplification. This semidestructive approach was conducted in order to allow further morphological analyses of each specimen. Polymerase chain reactions (PCR) were performed on all specimens for 16S, COI, 18S, and H3 (see Table 1 for a list of the primers used). However, it was not possible to obtain sequences from all four loci for all the specimens, even after several rounds of PCR optimization (see Table 2 for a list of available sequences and GenBank accession numbers of COI, 16S, 18S, and H3). The extraction and PCR protocols for 16S, COI, and 18S followed the methods of Riehl et al. (2014) and Brix et al. (2011). Extractions of H3 followed the methods described by Riehl et al. (2014) and Brix et al. (2011). Polymerase chain reaction of H3 comprised an initial 5-min denaturation at 95 °C, followed by 4 cycles of 30 s at 94 °C, 45 s at 50 °C, 60 s at 72 °C, followed by 34 cycles of 30 s at 94 °C, 45 s at 47 °C, 60 s at 72 °C. The cycling ended with an 8-min extension at 72 °C. The H3 primers H3ar/H3af from Colgan et al. (1998) were used for amplification. ExoSap-IT (USB) was used for purification of PCR products. Cycle sequencing of purified products was performed with BigDye chemistry (Perkin-Elmer) by 30 cycles of 30 s at 95 °C, 30 s at 50 °C, and 4 min at 60 °C. Sequences were obtained with an ABI 3730xl 96-well capillary sequencer. All the sequencing of the individuals used in this study was conducted at the Laboratories of Analytical Biology (LAB), Smithsonian National Museum of Natural History, Washington, DC, USA.

Location of the sampled stations of the IceAGE1 and IceAGE2 cruises used in the current study
All individuals of the two putative species complexes were analyzed morphologically. Drawings were created following the guidelines of Wilson (2008) and Hessler (1970). Adobe Illustrator CS6 (http://www.adobe.com/products/illustrator.html) was used for finalizing the drawings following the guidelines of Coleman (2003, 2009). Only characters needed for determination of the species are presented within this study.
Specimens used in this study are stored at the Zoological Museum of Hamburg (ZMH K-45583–K-45765; Table 2).
Genetic analyses
The forward and reverse sequences of each individual were assembled using Geneious v. 7.0.4 (Biomatters; available from www.geneious.com). All consensus sequences were manually edited and checked. The COI and H3 consensus sequences were translated into amino-acid sequences in order to prevent the inclusion of pseudogenes (Buhay 2009). Further, all consensus sequences were compared against the GenBank nucleotide database by using BLASTN (Altschul et al. 1990). Afterwards, the edited consensus sequences of 16S, COI, 18S, and H3 were aligned using the default settings of MAFFT v. 7.017 (Katoh et al. 2002) under the E-INS-i option and alignments were manually edited, if needed. Eurycope complanata Bonnier, 1896 (GenBank accession no: 16S: MH101741; COI: EF682281; 18S: EF682256) and Eurycope elianae Schnurr and Malyutina, 2014 (GenBank accession no: 16S: KJ716799; COI: MH056597; 18S: KJ716804) were used as an outgroup for E. producta and E. inermis, respectively. All sequences produced for this project can be retrieved from GenBank (see Table 2 for accession numbers). The final alignments of the E. producta complex (18S, 73 sequences, with an alignment length of 2142 bp; 16S, 66 sequences, with an alignment length of 421 bp; COI, 33 sequences, with an alignment length of 601 bp) and the E. inermis complex (18S, 98 sequences, with an alignment length of 2113 bp; 16S, 76 sequences, with an alignment length of 435 bp; COI, 47 sequences, with an alignment length of 600 bp) can be retrieved from TreeBase (http://purl.org/phylo/treebase/phylows/study/TB2:S22443). Because nodal support of H3 analyses was low in both species complexes (although respective species clades appeared to cluster together), H3 sequences were only used in the concatenated dataset. Thus, concatenated alignments of 16S, COI, 18S, and H3 were created for each species complex, using SequenceMatrix (Meier et al. 2006), and were used to reconstruct species trees in addition to the six single gene alignments.
Bayesian inference (BI) and maximum likelihood (ML) tree construction methods were used in order to identify possible clades within the two putative species complexes. The best-fitting substitution model of DNA sequence evolution was identified with MrAIC (Nylander 2004) for each alignment under the Akaike’s information criterion (AIC). Bayesian trees were obtained with MrBayes v. 3.2 (Ronquist et al. 2012). Two independent runs were conducted for 100 million generations each, where every 2000th generation was sampled (resulting in 50,000 trees), using three heated and one cold chains. The program Are We There Yet (AWTY) (Wilgenbusch et al. 2004) was used to determine if stable posterior probabilities had been reached. Consensus trees of single loci datasets as well as concatenated partitioned datasets were calculated with MrBayes, considering the model of nucleotide substitution estimated by MrAIC, with a burn-in of 15,000 generations. The models for the single loci datasets and partitions of the concatenated datasets were GTR+G+I for 18S and GTR+G for 16S, COI, and H3 for the E. producta complex datasets and GTR+G+I for 18S, 16S, and COI and GTR+G for H3 for the E. inermis complex datasets. Posterior probabilities of < 0.9 were collapsed into polytomies.
Maximum likelihood trees were obtained using RAxML v. 7.2.8 (Stamatakis et al. 2008) using a total of 10,000 replicates for bootstrap calculations (Felsenstein 1985). All trees were visualized with FigTree v1.3.1 (http://tree.bio.ed.ac.uk/software/figtree/) and prepared for publication with Adobe Illustrator. Bootstrap percentages of < 75 were collapsed into polytomies.
Relationships between haplotypes of 16S, COI, and 18S datasets were explored for each species complex with TCS v. 1.21 (Clement et al. 2000). Gaps were treated as fifth states and the probability threshold was set to 95% (Clement et al. 2000; Templeton 2001). Haplotype networks are not displayed, but shared haplotypes are indicated in the tree figures (Figs. 2, 3, 4, and 5).

Consensus Bayesian tree for E. producta a 16S and b COI datasets. The branch lengths are proportional to the number of substitutions per site considering the models of nucleotide substitution estimated by MrAIC for the respective loci. Posterior probabilities (> 0.9) from Bayesian analyses and bootstrap percentages (> 70) from maximum likelihood trees are indicated at the nodes. The colored vertical bars represent different species clades supported by ABGD at different thresholds a for 16S (aa) 0.00100–0.001668, (ab) 0.002783–0.007743, (ac) 0.012915–0.035938, and (ad) 0.059948 and b for COI (ba) 0.001000–0.001668, (bb) 0.002783, and (bc) 0.004642–0.100000 as well as species clades supported by NDT, GMYC, bPTP, and morphology. The different clusters within the dataset are named Ep_1–8

Consensus Bayesian tree for E. producta a 18S and b the consensus of the concatenated four gene loci dataset (16S, COI, 18S, and H3). The branch lengths are proportional to the number of substitutions per site considering the models of nucleotide substitution estimated by MrAIC for the respective loci or partition. Posterior probabilities (> 0.9) from Bayesian analyses and bootstrap percentages (> 70) from maximum likelihood trees are indicated at the nodes. The colored vertical bars represent different species clades supported by ABGD at different thresholds a for 18S (aa) 0.001000–0.001668, (ab) 0.002783, and (ac) 0.004642 as well as species clades supported by NDT, GMYC, bPTP, and morphology. b E. producta consensus of the concatenated dataset. Depth ranges of each species cluster and sampling region are included. The different clusters within the dataset are named Ep_1–8

Consensus Bayesian tree for E. inermis a 16S and b COI datasets. The branch lengths are proportional to the number of substitutions per site considering the models of nucleotide substitution estimated by MrAIC for the respective loci. Posterior probabilities (> 0.9) from Bayesian analyses and bootstrap percentages (> 70) from maximum likelihood trees are indicated at the nodes. The colored vertical bars represent different species clades supported by ABGD at different thresholds a for 16S (aa) 0.001000–0.001668, (ab) 0.002783–0.021544, and (ac) 0.035938–0.059948 and b for COI (ba) 0.001–0.001668 and (bb) 0.002783–0.100000, as well as species clades supported by NDT, GMYC, bPTP, and morphology. The different clusters within the dataset are named Ei_A–E

Consensus Bayesian tree for E. inermis a 18S and b the consensus of the concatenated four gene loci dataset (16S, COI, 18S, and H3). The branch lengths are proportional to the number of substitutions per site considering the models of nucleotide substitution estimated by MrAIC for the respective loci. Posterior probabilities (> 0.9) from Bayesian analyses and bootstrap percentages (> 70) from maximum likelihood trees are indicated at the nodes. The colored vertical bars represent different species clades supported by ABGD at different thresholds a for 18S (aa) 0.001000–0.007743 and (ab) 0.012915–0.035938, as well as species clades supported by NDT, GMYC, bPTP, and morphology. b E. inermis consensus of the concatenated dataset. Depth ranges of each species cluster and sampling region are included. The different clusters within the dataset are named Ei_A–E
Uncorrected p-distances of the 16S, COI, and 18S single gene datasets were calculated with MEGA v.6.06 (Tamura et al. 2013) and used for comparing the genetic variability within clades (Tables 3 and 4; Online resources 1–2).
Species delimitation
Four different methods of species delimitation were conducted on 16S, COI, and 18S alignments for each species complex in order to delimit species within the complexes: ABGD, nucleotide divergence threshold (NDT), generalized mixed Yule coalescent (GMYC) model, and the Poisson tree process (PTP) model.
The ABGD by Puillandre et al. (2012) is an automated iterative method, which groups specimens based on pairwise distance measures. Sequences are automatically grouped by assuming that the distance between different species is always larger than within species. Thus, the sequences are grouped on the basis of the automatically determined significant differences, the barcoding gap. Alignments of 16S, COI, and 18S were uploaded to the online server of ABGD (http://wwwabi.snv.jussieu.fr/public/abgd/abgdweb.html) without outgroups by using the default settings and the Kimura (K80) mutational model.
The NDT analysis after Tang et al. (2012) clusters sequences in an alignment based on an uncorrected distance matrix and a threshold, which must be defined by the user. We used a threshold of 97% for the alignments of the three different gene loci. The R script of the NDT analysis by Tang et al. (2012) was run in RStudio v.0.97.318.
The GMYC approach by Monaghan et al. (2009) and Pons et al. (2006) is a maximum likelihood method that identifies the significant shift in a gene tree from within-species (e.g., coalescence) events to between-species events (e.g., speciation) on an ultrametric phylogenetic tree without an outgroup. The branching rates between and within species are used to identify where the most likely point of shift is, compared to a null model (all specimens derived from a single species). The analysis was run in RStudio using the package ‘splits’ (Edzard et al. 2009). Prior to the analysis, an ultrametric input tree was generated with BEAST v.1.8 (Drummond et al. 2012), using a relaxed lognormal clock with a coalescent prior. MCMC analyses were run for 100 million generations, with every 2000th step sampled. The burn-in was set to 0.25%. The MCMC output was analyzed with AWTY and trees were assembled with Tree Annotator (Rambaut and Drummond 2007).
The PTP model by Zhang et al. (2013) models speciation or branching events in terms of substitutions. We used the Bayesian (bPTP) implementation within our study, which also accepts multifurcating phylogenetic trees (and even zero branch lengths). The branch lengths of the phylogenetic input tree have to represent the number of substitutions. Unrooted phylogenetic trees, without an outgroup, created by MrBayes were uploaded to the online server of bPTP (http://species.h-its.org/ptp/). The following parameters were used: MCMC, 500,000 generations; thinning, 100; burn-in, 0.25; and seed, 123. Further, convergence was always checked in order to be sure that sufficient generations had been conducted.
Species distribution modeling
The assumption of SDM is to predict spatial distributions of (for instance) species by using presence and (if available) absence data. The datasets are then combined with predictor variables (which cover the whole research area). Random forest (RF) is a machine learning method (Breiman 2001), that uses recursive partitioning to create decision trees. A great number of subtrees are created using a random selection of variables and observations. The best splits of all the subtrees are then merged into a final ensemble tree.
Nine layers of environmental predictors recorded from across the full research area were used for the creation of SDMs (see Meißner et al. 2014, Table 1). The predictors used within this study were bottom depth (ETOPO2v2 2006); near-bottom temperature, temperature difference, and salinity (Nilsen et al. 2008; Jochumsen et al. 2016); bottom oxygen (Seiter et al. 2005); seasonal variation index (SVI; Lutz et al. 2007); particulate organic carbon flux (POC; Lutz et al. 2007); bottom roughness (Whittaker et al. 2008); and sediment thickness (Divins 2003). Only species clades obtained from at least two stations were used for the creation of SDMs. Data were imported into QGIS v.2.0.1 (http://qgis.osgeo.org). The values associated with the different layers were then extracted using the ‘point sampling’ tool of QGIS. Further, a total of 22,139 points regularly distributed throughout the research area were generated with QGIS, and the corresponding predictor values were extracted to be used for generating the SDMs with random forest (Breiman 2001). Random forest models were calculated in RStudio with the package ‘randomForest’ (Liaw and Wiener 2002). A total of 6000 random trees (‘ntree’ option) and 4 randomly chosen predictors (‘mtry option’) were chosen for all the species. The values of the ‘sampsize’ option were adjusted to the number of presence records of each species, so that the same number of presences and absences were always used for each randomly created tree, in order to avoid biased accuracy of the ‘absent’ class in the model.
Final prediction maps were generated with GMT v. 5.1.0 (Generic Mapping Tool; SOEST; http://gmt.soest.hawaii.edu/doc/5.1.0/). Interpolation was conducted with the ‘surface’ function, using a tension factor of 0.5 and a gridding space of 0.005. Predictions higher than 0.5 most likely represent the actual distribution of the respective species. Finally, positions of the presence records of the respective species were plotted on top of the interpolated SDMs.
Results
Genetic analyses
ML and BI tree reconstruction revealed identical tree topologies, with mostly comparable node support in both approaches; therefore, only BI trees are shown. Eight clades could be observed within the E. producta complex datasets (Figs. 2 and 3). Analysis of the E. inermis complex yielded five or four different clades, depending on the locus (Figs. 4 and 5). However, some discrepancies in node support between the two approaches were apparent in the 16S and COI datasets of the E. producta complex, wherein on some branches good support was obtained in the BI tree, but lower support in the ML tree (Figs. 4 and 5). The 16S and 18S alignments of the E. producta complex contained sequences for all eight clades, and those for the E. inermis complex contained five clades in the 16S dataset and four in the 18S dataset. The sequencing success of COI was lowest, even after several rounds of PCR optimization. Therefore, the COI alignment of E. producta complex lacks sequences for Ep_1, Ep_6, and Ep_7 and for Ei_B of the E. inermis complex (Table 2).
The E. producta complex 16S dataset contained 66 sequences with 27 haplotypes (Fig. 2a). The number of potential species predicted by ABGD varied with different prior thresholds, which ranged from 0.001000 to 0.059948. A total of 23 potential species were predicted under the lowest threshold (0.001000–0.001668), at which almost every haplotype clustered as a separate species. The prior threshold from 0.012915 to 0.035938 led to eight clusters, which corresponded with the number of clusters achieved by BI, ML, NDT, GMYC, bPTP, and morphology. The E. producta complex COI dataset contained 33 sequences with 20 haplotypes (Fig. 2b). The prior thresholds of ABGD ranged from 0.001 to 0.1. The results of the highest threshold (0.004642–0.1) showed the same species clusters determined by BI, ML, NDT, GMYC, bPTP, and morphology. The E. producta complex 18S dataset consisted of 73 sequences and featured 16 haplotypes (Fig. 3a). The prior thresholds ranged from 0.001 to 0.004642 and the lowest threshold (0.001–0.001668) showed the same species clusters as produced by BI, ML, GMYC, bPTP, and morphology. Results of NDT at a level of 97% similarity were not conclusive. The concatenated dataset including all four loci (16S, COI, 18S, and H3) revealed the same species clusters observed in the single gene analyses (Fig. 3b). All mtDNA haplotype networks of individual species clades were unconnected to each other at a similarity level of 95%. The 16S network of Ep_1 was further split into two unconnected networks. The nuDNA networks of Ep_1, Ep_2, Ep_5, Ep_6, and Ep_7 were connected at a similarity level of 95%.
The E. inermis complex 16S dataset consisted of 75 sequences, featuring 20 haplotypes (Fig. 4a). The number of potential species clusters predicted by ABGD varied among different prior thresholds, which ranged from 0.001 to 0.059948. The same clusters predicted by BI, ML, NDT, and GMYC were recovered at the intermediate threshold (0.002783–0.021544). The bPTP approach split Ei_A into two species. The E. inermis complex COI dataset contained 46 sequences and a total of 24 different haplotypes (Fig. 4b). The prior threshold of ABGD ranged from 0.001 to 0.1. Almost every haplotype was predicted to be a species at the lowest threshold. The clustering results of the threshold from 0.002783 to 0.1 coincided with BI, ML, NDT, GMYC, and bPTP results. The E. inermis complex 18S dataset consisted of 97 sequences with 10 haplotypes (Fig. 5a). The prior threshold ranged from 0.001 to 0.035938. Specimens of E. inermis complex Ei_B and Ei_C could not be distinguished from each other by ABGD and GMYC based on 18S data. The highest prior threshold (0.012915–0.035938) predicted only two species clusters. Only bPTP revealed five species clusters. Moreover, analysis of the concatenated dataset revealed the same species clusters as observed within the single locus 16S tree (Fig. 5b). All the mtDNA haplotype networks of the E. inermis complex species clades were unconnected at a similarity level of 95%. Only the nuDNA networks of Ei_B and Ei_C were connected on a similarity level of 95%.
A clear gap between intra- and interclade divergences was observed for all loci of both species complexes, with one exception within the 18S dataset of the E. inermis complex (Fig. 6), where no barcoding gap was detected. However, the barcoding gap became visible when specimens of Ei_B and Ei_C of the E. inermis complex were combined into one species (data not shown).

Histograms show the percentage of the p-distances within and between the specimens of the E. producta and E. inermis datasets. The barcoding gap between intraspecific (dark gray bars) and interspecific (light gray bars) variability is indicated by a black arrow. Barcoding gap histogram of 16S a E. producta and b E. inermis, of COI c E. producta and d E. inermis, and of 18S e E. producta and f E. inermis
Analysis of uncorrected p-distances of mtDNA and nuDNA sequences of both species complexes also supported the existence of eight and five different species within the E. producta (Table 3) and the E. inermis complex (Table 4) datasets, respectively (see also Online resources 1–2 for a detailed documentation of uncorrected pairwise p-distances of the 16S gene of E. producta and E. inermis). Intraclade divergences were low in the E. producta complex dataset (16S, 0.0–2.50%; COI, 0.0–1.88%; 18S, 0.0–0.10%) as well as within the E. inermis complex dataset (16S, 0.0–1.20%; COI, 0.0–1.95%; 18S, 0.0–0.14%). Intraclade variation for the E. producta complex was highest at 16S in Ep_1 (2.5%) as well as for the E. inermis complex in Ei_B (1.19%) and Ei_C (1.20%). Interclade divergences were higher than intraclade divergences in the E. producta complex dataset (16S, 4.90–23.40%; COI, 19.06–30.31%; 18S, 0.20–4.10%) as well as within the E. inermis complex dataset (16S, 2.83–25.41%; COI, 17.5–27.11%; 18S, 0.10–4.01%). Interclade distances for the E. producta complex were lowest at 16S in Ep_2 and Ep_3 (both 4.90%) and for E. inermis complex Ei_B and Ei_C (both 2.83%; Tables 3 and 4). However, when fusing Ei_B and Ei_C to one potential species clade, the interclade distances of 16S range from 8.96 to 25.41%.
Morphological analyses
Morphological evaluation of the samples of this study revealed small, but visible differences between the different genetically delimited species clades within the two complexes, with one exception: specimens of E. inermis complex Ei_B and Ei_C appeared to be morphologically identical. Males and females were present and studied for all species clades, except for E. producta complex Ep_8, where only females were present within the evaluated specimens. Examples of some morphological interclade differences of the two species complexes are shown in Figs. 7 and 8 for the E. producta complex and the E. inermis complex, respectively.

Habitus drawings of E. producta Ep_1–8 (upper row), magnification of the rostrum (middle row), and male pleopod 1 (lower row). The relative size and shape of the rostrum (r) compared to the size and shape of article 1 of the first antenna (art 1) in combination with the shape of the distal margin of the male pleopod 1 are useful characters to distinguish species within the E. producta complex. Scale bar habitus 1 mm and pleopod 0.1 mm

Habitus drawings of E. inermis Ei_A–E (upper row), magnification of the rostrum (middle row), and male pleopod 1 (lower row). The relative size and shape of the rostrum (r) compared to the size and shape of article 1 of the first antenna (art 1) in combination with the shape of the distal margin of the male pleopod 1 are useful characters to distinguish species within the E. inermis complex. No drawings are presented for Ei_C, since there were no morphological differences to specimens of Ei_B. Scale bar habitus 1 mm and pleopod 0.1 mm
Characters potentially useful in distinguishing species within each complex were (1) the relative size and shape of the rostrum compared to the size and shape of article 1 of the first antenna and (2) the shape of the distal margin of the male pleopod 1. Specimens of the E. producta complex species clades Ep_1–4 have a rostrum of comparable size or smaller than article 1 (r < art1) of the first antenna, and the distal margin of the male pleopod 1 is broad and blunt cut with inner lobes not projected. In contrast, in E. producta species clades Ep_5–8, the article 1 of the first antenna is longer than the rostrum, and the male pleonite 1 tapers apically. The tip is narrow and has projected inner lobes. Some differences in morphological characters can also be observed within the evaluated E. inermis complex specimens. The species clades Ei_A and Ei_B_C have a narrower rostrum, and article 1 of the first antenna has an extended distomedial lobe, which is visibly longer than article 2. Additionally, the male pleopod 1 is tapering with inner lobes projected apically in Ei_A and Ei_B_C. Specimens of Ei_D and Ei_E feature broader rostrums, and the distomedial lobe of the first antenna is shorter, which is subequal or shorter than article 2. Similarly, the inner lobes of the male pleopod 1 distal margins are curved on the outside.
A unique combination of morphological character states could be observed between the putative species clades of both species complexes, including more morphological differences than presented herein. A thorough taxonomic description of the evaluated specimens will be part of a different study. A total of eight and four morphospecies were present within the E. producta complex and E. inermis complex datasets, respectively, including the previously described and morphologically similar species E. dahli, E. hanseni, and E. cornuta.
Species distribution modeling and bathymetric ranges of the species clades
Putative distributions using SDMs were developed for all clades of the E. producta complex (Ep_1–8; Fig. 9) and the E. inermis complex (Ei_A–E; Fig. 10), with the exception of Ep_3, since specimens belonging to this group were only sampled at a single station. Some species occur in partial sympatry (E. producta: Ep_1, Ep_2, Ep_5, and Ep_6; E. inermis: Ei_A with Ei_C and Ei_D with Ei_E). Predictions above the probability threshold of 0.5 are considered to indicate the most likely distribution potential of the respective species. The best model fit was observed for E. producta: Ep_4, Ep_5, Ep_7, and Ep_8 and for E. inermis: Ei_A and Ei_B, where the presence class error was 0% (Table 5).

a–g Species distribution modeling for the species clades of the E. producta complex. No species distribution model was created for Ep_ 3, since specimens belonging to this group were only sampled at a single station. Color scales refer to probability of occurrence, black dots indicate the presence sites of each species clade, and white dots indicate the absence of the respective species clade. Values above 0.5 are considered to indicate the most likely distribution potential of the respective species clade

a–f Species distribution modeling for the four species clades of the E. inermis complex. Clades Ei_B and Ei_C are modeled separately and together (Ei_B_C), in order to demonstrate their geographic separation by the Greenland-Scotland Ridge. Color scales refer to probability of occurrence, black dots indicate the presence sites of each species clade, and white dots indicate the absence of the respective species clade. Values above 0.5 are considered to indicate the most likely distribution potential of the respective species clade
Species could be grouped into three main categories after Schnurr et al. (2014):
-
Group 1. Northern species: Ep_2, Ei_A, Ei_C, and Ei_D; species occurring on the northern side of the GSR and across the Iceland-Faroe Ridge: Ep_6 and Ei_E
-
Group 2. Trans-GSR species: Ep_1; species occurring only across the IFR: Ep_5
-
Group 3. Southern species: Ep_3, Ep_4, Ep_7, Ep_8 and Ei_B
Eight species clades feature depth spans of less than 400 m, occur either only north of the GSR (Ep_2, Ep_6, Ei_A, and Ei_D) or south of the ridge (Ep_3, Ep_4, Ep_7, and Ep_8), and feature depth ranges that are either below or above the deepest depression of the GSR (Fig. 11). Two species clades feature a range of less than 650 m (Ep_5 and Ei_E). Both of these clades feature a depth range that includes the deepest depth of the GSR. However, only one of them (Ep_5) was present in samples across the IFR. The remaining three species feature depth spans between 1000 and 1500 m. Two of them (Ep_1 and Ei_B) feature depth ranges that include the maximum depression of the GSR, though only Ep_1 occurs north and south of the ridge. Eurycope inermis C was restricted to areas north of the ridge (Fig. 11).

Observed depth ranges of all the evaluated specimens used in this study of the species clades belonging to the two species complexes E. producta and E. inermis. The geographic distribution of the species clades is visualized in light gray bars (northern species clades; group 1), dark gray bars (northern and southern species clades; group 2), and black bars (southern species clades; group 3). Clades E. inermis B and C are visualized separately, in order to demonstrate their different depth distributions. Asterisk indicates the maximum depth of the Greenland-Scotland Ridge
Discussion
Multiple species within both species complexes
Molecular analyses of deep-sea isopods have so far been mostly restricted to maximum parsimony analyses (e.g., Raupach and Wägele 2006) or BI and ML analyses (e.g., Brix et al. 2011). Only very recently submitted work also used species delimitation methods (Kaiser et al. 2017; Brix et al. 2018). However, a combination of the four different species delimitation methods (ABGD, GMYC, NDT, and bPTP) with morphology and species distribution modeling, as used within this study, has thus far not been applied to benthic isopods. The current study provides strong molecular evidence for multiple species within the two species complexes E. producta and E. inermis, which were mostly congruent among mtDNA and nuDNA analyses.
The existence of species complexes and cryptic species has been observed in different isopod families, such as the Janiridae (Carvalho and Piertney 1997), Munnopsidae (Wilson 1982; Raupach and Wägele 2006), Parammunidae (Just and Wilson 2004), Haploniscidae (Brökeland and Raupach 2008; Brix et al. 2011), Serolidae (Held 2003; Leese et al. 2008), and Chaetiliidae (Held and Wägele 2005), potentially Desmosomatidae (Brix et al. 2014b), as well as in other peracarid crustaceans, for instance amphipods (Baird et al. 2011; Lörz et al. 2012; Havermans et al. 2013). Thus, overlooked morphologically similar species and the presence of cryptic speciation can lead to an underestimation of biodiversity (Vrijenhoek 2009).
The existence of different species within both species complexes is suggested by high statistical support for each potential species cluster (posterior probabilities > 0.95 and bootstrap values > 70) according to our multilocus analyses of mtDNA and nuDNA. Single locus as well as concatenated datasets revealed similar tree topologies indicating that gene and species trees do not differ. The results of the different species delimitation methods were largely congruent. All four delimitation methods (ABGD, NDT, GMYC, and bPTP) revealed multiple species clades within each of the two complexes, although intraclade sampling for some of the species was small (e.g., Ep_7, Ep_8, and Ei_A).
Congruence between mtDNA and nuDNA
Classic DNA barcoding (Hebert et al. 2003) is based on a distinct gap between intraspecific variability and interspecific variability in genetic distances of COI, for which a threshold of 3% for delineating species is generally recommended. However, thresholds are sometimes not applicable to all taxonomic groups and thus have to be applied carefully across taxa. Schwentner et al. (2011) determined a 5–6% threshold between intra- and interspecific divergence in branchiopods, and Radulovici et al. (2009) detected intraspecific divergence between 3.78 and 13.6% in amphipods. However, Radulovici et al. (2009) supposed that especially the larger distances can be an evidence for cryptic species in amphipods. Thus far, only a limited amount of genetic data are available for isopods and we are still at a stage of finding a recommendable threshold for this group, and therefore, we, as have recent studies, applied a threshold of 3% (e.g., Brix et al. 2018).
Species delimitation based on a single locus can lead to an under- or overestimation of the number of species, for instance due to incomplete lineage sorting or pseudogenes (Song et al. 2008). Thus, inclusion of a nuDNA marker with a different level of gene flow in combination with mtDNA markers is useful to confirm the existence of putative species (Hare 2001; Petit and Excoffier 2009). It is known that mtDNA is more sensitive to recent divergence than nuDNA (Wilson et al. 1985; Barrowclough and Zink 2009). Discordance between nuDNA and mtDNA is a sign for recent or ongoing speciation (e.g., Shaw 2002; Johnson et al. 2006), which has also been recently observed within marine taxa (e.g., Eytan et al. 2009; Reveillaud et al. 2010; Baird et al. 2011; Schüller 2011; Jennings et al. 2013; Marlétaz et al. 2017).
Intraspecific genetic divergence of mtDNA and nuDNA was low in our study in comparison to interspecific divergences (Tables 3 and 4), a finding congruent with previous studies on isopods (e.g., Raupach et al. 2009; Brix et al. 2011, 2014a, b). For instance, haploniscid isopods featured interspecific divergences of 9–20% and intraspecific divergences below 1.8% in COI (Brix et al. 2011). Interspecific divergences in macrostylid isopods based on 16S ranged between 23 and 31%, whereas intraspecific divergences were close to zero (Riehl and Brand 2013). Similar examples exist for instance for Desmosomatidae (16S data; Brix et al. 2018), Macrostylidae (16S, 18S data; Riehl et al. 2017), and Nannoniscidae (COI, 16S, 18S data; Kaiser et al. 2017). Thus, the distances observed within our dataset fall within the ranges that were previously observed in other isopod families. Interestingly, all these isopod studies as well as our dataset have one thing in common: low intraspecific divergence and high interspecific divergence.
The ‘4×’ criterion (Birky et al. 2005) was fulfilled for the three loci of both species complexes (except for the 16S and 18S dataset of E. inermis Ei_B and Ei_C, where the difference was only 2×). Further, a distinct barcoding gap could be observed in all mtDNA datasets as well as in the nuDNA datasets, except for E. inermis Ei_B and Ei_C in 18S (Fig. 6), which became visible when Ei_B and Ei_C were considered as one species. In contrast to our expectations, groups with the lowest interspecies divergences did not occur in sympatry but were either separated by the GSR (e.g., 16S and 18S in E. inermis Ei_B and Ei_C), or by depth (e.g., 16S and COI in E. producta Ep_2 and Ep_3).
The mtDNA networks of this study were all unconnected at 95% similarity (networks not shown). Particularly, the formation of unconnected parsimony haplotype networks supports the existence of separate species (Hart and Sunday 2007). It is not surprising that some of the determined nuDNA haplotype networks were connected to each other at a level of 95%, since we were examining relationships within two complexes of closely related species. However, two discordant observations were made between the 16S and 18S networks: E. producta 1 and E. inermis B and C were each split into two independent networks in the 16S datasets, but not the 18S datasets. In contrast, Ep_1 as well as Ei_B together with Ei_C formed a connected network within the nuDNA network. We assume that E. producta 1 could be at the beginning of species formation and that part of the group might be only successful in shallow waters (down to 330 m), whereas the other part was present from 288 to 1372 m, although the signal is still very weak. Topographic barriers can potentially hinder gene flow between populations (Etter et al. 2011). Separation by the GSR or factors related to the physical barrier could be observed between populations of E. inermis Ei_B and Ei_C. Those two populations have thus far not been isolated long enough to diverge in the slow evolving nuDNA 18S gene locus. However, until now, there has not been enough evidence to support that there are two populations diverging into different species either in E. producta 1 nor in E. inermis B_C. Further sampling and also further genetic information are needed to draw robust conclusions. Apart from those two exceptions, results of mtDNA and nuDNA were congruent and supported the likely existence of eight and four species within the E. producta and the E. inermis datasets, respectively.
Morphological findings
Geographically widespread species tend to exhibit variation in species-level morphological characters. Thus, elucidation of the variation within the species characters can lead to discovery of new species and better knowledge of species boundaries and their distributions and improve our knowledge of deep-sea biogeography (Wilson 1985).
Application of a combined morphological and molecular approach helped to identify multiple morphospecies within both species complexes. Specimens of both species complexes evaluated within this study feature small, but visible morphological differences, which are congruent with mtDNA and nuDNA species delimitations. One exception occurred between the E. inermis groups Ei_B and Ei_C, which could not be distinguished from each other morphologically.
Some of the species clades could be linked to species already known to science. Overall, a total of eight putative morphospecies could be observed within the E. producta dataset; specimens of E. producta Ep_1 were most similar to the original description of E. producta sensu stricto (type locality: Norwegian Sea), whereas specimens of Ep_2 belong to the known species Eurycope dahli (type locality: Norwegian Sea). Eurycope producta 3–8 are not yet described. Similarly, a total of three species of E. inermis evaluated herein are already known to science. Specimens of Ei_A resemble Eurycope hanseni (type locality: NW Atlantic) and specimens of the Ei_B and Ei_C group are most similar to E. inermis sensu stricto (type locality: NW Atlantic, Ingolf St. 120, NE of Iceland). Eurycope inermis Ei_D resembles E. cornuta (type locality: Drøbak Strait, Oslofjord, Norway), the type species of the genus; thus, one species within this complex (E. inermis E) is new to science.
Putative species are geographically and bathymetrically isolated
Environmental factors, for instance topographic barriers and hydrographic conditions, are factors known to have an impact on organism dispersal; however, these barriers are often semipermeable (McClain and Hardy 2010). Long-range dispersal across oceanic ridges has been observed in smaller, meiofaunal organisms and also in macrofaunal groups that feature dispersal stages such as larvae or adult swimmers (Zardus et al. 2006; Bik et al. 2010; Menzel et al. 2011; Schüller and Hutchings 2012). Benthic isopods are brooders without a larval life stage; thus, their dispersal ability seems to be more restricted by submarine ridges (Schnurr et al. 2014; Kaiser et al. 2017; Riehl et al. 2017; Bober et al. 2018). Further, the speciation potential of marine brooders is assumed to be increased due to their low vagility and their small body size (Teske et al. 2007). Previous studies on putatively widespread isopods with similar morphology established the existence of distinct species with the original species based on genetic analysis (e.g., Betamorpha fusiformis (Barnard 1920); Raupach et al. 2007), Atlantoserolis vemae ((Menzies 1962); Brandt et al. 2014). However, munnopsid isopods have an enhanced potential for dispersal (Wilson 1983b), since they have secondarily evolved natatory adaptations (Wilson 1989) and can swim off the bottom using their natatory legs (Hessler and Strömberg 1989; Marshall and Diebel 1995); thus, some of them are able to traverse larger distances (Raupach et al. 2007) likely with some help from near-bottom currents once up off the sea floor.
Specimens from each species complex evaluated herein were reported in former studies to occur on both sides of the GSR and to exhibit depth ranges from 103 to 2029 m depth (E. producta) and from 302 to 2137 m depth (E. inermis; Schnurr et al. 2014). However, delimiting species within the two complexes based on our current dataset revealed that most component species are not only geographically more restricted than the whole complex, but also bathymetrically more restricted (Figs. 9, 10, and 11) than previously assumed. Differences in previously recorded depth ranges could be observed in comparison to the results of Schnurr et al. (2014). Thus, the genetically and morphologically identified species clades feature much smaller depth ranges than previously assumed: for instance, specimens of Ep_2 (E. dahli; former depth range, 1624–2590 m; observed depth range within this study, 2130–2346 m), Ei_A (E. hanseni; former depth range, 893–2410 m; observed depth range within this study, 2134–2410 m), and Ei_D (E. cornuta; former depth range, 229–1320 m; observed depth range within this study, 833–1225 m). Most species clades feature a depth range spanning less than 400 m (e.g., E. producta clades Ep_2, Ep_4, Ep_6, Ep_7, and Ep_8). Only four species clades (E. producta: Ep_1 and Ep_3 and E. inermis: Ei_B and Ei_C) feature depth ranges spanning 1000 to 1500 m. Thus, a vertical zonation of species was observed. This is in line with the findings of Brix et al. (2014b) for different lineages within Chelator insignis (Hansen, 1916) south of Iceland. The observed genetic differences of the putative species from different depths suggest that bathymetry has an effect on the speciation process of the examined species complexes. Similar observations have previously been made in various taxa (France and Kocher 1996; Rogers 2003; Schüller 2011; Havermans et al. 2013; Brix et al. 2014b). Depth or factors related to depth can increase the genetic differentiation in benthic organisms (e.g., Held 2003; Rex and Etter 2010; Havermans et al. 2013; Jennings et al. 2013; Eustace et al. 2016). Further, depth has been shown to influence distributional patterns of munnopsid isopods (Schnurr et al. 2014) and ampeliscid amphipods (Dauvin et al. 2012). However, the depth is correlated with several other factors such as hydrostatic pressure (Somero 1992), dissolved oxygen concentration (Watling et al. 2013), total organic carbon within the sediment, and availability of food (Altabet et al. 1991), making it unclear which factor is the ultimate driver of divergence.
Only two species, E. producta (Ep_1) and E. inermis (Ei_B_C), were present on both sides of the GSR. However, Eurycope inermis Ei_B and Ei_C were clearly separated from each other by the GSR. Our 16S results show tendencies of incipient speciation. However, this evidence is not enough to support that there are two populations diverging into different species, without analyzing further specimens. The remaining species were either restricted to the deep areas north of the ridge (Ep_2, Ei_A, Ei_D), to the deep areas south of the ridge (Ep_4, Ep_7, Ep_8), along the GSR itself (Ep_8, Ei_E), or along the IFR (Ep_5). Thus, the GSR or factors related to this extensive submarine ridge might affect the distribution of most of the species evaluated herein (except for E. producta Ep_1). However, the bathymetric distribution of this species (288–1372 m) encompasses depths shallower than the deepest depression of the GSR (840 m). Thus, crossing the ridge should be possible for this species, since the depth of the passageways falls within the bathymetric range of this species (Fig. 11).
The topography of the Reykjanes Ridge differs from other oceanic ridges. This ridge is more a chain of seamounts than a continuous ridge and does not necessarily prevent gene flow between the Irminger Basin and the Icelandic Basin. Thus, the Reykjanes Ridge does not always act as a barrier for the southern distributed species evaluated here, as seen in the distribution of E. producta 7 and E. inermis B. This distributional pattern has also been observed within other isopods south of Iceland (Brix et al. 2014b).
Species distribution modeling and limitations of our dataset
Species distribution models are a helpful tool for illustrating potential distributional patterns of species. Implementation of SDMs on datasets allows more generalized assumptions on distributional patterns of species. The use of SDMs within the marine environment is still in its initial stage (Degraer et al. 2008), especially, since data collection within the marine environment relies on point data only, requires a lot of effort, and is expensive. Studies on benthic invertebrates are so far mainly modeled over local scales (e.g., Meißner et al. 2008), but also some on larger scales as, e.g., the Baltic Sea (Gogina and Zettler 2010), the North Sea (Reiss et al. 2011), or Icelandic waters (Meißner et al. 2014). Random forest works with presence and absence data, and the prediction accuracy of RF is known for its high performance (e.g., Iverson et al. 2008).
This study is the first known attempt of modeling the distributions of marine benthic isopods based on a combination of genetic and morphological data. We are aware that the SDMs presented here are based only on a small dataset, which should be expanded in the future. However, our dataset was well resolved using RF. The SDMs give an insight on the potential distribution and the limits of the resolved species clades.
Conclusion
A solid knowledge on species is essential for taxonomists, evolutionary biologists, ecologists, and conservationists (Harrison 1998; Kunz 2001). However, biodiversity can be underestimated by overlooked morphologically similar species and the existence of cryptic species (Vrijenhoek 2009). For several years, the two species E. producta and E. inermis were considered to be species complexes. No attempts at resolving these species complexes had yet been undertaken, and thus, it was not possible to determine the number and also the potential distribution of candidate species in previous studies (e.g., Meißner et al. 2014; Schnurr et al. 2014). As hypothesized, samples from the two putative species complexes within Icelandic waters represent not only genetically, but also morphologically different species. Our BI and ML analyses of mtDNA and nuDNA loci, as well as species delimitation methods, support the existence of eight species within the E. producta complex (six new to science) and four species within the E. inermis complex (one new to science).
The elucidated species clades featured (based on our analyzed dataset) much smaller bathymetric ranges and were much more geographically restricted than previously assumed. Vertical zonation was observed, with eight species clades having a depth span of less than 400 m and four species clades having a depth span of 1000 to 1500 m (Fig. 11). Interestingly, E. producta 1 was present on both sides of the GSR. Thus far, there may not be enough evidence to suspect that this species clade is at the beginning of species formation, although discordant observations between the 16S and 18S datasets were made. However, we assume that part of the E. producta 1 group might be only successful in shallow waters down to 330 m depth, whereas the other part of the group was present from 288 to 1372 m depth. Eurycope inermis B_C were separated from each other by the Greenland-Scotland Ridge. We assume that they are two different populations, which might be at the beginning of species formation. However, we choose to take the conservative approach and suggest they are not yet separate species, that further sampling needs to be done in order to draw robust conclusions and confirm speciation for both E. producta 1 and E. inermis B_C.
Our integrative approach holistically supported the need of a taxonomic revision of the two species complexes. Further molecular research in combination with taxonomy and inclusion of SDM at the transition of the northern North Atlantic and the Nordic Seas will eventually enhance our knowledge of biodiversity, distribution, and dispersal of benthic organisms and, thus, will offer options on how to conserve the environment. Moreover, inclusion of climate-related variables into SDMs will enable us to predict responses to environmental changes.
Abbreviations
- ABGD:
-
Automated Barcoding Gap Discovery
- AWTY:
-
Are We There Yet
- BI:
-
Bayesian inference
- DZMB:
-
German Centre for Marine Biodiversity Research
- GMYC:
-
Generalized mixed Yule coalescent
- GSR:
-
Greenland-Scotland Ridge
- IFR:
-
Iceland-Faroe Ridge
- ML:
-
Maximum likelihood
- NDT:
-
Nucleotide divergence threshold
- OOB:
-
Out-of-the-box error
- PCR:
-
Polymerase chain reactions
- PTP:
-
Poisson tree process
- RF:
-
Random forest
- SDM:
-
Species distribution modeling
- SQ:
-
Sequencing
- ZMH:
-
Zoological Museum of Hamburg
References
Altabet MA, Deuser WG, Honjo S, Stienen C (1991) Seasonal and depth-related changes in the source of sinking particles in the North Atlantic. Nature 354:136–139. https://doi.org/10.1038/354136a0
Altschul S, Gish W, Miller W, Myers E, Lipman D (1990) Basic local alignment search tool. J Mol Biol 215:403–410
Baird HP, Miller KJ, Stark JS (2011) Evidence of hidden biodiversity, ongoing speciation and diverse patterns of genetic structure in giant Antarctic amphipods. Mol Ecol 20:3439–3454
Barnard KH (1920) Contributions to the crustacean fauna of South Africa. 6. Further additions to the list of marine isopods. Ann S Afr Mus 17:319–438
Barrowclough GF, Zink RM (2009) Funds enough, and time: mtDNA, nuDNA and the discovery of divergence. Mol Ecol 18(14):2934–2936. https://doi.org/10.1111/j.1365-294X.2009.04271.x
Bik HM, Thomas WK, Lunt DH, Lambshead PJ (2010) Low endemism, continued deep-shallow interchanges, and evidence for cosmopolitan distributions in free-living marine nematodes (order Enoplida). BMC Evol Biol 10:389. https://doi.org/10.1186/1471-2148-10-389
Birky CW Jr, Wolf C, Maughan H, Herbertson L, Henry E (2005) Speciation and selection without sex. Hydrobiologia 546:29–45. https://doi.org/10.1007/1-4020-4408-9_3
Birstein JA (1971) Additions to the fauna of isopods (Crustacea, Isopoda) of the Kurile-Kamtchatka Trench. Part II. Asellota 2. J Inst Oceanol Russ Acad Sci 92:162–198
Bober S, Brix S, Riehl T, Schwentner M, Brandt A (2018) Does the Mid-Atlantic Ridge affect the distribution of abyssal benthic crustaceans across the Atlantic Ocean? Deep Sea Res PT II. https://doi.org/10.1016/j.dsr2.2018.02.007
Bonnier J (1896) Edriophthalmes. In: Koehler R (ed) Résultats Scientifiques de la Campagne du “Caudan” dans le Golfe de Gascogne: Annelides, Poissons, Edriophthalmes, Diatomees, Debris Vegetaux et Roches, Liste des especes recueillies. Annales de l’Université de Lyon, pp 527–689
Brandt A (1992) Origin of Antarctic Isopoda (Crustacea, Malacostraca). Mar Biol 113(3):415–423. https://doi.org/10.1007/bf00349167
Brandt A, Gooday AJ, Brandao SN, Brix S, Brökeland W, Cedhagen T, Choudhury M, Cornelius N, Danis B, De Mesel I, Diaz RJ, Gillan DC, Ebbe B, Howe JA, Janussen D, Kaiser S, Linse K, Malyutina M, Pawlowski J, Raupach M, Vanreusel A (2007) First insights into the biodiversity and biogeography of the Southern Ocean deep sea. Nature 447(7142):307–311 http://www.nature.com/nature/journal/v447/n7142/suppinfo/nature05827_S1.html
Brandt A, Elsner N, Brenke N, Golovan OA, Riehl T, Schwabe E, Würzberg L (2013) Epifauna of the Sea of Japan collected via a new epibenthic sledge equipped with camera and environmental sensor systems. Deep Sea Res PT II 86-87:43–55
Brandt A, Brix S, Held C, Kihara TC (2014) Molecular differentiation in sympatry despite morphological stasis: deep-sea Atlantoserolis Wägele, 1994 and Glabroserolis Menzies, 1962 from the south-west Atlantic (Crustacea: Isopoda: Serolidae). Zool J Linnean Soc 172(2):318–359. https://doi.org/10.1111/zoj.12178
Breiman L (2001) Random forests. Mach Learn 45:5–32
Brenke N (2005) An epibenthic sledge for operations on marine soft bottom and bedrock. Mar Technol Soc J 39(2):10–19
Brix S, Svavarsson J (2010) Distribution and diversity of desmosomatid and nannoniscid isopods (Crustacea) on the Greenland–Iceland–Faeroe Ridge. Polar Biol 33(4):515–530. https://doi.org/10.1007/s00300-009-0729-8
Brix S, Riehl T, Leese F (2011) First genetic data for species of the genus Haploniscus Richardson, 1908 (Isopoda: Asellota: Haploniscidae) from neighbouring deep-sea basins. Zootaxa 2838:79–84
Brix S, Leese F, Riehl T, Kihara T (2014a) A new genus and new species of Desmosomatidae Sars, 1897 (Isopoda) from the eastern South Atlantic abyss described by means of integrative taxonomy. Mar Biodiv:1–55. doi: 10.1007/s12526-014-0218-3
Brix S, Svavarsson J, Leese F (2014b) A multi-gene analysis reveals multiple highly divergent lineages of the isopod Chelator insignis (Hansen, 1916) south of Iceland. Pol Polar Res 35(2):225–242
Brix S, Bober S, Tschesche C, Kihara T-C, Driskell A, Jennings RM (2018) Molecular species delimitation and its implications for species descriptions using desmosomatid and nannoniscid isopods from the VEMA fracture zone as example taxa. Deep-Sea Res PT II. https://doi.org/10.1016/j.dsr2.2018.02.004
Brökeland W, Raupach MJ (2008) A species complex within the isopod genus Haploniscus (Crustacea: Malacostraca: Peracarida) from the Southern Ocean deep sea: a morphological and molecular approach. Zool J Linnean Soc 152(4):655–706. https://doi.org/10.1111/j.1096-3642.2008.00362.x
Buhay JE (2009) “COI-like” sequences are becoming problematic in molecular systematic and DNA barcoding studies. J Crustacean Biol 29:96–110
Carvalho GR, Piertney SB (1997) Interspecific comparisons of genetic population structure in members of the Jaera albifrons species complex. J Mar Biol Ass UK 77:77–93
Claridge MF, Dawah HA, Wilson MR (1997) Species: the units of biodiversity. Chapman & Hall, London
Clement M, Posada D, Crandall KA (2000) TCS: a computer program to estimate gene genealogies. Mol Ecol 9:1657–1659
Coleman CO (2003) “Digital inking”: how to make perfect line drawings on computers. Org Divers Evol 3(4):303–304. https://doi.org/10.1078/1439-6092-00081
Coleman CO (2009) Drawing setae the digital way. Zoosyst Evol 85(2):305–310. https://doi.org/10.1002/zoos.200900008
Colgan DJ, McLauchlan A, Wilson GDF, Livingston SP, Edgecombe GD, Macaranas J, Cassis G, Gray MR (1998) Histone H3 and U2 snRNA DNA sequences and arthropod molecular evolution. Aus J Zool 46(5):419–437. https://doi.org/10.1071/ZO98048
Dauvin J-C, Alizier S, Weppe A, Guðmundsson G (2012) Diversity and zoogeography of Icelandic deep-sea Ampeliscidae (Crustacea: Amphipoda). Deep Sea Res PT I 68:12–23. https://doi.org/10.1016/j.dsr.2012.04.013
Dayrat B (2005) Towards integrative taxonomy. Biol J Linn Soc 85(3):407–415. https://doi.org/10.1111/j.1095-8312.2005.00503.x
Degraer S, Verfaillie E, Willems W, Adriaens E, Vincx M, Van Lancker V (2008) Habitat suitability modelling as a mapping tool for macrobenthic communities: an example from the Belgian part of the North Sea. Cont Shelf Res 28(3):369–379. https://doi.org/10.1016/j.csr.2007.09.001
Dijkstra HH, Warén A, Gudmundsson G (2009) Pectinoidea (Mollusca: Bivalvia) from Iceland. Mar Biol Res 5:207–243
Divins DL (2003) Total sediment thickness of the world’s oceans & marginal seas. Ver 1. World Data Service for Geophysics. http://www.ngdc.noaa.gov/mgg/sedthick/sedthick.html
Dreyer H, Wägele JW (2001) Parasites of crustaceans (Isopoda: Bopyridae) evolved from fish parasites: molecular and morphological evidence. Zool 103:157–178
Drummond AJ, Suchard MA, Xie D, Rambaut A (2012) Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol 29(8):1969–1973. https://doi.org/10.1093/molbev/mss075
Edzard T, Fujisawa T, Barraclough TG (2009) SPLITS: SPecies’ LImits by Threshold Statistics. R package version 1.0-18/r45
Elith J, Graham CH (2009) Do they? How do they? Why do they differ? On finding reasons for differing performances of species distribution models. Ecography 32(1):66–77. https://doi.org/10.1111/j.1600-0587.2008.05505.x
ETOPO2v2 (2006) 2-minute gridded global relief data ETOPO2v2. U.S. Department of Commerce. doi: 10.7289/V5J1012Q
Etter RJ, Rex MA, Chase MC, Quattro JM (1999) A genetic dimension to deep-sea biodiversity. Deep Sea Res PT I 46(6):1095–1099. https://doi.org/10.1016/S0967-0637(98)00100-9
Etter RJ, Boyle EE, Glazier A, Jennings RM, Dutra E, Chase MR (2011) Phylogeography of a pan-Atlantic abyssal protobranch bivalve: implications for evolution in the Deep Atlantic. Mol Ecol 20(4):829–843. https://doi.org/10.1111/j.1365-294X.2010.04978.x
Eustace RM, Ritchie H, Kilgallen NM, Piertney SB, Jamieson AJ (2016) Morphological and ontogenetic stratification of abyssal and hadal Eurythenes gryllus sensu lato (Amphipoda: Lysianassoidea) from the Peru–Chile Trench. Deep Sea Res PT I 109(Supplement C):91–98. https://doi.org/10.1016/j.dsr.2015.11.005
Eytan RI, Hayes M, Arbour-Reily P, Miller M, Hellberg ME (2009) Nuclear sequences reveal mid-range isolation of an imperilled deep-water coral population. Mol Ecol 18(11):2375–2389. https://doi.org/10.1111/j.1365-294X.2009.04202.x
Felsenstein J (1985) Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39(4):783–791. https://doi.org/10.2307/2408678
Folmer O, Black M, Hoeh W, Lutz R, Vrijenhoek R (1994) DNA primers for amplification of mitochondiral cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol Mar Biol Biotechnol 5(3):294–299
France SC, Kocher TD (1996) Geographic and bathymetric patterns of mitochondrial 16S rRNA sequence divergence among deep-sea amphipods, Eurythenes gryllus. Mar Biol 126(4):633–643. https://doi.org/10.1007/bf00351330
Fujita MK, Leaché AD, Burbrink FT, McGuire JA, Moritz C (2012) Coalescent-based species delimitation in an integrative taxonomy. Trends Ecol Evol 27(9):480–488
Galtier N, Nabholz B, Glemin S, Hurst GD (2009) Mitochondrial DNA as a marker of molecular diversity: a reappraisal. Mol Ecol 18(22):4541–4550. https://doi.org/10.1111/j.1365-294X.2009.04380.x
Gogina M, Zettler ML (2010) Diversity and distribution of benthic macrofauna in the Baltic Sea: data inventory and its use for species distribution modelling and prediction. J Sea Res 64(3):313–321. https://doi.org/10.1016/j.seares.2010.04.005
Hansen HJ (1916) Crustacea Malacostraca: the Order Isopoda. Dan Ingolf-Exp 3:1–262
Hansen B, Østerhus S (2000) North Atlantic-Nordic Seas exchanges. Prog Oceanogr 45(2):109–208
Hare MP (2001) Prospects for nuclear gene phylogeography. Trends Ecol Evol 16(12):700–706. https://doi.org/10.1016/S0169-5347(01)02326-6
Harrison RG (1998) Linking evolutinary pattern and process. The relevance of species concepts fo the study of speciation. In: Howard DJ, Berlocher S (eds) Endless forms: species and speciation. Oxford University Press, New York, pp 19–31
Hart MW, Sunday J (2007) Things fall apart: biolgical species from unconnected parsimony networks. Biol Lett 3:509–512
Havermans C, Sonet G, d’Udekem d’Acoz C, Nagy ZT, Martin P, Brix S, Riehl T, Agrawal S, Held C (2013) Genetic and morphological divergences in the cosmopolitan deep-sea amphipod Eurythenes gryllus reveal a diverse abyss and a bipolar species. PLoS One 8(9):e74218. https://doi.org/10.1371/journal.pone.0074218
Hebert PDN, Ratnasingham S, deWaard JR (2003) Barcoding animal life: cytochrome c oxidase subunit 1 divergences among closely related species. P Roy Soc B Biol Sci 270(Suppl 1):S96–S99. https://doi.org/10.1098/rsbl.2003.0025
Held C (2003) Molecular evidence for cryptic speciation within the widespread Antarctic crustacean Ceratoserolis trilobitoides (Crustacea, Isopoda). In: Antarctic biology in a global context. pp 135–139
Held C, Wägele J-W (2005) Cryptic speciation in the giant Antarctic isopod Glyptonotus antarcticus (Isopoda, Valvifera, Chaetiliidae). Sci Mar 69:175–181
Hessler RR (1970) The Desmosomatidae (Isopoda, Asellota) of the gay head-bermuda transect. Bull Scripps Inst Oceanogr 15:1–185
Hessler RR (1981) Evolution of Arthropod locomotion: a crustacean model. In: Herraid CF, Fourtner CR (eds) Locomotion and exercise in arthropods. Plenum, New York, pp 9–29
Hessler RR, Strömberg JO (1989) Behavior of janiroidean isopods (Asellota), with special reference to deep-sea genera. Sarsia 74:145–159
Iverson LR, Prasad AM, Matthews SN, Peters M (2008) Estimating potential habitat for 134 eastern US tree species under six climate scenarios. For Ecol Manag 254(3):390–406. https://doi.org/10.1016/j.foreco.2007.07.023
Jennings RM, Etter RJ, Ficarra L (2013) Population differentiation and species formation in the deep sea: the potential role of environmental gradients and depth. PLoS One 8(10):e77594. https://doi.org/10.1371/journal.pone.0077594
Jochumsen K, Schnurr SM, Quadfasel D (2016) Bottom temperature and salinity distribution and its variability around Iceland. Deep Sea Res PT I 111:79–90. https://doi.org/10.1016/j.dsr.2016.02.009
Johnson NK, Cicero C (2002) The role of ecologic diversification in sibling speciation of Empidonax flycatchers (Tyrannidae): multigene evidence from mt DNA. Mol Ecol 11:2065–2081
Johnson SB, Young CR, Jones WJ, Warén A, Vrijenhoek RC (2006) Migration, isolation, and speciation of hydrothermal vent limpets (Gastropoda; Lepetodrilidae) across the Blanco Transform Fault. Biol Bull 210(2):140–157
Just J, Wilson GDF (2004) Revision of the Paramunna complex (Isopoda: Asellota: Paramunnidae). Invertebr Syst 18(4):377–466. https://doi.org/10.1071/IS03027
Kaiser S, Brix S, Kihara TC, Janssen A, Jennings RM (2017) Integrative species delimitation in the deep-sea genus Thaumastosoma Hessler, 1970 (Isopoda, Asellota, Nannoniscidae) reveals a new genus and species from the Atlantic and central Pacific abyss. Deep Sea Res PT II. https://doi.org/10.1016/j.dsr2.2017.05.006
Katoh K, Misawa K, Kuma K-i, Miyata T (2002) MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res 30(14):3059–3066
Kunz W (2001) Taking more care in using different species concepts—an opinion. Parasitol Res 87(5):413–416. https://doi.org/10.1007/s004360000372
Kussakin OG (2003) Marine and brackishwater like-footed Crustacea (Isopoda) from the cold and temperate waters of the Northern hemisphere. III. Suborder Asellota. Part 3. Family Munnopsidae. Opredeliteli po faune, izdavaemie Zoologicheskim Institutom Rossiyskoy Academii Nauk
Leaché AD, Koo MS, Spencer CL, Papenfuss TJ, Fisher RN, McGuire JA (2009) Quantifying ecological, morphological, and genetic variation to delimit species in the coast horned lizard species complex (Phrynosoma). Proc Natl Acad Sci 106(30):12418–12423. https://doi.org/10.1073/pnas.0906380106
Leese F, Kop A, Wägele J-W, Held C (2008) Cryptic speciation in a benthic isopod from Patagonian and Falkland Island waters and the impact of glaciations on its population structure. Front Zool 5(1):19
Liaw A, Wiener M (2002) Classification and regression by randomForest. R News 2(3):18–22
Lilljeborg W (1864) Bidrag till Kannedomen om de inom Sverige och Norrige frekommende Crustaceen af Isopodernas underordning och Tanaidernas familj. Inbjudningsskrift till hrande af de Offentliga Frelsninger 1:1–32
Lörz A-N, Smith P, Linse K, Steinke D (2012) High genetic diversity within Epimeria georgiana (Amphipoda) from the southern Scotia Arc. Mar Biodivers 42(2):137–159. https://doi.org/10.1007/s12526-011-0098-8
Lutz MJ, Caldeira K, Dunbar RB, Behrenfeld MJ (2007) Seasonal rhythms of net primary production and particulate organic carbon flux to depth describe the efficiency of biological pump in the global ocean. J Geophys Res 112(C10):C10011. https://doi.org/10.1029/2006jc003706
Malyutina M, Brandt A (2006) A revaluation of the Eurycopinae (Crustacea, Isopoda, Munnopsidae) with a description of Dubinectes gen. nov. from the southern Atlantic deep sea. Zootaxa 1272:1–44
Marlétaz F, Le Parco Y, Liu S, Peijnenburg KTCA (2017) Extreme mitogenomic variation in natural populations of chaetognaths. GBE 9(6):1374–1384. https://doi.org/10.1093/gbe/evx090
Marshall N, Diebel C (1995) Deep-sea spiders’ that walk through the water. J Exp Biol 198(Pt 6):1371–1379
McCallum KP, Guerin GR, Breed MF, Lowe AJ (2014) Combining population genetics, species distribution modelling and field assessments to understand a species vulnerability to climate change. Austral Ecol 39(1):17–28. https://doi.org/10.1111/aec.12041
McClain CR, Hardy SM (2010) The dynamics of biogeographic ranges in the deep sea. P Roy Soc B Biol Sci 277:3533–3546
Meier R, Kwong S, Vaidya G, Ng PKL (2006) DNA barcoding and taxonomy in Diptera: a tale of high intraspecific variability and low identification success. Syst Biol 55:715–728
Meißner K, Darr A, Rachor E (2008) Development of habitat models for Nephtys species (Polychaeta: Nephtyidae) in the German Bight (North Sea). J Sea Res 60(4):276–291. https://doi.org/10.1016/j.seares.2008.08.001
Meißner K, Fiorentino D, Schnurr S, Martinez Arbizu P, Huettmann F, Holst S, Brix S, Svavarsson J (2014) Distribution of benthic marine invertebrates at northern latitudes―an evaluation applying multi-algorithm species distribution models. J Sea Res 85:241–254. https://doi.org/10.1016/j.seares.2013.05.007
Menzel L, George KH, Arbizu PM (2011) Submarine ridges do not prevent large-scale dispersal of abyssal fauna: a case study of Mesocletodes (Crustacea, Copepoda, Harpacticoida). Deep Sea Res PT I 58(8):839–864. https://doi.org/10.1016/j.dsr.2011.05.008
Menzies RJ (1962) The isopods of the abyssal depths in the Atlantic Ocean. Vema Res Ser 1:79-206
Monaghan MT, Wild R, Elliot M, Fujisawa T, Balke M, Inward DJG, Lees DC, Ranaivosolo R, Eggleton P, Barraclough TGB, Vogler AP (2009) Accelerated species inventory on Madagascar using coalescent-based models of species delineation. Syst Biol 58(3):298–311
Nilsen J, Hátún H, Mork K, Valdimarsson H (2008) The NISE dataset. Technical Report 08-07. Tórshavn, Faroe Islands
Nylander JAA (2004) MrAIC.pl. Program distributed by the author. Evolutionary Biology Centre, Uppsala University, Uppsala
Ohlin A (1901) Arctic Crustacea collected during the Swedish Arctic Expeditions 1898 and 1899 under the Direction of Professor A G Nathorst. Leptostraca, Isopoda, Cumacea. Bihang till Kungliga Svenska Vetenskaps-Akademiens Handlingar 26:1–54
Osborn KJ (2009) Relationships within the Munnopsidae (Crustacea, Isopoda, Asellota) based on three genes. Zool Scr 38(6):617–635. https://doi.org/10.1111/j.1463-6409.2009.00394.x
Padial JM, Miralles A, DeLaRiva I, Vences M (2010) The integrative future of taxonomy. Front Zool 7(1):16. https://doi.org/10.1186/1742-9994-7-16
Palumbi SR, Martin A, Romanos S, McMillan WO, Stice L (1991) The simple fool’s guide to PCR. Version 2. University of Hawaii Press, Hawaii, Honolulu
Petit RJ, Excoffier L (2009) Gene flow and species delimitation. Trends Ecol Evol 24(7):386–393. https://doi.org/10.1016/j.tree.2009.02.011
Pons J, Barraclough TG, Gomez-Zurita J, Cardoso A, Duran DP, Hazell S, Kamoun S, Sumlin WD, Vogler AP (2006) Sequence-based species delimitation for the DNA taxonomy of undescribed insects. Syst Biol 55(4):595–609
Puillandre N, Lambert A, Brouillet S, Achaz G (2012) ABGD, Automatic Barcode Gap Discovery for primary species delimitation. Mol Ecol 21(8):1864–1877. https://doi.org/10.1111/j.1365-294X.2011.05239.x
Radulovici AE, Sainte-Marie B, Dufresne F (2009) DNA barcoding of marine crustaceans from the Estuary and Gulf of St Lawrence: a regional-scale approach. Mol Ecol Resour 9(Suppl s1):181–187. https://doi.org/10.1111/j.1755-0998.2009.02643.x
Rambaut A, Drummond AJ (2007) Tracer v1.5.0
Raupach MJ, Wägele JW (2006) Distinguishing cryptic species in Antarctic Asellota (Crustacea: Isopoda)—a preliminary study of mitochondrial DNA in Acanthaspidia drygalskii. Antarct Sci 18:191–198. https://doi.org/10.1017/S0954102006000228
Raupach MJ, Malyutina M, Brandt A, Wägele J-W (2007) Molecular data reveal a highly diverse species flock within the munnopsoid deep-sea isopod Betamorpha fusiformis (Barnard, 1920) (Crustacea: Isopoda: Asellota) in the Southern Ocean. Deep-Sea Res PT II 54(16–17):1820–1830
Raupach MJ, Mayer C, Malyutina M, Wägele JW (2009) Multiple origins of deep-sea Asellota (Crustacea: Isopoda) from shallow waters revealed by molecular data. Proc Biol Sci 276(1658):799–808
Reiss H, Cunze S, König K, Neumann H, Kröncke I (2011) Species distribution modelling of marine benthos: a North Sea case study. Mar Ecol Prog Ser 442:71–86
Reveillaud J, Remerie T, van Soest R, Erpenbeck D, Càrdenas P, Derycke S, Xavier JR, Rigaux A, Vanreusel A (2010) Species boundaries and phylogenetic relationships between Atlanto-Mediterranean shallow-water and deep-sea coral associated Hexadella species (Porifera, Ianthellidae). Mol Phylogenet Evol 56:104–114
Rex MA, Etter RJ (2010) Deep-sea biodiversity: pattern and scale. Harvard University Press, Cambridge
Riehl T, Brand A (2013) Southern Ocean Macrostylidae reviewed with a key to the species and new descriptions from Maud Rise. Zootaxa 3692(1):160–203. https://doi.org/10.11646/zootaxa.3692.1.10
Riehl T, Kaiser S (2012) Conquered from the deep sea? A new deep-sea isopod species from the Antarctic shelf shows pattern of recent colonization. PLoS One 7(11):e49354. https://doi.org/10.1371/journal.pone.0049354
Riehl T, Brenke N, Driskell A, Kaiser S, Brand A (2014) Field and laboratory methods for DNA studies on deep-sea isopod crustaceans. Pol Polar Res 35(2):203–224
Riehl T, Lins L, Brandt A (2017) The effects of depth, distance, and the Mid-Atlantic Ridge on genetic differentiation of abyssal and hadal isopods (Macrostylidae). Deep Sea Res PT II. https://doi.org/10.1016/j.dsr2.2017.10.005
Rogers AD (2003) Molecular ecology and evolution of slope species. In: Wefer G, Billett D, Hebbeln D, Jørgensen B, Schlüter M, van Weering TE (eds) Ocean margin systems. Springer, Berlin, pp 323–337. https://doi.org/10.1007/978-3-662-05127-6_20
Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Höhna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP (2012) MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. https://doi.org/10.1093/sysbio/sys029
Rothlisberg PC, Pearcy WG (1977) An epibenthic sampler used to study the ontogeny of vertical migration of Pandalus jordani (Decapoda, Caridea). Fish Bull 74:994–997
Rubinoff D, Holland BS (2005) Between two extremes: mitochondrial DNA is neither the panacea nor the nemesis of phylogenetic and taxonomic inference. Syst Biol 54(6):952–961. https://doi.org/10.1080/10635150500234674
Sanders HL, Hessler RR (1969) Ecology of the deep-sea benthos. Science 163(3874):1419–1424
Sanders HL, Hessler RR, Hampson GR (1965) An introduction to the study of deep-sea benthic faunal assemblages along the Gay Head-Bermuda transect. Deep Sea Res Oceanogr Abstr 12(6):845–867
Sars GO (1864) On en anomal Gruppe af Isopoder. Forh Vidensk Selsk Kristiania 1863:1–16
Sars GO Beretning om en i Sommeren 1865 foretagan zoologisk Reise ved Kysterne af Christianias og Christiansands Stifter-Crustaceer. Forh Vidensk Selsk Kristiania 1868, 1868:1–47
Schnurr S, Malyutina M (2014) Two new species of the genus Eurycope (Isopoda, Munnopsidae) from Icelandic waters. Pol Polar Res 35(2):361–388
Schnurr S, Brandt A, Brix S, Fiorentino D, Malyutina M, Svavarsson J (2014) Composition and distribution of selected munnopsid genera (Crustacea, Isopoda, Asellota) in Icelandic waters. Deep Sea Res PT I 84:142–155. https://doi.org/10.1016/j.dsr.2013.11.004
Schüller M (2011) Evidence for a role of bathymetry and emergence in speciation in the genus Glycera (Glyceridae, Polychaeta) from the deep Eastern Weddell Sea. Polar Biol 34(4):549–564. https://doi.org/10.1007/s00300-010-0913-x
Schüller M, Hutchings PA (2012) New species of Terebellides (Polychaeta: Trichobranchidae) indicate long-distance dispersal between western South Atlantic deep-sea basins. Zootaxa 3254:1–31
Schwentner M, Timms BV, Richter S (2011) An integrative approach to species delineation incorporating different species concepts: a case study of Limnadopsis (Branchiopoda: Spinicaudata). Biol J Linn Soc 104(3):575–599. https://doi.org/10.1111/j.1095-8312.2011.01746.x
Seiter K, Hensen C, Zabel M (2005) Benthic carbon mineralization on a global scale. Glob Biogeochem Cy 19(1):GB1010. https://doi.org/10.1029/2004gb002225
Shaw KL (2002) Conflict between nuclear and mitochondrial DNA phylogenies of a recent species radiation: what mtDNA reveals and conceals about modes of speciation in Hawaiian crickets. Proc Natl Acad Sci 99(25):16122–16127. https://doi.org/10.1073/pnas.242585899
Sites JW, Marshall JC (2004) Operational criteria for delimiting species. Annu Rev Ecol Syst 35:199–227. https://doi.org/10.2307/30034115
Somero GN (1992) Adaptations to high hydrostatic pressure. Annu Rev Physiol 54:557–577
Song H, Buhay JE, Whiting MF, Crandall KA (2008) Many species in one: DNA barcoding overestimates the number of species when nuclear mitochondrial pseudogenes are coamplified. Proc Natl Acad Sci 105(36):13486–13491. https://doi.org/10.1073/pnas.0803076105
Stamatakis A, Hoover P, Rougemont J (2008) A rapid bootstrap algorithm for the RAxML Web servers. Syst Biol 57(5):758–771. https://doi.org/10.1080/10635150802429642
Svavarsson J (1987) Eurycopidae (Isopoda, Asellota) from bathyal and abyssal depths in the Norwegian, Greenland, and North Polar Seas. Sarsia 72:183–196
Svavarsson J (1997) Diversity of isopods (Crustacea): new data from the Arctic and Atlantic Oceans. Biodivers Conserv 6:1571–1579
Svavarsson J, Brattegard T, Strömberg JO (1990) Distribution and diversity patterns of asellote isopods (Crustacea) in the deep Norwegian and Greenland Seas. Prog Oceanogr 24:297–310
Svavarsson J, Strömberg JO, Brattegard T (1993) The deep-sea asellote (Isopoda, Crustacea) fauna of the Northern Seas: species composition, distributional patterns and origin. J Biogeogr 20(5):537–555
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30:2725–2729
Tang CQ, Leasi F, Obertegger U, Kieneke A, Barraclough TG, Fontaneto D (2012) The widely used small subunit 18S rDNA molecule greatly underestimates true diversity in biodiversity surveys of the meiofauna. Proc Natl Acad Sci 109(40):16208–16212. https://doi.org/10.1073/pnas.1209160109
Templeton AR (2001) Using phylogeographic analyses of gene trees to test species status and processes. Mol Ecol 10(3):779–791. https://doi.org/10.1046/j.1365-294x.2001.01199.x
Teske PR, Papadopoulos I, Zardi GI, McQuaid CD, Edkins MT, Griffiths CL, Barker NP (2007) Implications of life history for genetic structure and migration rates of southern African coastal invertebrates: planktonic, abbreviated and direct development. Mar Biol 152(3):697–711. https://doi.org/10.1007/s00227-007-0724-y
Tsang LM, Chan BKK, Shih F-L, Chu KH, Allen Chen C (2009) Host-associated speciation in the coral barnacle Wanella milleporae (Cirripedia: Pyrgomatidae) inhabiting the Millepora coral. Mol Ecol 18(7):1463–1475. https://doi.org/10.1111/j.1365-294X.2009.04090.x
Vrijenhoek RC (2009) Cryptic species, phenotypic plasticity, and complex life histories: assessing deep-sea faunal diversity with molecular markers. Deep Sea Res PT II 56(19–20):1713–1723. https://doi.org/10.1016/j.dsr2.2009.05.016
Watling L, Guinotte J, Clark MR, Smith CR (2013) A proposed biogeography of the deep ocean floor. Prog Oceanogr 111:91–112. https://doi.org/10.1016/j.pocean.2012.11.003
Whittaker JM, Muller RD, Roest WR, Wessel P, Smith WHF (2008) How supercontinents and superoceans affect seafloor roughness. Nature 456 (7224):938–941. doi:http://www.nature.com/nature/journal/v456/n7224/suppinfo/nature07573_S1.html
Wilgenbusch JC, Warren DL, Swofford DL (2004) AWTY: a system for graphical exploration of MCMC convergence in Bayesian phylogenetic inference. http://ceb.csit.fsu.edu/awty
Wilson GDF (1982) Systematics of a species complex in the deep-sea genus Eurycope, with a revision of six previously described species (Crustacea, Isopoda, Eurycopidae). Bull Scripps Inst Oceanogr 25:1–64
Wilson GDF (1983a) An unusual species complex in the genus Eurycope (Crustacea: Isopoda: Asellota) from the deep north Atlantic Ocean. P Biol Soc Wash 96(3):452–467
Wilson GDF (1983b) Variation in the deep-sea isopod Eurycope iphthima (Asellota, Eurycopidae): depth related clines in rostral morphology and in populations structure. J Crustacean Biol 3(1):127–140
Wilson GDF (1985) The distribution of eurycopid species complexes (Crustacea: Isopoda: Asellota). In: Laubier L, Monniot C (eds) Peuplements profonds du Golfe de Gascogne. Brest, pp 630–468
Wilson GDF (1989) A systematic revision of the deep-sea subfamily Lipomerinae of the isopod crustacean family Munnopsidae. Bull Scripps Inst Oceanogr 27:1–138
Wilson GDF (2008) A review of taxonomic concepts in the Nannoniscidae (Isopoda, Asellota), with a key to the genera and a description of Nannoniscus oblongus Sars. Zootaxa 1680:1–24
Wilson GDF, Hessler RR (1981) A revision of the genus Eurycope (Isopoda, Asellota) with descriptions of three new genera. J Crustacean Biol 1(3):401–423
Wilson GDF, Hessler RR (1987) Speciation in the deep sea. Annu Rev Ecol Syst 18:185–207
Wilson GDF, Schotte M (2017) Munnopsidae Lilljeborg, 1864. http://www.marinespecies.org/aphia.php?p=taxdetails&id=118264. Accessed 2017-12-14 2017
Wilson AC, Cann RL, Carr SM, George M, Gyllensten UB, Helm-Bychowski KM, Higuchi RG, Palumbi SR, Prager EM, Sage RD, Stoneking M (1985) Mitochondrial DNA and two perspectives on evolutionary genetics. Biol J Linn Soc 26(4):375–400. https://doi.org/10.1111/j.1095-8312.1985.tb02048.x
Wolff T (1962) The systematics and biology of bathyal and abyssal Isopoda Asellota. Galathea Rep 6:1–320
Zardus JD, Etter RJ, Chase MR, Rex MA, Boyle EE (2006) Bathymetric and geographic population structure in the pan-Atlantic deep-sea bivalve Deminucula atacellana (Schenck, 1939). Mol Ecol 15(3):639–651. https://doi.org/10.1111/j.1365-294X.2005.02832.x
Zhang J, Kapli P, Pavlidis P, Stamatakis A (2013) A general species delimitation method with applications to phylogenetic placements. Bioinformatics 29(22):2869–2876. https://doi.org/10.1093/bioinformatics/btt499
Acknowledgements
We would like to thank the crew and participants of the IceAGE expeditions, as well as all pickers and sorters at the DZMB in Hamburg, Germany, and the Nature Centre in Sandgerði, Iceland. Special thanks go to Dr. Herman Wirshing for his help during S. Schnurr’s stays at the facilities of the Laboratories of Analytical Biology (LAB) at the Smithonian National Museum of Natural History, Washington, DC. We thank Karen Jeskulke for introducing lab workflow, Lukas Rischke for solving hardware problems, and Falk Huettmann for his introduction to random forest models.
Funding
S. Schnurr was partly funded by the German Science Foundation (DFG) under contract 2843/4-1 and BR3843/6-1, as well as by the Census of the Diversity of Abyssal Marine Life (CeDAMar), the Smithsonian’s Rathbun Fund for Crustacean Research, and the Annette Barthelt Stiftung. The IceAGE cruises were funded by the DFG contract BR3843/3-1 (IceAGE1) and 4-1 (IceAGE2).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed by the authors.
Sampling and field studies
All necessary permits for sampling and observational field studies have been obtained by the authors from the competent authorities and are mentioned in the acknowledgements, if applicable.
Additional information
Communicated by K. Halanych
Electronic supplementary material
Online Resource 1
Uncorrected pairwise p-distances for the 16S gene of all evaluated E. producta complex specimens. (XLSX 26.8 kb)
Online Resource 2
Uncorrected pairwise p-distances for the 16S gene of all evaluated E. inermis complex specimens. (XLSX 33.7 kb)
Rights and permissions
About this article

Cite this article
Schnurr, S., Osborn, K.J., Malyutina, M. et al. Hidden diversity in two species complexes of munnopsid isopods (Crustacea) at the transition between the northernmost North Atlantic and the Nordic Seas. Mar Biodiv 48, 813–843 (2018). https://doi.org/10.1007/s12526-018-0877-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12526-018-0877-6