
Abstract
Baculovirus-Bombyx mori protein expression system has mainly been used for translation of eukaryotic proteins. In contrast, information pertaining to bacterial protein expression using this system is not sufficient. Therefore, recombinant nucleases from Serratia liquefaciens (rSlNucAs) were expressed in a Baculovirus-B. mori protein expression system. rSlNucAs containing the native signal peptide (rSlNucA-NSP) or silkworm 30-K signal peptide (rSlNucA-30K) at the NH2-terminus were constructed to enable secretion into the extracellular fraction. Both rSlNucA-30K and rSlNucA-NSP were successfully secreted into hemolymph of B. mori larvae. Affinity-purified rSlNucAs showed high nuclease activity. Optimum pH was 7.5 and half of maximum activity was maintained between pH 7.0 and 9.5. Optimum temperature was 35 °C. rSlNucAs showed sufficient activity in twofold-diluted radioimmunoprecipitation assay buffer and undiluted, mild lysis buffer. Genomic DNA of Escherichia coli was efficiently digested by rSlNucAs in the bacterial lysate. The results in this study suggest that rSlNucAs expressed by the Baculovirus-B. mori protein expression system will be a useful tool in molecular biology. Functional recombinant protein of bacteria was produced by Baculovirus-B. mori protein expression system. This system may be highly suitable for bacterial extracellular protein secreted via Sec pathway.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The genus Serratia, a member of Family Enterobacteriaceae, comprises a group of bacteria that are phenotypically and genetically related [16]. Within the genus Serratia, S. marcescens is one of the most studied species. S. marcescens secretes a number of enzymes including chitinase [35], proteases [5, 6], lipase [21], and nuclease [9]. S. marcescens is an opportunistic pathogen and infects a wide range of hosts. The proteins secreted by this species have been studied with regard to pathology and toxicology, and some have been shown to function as virulence factors.
The virulent strain of S. marcescens produces ~10 times more protease in culture than the less-virulent strain [28]. When injected into the corneas, the protease has been demonstrated to induce severe lesions, similar to those caused by live bacteria. Lysenko [30] purified chitinase from S. marcescens culture, which was then toxic to Galleria mellonella larvae. To the best of our knowledge, there is no experimental evidence to show that serratial lipase and nuclease are virulence factors; however, it is suggested that microbial lipases may function as virulence factors [43]. Furthermore, Streptococcus pyogenes nuclease A functions as a virulence factor in a mouse infection model [19]. Recently, nuclease from Aeromonas hydrophila evades innate immune defenses of the host [25]. Thus, certain extracellular proteins produced by S. marcescens may be potential virulence factors.
Several enzymological studies have been conducted to clarify the pathological and biological significance of these enzymes. The results showed that some bacterial enzymes also have industrial applications or are useful as a tool in other scientific studies [18]. Among serratial enzymes, S. marcescens nuclease (NucA) is widely used in scientific study. NucA is often used to eliminate nucleic acid contamination from purified proteins, commonly from recombinant DNA products, or to reduce viscosity for subsequent processing steps. High viscosity of bacterial lysate caused by released nucleic acid would otherwise hinder subsequent purification steps. Use of nuclease is an inexpensive and effective method to remove the contaminating nucleic acid [3].
It is known that Serratia species, with the exception of S. fonticola, produce nuclease [16]. Study of other serratial nucleases, however, is insufficient in comparison with that on S. marcescens NucA. In a previous study, Serratia liquefaciens isolated from an antlion has been shown to be pathogenic to insects, Bombyx mori, and Periplaneta americana [10]. S. liquefaciens is also shown to be closely related to Serratia proteamaculans and Serratia grimesii [17], and relatively far from S. marcescens in phylogenetic trees based on 16S rRNA gene [1, 8] and housekeeping genes [1].
While studying the pathogenesis of S. liquefaciens Kuo1-1, we previously determined the draft genome sequence using strain FK01, a nalidixic acid mutant of Kuo1-1 [27, 45]. In the sequence (BAZB00000000), nucA gene homolog (slnucA, locus tag: SLIQ_04890) was found in scaffold 01 (DF820427). Subsequently, we planned to demonstrate the expression of recombinant S. liquefaciens nuclease (rSlNucA).
In this study, rSlNucA was produced using the Baculovirus-B. mori protein expression system. This system is widely used for expression of the recombinant protein [29]. This system previously enabled the expression of several useful proteins such as bacterial peptide-N-glycosidase F [31], urease B subunit and heat shock protein A subunit [47], fungal peroxidase fusion protein [20], arthropodic endo-β-1,4-glucanase [24], insect ferritin light chain [23], endo-β-N-acetylglucosaminidase H [33], DNA methyltransferase DNMT-1 [32], fish tributyltin-binding protein type 1 [41] and type 2 [40], plant mismatch endonuclease [34], and human defensins [14]. S. marcescens NucA is also expressed by E. coli [13]. Although experimental evidence is unavailable, rSlNucA can be successfully expressed in the E. coli system owing to its sequence similarity. The expressed rSlNucA is found in the E. coli inclusion body, which is dissolved by urea to obtain the S. marcescens NucA enzyme. The purification procedure for soluble protein is simpler and faster than that for insoluble protein and is subsequently, cost effective. Therefore, in this study, we attempted to express rSlNucA as a soluble protein in hemolymph using the Baculovirus-B. mori protein expression system.
Baculovirus-B. mori protein expression system is mainly used for production of eukaryotic proteins; however, information regarding bacterial protein expression is limited [31, 47]. Although the E. coli expression system is a useful and widely used method, certain drawbacks remain, even with regard to bacterial protein expression. For example, if the expressed recombinant protein is toxic to E. coli, it may result in low yield or low activity by mutation. In this study, rSlNucA was expressed using this system as an example of bacterial proteins and was subsequently characterized.
Materials and Methods
Cloning of S. liquefaciens Nuclease (slnucA) Gene
Nuclease gene (slnucA) was amplified from genomic DNA of S. liquefaciens FK01 using the primer pair, SlnucA-5-NOATG (cgttttaacaagatgttagctctggtaacc) and SlnucA-3-XhoI (ggggctcgagctcttgcaacccatcagctc), and KOD-Plus-Neo DNA polymerase (TOYOBO, Tokyo, Japan). The amplified, complete coding region of slnucA (residues 1–265), including the native signal peptide (NSP) region, was cloned into the pENTRL21TEVH8STREP vector [33] that contained the attL1–attL2 sequence for Gateway technology and C-terminal His and STREP-tag.
The coding region of SlNucA (residues 21–265), not including the native signal peptide was amplified with the primer pair, SlnucA-5-NOSP (gaggcattggaatctatcgacaactgcgcg) and SlnucA-3-XhoI (ggggctcgagctcttgcaacccatcagctc), and cloned into a pENTRL2130KTEVH8STREP vector that contained the silkworm 30-K (MRLTLFAFVLAVCALASNA) signal peptides at its NH2-terminus and COOH-terminal His and STREP-tag.
Generation of Recombinant Baculoviruses
Above two recombinant entry plasmids (pENTRL21NSP-SlnucA-TEVH8STREP and pENTRL2130K-SlnucA-TEVH8STREP) were transposed to the destination vector (pDEST8, Invitrogen, USA) with Gateway LR reaction as per the manufacturer protocols. The obtained transfer plasmids were transformed into E. coli BmDH10Bac (BmNPV expression system [36]). The SlNucA expression cassette was transferred into a mini-attTn7 target site of a Baculovirus shuttle vector (bacmid) via in vivo transposition mediated by Tn7 transposase.
The recombinant bacmid DNA was transfected into NIAS-Bm-oyanagi2 cells (kindly provided by Dr. Imanishi) by lipofection using the FuGENE HD transfection reagent (Promega, USA). Generation and large-scale harvest of recombinant baculoviruses were carried out as per the manufacturer protocols (Invitrogen). The viral titers were determined by the end-point dilution method [38].
Expression of rSlNucA
The expression level of rSlNucA by viral infection in NIAS-Bm-oyanagi2 cells was measured as follows: 1 × 106 cells per well in six-well tissue culture plates were infected with recombinant BmNPVs at a multiplicity of infection (MOI) values of 1. The infected cells and culture medium were collected at 4 days post-infection (dpi). After centrifugation at 1000×g for 10 min at 4 °C, the culture medium (extracellular proteins) and cells (intracellular proteins) were diluted with phosphate-buffered saline (PBS, 8.1 mM Na2HPO4, 1.47 mM KH2PO4, 136.9 mM NaCl, 2.7 mM KCl) and solubilized in Laemmli sample buffer, respectively. Extracellular and intracellular samples (4.5-µl culture equivalent) were separated with 10 % sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). After electroblotting, the recombinant proteins were detected using HisProbe-HRP conjugate (1:2000, Thermo Fisher Scientific Inc., MA, USA) as previously described [33].
With regard to rSlNucA expression levels in silkworm hemolymph, recombinant viruses were carefully injected into the hemocoel of silkworm larvae (d17 strain) on Day 2 of the 5th instar at the dose of 1 × 105 pfu per larva using a microliter™ syringe with a 30-gage needle (Hamilton Co., USA). After 4 dpi, hemolymph was collected. After centrifugation at 10,000×g for 30 min at 4 °C to remove insoluble matter, hemolymph was diluted with PBS. rSlNucA (1-µl hemolymph equivalent) was separated by 15 % SDS-PAGE and recombinant proteins were detected with HisProbe-HRP conjugate. Hemolymph collected from mock-inoculated (empty vector), and noninfected larvae were used as negative control. The experiments were repeated three times.
Purification of rSlNucA
After 4 dpi, 10 ml hemolymph, from approximately 25 silkworm larvae, was purified by two-step affinity purification using HisTrap excel (GE Healthcare Bioscience, Piscataway, NJ, USA) and StrepTrap HP columns (IBA GmbH, Germany) as previously described [33].
Concentrations of purified rSlNucA were determined using Pierce BCA protein assay kit (Thermo Fisher Scientific Inc., MA, USA), with bovine serum albumin as a standard. The proteins were suspended in storage buffer containing PBS and then stored at −80 °C until use.
In order to confirm nuclease activity, a 10-µl reaction mixture containing rSlNucA, 20 mM NaH2PO4–Na2HPO4 buffer (pH 7.5), 2 mM MgCl2, and 100 ng substrate was incubated at 37 °C for 30 min. λDNA and B. mori total RNA were used as substrates in DNase and RNase activity tests, respectively. The reaction was stopped by adding 2 µl loading buffer containing 100 mM EDTA. Degradation of DNA and RNA was analyzed by agarose gel electrophoresis. Benzonase® (Merck Millipore Co., Darmstadt, Germany) was quantified by ImageJ using Coomassie Brilliant Blue R-250-stained SDS-PAGE gel, and was used as a positive control. The experiments were repeated at least twice.
Homomultimer Formation of rSlNucA
Ten µl of rSlNucA in storage buffer (400 ng/µl) was mixed with an equal volume of glutaraldehyde and the mixture was incubated at 30 °C for 30 min. The cross-linking reaction was quenched by adding 2 µl of 1 M Tris–Cl (pH 8.0). The samples were added to an equal volume of 2× Laemmli sample buffer and a 500-ng protein aliquot was analyzed by 10 % SDS-PAGE. The experiments were repeated twice.
Characterization of rSlNucA
Standard DNase Assay
An aliquot of 100-µl reaction mixture containing 1 ng rSlNucA, 20 mM NaH2PO4–Na2HPO4 buffer (pH 7.5), 2 mM MgCl2, and 50 µg DNA (salmon testes) was incubated at 37 °C for 30 min. The reaction was stopped by adding an equal volume of 5 % perchloric acid and then chilled for 20 min on ice. After centrifugation at 20,630×g, at 4 °C for 5 min, an aliquot of supernatant was diluted with nine volumes of water. Absorbance at 260 nm of diluent was measured. Sample without rSlNucA was used as blank, measured in the same manner.
To characterize rSlNucA, composition of reaction or reaction conditions were changed as needed, described below. The experiments were repeated three times.
Optimum pH
The reaction was carried out using different buffer systems, including acetic acid–sodium acetate (pH 4.0–5.5), NaH2PO4–Na2HPO4 (pH 6.0–8.0), boric acid–NaOH (pH 8.5–9.0), and NaHCO3–NaOH (pH 9.5–10.0).
pH Stability
rSlNucA (400 ng/µl) and 100 mM buffer (acetic acid–sodium acetate at pH 4.0–5.5, NaH2PO4–Na2HPO4 at pH 6.0–8.0, boric acid–NaOH at pH 8.5–9.0, NaHCO3–NaOH at pH 9.5–10.0) were mixed in a ratio of 1:3 and incubated at 4 °C for 18 h. An aliquot was diluted with 99-fold volume of storage buffer. The diluent (1 ng protein/µl) was used as enzyme solution.
Optimum Temperature
DNase assay was carried out at 5, 10, 15, 20, 25, 30, 35, 37, 40, 45, 50, 55, and 55 °C for determination of optimum temperature.
Heat Stability
rSlNucA in storage buffer was incubated at 30, 35, 40, 45, 50, 55, 60, 65, and 70 °C for 30 min. The heat-treated protein was used as enzyme solution.
Effect of Divalent ion on DNase Activity
To determine optimum concentration of MgCl2, reaction was supplemented with 0.125, 0.25, 0.5, 1, 2, 4, 8, and 16 mM of MgCl2.
Various metal ions including MgCl2, CaCl2, MnCl2, CoCl2, ZnCl2, and BaCl2 were added to the reaction at concentrations of 4 and 16 mM.
Effect of NaCl and KCl on DNase Activity
Reactions were supplemented with additional NaCl and KCl to be final concentrations of 6.25, 12.5, 25, 50, 100, 200, and 400 mM. Since storage buffer contained NaCl and KCl, traces of these salts (1.369 mM NaCl, 0.027 mM KCl) were also contributed by the storage buffer.
Effect of Detergents on DNase Activity
Detergents including Brij 35, Brij 58, Nonidet P-40, Triton X-100, Tween 20, Tween 80, 3-[(3-cholamidopropyl)dimethylammonio]propanesulfonate, sodium deoxycholate, sodium N-lauroylsarcosinate, and sodium dodecyl sulfate were added to reactions at the final concentrations of 0.1 and 1 %.
DNase Activity in Lysis Buffers
Nuclease is often used to reduce viscosity of the lysate during bacterial protein extraction. Therefore, two different lysis buffers: strong (RIPA buffer, 50 mM Tris–Cl, pH 8.0, 150 mM NaCl, 0.5 % sodium deoxycholate, 1 % Nonidet P-40) and mild (1.5 % Triton X-100, 0.4 % sodium deoxycholate in PBS) lysis buffer, were used for protein extraction from whole cell and inclusion body purification, respectively, for DNase activity of rSlNucA.
The reaction mixture (100 µl) containing 1 ng rSlNucA, 2 mM MgCl2, 50 µg DNA (salmon testes), and lysis buffer (series of twofold dilutions) was incubated at 37 °C for 30 min.
Analyses of DNA and Protein Prepared from Bacterial Lysate after rSlNucA Treatment
E. coli BL21 DE3 (pLysS, pET16b) was cultured in LB broth containing appropriate antibiotics at 37 °C for 16 h. After centrifugation of 25 µl bacterial culture, the pellet was resuspended with half-volume of water (12.5 µl). An equal volume of 2× RIPA buffer was added to the suspension and mixed well. Subsequently, 25 µl of 4 mM MgCl2 and 1 µl rSlNucA were added. The samples were incubated at 37 °C for 30 min.
For DNA extraction, the reaction was stopped by phenol/chloroform extraction. An aliquot (10-µl culture equivalent) of the water layer after centrifugation was analyzed by agarose gel electrophoresis.
For protein extraction, reaction was stopped by adding an equal volume of 2× Laemmli sample buffer and immediately incubated at 90 °C for 3 min. The protein sample (2.5-µl culture equivalent) was analyzed by SDS-PAGE. These experiments were repeated twice.
Results
Expression and Purification of rSlNucA
A phylogenetic tree based on the amino acid sequences of nucleases and multiple alignment is shown in Fig. 1. The amino acid residues contributing magnesium ion (Asn) and involved in substrate (Asp, Arg, Arg) binding were conserved in all sequences analyzed. rSlNucA constructs, as shown in Fig. 2a, contained signal peptide sequence (native or 30K) at the NH2 terminal and TEV cleavage, His-tag, and strep-tag sequences at the COOH-terminal.

Phylogenetic analysis and multiple alignments based on amino acid sequences of nucleases. In the phylogenetic tree, the number for each interior branch is the percent bootstrap value (1000 resamplings). Pseudomonas fuscovaginae nuclease was used as an outgroup. In alignment, underlined letters indicate three upstream and two downstream amino acid residues of putative cleavage sites. Arrowheads indicate conserved residues observed in Nuc domain. The numbers in brackets indicate the number of amino acids. Abbreviations for species (accession numbers): Sli, Serratia liquefaciens (GAK25990); Spr, S. proteamaculans (WP_012006235); Eco, Escherichia coli (KLX16257); Sma, S. marcescens (M19495); Eae, Enterobacter aerogenes (WP_020079743); Yru, Yersinia ruckeri (WP_038251463); Pfu, P. fuscovaginae (WP_010451627)

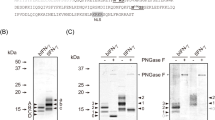
Expression and purification of rSlNucA. Two nuclease genes harboring different signal peptides (a). Expression of rSlNucA was carried out using cultured cells (b, left panel) and B. mori larvae (b, right panel). Closed triangles indicate recombinant nuclease. Open triangle indicates nonspecific band originated from hemolymph protein that reacts with HisProbe. Purification of rSlNucA containing native signal peptide (c, upper panel; rSlNucA-NSP) and 30K signal peptide (c, lower panel; rSlNucA-30K). First, rSlNucAs were purified by His-tag, subsequently by Strept-tag. To detect contaminating protein, more than adequate quantity was loaded in the analysis
Recombinant baculoviruses constructed for rSlNucA expression were inoculated into NIAS-Bm-oyanagi2 cell culture. Both rSlNucAs were detected in intracellular fraction but not in extracellular fraction (Fig. 2b, left). When the baculoviruses were used to infect silkworm larvae, recombinant proteins were secreted into the hemolymph (Fig. 2b, right). Fractions were analyzed by SDS-PAGE (Fig. 2c) at each purification step. As a result, rSlNucAs were highly purified by two-step affinity purification.
rSlNucA digested double-stranded DNA (λDNA) and total RNA (B. mori) in a dose-dependent manner (Fig. 3). At 37 °C, 100 pg rSlNucA adequately digested 100 ng DNA and RNA within 30 min. DNase and RNase activities were approximately 10 times lower than those of commercially available recombinant S. marcescens NucA, Benzonase®, the reason for which is unclear. This result indicated that SlNucA is a nonspecific nuclease found in a wide range of organisms such as Bacillus subtilis, Azotobacter agilis, S. pyogenes, Streptomyces antibioticus, Drosophila melanogaster, and Nicotiana tabacum [39].

Nuclease activity of rSlNucA. λDNA (upper panel) and B. mori total RNA (lower panel) were incubated with different doses of recombinant nuclease at 37 °C for 30 min. Benzonase® was used as a control. In the lower panel, values of molecular weight are indicated in parentheses, since DNA marker was used
Homomultimer Formation of rSlNucA
To determine whether rSlNucA exists as a monomer or multimer, the protein cross-linked by glutaraldehyde was analyzed by SDS-PAGE (Fig. 4). The intensity of the dimeric band increased while that of the monomeric band decreased with an increase in glutaraldehyde concentration. This result indicates that rSlNucA forms homodimers under the conditions studied.

Homomultimer formation of rSlNucA. The recombinant proteins were cross-linked with glutaraldehyde. After quenching, the proteins were analyzed by SDS-PAGE
Characterization of rSlNucA
Enzymatic characteristics of rSlNucAs were determined. In general, the difference in native (rSlNucA-NSP) or 30-K B. mori (rSlNucA-30K) signal peptides did not affect the characteristics to a large extent.
The optimum pH for rSlNucAs was 7.5, with 50 % relative activity between pH 7 and 9.5 (Fig. 5a). At pH 5.0 or lower, nuclease activity was not detected. These enzymes were highly stable within a wide range of pH at 4 °C for 18 h (Fig. 5b). They retained at least 72 % activity (rSlNucA-30K at pH 4.0) or more within this range.

Optimum pH and pH stability of rSlNucA. Relative deoxyribonuclease activities under different pH conditions (a) and stability after incubation at different pH values, at 4 °C for 18 h (b). Maximum activity in each test was defined as 100 % (arrow). Data obtained from the same pH buffer system are connected. The plots indicate average ± standard error (n = 3). Closed and open circles indicate rSlNucA-NSP and rSlNucA-30K, respectively
Optimum temperature was 35 °C (Fig. 6a). At 15 °C or lower, and 50 °C or higher, the relative activity was less than 50 %. Heat stability testing revealed that the enzymes were inactivated as temperature increased (Fig. 6b). At 55 °C or higher, the activity was less than 50 %.

Optimum temperature and heat stability of rSlNucA. Relative deoxyribonuclease activities under different temperatures (a) and stability after incubation in different temperatures for 30 min. Maximum activity in the test (a, arrow) or activity of nontreated nuclease (b) are defined as 100 %. The plots indicate average ± standard error (n = 3). Closed and open circles indicate rSlNucA-NSP and rSlNucA-30K, respectively
When MgCl2 was omitted from the reaction, nuclease activity was not detected (Fig. 7a). Until 4 mM, the activity increased in a dose-dependent manner. In the range of 1–16 mM, the relative activity was 76 % (rSlNucA-30K with 1 mM MgCl2) or more. Although divalent metal ions also promoted the activity partially, the effect was approximately 50 % at best (rSlNucA-30K with 4 mM CaCl2), in case of MgCl2 (Fig. 7b).

Effect of divalent metal ions on deoxyribonuclease activity of rSlNucA. Relative deoxyribonuclease activities with different concentrations of MgCl2 (a) and various metal ions (b). Maximum activity in the tests was defined as 100 % (arrows). In panel A, data shown in logarithmic scale are connected, and closed and open circles indicate rSlNucA-NSP and rSlNucA-30K, respectively. In panel B, closed and open bars indicate 4 and 16 mM, respectively. ND means “not detected.” The data indicate average ± standard error (n = 3)
Higher concentrations of NaCl (Fig. 8a) and KCl (Fig. 8b) inhibited nuclease activity. Since the reaction contained at least 1.369 mM NaCl and 0.027 mM KCl contributed by the storage buffer, the activity in the complete absence of these salts was not determined in this study.

Effect of NaCl and KCl on deoxyribonuclease activity of rSlNucA. Relative deoxyribonuclease activities with different concentrations of NaCl (a) and KCl (b). The sample in which salt was not supplemented was defined as 100 % (arrows). Small amount of salts (1.369 mM NaCl, 0.027 mM KCl) was contributed by the enzyme stock solution. Closed and open circles indicate rSlNucA-NSP and rSlNucA-30K, respectively. The data indicate average ± standard error (n = 3)
Activity was promoted by supplementation of nonionic detergent at 0.1 % (Fig. 9). Although most nonionic detergent did not inhibit the activity at 1 %, Triton X-100 and Tween 20 inhibited at this concentration (less than approximately 50 %). A twitter ionic detergent, CHAPS inhibited activity of rSlNucA-NSP to 50 %, but did not affect rSlNucA-30K activity. Among anionic detergents, no activity was detected when 1 % sodium deoxycholate (DOC) or 0.1 and 1 % SDS were used.

Effect of detergents on deoxyribonuclease activity of rSlNucA. Relative deoxyribonuclease activities with 0.1 % (open bar) and 1 % (closed bar) detergent. The sample in which detergent was not supplemented is defined as 100 % (arrows). Abbreviations for detergents: B-35 Brij 35, B-58 Brij 58, N-40 Nonidet P-40, TX100 Triton X-100, T-20 Tween 20, T-80 Tween 80, CHAPS 3-[(3-cholamidopropyl)dimethylammonio]propanesulfonate; DOC, sodium deoxycholate, LS sodium N-laurylsarcosinate, SDS sodium dodecyl sulfate. The data indicate average ± standard error (n = 3)
The optimum pH and temperature for S. marcescens nuclease are 8.0–8.5 and 35–44 °C, respectively, and require Mg2+, Mn2+, or Co2+ for activity [39]. Additionally, as per the Benzonase instruction manual, values for optimal Mg2+ concentration, pH, temperature, and monovalent cation concentration are 1–2 mM (effective: 1–10 mM), pH 8.0–9.0 (pH 6.0–10.0), 37 °C (0–42 °C), and 0–20 mM (0–150 mM), respectively. The enzymatic characteristics of rSlNucA were similar to these of Benzonase®. Although the activity was slightly low, rSlNucA is comparable to Benzonase® as a molecular biology tool.
DNase Activity in Lysis Buffers
DNase activity of rSlNucA in lysis buffers was measured (Fig. 10). Although relative activities in nondiluted RIPA buffer were approximately 10, 89 % or more activity was detected when RIPA buffer was diluted two- or fourfold. Nuclease activity was not inhibited in a mild lysis buffer containing Triton X-100 and DOC.

Effect of lysis buffers on deoxyribonuclease activity of rSlNucA. Relative deoxyribonuclease activities in different concentrations of strong and mild lysis buffers. The deoxyribonuclease activity of the sample in which lysis buffer was not supplemented, was defined as 100 % (arrows). Closed and open columns indicate rSlNucA-NSP and rSlNucA-30K, respectively. The data indicate average ± standard error (n = 3)
Analyses of DNA and Protein Prepared from Bacterial Lysate after rSlNucA Treatment
Nuclease is often used to digest genomic DNA during purification of recombinant protein expressed in E. coli. Therefore, nuclease activity was assessed in E. coli lysate. Since rSlNucA was active in twofold-diluted RIPA buffer, as described above (Fig. 10), E. coli lysate prepared with RIPA buffer was diluted. Obvious DNA fragmentation was observed when 10 pg rSlNucA was added to 50 µl lysate (Fig. 11a). Higher dose of rSlNucA caused further DNA degradation. Protein profiles were not affected by addition of excessive rSlNucA (Fig. 11b). This result indicates that the purified rSlNucA solution did not contain contamination by proteinase.

Electrophoretic profiles of DNA and protein prepared from E. coli lysate after rSlNucA treatment. Lysate of E. coli BL21 DE3 (pLysS, pET16b) with RIPA buffer was diluted with an equal volume of 4 mM MgCl2. After incubation with rSlNucA, DNA (a) and protein (b) were separately extracted and analyzed
Discussion
Baculovirus-silkworm system is widely used for the production of recombinant eukaryotic protein. We expressed recombinant nuclease from S. liquefaciens to ascertain if this system is also useful for bacterial protein expression. Previously, the signal peptide of a 30-kDa protein from B. mori was shown to exhibit efficient secretion of recombinant proteins produced by the Baculovirus-silkworm expression system [15, 42]. Therefore, the recombinant protein, harboring not only native signal peptide (rSlNucA-NSP) but also the 30-K signal peptide (rSlNucA-30K), was expressed.
It is known that the endoplasmic reticulum (ER) and Golgi body, which are necessary apparatus for protein secretion in eukaryotic cells, are present in the cultured cell. Therefore, we expected that at least rSlNucA-30K will be detected in the extracellular fraction of cultured cells. However, neither rSlNucA was detected nor abundant recombinant proteins were detected in hemolymph when B. mori larvae were used as the host. Although the reason is unclear, we speculate that rSlNucAs may have been secreted at lower levels than the detection limit when cultured cells were used as host. In a complex multicellular organism, different cell types engage in specific functions and as a result, the secretory output of cells and tissues varies widely [4]. rSlNucAs are efficiently secreted only in B. mori. The difference in cellular localization of rSlNucAs between cultured cells and B. mori larvae may be due to the systematically controlled secretion system of a multicellular organism. Therefore, using B. mori larvae to produce recombinant proteins may be advantageous.
rSlNucA-NSP harboring native signal peptide was efficiently secreted to B. mori hemolymph. In gram-negative bacteria, proteins are secreted via two membranes, cytoplasmic (inner) and outer membranes. To secrete protein from cytoplasmic to the extracellular space, gram-negative bacteria possess several secretion systems, categorized Type I to VI [7]. In S. marcescens, NucA is secreted via Type II secretion system, a two-step process. In the first step, NucA is rapidly translocated across the cytoplasmic membrane and the signal peptide is rapidly processed. Subsequently, mature NucA is slowly secreted to the extracellular space, the second step [44]. Sec and Tat pathways are two known pathways in the first of the two-step process, and NucA is transferred by the Sec pathway [44] from the cytoplasmic to the periplasmic space. The signal peptide for Sec pathway typically has an average length of 20 amino acid residues with tripartite structure, i.e., a positively charged amino-terminal (n-region), a hydrophobic core (h-region), and a polar carboxyl-terminal. The overall tripartite structure is recognized by Sec components [37]. The bacterial Sec signal peptide is functionally interchangeable with the signal sequences that direct protein to the Sec systems of the thylakoid membrane and the ER [37, 46]. Based on the information available in the literature, it was estimated that signal peptide of rSlNucA-NSP was functional in B. mori larvae; therefore, rSlNucA-NSP was successfully secreted to hemolymph.
In addition, the results from this study suggest that bacterial protein secreted through Sec pathway can be secreted into hemolymph without additional signal peptide in the Baculovirus-silkworm system. Bagos et al. [2] reported prediction of Tat and Sec signal peptides (READ-TAT, http://www.compgen.org/tools/PRED-TAT). Since it was inferred that this program is useful to estimate localization of recombinant bacterial protein expressed by Baculovirus-silkworm system, rSlNucAs were analyzed by READ-TAT program. The prediction was as follows: rSlNucA-NSP, Sec signal peptide (0.996); rSlNucA-30K, Sec signal peptide (0.999). The results suggest that READ-TAT possibly predicts localization of bacterial recombinant protein expressed in the Baculovirus-silkworm system, and bacterial extracellular proteins secreted through Sec pathway may be highly compatible with the protein expression system. It is necessary to investigate correlation between the prediction and experimental evidence of localization using several other bacterial proteins.
S. marcescens NucA forms a dimer. Although the two subunits are functionally independent and dimer formation is not essential for its activity, dimeric forms of NucA were relatively more active than monomeric forms [12]. Variants (H184A, H184N, H184T, and H184R) of S. marcescens NucA were monomers, have the same secondary structure and activity as the wild-type enzyme [11]. In contrast, amino acid substitution that alters the secondary structure (S179C) is suggested to cause decrease in activity. Thus, secondary structure and dimer formation are important for optimal nuclease activity.
In the present study, artificial sequences for His-, Strep-tags, and TEV cleavage site were added at the COOH-terminus of rSlNucA for effective and simple purification. It was important to ascertain whether this artificial sequence affected secondary structure and dimer formation. It was predicted by PSIPRED V3.3 [26] that the addition of the artificial sequence would not affect the intramolecular secondary structure of the other regions (data not shown). In addition, dimer formation of rSlNucAs was experimentally demonstrated. Therefore, the activity of rSlNucA may not presumably decrease in comparison with native SlNucA. It was shown that these tags help effective and rapid purification of rSlNucA. When rSlNucAs are used to reduce viscosity of the solution during purification of recombinant proteins, these tags enable to separate rSlNucA from other proteins. When the tags are identical between rSlNucA and other proteins, the tags can be eliminated by TEV digestion.
In conclusion, rSlNucA was expressed by the Baculovirus-silkworm system. Purified rSlNucA digested DNA and RNA. Enzymatic characteristics were found to be similar to those of S. marcescens NucA, and it could digest E. coli DNA in diluted RIPA buffer. These results indicate that rSlNucA could be a useful tool in molecular biology.
Although biological function of serratial nuclease is not yet well characterized, it has been shown that Shewanella oneidensis nuclease utilizes extracellular DNA as a source of nutrients such as carbon, nitrogen, and phosphorus [22]. In addition, nucleases of some bacterial species were shown to be potential virulence factors [19, 25]. Because it is possible that S. liquefaciens nuclease contributes to pathogenesis or contributes to bacterial survival in the host, this enzyme may be a potential target molecule for drug development. rSlNucAs produced by the Baculovirus-silkworm system utilized in this study will therefore contribute to such fundamental studies. However, determination of biological and pathological function of rSlNucA in S. liquefaciens needs further study.
References
Abebe-Akele, F., Tisa, L. S., Cooper, V. S., Hatcher, P. J., Abebe, E., & Thomas, W. K. (2015). Genome sequence and comparative analysis of a putative entomopathogenic Serratia isolated from Caenorhabditis briggsae. BMC Genomics, 16, 531.
Bagos, P. G., Nikolaou, E. P., Liakopoulos, T. D., & Tsirigos, K. D. (2010). Combined prediction of Tat and Sec signal peptides with hidden Markov models. Bioinformatics, 26, 2811–2817.
Benedik, M. J., & Strych, U. (1998). Serratia marcescens and its extracellular nuclease. FEMS Microbiology Letters, 165, 1–13.
Benham, A. M. (2012). Protein secretion and the endoplasmic reticulum. Cold Spring Harbor Perspectives in Biology, 4, a012872.
Braun, V., & Schmitz, G. (1980). Excretion of a protease by Serratia marcescens. Archives of Microbiology, 124, 55–61.
Bromke, B. J., & Hammel, J. M. (1979). Regulation of extracellular protease formation by Serratia marcescens. Canadian Journal of Microbiology, 25, 47–52.
Costa, T. R., Felisberto-Rodrigues, C., Meir, A., Prevost, M. S., Redzej, A., Trokter, M., & Waksman, G. (2015). Secretion systems in Gram-negative bacteria: structural and mechanistic insights. Nature Reviews Microbiology, 13, 343–359.
Dauga, C., Grimont, F., & Grimont, P. A. (1990). Nucleotide sequences of 16S rRNA from ten Serratia species. Research in Microbiology, 141, 1139–1149.
Eaves, G. N., & Jeffries, C. D. (1963). Isolation and properties of an exocellular nuclease of Serratia marcescens. Journal of Bacteriology, 85, 273–278.
Egami, I., Iiyama, K., Zhang, P., Chieda, Y., Ino, N., Hasegawa, K., et al. (2009). Insecticidal bacterium isolated from an ant lion larva from Munakata, Japan. Journal of Applied Entomology, 133, 117–124.
Franke, I., Meiss, G., Blecher, D., Gimadutdinow, O., Urbanke, C., & Pingoud, A. (1998). Genetic engineering, production and characterisation of monomeric variants of the dimeric Serratia marcescens endonuclease. FEBS Letters, 425, 517–522.
Franke, I., Meiss, G., & Pingoud, A. (1999). On the advantage of being a dimer, a case study using the dimeric Serratia nuclease and the monomeric nuclease from Anabaena sp. strain PCC 7120. Journal of Biological Chemistry, 274, 825–832.
Friedhoff, P., Gimadutdinow, O., Rüter, T., Wende, W., Urbanke, C., Thole, H., & Pingoud, A. (1994). A procedure for renaturation and purification of the extracellular Serratia marcescens nuclease from genetically engineered Escherichia coli. Protein Expression and Purification, 5, 37–43.
Fukushima, M., Iiyama, K., Yamashita, J., Furue, M., Tsuji, G., Imanishi, S., et al. (2013). Production of small antibacterial peptides using silkworm-baculovirus protein expression system. Preparative Biochemistry & Biotechnology, 43, 565–576.
Futatsumori-Sugai, M., & Tsumoto, K. (2010). Signal peptide design for improving recombinant protein secretion in the baculovirus expression vector system. Biochemical and Biophysical Research Communications, 91, 931–935.
Grimont, F., & Grimont, P. A. (2006). The genus Serratia. Prokaryotes, 6, 219–244.
Grimont, P. A., Irino, K., & Grimont, F. (1982). The Serratia liquefaciens–S. proteamaculans–S. grimesii complex: DNA relatedness. Current Microbiology, 7, 63–67.
Gurung, N., Ray, S., Bose, S., & Rai, V. (2013). A broader view: microbial enzymes and their relevance in industries, medicine, and beyond. Biomedical Research International, 2013, 329121.
Hasegawa, T., Minami, M., Okamoto, A., Tatsuno, I., Isaka, M., & Ohta, M. (2010). Characterization of a virulence- associated and cell-wall-located DNase of Streptococcus pyogenes. Microbiology, 156, 184–190.
Hayashi, K., Lee, J. M., Tomozoe, Y., Kusakabe, T., & Kamiya, N. (2015). Heme precursor injection is effective for Arthromyces ramosus peroxidase fusion protein production by a silkworm expression system. Journal of Bioscience and Bioengineering, 120, 384–386.
Heller, K. B. (1979). Lipolytic activity copurified with the outer membrane of Serratia marcescens. Journal of Bacteriology, 140, 1120–1122.
Heun, M., Binnenkade, L., Kreienbaum, M., & Thormann, K. M. (2012). Functional specificity of extracellular nucleases of Shewanella oneidensis MR-1. Applied and Environment Microbiology, 78, 4400–4411.
Hong, S. M., Mon, H., Lee, J. M., & Kusakabe, T. (2014). Characterization and recombinant protein expression of ferritin light chain homologue in the silkworm, Bombyx mori. Insect Science, 21, 135–146.
Hong, S. M., Sung, H. S., Kang, M. H., Kim, C. G., Lee, Y. H., Kim, D. J., et al. (2014). Characterization of Cryptopygus antarcticus endo-β-1,4-glucanase from Bombyx mori expression systems. Molecular Biotechnology, 56, 878–889.
Ji, Y., Li, J., Qin, Z., Li, A., Gu, Z., Liu, X., et al. (2015). Contribution of nuclease to the pathogenesis of Aeromonas hydrophila. Virulence, 6, 515–522.
Jones, D. T. (1999). Protein secondary structure prediction based on position-specific scoring matrices. Journal of Molecular Biology, 292, 195–202.
Kaibara, F., Iiyama, K., Chieda, Y., Lee, J. M., Kusakabe, T., Yasunaga-Aoki, C., & Shimizu, S. (2012). Construction of serralysin-like metalloprotease-deficient mutants of Serratia liquefaciens and their virulence in the silkworm, Bombyx mori. Journal of Insect Biotechnology and Sericology, 81, 55–61.
Kamata, R., Matsumoto, K., Okamura, R., Yamamoto, T., & Maeda, H. (1985). The serratial 56K protease as a major pathogenic factor in serratial keratitis. Clinical and experimental study. Ophthalmology, 92, 1452–1459.
Lee, J. M., Mon, H., Banno, Y., Iiyama, K., & Kusakabe, T. (2012). Bombyx mori strains useful for efficient recombinant protein production using a baculovirus vector. Journal of Biotechnology & Biomaterials, S9, 003.
Lysenko, O. (1976). Chitinase of Serratia marcescens and its toxicity to insects. Journal of Invertebrate Pathology, 27, 385–386.
Masuda, A., Xu, J., Mitsudome, T., Nagata, Y., Morokuma, D., Mon, H., et al. (2015). Mass production of an active peptide-N-glycosidase F using silkworm-baculovirus expression system. Molecular Biotechnology, 57, 735–745.
Mitsudome, T., Mon, H., Xu, J., Li, Z., Lee, J. M., Patil, A. A., et al. (2015). Biochemical characterization of maintenance DNA methyltransferase DNMT-1 from silkworm, Bombyx mori. Insect Biochemistry and Molecular Biology, 58, 55–65.
Mitsudome, T., Xu, J., Nagata, Y., Masuda, A., Iiyama, K., Morokuma, D., et al. (2014). Expression, purification, and characterization of endo-β-N-acetylglucosaminidase H using baculovirus-mediated silkworm protein expression system. Applied Biochemistry and Biotechnology, 172, 3978–3988.
Mon, H., Lee, J. M., Fukushima, M., Nagata, Y., Fujii, M., Xu, J., et al. (2013). Production and characterization of the celery mismatch endonuclease CEL II using baculovirus/silkworm expression system. Applied Microbiology and Biotechnology, 97, 6813–6822.
Monreal, J., & Reese, E. T. (1969). The chitinase of Serratia marcescens. Canadian Journal of Microbiology, 15, 689–696.
Motohashi, T., Shimojima, T., Fukagawa, T., Maenaka, K., & Park, E. Y. (2005). Efficient large-scale protein production of larvae and pupae of silkworm by Bombyx mori nuclear polyhedrosis virus bacmid system. Biochemical and Biophysical Research Communications, 326, 564–569.
Natale, P., Brüser, T., & Driessen, A. J. (2008). Sec- and Tat-mediated protein secretion across the bacterial cytoplasmic membrane—distinct translocases and mechanisms. Biochimica et Biophysica Acta, 1778, 1735–1756.
O’Reilly, D. R., Miller, L., & Luckow, V. A. (1992). Baculovirus expression vectors: a laboratory manual. New York: Oxford University Press.
Rangarajan, E. S., & Shankar, V. (2001). Sugar non-specific endonucleases. FEMS Microbiology Reviews, 25, 583–613.
Satone, H., Akahoshi, E., Nakamura, A., Lee, J. M., Honda, M., Shimasaki, Y., et al. (2013). Expression and functional characterization of recombinant tributyltin-binding protein type 2. Journal of Toxicological Sciences, 38, 885–890.
Satone, H., Lee, J. M., Oba, Y., Kusakabe, T., Akahoshi, E., Miki, S., et al. (2011). Tributyltin-binding protein type 1, a lipocalin, prevents inhibition of osteoblastic activity by tributyltin in fish scales. Aquatic Toxicology, 103, 79–84.
Soejima, Y., Lee, J. M., Nagata, Y., Mon, H., Iiyama, K., Kitano, H., et al. (2013). Comparison of signal peptides for efficient protein secretion in the baculovirus-silkworm system. Central European Journal of Biology, 8, 1–7.
Stehr, F., Kretschmar, M., Kröger, C., Hube, B., & Schäfer, W. (2003). Microbial lipases as virulence factors. Journal of Molecular Catalysis. B, Enzymatic, 22, 347–355.
Suh, Y., Jin, S., Ball, T. K., & Benedik, M. J. (1996). Two-step secretion of the Serratia marcescens extracellular nuclease. Journal of Bacteriology, 178, 3771–3778.
Taira, E., Iiyama, K., Mon, H., Mori, K., Akasaka, T., Tashiro, K., et al. (2014). Draft genome sequence of entomopathogenic Serratia liquefaciens strain FK01. Genome Announcements, 2, e00609–e00614.
Wickner, W., Driessen, A. J., & Hartl, F. U. (1991). The enzymology of protein translocation across the Escherichia coli plasma membrane. Annual Review of Biochemistry, 60, 101–124.
Zhang, X., Shen, W., Lu, Y., Zheng, X., Xue, R., Cao, G., et al. (2011). Expression of UreB and HspA of Helicobacter pylori in silkworm pupae and identification of its immunogenicity. Molecular Biology Reports, 38, 3173–3180.
Acknowledgments
The cost of publication was supported in part by a Research Grant for Young Investigators from the Faculty of Agriculture, Kyushu University. We would like to thank Editage (www.editage.jp) for English language editing.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
No conflict of interest declared.
Additional information
Kazuhiro Iiyama and Jae Man Lee contributed equally to this manuscript.
Rights and permissions
About this article

Cite this article
Iiyama, K., Lee, J.M., Tatsuke, T. et al. Expression and Characterization of Recombinant Serratia liquefaciens Nucleases Produced with Baculovirus-mediated Silkworm Expression System. Mol Biotechnol 58, 393–403 (2016). https://doi.org/10.1007/s12033-016-9937-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12033-016-9937-y