
Abstract
Endo-β-1,4-glucanase (CaCel) from Antarctic springtail, Cryptopygus antarcticus, a cellulase with high activity at low temperature, shows potential industrial use. To obtain sufficient active cellulase for characterization, CaCel gene was expressed in Bombyx mori-baculovirus expression systems. Recombinant CaCel (rCaCel) has been expressed in Escherichia coli (Ec-CaCel) at temperatures below 10 °C, but the expression yield was low. Here, rCaCel with a silkworm secretion signal (Bm-CaCel) was successfully expressed and secreted into pupal hemolymph and purified to near 90 % purity by Ni-affinity chromatography. The yield and specific activity of rCaCel purified from B. mori were estimated at 31 mg/l and 43.2 U/mg, respectively, which is significantly higher than the CaCel yield obtained from E. coli (0.46 mg/l and 35.8 U/mg). The optimal pH and temperature for the rCaCels purified from E. coli and B. mori were 3.5 and 50 °C. Both rCaCels were active at a broad range of pH values and temperatures, and retained more than 30 % of their maximal activity at 0 °C. Oligosaccharide structural analysis revealed that Bm-CaCel contains elaborated N- and O-linked glycans, whereas Ec-CaCel contains putative O-linked glycans. Thermostability of Bm-CaCel from B. mori at 60 °C was higher than that from E. coli, probably due to glycosylation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
As global warming emerges as a serious problem, there is an increasing demand for bioethanol produced from cellulosic materials. Cellulosic material from plant cell walls is an abundant and renewable carbon source for the bioenergy industry. Cellulose can be degraded by cellulases encoded in microorganisms, plants, and animals [1–3]. Cellulases are glycosyl hydrolases that can break the β (1 → 4) bonds in cellulose and are the key enzymes in the production of cellulosic ethanol from plant biomass. There are three different types of cellulases, i.e., endoglucanase (1,4-β-d-glucanohydrolase), exocellobiohydrolase (1,4-β-d-glucan glucohydrolase), and β-glucosidase (β-d-glucoside glucohydrolase) [4]. Cellulases have a wide range of potential applications. There is considerable interest in the biotechnological application of cellulase to food, detergent, and textile industries; and bioremediation of polluted soils and wastewater, as well as production of bioenergy [5–8]. In the food industry, cellulases are being used in the clarification of fruit juices, to increase homogeneous water absorption of cereal, for the gelatinization of seaweeds to increase digestibility, in the filtration of beer, and to increase the aroma in wines [9]. In the textile and detergent industries, cellulases have been used for biostoning of denim, for minimizing loss of tensile strength in textile production processes [10], and for removing microfibrils from cotton fabrics and stains from laundry [6, 9]. For use as a food additive, in the detergent industry, and in the stonewashing process, cellulase should be active at low and moderate temperatures. Although a large number of cellulase-producing psychrotrophic microorganisms have been isolated from various environments and characterized in the last one and a half decades [11–13], few studies have been directed at isolating cold-active enzymes from animals.
The Antarctic is recognized as an important resource for isolating cold-adapted microorganisms and enzymes. The freezing-intolerant Antarctic springtail, Cryptopygus antarcticus Willem (Collembola, Isotomidae), is the most abundant and widespread terrestrial micro-arthropod in the maritime Antarctic region. Collembola species found in topsoil and decaying material are known to possess several carbohydrases, such as cellulase, trehalase, and chitinase [14]. In a previous study, cDNAs encoding cold-active cellulase, mannanase, and laminarinase from C. antarcticus were isolated, and the recombinant enzymes were expressed in Escherichia coli cells [15–17]. A small amount of active C. antarcticus cellulase (CaCel) was produced in the E. coli expression system by lowering the cultivation temperature to the minimal growth condition [17]. Establishment of a high-yield and low-cost cellulase expression system is required for commercial utilization.
The baculovirus-silkworm expression system is very efficient for the large-scale production of functional eukaryotic proteins and glycoproteins [18, 19]. Glycosylation is one of the most common and important post-translational modifications of proteins and is known to play an essential role in the function, structural folding, and stability of proteins [20]. Two major types of glycosylation, N-linked and O-linked, are frequently observed in cellulases [21]. The most extensively studied cellulase, cellobiohydrolase I (Cel7A) of Trichoderma reesei, contains 4 N-linked oligosaccharides that play important roles in catalytic activity and cellulose-binding affinity, and 29 O-linked oligosaccharides, which protect the enzyme from proteolytic degradation [22–24]. Furthermore, Zhao et al. [25] reported that the thermal stability of recombinant cellulase produced in Pichia pastoris can be increased after glycosylation. Biologically active recombinant cellulases have been produced using various expression systems [17, 26–29]. In general, it is difficult to generate glycosylated proteins in the E. coli system, and few proteins can be secreted into the culture medium. In addition, there is little information available on whether glycosylation of recombinant cellulase affects the enzyme’s properties.
As previously stated, a CaCel had previously been purified from E. coli and characterized, and the purified enzyme was successfully applied to the hydrolysis of glycosides in vitro [17]. However, enzyme production was insufficient for commercial applications. In the present study, we attempted to produce recombinant CaCel using the baculovirus-silkworm pupae system, and we assayed the resulting enzyme’s biological activity. In addition, we compared the expression level, glycosylation, and properties of the cold-adapted CaCel synthesized in E. coli Rosetta [17] and in Bombyx mori pupae.
Materials and Methods
Materials
PCR-amplified cDNA encoding the CaCel of an Antarctic springtail (accession no. Fj648735) was provided by one of the authors and was subcloned into the vector pET28(a) for E. coli expression (Novagen, USA) and pENTR11 (modified by adding signal peptide and 8× His sites) and pDEST8 for B. mori expression (Invitrogen, USA). Taq DNA polymerase and pfu DNA polymerase were purchased from Toyobo (Japan) and Promega (USA). All the restriction endonucleases and T4 DNA ligase were purchased from Takara (Japan). DNA purification kits were purchased from Qiagen (USA).
Silkworm strains jam125 × jam126 (Kangwon wonjongjang, Korea) were routinely reared on fresh mulberry leaves at 25 °C, and pupae were maintained at 25–27 °C. The B. mori cell line BmN4 was cultured in TNM-FH insect medium (WillGene, Korea) supplemented with 10 % fetal bovine serum (GIBCO, USA) at 25 °C.
Expression and Purification of CaCel
Previously, recombinant CaCel (rCaCel) was produced in E. coli Rosetta gami (DE3) and purified as described by Song et al. [17]. The deduced mature sequence without signal sequence was amplified from C. antarcticus cDNA using primers P1 and P2 (P1, 5′-CCATGGCTAACATCTGGCTCCGGTG-3′; P2, 5′-CTCGAGAGGTCCAGGAGTTCTGACGC-3′; Fig. 1a). The underlined nucleotides in the primers are the restriction enzyme sites for NcoI and XhoI, respectively. The rCaCel supernatant was purified using His-Bind resin, according to the manufacturer’s instructions (Novagen, USA). The final purification was performed by size exclusion chromatography on a Superdex G-75 column (GE Healthcare, USA) equilibrated with 0.02 M Tris–HCl (pH 7.9).

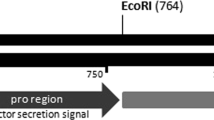
Production of CaCel in E. coli Rosetta-gami (DE-3) and B. mori pupae. a Schematic structure of the pET28a(+)-CaCel expression cassette. T7 T7lac promoter, Trx thioredoxin, 6× His hexa histidine residues. b Schematic structure of the BmNPVD8-30 kCa·Cel expression cassette. polH, polyhedrin promoter sequences, B1 and B2 site-specific recombinant sequences after LR reaction; 30 k signal peptide originating from silkworm. TEV TEV protease recognition sequences, 8× His octa histidine residues. P1, P2, P3, and P4 primers used to amplify C. antarcticus cDNA. c Purification of the CaCel expressed in E. coli Rosetta-gami (DE3) at 10 °C for 5 days. d Purification of the CaCel expressed in B. mori pupae at 25 °C for 4 days. Coomassie Brilliant Blue staining (CBB) and immunoblot analysis (IB, His-tag use) of the recombinant CaCel purified on the Ni2+ His-tag immunoaffinity column; M molecular weight standard marker (BioRad), CL cell lysate, H hemolymph, Fb fatty body lysate. The arrows indicate the position of recombinant CaCel. e IEF analysis on silver-stained gel for determination of the isoelectric point of the purified Ec-CaCel and Bm-CaCel. M broad pI calibration markers. The arrows indicate the position of each CaCel
For the expression and purification of CaCel in B. mori, PCR amplification was performed on pET28(a)/CaCel using a pair of primers, P3 and P4 (P3, 5′-GATATCCTAACATCTGGCTCCGGTG-3′; P4, 5′-GCGGCCGCAGGTCCAGGAGTTCTGACGC-3′; Fig. 1b) differing from the P1 and P2 primers only in the restriction enzyme sites. The underlined nucleotides in the primers are the restriction enzyme sites for EcoRV and NotI, respectively. The PCR reaction was conducted with KOD DNA polymerase (Toyobo, Japan) under the following conditions: an initial denaturation at 95 °C for 5 min, 30 cycles of 95 °C for 20 s, 57 °C for 20 s, and 68 °C for 1 min, with a final extension step at 72 °C for 5 min. Next, the PCR product was inserted between the EcoRV and NotI (Takara, Japan) sites of pENTR11C (Invitrogen, USA), and a 30 k (MRLTLFAFVLAVCALASNADI; originating from silkworm) signal peptides and an 8× His-tag were introduced at its N- and C-terminus, respectively. The transfer vectors were constructed by performing the LR recombination reaction with the destination vector (pDEST8, Invitrogen, USA) and the pENTR11-CaCel. To yield the recombinant bacmid, BmNPVD8/30 k CaCel, the BmDH10Bac E. coli strain was used to host the transfer vector in which the recombinant cellulase was to be expressed under the polyhedrin promoter (Fig. 1b). After the transformation, the large recombinant baculovirus DNA was isolated by the alkaline lysis method [30]. The recombinant bacmid DNA was transfected in 1.5 × 106 BmN4 cells for 48 h using FuGENE HD transfection reagent (Promega, USA). To generate the previously described recombinant baculovirus [31], the transfected cells were cultivated in TNM-FH medium at 25 °C for 4 days, and the resulting recombinant virus was stored at 4 °C.
The recombinant baculovirus (approximately 0.5 × 105 PFU) was inoculated into the body cavity of the pupae with a syringe. The fatty body and hemolymph were collected from silkworm pupae at 4–5 days post-infection as cytosol and secretory proteins, respectively. The fatty body (~9 g fatty body can be harvested from twenty pupae) was added to 500 ml of homogenization buffer (PBS containing 10 mM 2-mercaptoethanol, 1 mM PMSF, 10 % complete, pH 7.0) and homogenized using a Vibra-Cell homogenizer (Sonics, USA) for 5 min at 4 °C. The homogenate was ultracentrifuged at 10,000 rpm for 30 min at 4 °C, and the soluble fraction was filtered using a MILLEX AA column (Millipore). The recovered hemolymph (about 2 ml can be harvested from twenty pupae) was ultracentrifuged at 10,000 rpm for 30 min at 4 °C and was filtered using a MILLEX AA column (Millipore, USA). Fatty body supernatants and hemolymph in which recombinant CaCel protein was detected were purified using HiTrap TALON (GE Healthcare, USA). The column was equilibrated by successive passes of Lysis–Equilibration–Wash buffer (LEW, 50 mM sodium phosphate, 300 mM NaCl, pH 8.0). The fatty body supernatant and hemolymph filtrate were loaded onto the column at 1 ml/min. After washing three times, column-bound protein was twice eluted by rinsing with LEW and 250 mM imidazole, and collected in 10 ml fractions. The final purification was performed using a Superdex G-75 column (GE Healthcare, USA).
The β-1,4-mannanase of C. antarcticus (CaMan) deposited in GenBank under the accession numberABV68808.1 was used as a negative control and purified in E. coli and B. mori, as described above. The CaMan did not present either an N- or an O-glycosylation site in the analysis with NetOGlyc3.1 and NetNGly 1.0 (http://www.cbs.dtu.dk/services/).
Characterization of Recombinant Proteins
The concentration of the purified proteins was determined using the Bradford method, using bovine serum albumin as a standard [32] and a Qubit 2.0 Fluorometer (Invitrogen). The recombinant proteins were resolved by SDS-PAGE in 12 % gel, and the proteins were visualized by Coomassie Brilliant Blue R-250 (CBB R-250) staining, and a Glycoprotein Staining Kit (Pierce, USA), using the periodic acid-Schiff (PAS) method. The separated proteins were transferred onto a PVDF membrane (Millipore). After blocking in TBST buffer (20 mM Tris–buffered saline, pH 7.4; 0.5 M NaCl, and 0.1 % Tween-20), the recombinant CaCel protein was identified by western blot analysis using anti-His (Rockland, USA) antibody for 1 h. Subsequently, filters were reacted with AP-conjugated anti-mouse IgG secondary antibody (Amersham, USA) for 1 h at room temperature (RT), and then visualized using the ECL system (Amersham, USA). Isoelectric focusing (IEF) was performed using a Novex® Isoelectric Focusing Electrophoresis System (Invitrogen, USA), according to the method of Righetti and Dryssale [33], and proteins were visualized using CBB R-250 stain.
The amino-terminal sequences of rCaCel were determined by automated Edman degradation performed on an ABI492 sequencer (Applied Biosystems, USA).
Glycosylation Analysis of Recombinant Proteins
The CaCels purified from B. mori and E. coli were subjected to deglycosylation with peptide-N-glycosidase F (PNGaseF) and endoglycosidase H (EndoH), according to the manufacturer’s instructions (New England Biolabs, Japan) and visualized with CBB R-250 stain. CaCels before and after deglycosylation were analyzed by SDS-PAGE and immunological assays with anti-His.
Sugar chains attached to proteins were analyzed by lectin blot using ConA [34], LCA [35], PHA-E4 [36], WGA [37, 38], RCA120 [39], DBA [40], UEA-I [38], PNA [38], MAM [41], and SSA [42]. Subsequently, the membranes were reacted with HRP-conjugated streptavidin (EN-N100; Thermo) for 1 h at RT, washed in TBST, and then detected using DAB solution (Thermo, USA).
Monosaccharide Analysis
The CaCels purified from both systems were analyzed using a p-aminobenzoic ethyl ester (ABEE) Labeling Kit Plus S (J-Oil Mills Inc., Japan), according to the manufacturer’s instructions [43]. Briefly, the CaCels were hydrolyzed by 8 M trifluoroacetic acid (TFA) at 100 °C for 3 h, and the monosaccharides thus obtained were N-acetylated and labeled with ABEE. ABEE-derivatized saccharides were subjected to HPLC on a 4.5 × 75 mm2 Honenpak C18 column (J-Oil Mills Inc., Japan) and were eluted with 0.2 M potassium borate buffer (pH 8.9) containing 7 % acetonitrile or 0.02 % TFA containing 10 % acetonitrile at a flow rate of 1.0 ml/min at 30 °C. The effluent was monitored by a Tosho FS-8010 fluorescence spectrophotometer with an excitation wavelength of 305 nm and an emission wavelength of 360 nm. A mixture of Gal (galactose), Man (mannose), GlcNaC (N-acetylglucosamine), GalNAc (N-acetylgalactosamine), ManNAc (N-acetylmannosamine), and Fuc (fucose) was treated as described above, and each was then employed as a standard substance (J-Oil Mills Inc., Japan).
Enzyme Assay of Recombinant Protein
The β-1,4-glucanase activity was measured using the 3,5-dinitrosalicylic acid (DNS) method [44], through the determination of the amount of reducing sugars liberated from 0.5 % carboxymethylcellulose (CMC) solubilized in 50 mM sodium citrate buffer (pH 3.5) and 0.5 µg of CaCel. The mixture was incubated for 30 min at 50 °C, and the reaction was stopped by the addition of DNS solution. The treated samples were boiled for 5 min, cooled to RT, and the extent of enzymatic hydrolysis of CMC was determined by measuring the absorbance at 540 nm. One unit of enzyme activity was defined as the amount of enzyme that released 1 µM of d-glucose per minute. The enzyme assay was repeated three times for each of the three replicates.
Results
Expression of CaCel in B. mori Pupae
Previously, recombinant endo-β-1,4-glucanase, endo-β-1,3-glucanase (laminarinase), and β-1,4-d-mannanase, lacking signal sequences but fused to histidine-tagged thioredoxin, were expressed using the pET-28a (+)-E. coli Rossetta-gami2 (DE3) system by incubating at 6–10 °C Song [15–17, 45]. In these studies, the active enzymes with proper disulfide bonds were refolded from inclusion bodies. Several attempts were made to obtain recombinant carbohydrases, but the amount of protein purified using our system was lower than that obtained using fungal secretory expression systems (31 mg/l or more) [25, 26, 28]. In the present study, we chose endo-β-1,4-glucanase, which is an important glycosyl hydrolase and glycoprotein. Antarctic springtail CaCel has been produced and efficiently expressed in the E. coli system, using E. coli Rossetta-gami with histidine-tagged thioredoxin at low temperature, 10 °C (Ec-CaCel, Fig. 1a). The E. coli-expressed protein appeared at a molecular weight (MW) of 25 kDa in SDS–PAGE and immunoblotting with the sum of the MW of CaCel with hexa histidine (Fig. 1c). Approximately, 0.46 mg of the recombinant Ec-CaCel was obtained from 1 l of culture, and the specific activity of Ec-CaCel toward CMC was 35.8 U/mg (Table 1).
To overcome the problem of producing quantitatively and functionally active CaCel from E. coli expression systems, we turned to the baculovirus expression system for the production of CaCel using a secretion sequence originating from silkworm (30 k protein signal sequence) in B. mori pupae. Exploiting the fact that CaCel is a naturally secreted protein, we cloned CaCel cDNA, including the 30-kb signal sequence for secretion and eight histidine residues, into the pDEST8 vector. A recombinant BmNPV harboring CaCel was generated by recombination between plasmid pDEST8-30-kb CaCel DNA and the wild-type BmNPV genome following lipofection in BmN cells (Fig. 1b). RT-PCR with gene-specific primers, a plaque assay (data not shown), and immunoblotting with His-tag (Fig. 1c) confirmed that the BmNPVD8-30 k CaCel was successfully propagated into BmN cells. This recombinant virus was used to infect silkworm pupae. Hemolymph and fatty body were sampled at 4 days post-infection (d.p.i.) and subjected to CaCel expression and activity assays. After SDS-PAGE and immunoblotting of the hemolymph and fatty body containing the CaCel, a protein of similar MW could be visualized (Fig. 1d). The recombinant CaCel from pupae of B. mori (Bm-CaCel) was larger, at approximately 31 kDa, than the protein corresponding to 30 k CaCel-8×His (25 kDa). The identity of the protein to Bm-CaCel was confirmed by its specific reaction with anti-His antiserum in an immunoblot (Fig. 1d, right panels). The protein content and the enzyme activity toward CMC of the recombinant Bm-CaCel secreted to the hemolymph and fatty body by twenty infected pupae were 67 mg per 500 ml of lysate buffer and 48.5 U/mg, respectively (Table 1). This corresponds to an average yield of 3.35 mg of Bm-CaCel per infected pupa. The recovery yield and activity of the enzyme produced in 9 g of fatty body (in 500 ml buffer) from the pupae were about 10- and 3-fold higher, respectively, than those produced by E. coli in 1 L of culture broth. In addition, the CMCase-specific activity of Bm-CaCel was higher than that of the E. coli cell lysate (Table 1). After desalting in a Vivaspin 20 (10 kDa MWCO; Sartorius, Germany), 23.5 mg of the recombinant enzyme was obtained from twenty pupae, and the specific enzyme activity toward CMC was determined to be 24.5 µmol/min/mg.
To identify the isoelectric points of CaCel produced in both systems, IEF analysis was performed for Ec-CaCel and Bm-CaCel. The isoelectric point was measured using standard pH 3–10 markers (Serva, Germany). Results of CBB-R250 staining showed that the isoelectric point of Ec-CaCel and Bm-CaCel was approximately 7.0 (Fig. 1e), which differs from that of the calculated isoelectric point of 8.1 (DNASTAR Lasergene 7.1). Compared to the CaCel produced in both systems, CaCel was detected in similar isoelectric point 7.0 in silkworm pupae and E. coli.
Enzymatic Characterization of both Purified Recombinant CaCels
The CaCels purified from both systems showed optimal activity at 50 °C. More than 50 % activity was observed between 10 and 70 °C, and approximately 35–45 % activity was retained at 0–10 °C (Fig. 2a). Enzyme activity decreased markedly at temperatures above 70 °C, but that of the CaCel purified from pupae remained slightly higher. The optimum pH was 4, and both enzymes possessed more than 60 % activity at pH of 2–8 (Fig. 2b). More than 90 % of maximum activity was maintained at pH 3–5, and a slow decrease below pH 8 was observed. Thermal stability studies revealed that Ec-CaCel and Bm-CaCel were stable at 50 °C for 30 min, but lost 74–43 and 83–77 % of their activity after incubation for 1 and 8 h, respectively (Fig. 2c). Although the thermostability during the incubation of Bm-CaCel was higher than that of E. coli CaCel, there were no significant differences in activity, optimal temperature, or pH.

Effect of temperature and pH on activity and thermostability of CaCel purified in E. coli and B. mori. a The influence of temperature on enzyme activity was determined in 50 mM sodium citrate buffer, pH 3.5 at various temperatures for 30 min. b The influence of pH on enzyme activity was determined at 50 °C in the following 50 mM buffers: sodium citrate (pH 2–4), sodium acetate (pH 4–5), sodium phosphate (pH 5–7), Tris–HCl (pH 7–9), and glycine–NaOH (pH 9–10). c In the thermostability test, enzymes were incubated at 60 °C in 50 mM sodium citrate buffer, pH 3.5 each time. Data were obtained from three independent experiments, each conducted in triplicate. The triangle and square indicate the enzyme activity of CaCel purified in E. coli and B. mori, respectively
Glycosylation of both Types of Purified Recombinant CaCel
Most cellulases are glycoproteins, and proper glycosylation, phosphorylation, and acetylation are very important for their activities [46]. From the amino acid sequence of Antarctic springtail endo-β-1,4-glucanase, only one putative N-glycosylation and two putative O-glycosylation sites were confirmed. The only N-glycosylation site (N-X-S/T motif) present in CaCel is NTT (60–62), and the two residues corresponding to O-glycosylation (S/T) are Thr23 and Thr227 (Fig. 3a).

Glycosylation of recombinant CaCel purified in E. coli and B. mori. a The Antarctic springtail endo-β-1,4-glucanase sequence from GenBank, Accession no. ACV50414. The 30 k signal peptides, putative N-glycosylation, and putative O-glycosylation sites are shown with underlining, a box, and an asterisk, respectively. (NetNGlyc 1.0 Server, http://www.cbs.dtu.dk/services/NetNGlyc/). Peptides identified by N-terminal sequencing are double underlined. b N-terminal amino acid sequence analysis of Bm-CaCel and Ec-CaCel using an ABI492N-terminal amino sequencer. Common amino acids obtained by N-terminal sequencing are double underlined. c Deglycosylation of recombinant CaCel by PNGaseF and Endo-H. Deglycosylation of Bm-CaCel (upper panel) and Ec-CaCel (lower panel) treated with PNGaseF and Endo-H were analyzed by CBB staining and immunoblot assays with anti-His after glycosidase enzyme treatment. ND no digest, PF PNGaseF, EH Endo-H. d Glycoprotein staining of CaCel proteins purified from B. mori and E. coli. PAS, periodic acid–Schiff staining. M molecular weight standard marker (BioRad), Ec CaCel purified from E. coli, Bm CaCel purified from B. mori pupae. Black arrows indicate the position of CaCel after CBB and PAS staining and immunoblotting with anti-His
To confirm that the recombinant proteins were correctly processed, their N-terminal sequences were determined (Fig. 3b). The sequence obtained from Bm-CaCel was ILTSGSGVTT (Fig. 3b), which was similar to the N-terminus sequence of the mature CaCel from Antarctic springtail, indicating the proper cleavage of the 30 k secretion signal, and matched the sequence from Ec-CaCel MVTSGSGVTT in all but the first two amino acids. The Bm-CaCel secreted in the pupal hemolymph (~31 kDa) and Ec-CaCel (~25 kDa) were larger than the estimated MW (~25 and 23 kDa, respectively) without the signal peptide.
Deglycosylation with PNGaseF or EndoH for release of N-linked glycans was performed to confirm the glycosylation of CaCel. As shown in Fig. 3c, the Bm-CaCel detected with immunoblotting or CBB staining showed a slight change in mobility after PNGaseF treatment, but not after EndoH treatment (Fig. 3c, upper panel). The PNGaseF specifically cleaves the β1 → N linkage between the innermost GlcNAc and the Asn residue of high mannose, and hybrid and complex oligosaccharides from the N-linked glycoprotein backbone [47, 48]. However, EndoH cleaves the GlcNAcβ1–4GlcNAc linkage within the second GlcNAc core of the high mannose of the N-glycoprotein backbone alone [49]. This suggests that the Bm-CaCel carried hybrid-type or more elongated N-linked glycans. The fact that Ec-CaCel did not change after treatment with PNGaseF and EndoH (Fig. 3c, lower panel) suggests that it did not contain N-linked glycans.
In addition, PAS staining for N-glycan (Fig. 3d, center) suggested that Bm-CaCel was glycosylated. In contrast, PAS staining of Ec-CaCel did not show a band, indicating that Ec-CaCel cannot go through N-glycosylation. As stated above (Fig. 3a), CaCel has, at least, one potential N- and two potential (N60) O- glycosylation sites (T23, T227). The masses of the PNGaseF deglycosylated Bm- and Ec-CaCels show at about 27–28 (shift) and 25 kDa (no shift), respectively. In particular, the mass of the deglycosylated Bm-CaCel was slightly lower than before PNGaseF treatment, providing further evidence that it is most likely minimally populated at the N-linked glycosylation site. Subtracting the deglycosylated mass of 27–28 and 25 kDa of Bm- and Ec-CaCel, respectively, yields a mass contribution to CaCel by SDS-PAGE of approximately 2 kDa, suggesting the possibility of a diversity O-linked glycans in the CaCel expressed in both systems.
To understand the patterns of glycosylation of CaCel, carbohydrate-specific lectin staining was performed (Fig. 4a). To exclude the possibility of non-specific binding of lectin to the proteins produced in E. coli and B. mori, we used β-1,4-mannanase of C. antarcticus as a secreted nonglycoprotein in both systems. The MWs of Bm-CaMan and Ec-CaMan were approximately 43 and 41 kDa, respectively, without signal peptide.

Glycan structure analysis of recombinant CaCel purified from E. coli and B. mori. a Glycosylation of CaCel proteins purified from E. coli and B. mori was analyzed with anti-His and by lectin blotting with ConA, LCA, WGA, PHA-E4, RCA120, MAM, SSA, UEA-I, DBA, and PNA. m, β-1,4-mannanase (CaMan) served as the negative control (C. antarcticus β-1,4-d-mannanase purified from E. coli and B. mori). c C. antarcticus cellulase (CaCel); Ec CaCel purified from E. coli, Bm CaCel purified from B. mori pupae, M molecular weight standard marker (BioRad), white and black arrows indicate the positions of CaCel after immunoblotting with anti-His and lectin and CBB staining. b, c Chromatographic separation of ABEE-derivatized saccharides released from Ec-CaCel and Bm-CaCel, respectively
The carbohydrate-binding specificities of the different lectins used are indicated in Table 2. Purified CaCel and CaMan were compared using the same assays for binding to different plant lectins (Fig. 4a). Bm-CaCel interacted with ConA, LCA, WGA, PHA-E, RCA120, MAM, and SSA, which are known to bind to the terminal mannose, fucose, N-acetylglucosamine, N-acetylgalactosamine, galactose, and α2-3 and α2-6 sialic acid of N-glycans, and with UEA-I, DBA, and PNA, which are known to bind to the terminal fucose, N-acetylgalactosamine, and galactose of O-glycans, respectively. Contrary to expectation, Ec-CaCel weakly bound to ConA, LCA, WGA, PHA-E, RCA120, MAM, and SSA, suggesting the presence of minor monosaccharides in Ec-CaCel, as compared to CaMan purified from E. coli. Further, the presence of O-glycosidically linked carbohydrate chains were confirmed on Ec-CaCel using UEA-I, DBA, and PNA lectins to reveal the fucose, N-acetylgalactosamine, and galactose of O-glycans. These results suggest that the estimated MW of CaCel was higher than calculated, because of N- or O-glycosylation of the recombinant protein.
The monosaccharide composition of both CaCels was also investigated using HPLC (Fig. 4b, c). In Ec-CaCel (Fig. 4b), mannose, N-acetylglucosamine, and N-acetylgalactosamine were commonly found as minor components, concordant with our lectin result; N-acetylmannosamine was also detected. Thus, Ec-CaCel was partially N- or O-glycosylated, although it is likely that there was no shift after the PNGaseF, EndoH (Fig. 3c), and Endo-α-N-Acetylgalactosaminidase (O-glycosidase, data not shown) treatments. In Bm-CaCel (Fig. 4c), galactose, mannose, and N-acetylglucosamine were detected as minor components; N-acetylmannosamine was also detected. Indeed, Bm-CaCel, which is only N-glycosylated, showed a slightly higher MW, bound the specific lectin-carrying N-glycan, and showed the presence of monosaccharides.
As shown by the differences in relative monosaccharide compositions and lectin affinities, the structural profiles obtained for the two recombinant CaCels were not identical to that of the native CaCel. The differences in glycosylation between Bm-CaCel and Ec-CaCel resulted in the differences in thermostability discussed above (Fig. 2c), indicating that N-glycosylation could maintain the spatial structure of Bm-CaCel at high temperature.
Discussion
For the expression of glycoproteins such as cellulase, production and purification of recombinant cellulase proteins with biological activity in microorganisms such as E. coli that do not carry out post-translational modifications of proteins are technically difficult [50]. However, some endo-β-1,4-glucanases from bacteria, fungi, and insects have been successfully expressed in E. coli [15–17, 25, 51, 52], indicating that E. coli is a relatively suitable host for the heterologous expression of cellulase. Many industrially important enzymes are now produced by genetically engineered microorganisms. For commercial utilization of enzymes, higher enzyme yields are required at low capital cost. Cellulase-producing psychrotrophic microorganisms, which show broad biotechnological potential, offering numerous economic and ecological advantages over the use of enzymes that operate at higher temperatures, have been studied and produced by the bacteria Pseudoalteromonas haloplanktis, the fungus Acremonium alcalophilum, and the yeast Rhodotorula glutinis [11, 53, 54]. However, few cold-active cellulases from animals have been studied to date. Most cellulases produced by psychrophilic microorganisms show optimum activity at temperatures between 35 and 40 °C and between pH 4.5–7.0 [11, 53, 54]. The cellulase of C. antarcticus isolated in the present study showed maximal activity at 50 °C and pH 3.5. The enzyme was cold active, retaining more than 40 % activity at 0 °C, which is higher than that of the cellulase produced by the fungus A. alcalophilum, which retained 20 % activity at 0 °C [11]. C. antarcticus cellulase may be more useful for commercial purposes than other enzymes from psychrophiles because of its high catalytic activity at a broad range of temperatures, from 0 to 80 °C.
The silkworm B. mori is a successful system for the production of a variety of eukaryotic proteins because it performs post-translational processing similar to that found in mammals [55]. Scientists have long attempted to develop low-cost methods for high-level expression of recombinant proteins in their native form from silkworm [56, 57]. To date, heterologous proteins have been successfully produced in baculovirus-infected insect cells, and in silkworm larvae and pupae [57–59]. Since silkworm pupae can be used to express the protein of interest at any time, if they are kept in refrigeration, they potentially represent a more convenient and economical vehicle for the expression of heterologous proteins than silkworm larvae or insect cells. Silkworm pupae have the added advantage of no feeding, basically space for leaving pupae, reduced waste, lower production costs, and high-level expression compared to those in other eukaryotic and prokaryotic systems.
In the present study, we successfully produced cold-adapted endo-β-1,4-glucanase of Antarctic springtail, CaCel, in B. mori pupae. We also successfully expressed and produced CaCel in silkworm cells, BmN, using BmNPVD8-30 k CaCel (data not shown). The CaCel generated in silkworm pupae had a MW of 31 kDa, similar to that of the protein secreted by BmN cells. However, the CaCel produced in silkworm pupae showed a slightly different N-glycan pattern to the CaCel produced in silkworm cells (data not shown). Lectin analysis of CaCel expressed in silkworm cells showed a mannose-type structure that bound only with ConA, indicating that it underwent significantly less oligosaccharide processing (data not shown). Three problems to be resolved for usage of recombinant protein expressed in BmN cells are yield, contamination of FBS, and the apparent post-translational modification that recombinant proteins undergo in silkworm cells. Cellulase from Trichoderma viride has been successfully expressed and produced in B. mori larvae [29, 60], indicating that B. mori larvae are a suitable host for heterologous expression of cellulase. The silkworm expression system, both larvae and BmN cells, was shown to be a suitable host for cellulase expression. Of these systems, silkworm pupae were selected as superior, because it produced a higher yield than BmN cells and its retention period was longer than that of larvae. Calculated based on specific activity, the 5 d.p.i fatty body supernatants and hemolymph of silkworm pupae contained 31 mg of CaCel per 500 ml. This is the highest observed productivity of an animal cellulase produced in a heterologous host, including P. pastoris, Aspergillus oryzae, and E. coli [25, 27, 28, 61]; the activity level is also higher than that of CaCel produced in E. coli. The specific activity of Bm-CaCel was slightly higher than that of Ec-CaCel (approximately 0.3 times) possibly due to lack of N-glycosylation modification. Since post-translational modifications affect protein function, the glycosylation of recombinant proteins is one factor which must be considered in their production. Most proteins produced by the E. coli expression system are retained in the periplasmic space, and only a few proteins can be secreted into the culture medium. In the case of lectin-binding process, Ec-CaCel showed selective binding oligosaccharides. There is little available information on whether recombinant endoglucanases expressed in E. coli undergo glycosylation, or whether the glycosylation of endoglucanases expressed in E. coli has an effect on enzymatic properties. The glycosylation process is thought to occur in the bacterial periplasm, which is the functional equivalent of the ER in eukaryotes [62]. This may have resulted in the relatively high glycosylation of the protein generated by silkworm pupae. Bm-CaCel had a slightly higher molecular weight and carbohydrate-binding specificity, indicating that the oligosaccharides had been liberated from the peptide chain after PNGaseF treatment (Fig. 3c). In Bm-CaCel, the expected well-known oligomannose series was observed by carbohydrate recognition. Compared to N-glycosylated Bm-CaCel, Ec-CaCel was putatively O-glycosylated in E. coli Rossetta gami (DE3), because of its relatively strong interaction via lectin binding to O-glycans such as UEA-I, DBA, and PNA. Their differences resulted in increased thermostability as shown Fig. 2c, indicating that complex N-glycosylation could maintain the spatial structure of Bm-CaCel at high temperature. Jeoh et al. [23] have reported that the thermal stability of recombinant endoglucanases can either decrease or increase after glycosylation. In the present study, differences in the glycosylation of Bm-CaCel and Ec-CaCel did not affect the optimum pH or temperature, or the pH stability or substrate specificity, but did enhance the thermal stability of Bm-CaCel.
In summary, CaCel was successfully expressed in and purified from silkworm pupae, used for the first time as a bioreactor to express a cold-adapted glycosyl hydrolases protein. The endo-β-1,4-glucanase protein obtained from silkworm pupae displayed relatively high-level glycosylation and higher biological activity and yield than Ec-CaCel. These results suggest that the silkworm pupa expression system may be appropriate for the production of cold-active proteins with activities and stabilities over a broad range of pH values and temperatures.
References
Brummell, D. A., Catala, C., Lashbrook, C. C., & Bennett, A. B. (1997). A membrane-anchored E-type endo-1,4-beta-glycanase is localized on golgi and plasma membranes of higher plants. Proceedings of the National Academy of Sciences of the United States of America, 94, 4794–4799.
Crawford, A. C., Kricker, J. A., Anderson, A. J., Richardson, N. R., & Mather, P. B. (2004). Structure and function of a cellulase gene in redclaw crayfish Cherax quandricarinatus. Gene, 340, 267–274.
Tomme, P., Warren, R. A., & Gilkes, N. R. (1995). Cellulose hydrolysis by bacteria and fungi. Advances in Microbial Physiology, 37, 1–81.
Wilson, D. B., & Irwin, D. C. (1999). Genetics and properties of cellulases. Advances in Biochemical Engineering/Biotechnology, 65, 1–21.
Heikinheimo, L., Buchert, J., Miettinen-Oinonem, A., & Suominen, P. (2000). Treating denim fabrics with Trichoderma reesei cellulases. Textile Research Journal, 70, 969–973.
Ito, S. (1997). Alkaline cellulases from alkaliphilic Bacillus: Enzymatic properties, genetics, and application to detergents. Extremophiles, 1, 61–66.
Yu, P., Mckinnon, J. J., Maenz, D. D., Olkowski, A. A., Racz, V. J., & Christensen, D. A. (2003). Enzymic release of reducing sugars from oat hulls by cellulase, as influenced by Aspergillus ferulic acid esterase and Trichoderma xylanase. Journal of Agricultural and Food Chemistry, 1, 218–223.
Maki, M. L., Idrees, A., Leung, K. T., & Qin, W. (2012). Newly isolated and characterized bacteria with great application potential for decomposition of lignocellulosic biomass. Journal of Molecular Microbiology and Biotechnology, 22, 156–166.
Mandels, M. (1985). Applications of cellulases. Biochemical Society Transactions, 13, 414–415.
Lenting, H. B., & Warmoeskerken, M. M. (2001). Guidelines to come to minimized tensile strength loss upon cellulase application. Journal of Biotechnology, 89, 227–232.
Hayashi, K., Nimura, Y., Ohara, N., Uchimura, T., Suzuki, H., Komagata, K., et al. (1996). Low-temperature active cellulase produced by Acremonium alcalophilum JCM 7366. Seibutsu-kogaku kaishi, 74, 7–10.
Iyo, A. H., & Forsberg, C. W. (1999). A cold-active glucanase from the ruminal bacterium Fibrobacter succinogenes S85. Applied and Environment Microbiology, 65, 995–998.
Cristobal, H. A., Breccia, J. D., & Abate, C. M. (2008). Isolation and molecular characterization of Shewanella sp. G5, a producer of cold-active beta-d-glucosidases. Journal of Basic Microbiology, 48, 16–24.
Berg, M. P., Stoffer, M., & van den Heuvel, H. H. (2004). Feeding guilds in Collembola base on digestive enzymes. Pedobiologia, 48, 589–601.
Song, J. M., Nam, K. W., Kang, S. G., Kim, C. G., Kwon, S. T., & Lee, Y. H. (2008). Molecular cloning and characterization of a novel cold-active β-1,4-d-mannanase from the Antarctic springtail, Cryptopygus antarcticyus. Comparative Biochemistry and Physiology B, 151, 32–40.
Song, J. M., Nam, K. W., Sun, Y. U., Kang, M. H., Kim, C. G., Kwon, S. T., et al. (2010). Molecular and biochemical characterizations of a novel arthropod endo-β-1,3-glucanase from the Antarctic sprigtail, Cryptopygus antarcticus, horizontally acquired from bacteria. Comparative Biochemistry and Physiology B, 155, 403–412.
Song, J. M., An, Y. J., Kang, M. H., Lee, Y. H., & Cha, S. S. (2012). Cultivation at 6–10 °C is an effective strategy to overcome the insolubility of recombinant proteins in Escherichia coli. Protein Expression and Purification, 82, 297–301.
Maeda, S. (1994). Expression of foreign genes in insect cells using baculovirus vectors. Insect cell biotechnology (pp. 1–31). Boca Raton: CRC Press.
Miyajima, A., Schreurs, J., Otsu, K., Kondo, A., Arai, K., & Maeda, S. (1987). Use of the silkworm, Bombyx mori, and an insect baculovirus vector for high-level expression and secretion of biologically active mouse interleukin-3. Gene, 58, 273–281.
Imperiali, B., & O’Connor, S. E. (1999). Effect of N-linked glycosylation on glycopeptide and glycoprotein structure. Current Opinion in Chemical Biology, 3, 643–649.
Ingeborg, S., Koen, S., Steve, G., Roland, C., Jozef, V. B., & Marc, C. (2004). Factors influencing glycosylation of Trichoderma reesei cellulases. I: Postsecretorial changes of the O- and N-glycosylation pattern of Cel7A. Glycobiology, 14, 713–724.
Harrison, M. J., Nouwens, A. S., Jardine, D. R., Zachara, N. E., Gooley, A. A., Nevalainen, H., et al. (1998). Modified glycosylation of cellobiohydrolase I from a high cellulase-producing mutant strain of Trichoderma reesei. European Journal of Biochemistry, 15, 119–127.
Jeoh, T., Michener, W., Himmel, M. E., Decker, S. R., & Adney, W. S. (2008). Implications of cellobiohydrolase glycosylation for use in biomass conversion. Biotechnology for Biofuels, 1, 10–22.
MacLeod, A. M., Gilkes, N. R., Escote-Carlson, L., Warren, R. A., Kilburn, D. G., & Miller, R. C, Jr. (1992). Streptomyces lividans glycosylates an exoglucanase (Cex) from Cellulomonas fimi. Gene, 121, 143–147.
Zhao, X. H., Wang, W., Wang, F. Q., & Wei, D. Z. (2012). A comparative study of β-1,4-endoglucanase (possessing β-1,4-exoglucanase activity) from Bacillus subtilis LH expressed in Pichia pastoris GS115 and Escherichia coli Rosetta (DE3). Bioresource Technology, 110, 539–545.
Yan, Q., Hua, C., Yang, S., Li, Y., & Jiang, Z. (2012). High level expression of extracellular secretion of a β-glucosidase gene (PtBglu3) from Paecilomyces thermophile in Pichia pastoris. Protein Expression and Purification, 84, 64–72.
Ni, J., Takehara, M., & Watanabe, H. (2005). Heterologous overexpression of a mutant termite cellulase gene in Escherichia coli by DNA shuffling of four orthologous parental cDNA. Bioscience, Biotechnology, and Biochemistry, 69, 1711–1720.
Hirayama, K., Watanabe, H., Tokuda, G., Kitamoto, K., & Arioka, M. (2010). Purification and characterization of termite endogenous beta-1,4-endoglucanases produced in Aspergillus oryzae. Bioscience, Biotechnology, and Biochemistry, 74, 1680–1686.
Li, X. H., Zhang, P., Wang, M. X., Zhou, F., Malik, F. A., Yang, H. J., et al. (2011). Expression of Trichoderma viride endoglucanase III in the larvae of silkworm, Bombyx mori. L. and characteristic analysis of the recombinant protein. Molecular Biology Reports, 38, 3897–3902.
Sambrook, J., Fritsch, E. F., & Maniatis, T. (1989). Molecular cloning; a laboratory manual (2nd ed.). New York: Cold Spring Harbor Laboratory Press.
Katsuma, S., Daimon, T., Mita, K., & Shimada, T. (2006). Lepidopteran ortholog of Drosophila breathless is a receptor for the baculovirus fibroblast growth factor. Journal of Virology, 80, 5474–5481.
Bradford, M. M. (1976). A rapid and sensitive method for the quantification of microgram quantities of protein using the principle of protein-dye binding. Analytical Biochemistry, 72, 248–254.
Righetti, P. G., & Drysdale, J. W. (1974). Isoelectric focusing in gels. Journal of Chromatography, 98, 271–321.
Baenziger, J. U., & Fiete, D. (1979). Structural determinants of concanavalin A specificity for oligosaccharides. Journal of Biological Chemistry, 254, 2400–2407.
Tateno, H., Nakamura-Tsuruta, S., & Hirabayashi, J. (2009). Comparative analysis of core-fucose-binding lectins from Lens culinaris and Pisum Sativum using frontal affinity chromatograph. Glycobiology, 19, 527–536.
Green, E. D., & Baenziger, J. U. (1987). Oligosaccharide specificities of Phaseolus vulgaris leukoagglutinating and erythroagglutinating phytohemagglutinins. Interactions with N-glycanase released oligosaccharides. Journal of Biological Chemistry, 262, 12018–12029.
Yamamoto, K., Tsuji, T., Matsumoto, I., & Osawa, T. (1981). Structural requirements for the binding of oligosaccharides and glycopeptides to immobilized wheat germ agglutinin. Biochemistry, 20, 5894–5899.
Molin, K., Fredman, P., & Svennerholm, L. (1986). Binding specificities of the lectins PNA, WGA and UEA I to polyvinylchloride adsorbed glycosphingolipids. FEBS Letters, 205, 51–55.
Maljaars, C. E., Halkes, K. M., de Oude, W. L., Haseley, S. R., Upton, P. J., McDonnell, M. B., et al. (2006). Affinity determination of Ricinus communis agglutinin ligands identified from combinatorial O- and S-, N-glycopeptide libraries. Journal of Combinatorial Chemistry, 8, 812–819.
Farr, A. G., Anderson, S. K., Braddy, S. C., & Mejino, J. L, Jr. (1988). Selective binding of Dolichos biflorus agglutinin to L3T4-, Lyt-2-thymocytes. Expression of terminal alpha-linked N-acetyl-d-galactosamine residues defines a subpopulation of fetal and adult murine thymocytes. Journal of Immunology, 140, 1014–1021.
Shah, M. H., Telang, S. D., Shan, P. M., & Patel, P. S. (2008). Tissue and serum alpha 2-3- and alpha 2-6-linkage specific sialylation changes in oral carcinogenesis. Glycoconjugate Journal, 25(3), 279–290.
Yabe, R., Itakura, Y., Nakamura-Tsuruta, S., Iwaki, J., Kuno, A., & Hirabayashi, J. (2009). Engineering a versatile tandem repeat-type alpha2-6sialic acid-binding lectin. Biochemical and Biophysical Research Communications, 384(2), 204–209.
Yasuno, S., Murata, T., Kokubo, K., Yamaguchi, T., & Kamei, M. (1997). Two-mode analysis by high-performance liquid chromatography of p-aminobenzoic ethyl ester-derivatized monosaccharides. Bioscience, Biotechnology, and Biochemistry, 61, 1944–1946.
Stalbrand, H., Siika-aho, M., Tenkanen, M., & Viikari, L. (1993). Purification and characterization of two β-mananase from Trichoderma reesei. Journal of Biotechnology, 29, 229–242.
Bessette, P. H., Aslund, F., Beckwith, J., & Georgiou, G. (1999). Efficient folding of proteins with multiple disulfide bonds in the Escherichia coli cytoplasm. Proceedings of the National Academy of Sciences of the United States of America, 96, 13703–13708.
Zhou, F., Olman, V., & Xu, Y. (2009). Large-scale analyses of glycosylation in cellulases. Genomics, Proteomics & Bioinformatics, 7, 194–199.
Maley, F., Trimble, R. B., Tarentino, A. L., & Plummer, T. H. (1989). Characterization of glycoportins and their associated oligosaccharides through the use of endoglycosidases. Analytical Biochemistry, 180, 195–204.
Tretter, V., Altmann, F., & Marz, L. (1991). Peptide-N4-(N-acetyl-beta-glucosminyl) asparagine amidase F cannot release glycans with fucose attached alph 1, 3 to the asparagine-linked N-acetylglycosamine residue. European Journal of Biochemistry, 199, 647–652.
Naoyuki, T., Akemi, S., Yukishige, I., Hisashi, N., Toshisuke, K., & Sumihiro, H. (2008). Experimental glycoscience glycochemistry (pp. 182–185). Berlin: Springer.
Okumura, F., Kameda, H., Ojima, T., & Hatakeyama, S. (2010). Expression of recombinant sea urchin cellulase SnEG54 using mammalian cell lines. Biochemical and Biophysical Research Communications, 395, 352–355.
Nakazawa, H., Okada, K., Kobayashi, R., Kubota, T., Onodera, T., Ochiai, N., et al. (2008). Characterization of the catalytic domains of Trichoderma reesei endoglucanase I, II, and III, expressed in Escherichia coli. Applied Microbiology and Biotechnology, 81, 681–689.
Fan, H. X., Miao, L. L., Liu, Y., Liu, H. C., & Liu, Z. P. (2011). Gene cloning and characterization of a cold-adapted β-glucosidase belonging to glycosyl hydrolase family 1 from a psychrotolerant bacterium Micrococcus antarcticus. Enyzme and Microbial Technology, 49, 94–99.
Garsoux, G., Lamotte, J., Gerday, C., & Feller, G. (2004). Kinetic and structural optimization to catalysis at low temperatures in a psychrophilic cellulase from the Antarctic bacterium Pseudoalteromonas haloplanktis. Biochemical Journal, 384, 247–253.
Oikawa, T., Tsukagawa, Y., & Soda, K. (1998). Endo- β-glucanase secreted by a psychrotrophic yeast: Purification and characterization. Bioscience, Biotechnology, and Biochemistry, 62, 1751–1756.
Wu, X., Kamei, K., Sato, H., Sato, S. I., Takano, R., Ichida, M., et al. (2001). High-level expression of human acidic fibroblast growth factor and basic fibroblast growth factor in silkworm (Bombyx mori L.) using recombinant baculovirus. Protein Expression and Purification, 21, 192–200.
Sumaty, S., Palhan, V. B., & Gopinathan, K. P. (1996). Expression of human growth hormone in silkworm larvae through recombinant Bombyx mori nuclear polyhedrosis virus. Protein Expression and Purification, 7, 262–268.
Liu, A., Yang, G. Z., & Wu, X. F. (2004). Production of recombinant human osteoprotegrin from Trichoplusia ni cells and Bombyx mori larvae. Protein and Peptide Letters, 11, 317–323.
Kost, T. A., Condreay, J. P., & Jarvis, D. L. (2005). Baculovirus as versatile vectors for protein expression in insect and mammalian cells. Nature Biotechnology, 23, 567–575.
Chen, J., Wu, X. F., & Zhang, Y. Z. (2006). Expression, purification and characterization of human GM-CSF using silkworm pupae (Bombyx mori) as a bioreactor. Journal of Biotechnology, 123, 236–247.
Li, X. H., Zhang, P., Liang, S., Zhou, F., Wang, M. X., Bhaskar, R., et al. (2012). Molecular cloning and characterization of a putative cDNA encoding endoglucanase IV from Trichoderma viride and its expression in Bombyx mori. Applied Biochemistry and Biotechnology, 166, 309–320.
Zhang, D., Lax, A. R., Raina, A. K., & Bland, J. M. (2009). Differential cellulolytic activity of native-form and C-terminal tagged-form cellulase derived from Coptotermes formosanus and expressed in E. coli. Insect Biochemistry and Molecular Biology, 39, 516–522.
Weerapana, E., & Imperiali, B. (2006). Asparagine-linked protein glycosylation: from eukaryotic to prokaryotic systems. Glycobiology, 16, 91–101.
Acknowledgments
We thank Young-Sik Seok of the Institute of Kangwon-do Agricultural Pure Stock, Korea for the provision and maintenance of silkworms. This work was supported by the Bio-industry Technology Development Program, Ministry for Food, Agriculture, Forestry and Fisheries, Republic of Korea (No: 111062-03-1-HD110 and 111116–01–1-SB010) and partially by the National Fisheries Research and Development Institute (RP-2014-AQ-027).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Hong, S.M., Sung, H.S., Kang, M.H. et al. Characterization of Cryptopygus antarcticus Endo-β-1,4-Glucanase from Bombyx mori Expression Systems. Mol Biotechnol 56, 878–889 (2014). https://doi.org/10.1007/s12033-014-9767-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12033-014-9767-8