
Abstract
Baculovirus expression vector system (BEVS) is widely used for production of recombinant eukaryotic proteins in insect larvae or cultured cells. BEVS has advantages over bacterial expression system in producing post-translationally modified secreted proteins. However, for some unknown reason, it is very difficult for insects to secrete sufficiently for certain proteins of interest. To understand the reasons why insect cells fail to secrete some kinds of recombinant proteins, we here employed three mammalian proteins as targets, EPO, HGF, and Wnt3A, with different secretion levels in BEVS and investigated their mRNA transcriptions from the viral genome, subcellular localizations, and interactions with silkworm ER chaperones. Moreover, we observed that no significantly influence on the secretion amounts of all three proteins when depleting or overexpressing most endogenous ER chaperone genes in cultured silkworm cells. However, among all detected ER chaperones, the depletion of BiP severely decreased the recombinant protein secretion in BEVS, indicating the possible central role of Bip in silkworm secretion pathway.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Baculovirus expression vector system (BEVS) has served as powerful tool for mass production of secreted mammalian proteins that are required for proteolytic processing and post-translational modifications [1]. Due to the importance of the functional processing and modification of proteins, especially for secreted proteins, BEVS is supposed to be beneficial for various industrial and medical applications. The problem with these systems, however, is that it is difficult to produce some glycoproteins in sufficient quantities. In most of the previous studies, therefore, focused on the improvement of host glycosylation pathway and baculovirus vector to increase protein production in both quantity and quality [2–4].
Compared to intracellular proteins, secreted proteins are destined to translocate to the cell exterior through the endoplasmic reticulum (ER) and Golgi apparatus [5]. During these processes, the proteins are folded into the correct conformation and acquire post-translational modifications, such as N- or O-linked glycosylation. ER chaperones, like BiP, calnexin (CNX), calreticulin (CRT), ERp57, and protein disulfide isomerase (PDI), play critical roles in ER quality control and ER-associated degradation (ERAD) to determine the destination of target proteins [6]. For example, correctly folded proteins can be exported into the Golgi apparatus for further processing, whereas misfolded proteins are degraded by ERAD [7, 8].
Endoplasmic reticulum possesses a capacity to maintain the homeostasis of cells by a programmed process called unfolded protein response (UPR) [9]. In eukaryotes, UPR induces the accumulation of various ER chaperones and ERAD components in the ER, and when the UPR is initiated, the unfolded proteins can be resynthesized to form correctly folded proteins or degraded products to reduce ER stress for cells [10, 11]. Severe ER dysfunction caused by the accumulation of misfolded proteins, however, will trigger programmed cell death [12].
In this study, we first expressed three mammalian glycoproteins in secreted forms by using BEVS in silkworms, including human erythropoietin (EPO), cat hepatocyte growth factor (HGF), and mouse wingless-type mouse mammary tumor virus (MMTV) integration site family of member 3A (Wnt3A). It was demonstrated that recombinant EPO was highly secreted by BEVS, whereas HGF and Wnt3A were secreted at lower levels. In BEVS, a large amount of foreign proteins are synthesized and secreted within a short time of about 3 days post-infection of baculovirus. Furthermore, baculovirus infections take over the host cell system physiologically and structurally. Therefore, it is sometimes difficult to speculate upon the reasons for this inadequacy of recombinant protein secretion. To figure out the molecular processes of protein secretion during baculovirus infection, we investigated the roles of silkworm ER chaperones in the secretion of recombinant proteins using BEVS.
Materials and methods
Experimental animal and cultured cells
The silkworm strain f38, which is susceptible to BmNPV infection, used for recombinant protein expression was stored at the Institute of Genetic Resources, Graduate School of Agriculture, and Kyushu University, Japan. The silkworm larvae were regularly fed on fresh mulberry leaves. The cultured silkworm cell lines, BmN4, BmN4-SID1 [13], Bme21, Bme21-SID1 [14], and NIAS-Bm-oyanagi2, were maintained in IPL-41 medium (Sigma Chemical Co) supplemented with 10 % fetal bovine serum (FBS, GIBCO Invitrogen Co) at 27 °C.
Construction of baculoviruses
The cDNAs for human EPO [15], cat HGF [16], and mouse Wnt3A [17] were amplified by PCR using specific primers which are listed in Table 1, and further cloned into the EcoRV–XhoI site of pENTR11L21TEVH8 vector containing a His × 8 tag [18]. Plasmids for human EPO, cat HGF, and mouse Wnt3A were named as pENTR11L21-HsEPO-TEVH8, pENTR11L21-FcHGF-TEVH8, and pENTR11L21-MmWnt3A-TEVH8, respectively. Baculovirus transfer plasmids of pDEST8-HsEPO-TEVH8, pDEST8-FcHGF-TEVH8, and pDEST8-MmWnt3A-TEVH8 were further generated by a gateway LR reaction between pDEST8 vector and the entry plasmids described previously [19, 20]. These destination plasmids were subsequently transformed into BmT3-DH10Bac E. coli cells to generate recombinant baculovirus genomes (bacmids). The resulting bacmid DNAs were further transfected into NIAS-Bm-oyanagi2 cells by lipofectin methods to obtain recombinant baculoviruses. Three days after transfection, the medium was recovered and cells were re-infected to prepare high-titer viral stocks.
Expression of recombinant proteins
To assess the expression level of recombinant EPO, HGF, and Wnt3A, Bme21 cells were plated on 6-well plates at a density of 1 × 105 cells per well, and infected with recombinant baculovirus for each protein at a multiplicity of infection (MOI) of 1. The cells and culture media were harvested at 2, 3, 4, and 5 days after infection. The cells were washed with phosphate buffered saline (PBS) and suspended in ice-cold cell lysis buffer (20 mM Tris–HCl pH 7.5, 0.5 M NaCl, 10 mM 2-mercaptoethanol, 1 % Triton X-100 (SIGMA), 1 mM PMSF, and 10 % Complete EDTA-free protease inhibitor) to carry out lysis. After centrifugation at 1000g for 30 min at 4 °C, the supernatant was defined as soluble form and the precipitates were termed as insoluble form. Subsequently, those samples quantified by protein assay kit (Bio-Rad, USA) were subjected to Western blotting analysis.
SDS-PAGE and immunoblot analysis
All the samples were separated on a 10 % SDS-PAGE gel and further transferred onto a PVDF membrane (Millipore). Then the membrane was blocked by TBST (20 mM Tris–HCl pH 7.5, 150 mM NaCl, 0.1 % Tween20) containing 5 % skim milk for 1 h. After blocking, the membrane was incubated with HisProbe-HRP (Thermo Scientific) for 1 h and then visualized using the Super Signal West Pico Chemiluminescent Substrate (Thermo Scientific).
RT-PCR analysis
Total RNA or genomic DNA from BmN4-SID1 and Bme21-SID1 cells was extracted using ISOGEN (Nippon Gene). cDNAs were reverse-transcribed from total RNA using RevaTra Ace (Toyobo) according to manufacturer’s instruction. The transcription of the target and viral genes was analyzed by semi-quantitative polymerase chain reaction (semi-quantitative PCR) using gene-specific primers (Table 1).
Immunofluorescence staining
To determine subcellular localization of the recombinant proteins, immunostaining was performed. The cells infected with recombinant baculoviruses were cultured on a cover glass and fixed with 4 % paraformaldehyde in PBS. After fixation for 20 min, the cells were permeabilized with 0.1 % Triton X-100 in PBS for 10 min, blocked in 3 % skim milk for 1 h, and then incubated with mouse antibody against HA-tag (sc7392, Santa Cruz) for 1 h. After washing for five times with PBS, the cells were incubated with FITC-labeled anti-mouse IgG antibody (F0257, Sigma) for 1 h. Simultaneously, ER was stained by antibodies against PDI and visualized by incubating with Cy3-Linked anti-rabbit IgG (ab6939, GE Healthcare). The Golgi apparatus was stained by BODIPY TR ceramide complexed to BSA (Invitogen).
RNA interference for ER chaperone genes
Partial cDNA fragments for the silkworm genes were amplified using specific primers (Table 1) and cloned into a dual T7 promoter-driven vector of pLits [21]. The pLits vector containing the silkworm cDNAs was used to synthesize double-stranded RNA (dsRNA) by T7 RNA polymerase in vitro and the RNA interference (RNAi) treatments were performed in above described cell lines according to our previous studies [21, 22].
To knock down ER chaperone genes, BmN4-SID1 cells or Bme21-SID1 cells were soaked with 300 ng of dsRNA for 3 days as described previously [20]. Three days after soaking, their knockdown efficiencies were analyzed by RT-PCR (Table 1).
Establishment of stable Bme21 cell lines overexpressing silkworm ER chaperones
The cDNAs for silkworm BiP, CNX, and ERp57 were amplified by PCR using specific primers listed in Table 1, and cloned between NcoI and XbaI sites of pENTR11 vector. These plasmids were named as pENTR11-BmBiP, pENTR11-BmCNX, and pENTR11-BmERp57, respectively. The piggyBac-based transposition plasmids, pPBO-IE2-Flag-BmBip, pPBO-IE2-Flag-BmCNX, and pPBO-IE2-Flag-BmERp57 were generated for the overexpression of BmBiP, BmCNX, and BmERp57, using a gateway LR reaction between pPBO-IE2-Flag and the entry plasmids described above. Each vector was co-transfected into Bme21 cells with helper plasmid as described previously [20]. After co-transfection, stably transformed cells were selected in culture medium containing 5 µg/ml Puromycin (CalBiochem) for 2 weeks. The overexpression level of each chaperone was validated by Western blot using anti-Flag antibody (F3165, Sigma).
Results
Secretion of recombinant proteins in silkworms and cultured cells
To understand the reason why it is difficult for some recombinant proteins to be secreted at considerable quantities from baculovirus-infected cells, we employed three mammalian proteins, human EPO, cat HGF, and mouse Wnt3A, as target proteins. Recombinant baculoviruses expressing these proteins were generated as described in the “materials and methods” section, and used to infect cultured silkworm cells Bme21 or silkworm fifth instar larvae.
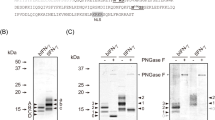
As shown in Fig. 1, the secretion level of human EPO was very high in both cultured cells and silkworm larvae, while the secretions of cat HGF and mouse Wnt3A were much lower. Mouse Wnt3A was secreted slightly into silkworm hemolymph but remained mainly insoluble in the cells. Compared to cat HGF and mouse Wnt3A, very little human EPO accumulated inside the cultured cells or fat body in the insoluble form. Interestingly, proteins remaining inside the cells were detected as several bands by Western blot, suggesting that these proteins may have different post-translational modifications in cells.

Expression analysis of recombinant proteins in cultured silkworm cells and silkworm individuals. The comparison of medium, cell lysate, and cell precipitates from Bme21 cells (a) and hemolymph of silkworm individuals (b) were isolated on SDS-PAGE and visualized by Western blotting using antibody against HisProbe. Cell lysate and cell precipitates were separated into soluble form and insoluble form. All fractions were collected after 2 (lanes 1, 4, and 7), 3 (lanes 2, 5, and 8), and 4 (lanes 3, 6, and 9) days infection to cultured cells (a) and collected after 2 (lanes 1, 5, and 9), 3 (lanes 2, 6, and 10), 4 (lanes 3, 7, and 11), and 5 (lanes 4, 8, and 12) days infection to silkworm individuals (b). The recombinant EPO was secreted into culture medium and hemolymph. However, neither the recombinant HGF nor Wnt3A were secreted
In order to confirm whether the recombinant baculoviruses expressing different proteins replicate at a similar efficiency and transcribe equal amounts of the target mRNAs, the viral DNAs and their transcripts in the baculovirus-infected cells were analyzed by semi-quantitative PCR. As for viral genomic DNA, the replication levels judged from ie1, p10, and each target protein region were similar (Fig. 2). The mRNA levels of EPO, HGF, and Wnt3A, however, were different when compared to each of their genomic DNAs, and the expression of EPO was much higher than that of HGF and Wnt3A, which may suggest distinct regulations for different proteins in BEVS.

RT-PCR analysis of the efficiency for recombinant baculovirus from genomic DNA and cDNA. The comparison of the recombinant virus efficiency, ie1, p10, EPO, HGF, Wnt3A, actin, and GAPDH were analyzed by RT-PCR using specific primers. Genomic DNA (lanes 1–3) and mRNAs (lanes 4–6) were isolated from silkworm fat body infected with virus. The expression level of each genomic DNA has no difference. However, the expression level of cDNA had slightly difference between each recombinant protein
Subcellular localization of the recombinant proteins
The mRNA expressions of HGF and Wnt3A mRNAs were lower than that of human EPO, but this fact alone does not suffice to explain the difference in the secretion amounts observed. Therefore, we next determined the subcellular localization of the target proteins in cultured silkworm cells by immunostaining as described in “materials and methods” section. All the three target proteins (FITC-labeled) were co-localized with Cy3-Linked PDI in ER, but not with the Golgi apparatus labeled with BODIPY TR ceramide (Fig. 3). Although secretion volumes were different, three target proteins remained in cells were localized at ER. This suggested that no secreted proteins were trapped and remained in ER.

Subcellular localization of recombinant proteins at viral infection phases. The localization of recombinant proteins expressed in cells at infection phase was analyzed by immunostaining. The HA-tagged recombinant proteins were stained with anti-HA antibody (green signals), and ER was stained by anti-PDI antibody (a) and Golgi apparatus was stained by BODIPY TR ceramide (b) (red signals). The merged figures showed the interaction of recombinant proteins with ER or Golgi marker. Three recombinant proteins were localized at ER but not at Golgi apparatus. (Color figure online)
Recombinant protein expression in ER chaperone-depleted cells
To investigate the roles of ER chaperones on secreted proteins made with the BEVS, dsRNAs for five ER chaperone genes were synthesized and their knockdown efficiencies were analyzed by RT-PCR. As shown in Fig. 4, soaking RNAi against ER chaperone genes could induce sequence-specific mRNA reduction in cultured silkworm cells, Bme21-SID1. It is interesting to observe that the mRNA expression levels of other chaperones, especially BiP and CRT, were also decreased (Fig. 4) in CNX-depleted cells, indicating the possible role of CNX in mediating the whole expression levels of other ER chaperones.

RT-PCR analysis of the knockdown efficiency of ER chaperone genes. RT-PCR was performed to analyze the knockdown efficiency from cells 7 days after incubating with dsRNAs specific for BiP, CNX, CRT, ERp57, and PDI. The GAPDH was used as loading control for normalization. Lanes 1–6 represented the samples of untreated cells, BiP dsRNA-treated cells, CNX dsRNA-treated cells, CRT dsRNA-treated cells, ERp57 dsRNA-treated cells, and PDI dsRNA-treated cells. The expression level of BiP and CRT mRNA was also decreased in CNX-depleted cells
Subsequently, recombinant baculoviruses for the target proteins were used to infect the ER chaperone-depleted cells. Four days after infection, the secretion level of each protein was analyzed by Western blot. Depletion of BiP reduced the secretion level of HGF and the insoluble fraction of EPO (Fig. 5). RNAi of CRT decreased the level of soluble EPO and increased the insoluble EPO amount, respectively. In contrast, CRT depletion decreased both soluble and insoluble HGF. Interestingly, silencing of PDI reduced the insoluble amounts of both EPO and HGF.

The recombinant protein expression in ER chaperone-depleted cells. His-tagged recombinant EPO, HGF, and Wnt3A were collected 4 days post-infection to ER chaperone gene-depleted cells. The resulting products were isolated by 10 % SDS-PAGE and visualized by Western blot using HisProbe. Lanes 1–4, and 13–15 showed secretory protein, lanes 5–8, and 16–18 showed soluble form of the protein, and lanes 9–12, and 19–21 showed insoluble form of the protein. The secretion volume of recombinant HGF was significantly reduced in BiP depletion cells
Recombinant protein expression in ER chaperone overexpressed cell lines
To further investigate whether an excess of each ER chaperone has effects on the production of recombinant proteins, we analyzed three cell lines overexpressing the ER chaperone genes, BiP, CNX, and ERp57, respectively (Fig. 6a).

Recombinant protein expressions in ER chaperone overexpressed cells. a N-terminal FLAG-fused BiP, CNX, or ERp57 was expressed in Bme21-SID1 cells and validated by Western blot with anti-FLAG antibody. The asterisk represents the location of each overexpressed chaperone protein. b His-tagged recombinant EPO and Wnt3A were collected 3 days after baculovirus infection in the BiP, CNX, and ERp57 overexpression cell lines, respectively. The resulting products were isolated on 10 % SDS-PAGE and visualized by Western blot using HisProbe. The recombinant baculovirus-infected Bme21 control cells (Lanes 1, 5, and 9), BiP overexpressed cells (Lanes 2, 6, and 10), CNX overexpressed cells (Lanes 3, 7, and 11), and ERp57 overexpressed cells (Lanes 4, 8, and 12)
The recombinant baculovirus was infected with the BiP-, CNX-, and ERp57-overexpressing cells, and 3 days after infection the secretion volume of recombinant EPO, HGF, and Wnt3A were analyzed by Western blot. It was shown that the secreted and soluble forms of recombinant EPO were increased in all three ER-chaperone-overexpressed cell lines, and both soluble and insoluble forms of recombinant HGF were also increased. In addition, the soluble form of Wnt3A was increased in the overexpression of BiP but not of CNX and ERp57 (Fig. 6b). In ER chaperone gene overexpressed cell lines, FITC-labeled target proteins were also localized at ER, not at Golgi apparatus like normal cells (Fig. 3). However, we also observed that overexpression of BiP, CNX, and ERp57 did not induce the secretion of recombinant Wnt3A.
Discussion
The secretion levels of recombinant proteins produced by BEVS often show significant difference depending on whether silkworms or cultured cells are used as hosts [23]. The viral replication and expression of viral mRNAs were not affected by insertion of distinct target genes but had different secretion efficiencies. Our current results presented that although the transcription of HGF or Wnt3A mRNAs from recombinant viruses was reduced by unknown mechanisms, this was not enough to explain the complete lack of secretion for Wnt3A. Moreover, recombinant HGF and Wnt3A were significantly accumulated inside of the cells in the insoluble form. As demonstrated by the immunostaining of recombinant baculovirus-infected cells, recombinant proteins showing difficulty in efficient secretion were trapped by the chaperones in the ER. In the ER, many ER chaperones assist proper protein folding and degradation of unfolded/misfolded proteins [24].
The three target proteins used in this study have different patterns of post-translational modifications, including N-linked glycosylation, lipidation, and disulfide bond formation [25–27]. It has been demonstrated that EPO has three predicted N-linked glycan modification sites and two disulfide bonds [25], HGF has four predicted N-linked glycan modification sites and a disulfide bond that connects alpha and beta chains [26], and Wnt3A has two N-linked glycan modification sites and a palmitoleic acid modification site. In addition, mammalian Wnt family protein is rich in cysteine residues, and its disulfide bonds were reported to play important roles in activation of gene expression functioning as a ligand [27]. This suggested that the secretion of recombinant Wnt3A might be related to correct formation of disulfide bond. Further experiments are needed to validate our hypothesis. Compared to EPO, HGF has two more N-linked glycan sites but fewer disulfide bonds, while Wnt3A has more disulfide bonds and palmitoleic acid modification than EPO. Although our findings suggest the possibility that the recombinant HGF and Wnt3A are trapped in the ER due to their incorrect disulfide bond formation or N-linked glycosylation, we could not elucidate the associations between the types and numbers of modifications with the secretion efficiency.
The secretion and expression of recombinant proteins were lower in cells depleted of BiP, CRT, or PDI. In contrast, overexpression of ER chaperones, BiP, CNX, and ERp57, increased the expression or secretion of the recombinant protein to some extent. To date, ER chaperones have been found playing relevant roles in many aspects of cellular responses. Among those, BiP has been reported as a member of the HSP70 family, which is involved in protein folding and degradation [28, 29], while PDI is known to catalyze the formation of correct disulfide bonds and modify the proteins under the condition of BiP being bound to the unfolded proteins [30]. On the other hand, CNX, CRT, and ERp57 were identified as separators to recognize glycoproteins [31]. Therefore, we presumed that the lack of ER chaperones would cause unusual folding of produced recombinant proteins and reduced their expressions. In contrast with depletion of ER chaperone genes, overexpression of ER chaperones, BiP, CNX, and ERp57, could increase the expression of recombinant proteins. These results suggested that overexpression of ER chaperones assists the formation of recombinant proteins, and then protein-folding capacity in ER was extended [32, 33].
However, in this study, we found that neither depletion nor overexpression of ER chaperones could significantly improve the secretion volume of recombinant proteins, especially Wnt3A. These results suggested that each of the ER chaperones may play a specialized role in the proper folding of recombinant proteins, and that the engineering of a single chaperone could not confer a significant increase in the secretion amounts of all products. It is our hypothesis that co-expression or co-depletion of several ER chaperons may give a better explanation to the protein secretion and degradation pathway. Thus, it is necessary to understand the entire molecular mechanisms of protein folding and modification during secretion in the insect ER and Golgi apparatus for the further improvement of BEVS.
References
Suzuki T, Kanaya T, Okazaki H et al (1997) Efficient protein production using a Bombyx mori nuclear polyhedrosis virus lacking the cysteine proteinase gene. J Gen Virol 78:3073–3080
Gómez-Sebastián S, López-Vidal J, Escribano JM (2014) Significant productivity improvement of the baculovirus expression vector system by engineering a novel expression cassette. PLoS One 9:e96562. doi:10.1371/journal.pone.0096562
Hong SM, Yamashita J, Mitsunobu H et al (2010) Efficient soluble protein production on transgenic silkworms expressing cytoplasmic chaperones. Appl Microbiol Biotechnol 87:2147–2156. doi:10.1007/s00253-010-2617-0
Jarvis DL (2003) Developing baculovirus-insect cell expression systems for humanized recombinant glycoprotein production. Virology 310:1–7. doi:10.1016/S0042-6822(03)00120-X
Ellgaard L, Molinari M, Helenius A (1999) Setting the standards: quality control in the secretory pathway. Science 286:1882–1888. doi:10.1126/science.286.5446.1882
Kleizen B, Braakman I (2004) Protein folding and quality control in the endoplasmic reticulum. Curr Opin Cell Biol 16:343–349. doi:10.1016/j.ceb.2004.06.012
Hampton RY (2002) ER-associated degradation in protein quality control and cellular regulation. Curr Opin Cell Biol 14:476–482. doi:10.1016/S0955-0674(02)00358-7
Ruggiano A, Foresti O, Carvalho P (2014) Quality control: ER-associated degradation: protein quality control and beyond. J Cell Biol 204:869–879. doi:10.1083/jcb.201312042
Shen X, Zhang K, Kaufman RJ (2004) The unfolded protein response–a stress signaling pathway of the endoplasmic reticulum. J Chem Neuroanat 28:79–92. doi:10.1016/j.jchemneu.2004.02.006
Schröder M, Kaufman RJ (2005) ER stress and the unfolded protein response. Mutat Res 569:29–63. doi:10.1016/j.mrfmmm.2004.06.056
Cao SS, Kaufman RJ (2012) Unfolded protein response. Curr Biol 22:R622–R626. doi:10.1016/j.cub.2012.07.004
Xu C, Bailly-Maitre B, Reed J (2005) Endoplasmic reticulum stress: cell life and death decisions. J Clin Investig 115:2656–2664. doi:10.1172/JCI26373.2656
Mon H, Li Z, Kobayashi I et al (2013) Soaking RNAi in Bombyx mori BmN4-SID1 cells arrests cell cycle progression. J Insect Sci 13:155. doi:10.1673/031.013.15501
Xu J, Mon H, Kusakabe T et al (2013) Establishment of a soaking RNA interference and Bombyx mori nucleopolyhedrovirus (BmNPV)-hypersensitive cell line using Bme21 cell. Appl Microbiol Biotechnol 97:10435–10444. doi:10.1007/s00253-013-5279-x
Delorme E, Lorenzini T, Giffin J (1992) Role of glycosylation on the secretion and biological activity of erythropoietin. Biochemistry 31:9871–9876
Kobayashi Y, Nakamura N, Ishizaka T et al (2001) Molecular cloning of feline hepatocyte growth factor (HGF) cDNA. J Vet Med Sci 63:211–214
Roelink H, Nusse R (1991) Expression of two members of the Wnt family during mouse development–restricted temporal and spatial patterns in the developing neural tube. Genes Dev 5:381–388. doi:10.1101/gad.5.3.381
Soejima Y, Lee JM, Nagata Y et al (2012) Comparison of signal peptides for efficient protein secretion in the baculovirus-silkworm system. Cent Eur J Biol 8:1–7. doi:10.2478/s11535-012-0112-6
Ono C, Nakatsukasa T, Nishijima Y et al (2007) Construction of the BmNPV T3 bacmid system and Its application to the functional analysis of BmNPV he65. J Insect Biotechnol Sericol 167:161–167
Mon H, Kobayashi I, Ohkubo S et al (2012) Effective RNA interference in cultured silkworm cells mediated by overexpression of Caenorhabditis elegans SID-1. RNA Biol 9:38–44. doi:10.4161/rna.9.1.18084
Li Z, Tatsuke T, Sakashita K et al (2012) Identification and characterization of Polycomb group genes in the silkworm, Bombyx mori. Mol Biol Rep 39:5575–5588. doi:10.1007/s11033-011-1362-5
Li Z, Cheng D, Mon H et al (2013) Cell cycle-dependent recruitment of polycomb proteins to the ASNS promoter counteracts C/ebp-mediated transcriptional activation in Bombyx mori. PLoS One 8:e52320. doi:10.1371/journal.pone.0052320
Imai S, Li Z, Iiyama K, Miyagawa Y (2013) Biologically active human bone morphogenetic protein 4 fused to collagen-binding domain produced in silkworm-baculovirus expression system. J Insect Biotechnol Sericol 44:39–44. doi:10.11416/jibs.82.2_039
Nishikawa S, Brodsky JL, Nakatsukasa K (2005) Roles of molecular chaperones in endoplasmic reticulum (ER) quality control and ER-associated degradation (ERAD). J Biochem 137:551–555. doi:10.1093/jb/mvi068
Takeuchi M, Kobata A (1991) Structures and functional roles of the sugar chains of human erythropoietins. Glycobiology 1:337–346
Nakamura T (1991) Structure and function of growth. Prog Growth Factor Res 3:67–85
MacDonald BT, Hien A, Zhang X et al (2014) Disulfide bond requirements for active wnt ligands. J Biol Chem 289:18122–18136. doi:10.1074/jbc.M114.575027
Hendershot LM, Valentine VA, Lee AS et al (1994) Localization of the gene encoding human BiP/GRP78, the endoplasmic reticulum cognate of the HSP70 family, to chromosome 9q34. Genomics 20:281–284. doi:10.1006/geno.1994.1166
Gething MJ (1999) Role and regulation of the ER chaperone BiP. Semin Cell Dev Biol 10:465–472. doi:10.1006/scdb.1999.0318
Mayer M, Kies U, Kammermeier R, Buchner J (2000) BiP and PDI cooperate in the oxidative folding of antibodies in vitro. J Biol Chem 275:29421–29425. doi:10.1074/jbc.M002655200
Ellgaard L, Frickel E (2003) Calnexin, calreticulin, and ERp57: teammates in glycoprotein folding. Cell Biochem Biophys 39:223–247. doi:10.1385/CBB:39:3:223
Zhang K, Kaufman RJ (2004) Signaling the unfolded protein response from the endoplasmic reticulum. J Biol Chem 279:25935–25938. doi:10.1074/jbc.R400008200
Walter P, Ron D (2006) The unfolded protein response: from stress pathway to homeostatic regulation. Mol Biotechnol 34:279–290. doi:10.1385/MB:34:2:279
Acknowledgments
The cost of publication was supported in part by the Research Grant for Young Investigators of Faculty of Agriculture, Kyushu University.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
None.
Rights and permissions
About this article

Cite this article
Imai, S., Kusakabe, T., Xu, J. et al. Roles of silkworm endoplasmic reticulum chaperones in the secretion of recombinant proteins expressed by baculovirus system. Mol Cell Biochem 409, 255–262 (2015). https://doi.org/10.1007/s11010-015-2529-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-015-2529-5