
Abstract
Purpose of Review
Mechanisms facilitating progression of hypertension via cross stimulation of the renin-angiotensin system (RAS) and inflammation have been proposed. Accordingly, we review and update evidence for regulation of RAS components by pro-inflammatory factors.
Recent Findings
Angiotensin II (Ang II), which is produced by RAS, induces vasoconstriction and consequent blood pressure elevation. In addition to this direct action, chronically elevated Ang II stimulates several pathophysiological mechanisms including generation of oxidative stress, stimulation of the nervous system, alterations in renal hemodynamics, and activation of the immune system. In particular, an activated immune system has been shown to contribute to the development of hypertension. Recent studies have demonstrated that immune cell-derived pro-inflammatory cytokines regulate RAS components, further accelerating systemic and local Ang II formation. Specifically, regulation of angiotensinogen (AGT) production by pro-inflammatory cytokines in the liver and kidney is proposed as a key mechanism underlying the progression of Ang II-dependent hypertension.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In its classic form, the renin-angiotensin system (RAS) consists of an enzymatic cascade beginning with liver-mediated production of angiotensinogen (AGT), the precursor of angiotensin (Ang) peptides. Ang II, the main effector of this system, results from successive enzymatic actions of renin and angiotensin-converting enzyme (ACE), and exerts most of its actions through activation of Ang II type 1 and type 2 receptors (AT1R and AT2R, respectively). In general, AT1R mediates the pathogenic actions of Ang II, whereas, activated AT2R elicits protective effects. In the last few decades, novel components of RAS including (pro)renin receptor, ACE2, other Ang peptides, and their receptors have been discovered [1]. Peptide hormones produced by RAS play crucial roles in controlling blood pressure and regulating electrolyte and body fluid homeostasis [2]. While originally described as a potent vasoconstrictor, accumulating evidence now indicates that both Ang II and RAS are key drivers of multiple physiological alterations and various disease pathologies. Of the wide-ranging actions of RAS, its role in immune-promoting hypertension development has come into focus [3,4,5]. Ang II induces differentiation of immune cells and augmentation of pro-inflammatory cytokine production, both contributing to elevated blood pressure and sustained hypertensive conditions. Pathological significance and mechanisms of Ang II-mediated immune activation leading to progression of inflammation and hypertension have been relatively well delineated. Moreover, there is an increasing evidence that pro-inflammatory factors enhance expression of RAS components. These findings suggest the immune system as a driver of RAS activation, thus amplifying systemic and local Ang II generation. In this brief review, the cross regulation between RAS and immune system and molecular mechanisms underlying regulation of RAS components by cytokines are summarized.
Immune System and Hypertension
Recent reports have described intimate links between inflammation and the regulation of hypertension in both humans and experimental animal models [6, 7]. In organ transplant recipients, treatment with immunosuppressive drugs such as sirolimus and cyclosporine to prevent graft-versus-host disease exhibited pro-hypertensive effects [8, 9]. In contrast, administration of immunosuppressive drugs to rodent models mitigated the development of hypertension in both lead induced and spontaneous hypertensive rats [10, 11]. Noteworthy findings demonstrate that elevated blood pressure induced by Ang II infusion and desoxycorticosterone acetate-salt are attenuated in RAG-1-deficient mice which do not produce mature B and T lymphocytes [12]. Additionally, Ang II infusion caused hypertension when RAG-1-deficient mice received adoptive transfer of T cells, further supporting a critical role for the adaptive immune system in driving hypertension. Furthermore, an elevated expression of CD70 in macrophages and dendritic cells coupled with exhaustion of effector memory T cells producing interleukin (IL)-17A and interferon γ (IFN- γ) were essential mechanisms to sustain high blood pressure in hypertension [13••]. Collectively, these studies incriminate adaptive immunity as a critical player in the development of hypertension. Likewise, roles of the innate immune system in the development of hypertension have been suggested [14]. Elevation of blood pressure by Ang II infusion was blunted in macrophage colony-stimulating factor null mice which do not produce mature macrophages [15] as well as in lysozyme M-positive monocyte-depleted mice [16]. Moreover, mineralocorticoid receptors are involved in regulation of monocyte/macrophages function, and gene deletion of these receptors in monocyte/macrophages prevented deoxycorticosterone-induced high blood pressure [17]. On the other hand, it has been reported that an anti-hypertensive role for macrophages has been demonstrated in skin through normalization of sodium concentrations and elevation of vascular endothelial growth factor-C expression [18]. Accordingly, while macrophages can be involved in anti-hypertensive mechanisms, the innate immune system including macrophages, in aggregate, contributes to the development of hypertension directly or through communication with cells of the adaptive immune system [19••].
Regulation of Immune Activity and Cytokine Production by Ang II
Immune cells including T lymphocytes, dendritic cells, and macrophages express AT1R [4]. A study using AT1R knockout mice demonstrated Ang II involvement in splenocyte proliferation [4]. In addition, Ang II increased the number of inflammatory monocytes by facilitating differentiation of hematopoietic stem cells [20]. Ang II directly stimulates adhesion molecules including intercellular adhesion molecule 1 and vascular cell adhesion molecule 1, and P-selectin which promote accumulation of immune cells in local tissues during progression of many diseases [5, 21]. It has been shown that Ang II induces increased leukocyte rolling flux, adhesion, and migration via augmentation of P-selectin without any vasoconstrictor action [22]. These findings indicate that chronically elevated Ang II promotes activation of the immune system and development of hypertension and inflammation. However, Ang II bound to AT1R induces M2 macrophage polarization, which has anti-inflammatory effects and protective actions in RAS-associated diseases [23,24,25]. Accordingly, the Ang II-immune axis may exert pathogenetic actions in early stage Ang II-dependent hypertension, triggering not only the development of hypertension but also associated end organ damages.
Animal studies have revealed that inappropriately elevated Ang II alters systemic and local levels of pro- and anti-inflammatory cytokines. Ang II infusion enhances plasma cytokine levels such as IL-6, IFN-γ, tumor necrosis factor α (TNF-α), and IL-1β [26]. Among plasma and aortic cytokines, IL-6 levels show the greatest increase following Ang II infusion, and plasma IFN-γ levels are substantially elevated as well [26, 27]. During the development of Ang II-dependent hypertension, immune cell infiltration into various tissues including the aorta, heart, brain, and kidneys is enhanced [28,29,30] leading to elevated local pro-inflammatory cytokine levels [28, 29, 31]. Results obtained from in vitro settings using cultured immune cells support these findings. For example, treatments of dendritic cells with Ang II resulted in augmentation of IL-6 and IFN-γ production [32]. Moreover, isolated peripheral blood T lymphocytes from Ang II-infused mice exhibited greater TNF-α and IFN-γ production than cells isolated from control mice [12]. Likewise, expression levels of TNF-α, IL-1β, and IL-6 in cultured macrophages were increased by Ang II [33, 34, 35••]. Evidently, Ang II promotes multi-hit activation of the immune system, stimulating not only immune cell proliferation, adhesion, and migration/infiltration but also production of cytokines ultimately facilitating Ang II-dependent hypertension. In contrast, Ang (1–7), ACE2, and MAS receptor, which have been regarded as anti-pathogenic RAS components [36], have been proposed to counteract with Ang II-induced activation of immune cells [37]. These findings suggest intimate but complicated regulation of immune system by RAS.
Regulation of RAS Components by Cytokines
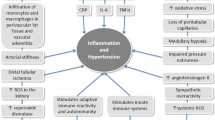
Because of the crucial roles that inflammation and the immune system play in the development of hypertension as described above, current research has focused on elucidating immune-mediated mechanisms underlying elevation of blood pressure. In a recent report, elevation of blood pressure and downregulation of sodium excretion rates by Ang II infusion were diminished, accompanied by abnormal regulation of renal transporter levels in both IL-17A- and IFN-γ-deficient mice [38]. This alteration in renal physiological function by elevated cytokines can be regarded as one of the immune-promoted mechanisms underlying elevated blood pressure. As AGT and renin are expressed in multiple tissues [39, 40], local RAS, which functions in individual organs in a tissue-specific manner, is increasingly being recognized as an independent entity from systemic RAS [39, 41, 42]. Since intrarenal Ang II is elevated in many forms of hypertension, renal RAS is acknowledged as a key target for clinical and biochemical studies. Importantly, administration of an immunosuppressive drug suppressed intrarenal Ang II elevation and mitigated the development of hypertension and renal injury [10], providing firm evidence that an activated immune system can regulate RAS activity (Fig. 1). We summarize the regulation of each RAS component by various cytokines in the following sections.

Proposed mechanism underlying systemic and local RAS activation elicited by a stimulated immune system during the development of hypertension. Ang II: angiotensin II and RAS: the renin-angiotensin system. Elevated Ang II in Ang II-dependent hypertension stimulates immune-mediated development of hypertension and associated tissue injury. Furthermore, Ang II enhances production of pro- and anti-inflammatory cytokines in immune cells, which up-regulates expression of RAS components and Ang II formation
AGT
As aforementioned, plasma and local cytokine levels including TNF-α, IL-1β, IL-6, and IFN-γ are elevated in Ang II-dependent hypertension. Regulation of AGT expression by these cytokines has been shown in multiple organs (Table 1). Hepatic AGT is the major source of circulating AGT protein. Because plasma AGT concentration is close to Km for its reaction with renin, AGT regulation in the liver together with plasma renin activity can dictate levels of circulating Ang II formation [43, 44]. Augmentation of AGT expression by IL-6 via signal transducer and activator of transcription 3 (STAT3) activation was shown in hepatocytes [45,46,47]. Thus, it is thought that this mechanism participates in amplification of circulating Ang II formation and the progression of atherosclerosis and vascular inflammation in Ang II-dependent hypertension [48]. Further molecular and biological characterization of AGT identified an acute phase-response element (APRE) in the rodent AGT-promoter region [49] in addition to three STATs binding sites within three APREs in the human AGT-promoter region [45, 50, 51]. In kidneys of Ang II-infused animals and human renin/human AGT double-transgenic mice, an elevated intrarenal AGT expression was observed [52,53,54] suggesting an intrarenal Ang II/AGT-amplifying mechanism in Ang II-dependent hypertension. In cultured renal proximal tubular cells (PTC), which predominantly express AGT in the kidney, Ang II alone increased AGT expression [55]. However, other studies failed to show augmentation of AGT expression in Ang II-treated PTCs [35, 56]. Thus, Ang II may be able to increase AGT expression in PCT under special conditions. In human PTC, Ang II stimulated AGT expression only in the presence of IL-6 [56] suggesting that Ang II-induced AGT upregulation in PTC requires co-factors or mediators such as pro-inflammatory cytokines. In an animal model of renal inflammation, elevated expression of intrarenal AGT was observed compared to control mice and treatment with an AT1R inhibitor resulted in suppressed AGT expression suggesting pro-inflammatory factors as mediators of Ang II-induced intrarenal AGT augmentation [57]. A sequential macrophage and PTC culture system demonstrated that IL-6 production was elevated in Ang II-treated macrophages, and that exposing PTC to culture medium of Ang II-treated macrophages resulted in augmentation of proximal tubular AGT expression which was attenuated by an IL-6 neutralizing antibody [35]. Taken together, these findings indicate elevated immune cell-derived IL-6 as a mediator of AGT augmentation via activation of STAT3, subsequently leading to further circulating and intrarenal Ang II formation in hypertension. These findings explain how Ang II-induced high blood pressure and the progression of kidney injury are prevented in IL-6 gene-deleted mice [58] and mice receiving STAT inhibitor treatment [59]. Interestingly, stimulatory effects of Ang II infusion on intrarenal AGT expression was observed in mice receiving a low dose of Ang II infusion (400 ng/kg/min), but not a high dose (1000 ng/kg/min) [53]. These differences may be due to induction of anti-inflammatory M2 macrophage polarization by high-dose Ang II, thus, limiting IL-6-induced intrarenal AGT augmentation. This supports the notion that activation of the Ang II-immune system-RAS cascade plays pathogenic roles in early stage Ang II-dependent hypertension.
IFN-γ is also a regulator of the Janus kinase (JAK)-STAT pathway and AGT expression. In hepatocytes, AGT expression was augmented by IFN-γ via activation of STAT1 but not STAT3 [51]. In renal PTC, IFN-γ exhibits biphasic effects on AGT regulation [60]. IFN-γ decreased AGT expression in early phase of treatment (6–12 h), with strong STAT1 activation and STAT3 suppression in PTC. In contrast, longer exposure (24 or 48 h) increased AGT expression accompanied by limited suppressor of cytokine signaling 1 (SOCS1, an endogenous suppressor of the JAK-STAT pathway) elevation and increased STAT3 activity. The switch between STAT1/STAT3 and biphasic regulation of AGT were caused by elevated SOCS1. These observations suggest that STAT1 and STAT3 have counteracting mechanisms in proximal tubular AGT regulation, and IFN-γ ultimately augments AGT expression in PTC.
Global gene deletion of TNF-α and administration of TNF-α neutralizing antibody attenuated the progression of Ang II-induced hypertension [61,62,63] suggesting that TNF-α is a key factor in the development of Ang II-dependent hypertension. In contrast to the pro-hypertensive actions of TNF-α, this cytokine has also shown suppressive effects on AGT expression in many tissues. Overexpression of TNF-α by genetic modification decreased AGT levels in the heart [64]. Moreover, TNF-α suppressed AGT expression in human adipocytes [65] which is an important source of circulating AGT. In renal PTC, TNF-α induced excessive formation of p50/p50 homodimer, a transcriptional repressor of NF-κB [66, 67] leading to the downregulation of AGT expression [68]. Accordingly, TNF-α is unlikely to participate in AGT amplification mechanisms in Ang II-dependent hypertension although it clearly contributes to the development of hypertension and associated tissue injury. In studies using TNF-α knockout mice [61,62,63], in which a high dose of Ang II (1000 ng/kg/min) was infused into the mice, intrarenal AGT was not stimulated [53]. Thus, a lower dose of Ang II infusion will be required to test effects of TNF-α on AGT regulation at least in kidneys in future studies.
Prorenin and Renin
In adults, prorenin/renin ((pro)renin) expression and secretion by juxtaglomerular apparatus (JGA) cells is the primary regulating mechanism determining circulating (pro)renin concentration. It has been shown that inflammatory signaling plays an important role in regulation of (pro)renin expression and activity. For example, IL-6 treatments decreased (pro)renin expression in As4.1 cells, an immortalized JGA-like cell line [69]. In this same cell line, IL-1β attenuated (pro)renin expression mediated via the p44/42 MAPK-STAT3 pathway [70]. Furthermore, oncostatin M, an endotoxin-responsive pro-inflammatory cytokine, was shown to inhibit (pro)renin expression via activation of STAT5 in As4.1 cells [71]. TNF-α is also known to downregulate (pro)renin expression in JGA cells via activated NF-κB targeting of a cAMP responsive element in the renin gene promoter [72, 73]. Therefore, activated inflammatory signaling and elevated pro-inflammatory cytokines in Ang II-dependent hypertension may serve as repressors of (pro)renin expression in JGA cells. Furthermore, because TNF-α suppresses both (pro)renin and AGT production, the rate-limiting reaction in RAS, TNF-α may exert pro-hypertensive actions in a RAS activation-independent manner through stimulation of pathophysiological factors including oxidative stress [63] in late stage hypertension [74] or in massive hypertension. In addition to JGA (pro)renin, studies have demonstrated important roles of (pro)renin production in renal connecting tubules and collecting ducts [2]. While chronic Ang II infusion suppresses (pro)renin expression and secretion in JGA cells [75], expression levels of collecting duct (pro)renin are increased [76], indicating that JGA (pro)renin and collecting duct (pro)renin are regulated by different mechanisms in hypertension. The importance of collecting duct (pro)renin activation in progression of hypertension has also been shown through gene deletion of (pro)renin receptor in collecting ducts which prevented Ang II-induced hypertension [77]. However, effects of pro-inflammatory cytokines on regulation of collecting duct (pro)renin in hypertension have not been established, warranting further investigation.
ACE
Information regarding regulation of ACE and ACE2, anti-hypertensive enzymes in RAS, by inflammatory factors is scarce. In an adjuvant-induced arthritis model, ACE levels in the heart and aorta were increased [78, 79] and expression levels of ACE2 were decreased in the heart [78], suggesting that pro-inflammatory conditions can induce local ACE/ACE2 imbalances and lead to accelerated RAS-associated tissue injury. Although TNF-α downregulates both JGA (pro)renin and AGT production, TNF-α stimulates local ACE expression. ACE expression levels and activity were enhanced in heart tissue of TNF-α transgenic mice [80] as well as in subfornical organ and paraventricular nucleus regions of TNF-α-injected brain tissue [81]. In addition to TNF-α-mediated transcriptional activity, the presence of NF-κB-binding sites [82] in human somatic and germline ACE promoter regions potentiates gene expression by other NF-κB-activating factors including IL-1β, IL-17A, and Ang II.
AT1R and AT2R
Roles of inflammatory factors on regulation of Ang II receptors by inflammatory factors in Ang II-dependent hypertension require special attention to the following issues. First, regulation of Ang II receptors in Ang II-dependent hypertension varies in tissue type- and cell type-specific manners [83]. Namely, these receptors exhibit both upregulation [84,85,86] and downregulation [87, 88] in disease conditions. Second, since commercially available anti-AT1R antibodies have low specificity [89], some previous conclusions regarding changes in AT1R protein levels might have to be re-evaluated. Additionally, rodents express two isoforms of AT1R gene, AT1a and AT1b, but humans do not have these isoforms. These mRNA isoforms are differentially regulated in hypertension [90], making evaluation of AT1R mRNA expression complicated in studies using rodent models or cells isolated from rodents. Furthermore, aging changes basal expression levels of AT1R and AT2R leading to altered Ang II-pressor responses [91]. This indicates diverse regulation of Ang II receptor expression by inflammatory factors among different age groups in experimental models. Greater expression of AT1R was shown in the aorta of an adjuvant-induced arthritis model, exacerbating hypertension, endothelial dysfunction, and vascular hypertrophy induced by Ang II infusion [79]. In human umbilical vein endothelial cells and vascular smooth muscle cells, treatment with serum obtained from patients with pregnancy-induced hypertension increased AT1R expression [92]. Administration of a TNF-α neutralizing antibody increased AT2R levels and attenuated the augmentation of AT1R levels [92]. These findings suggest that active inflammation can increase aortic AT1R levels and facilitate pathogenesis of Ang II in hypertension. In contrast, Ang II infusion suppressed AT1R in aorta, which was mediated by IL-6 [88]. Likewise, direct treatments with a cytokine cocktail containing TNF-α, IL-1β, and IFN-γ downregulated AT1R expression in vascular smooth muscle cells isolated from the aorta [93]. Thus, further studies will be required to delineate regulation of Ang II receptors in the aorta by an activated immune system. AT1R levels in skeletal muscle were reduced in Ang II-dependent hypertension [87]. In the brain, Ang II infusion increased AT1R expression in subfornical organ and paraventricular nucleus regions of the hypothalamus [94]. Since upregulation of AT1R expression was also observed in these brain regions of TNF-α-injected rats [81], elevated TNF-α may be involved in Ang II-induced cerebral AT1R augmentation. IL-1 treatment induced elevation of both AT1R and AT2R expression in chondrocytes isolated from articular cartilage of patients with rheumatoid arthritis and osteoarthritis [95], suggesting IL-1 can facilitate both pathogenic and protective actions in chondrocytes under inflammatory conditions. In kidneys, high Ang II levels induced by a low-salt diet decreased glomerular AT1R expression but increased tubular AT1R levels [86]. However, elevated glomerular AT1R levels in a glomerular nephritis model were reported [57]. Furthermore, in cultured renal PTC, neither Ang II nor IL-6 changed AT1R expression levels [56]. Therefore, pathological and protective mechanisms including inflammatory factors may be complexly intertwined in regulation of intrarenal Ang II receptor levels during the development of hypertension.
Conclusion
Inflammation has recently emerged as an important mechanism in progression of Ang II-dependent hypertension. In the process, inflammatory factors produced by the immune system and RAS intimately interplay and synergistically promote elevation of blood pressure and the development of RAS-associated tissue injury. Immune activation by elevated Ang II and roles of the activated immune system in hypertension have been relatively well described. Regulation of RAS components by elevated pro-inflammatory factors has been recently highlighted as inflammation-induced RAS activation provides a link between activated immune components and blood pressure elevation. Findings gleaned from these studies indicate that inflammatory factors regulate each RAS component through cytokine- and/or tissue-specific manners, which may ultimately induce further production of systemic or local Ang II in Ang II-dependent hypertension. Further studies to elucidate more detailed mechanisms underlying RAS regulation by inflammation and to disclose pathophysiological significance of the inflammation-RAS axis will contribute to the development of novel strategies to prevent and treat hypertension and RAS-associated tissue injury.
References
Papers of particular interest, published recently, have been highlighted as: •• Of major importance
Zhuo JL, Ferrao FM, Zheng Y, Li XC. New frontiers in the intrarenal renin-angiotensin system: a critical review of classical and new paradigms. Front Endocrinol. 2013;4:166. https://doi.org/10.3389/fendo.2013.00166.
Navar LG, Prieto MC, Satou R, Kobori H. Intrarenal angiotensin II and its contribution to the genesis of chronic hypertension. Curr Opin Pharmacol. 2011;11(2):180–6. https://doi.org/10.1016/j.coph.2011.01.009.
Rudemiller NP, Crowley SD. Interactions between the immune and the renin-angiotensin systems in hypertension. Hypertension. 2016;68(2):289–96. https://doi.org/10.1161/HYPERTENSIONAHA.116.06591.
Nataraj C, Oliverio MI, Mannon RB, Mannon PJ, Audoly LP, Amuchastegui CS, et al. Angiotensin II regulates cellular immune responses through a calcineurin-dependent pathway. J Clin Invest. 1999;104(12):1693–701. https://doi.org/10.1172/JCI7451.
Suzuki Y, Ruiz-Ortega M, Gomez-Guerrero C, Tomino Y, Egido J. Angiotensin II, the immune system and renal diseases: another road for RAS? Nephrol Dial Transplant. 2003;18(8):1423–6.
Harrison DG, Guzik TJ, Lob HE, Madhur MS, Marvar PJ, Thabet SR, et al. Inflammation, immunity, and hypertension. Hypertension. 2011;57(2):132–40. https://doi.org/10.1161/HYPERTENSIONAHA.110.163576.
Rodriguez-Iturbe B, Pons H, Johnson RJ. Role of the immune system in hypertension. Physiol Rev. 2017;97(3):1127–64. https://doi.org/10.1152/physrev.00031.2016.
Reis F, Parada B, Teixeira de Lemos E, Garrido P, Dias A, Piloto N, et al. Hypertension induced by immunosuppressive drugs: a comparative analysis between sirolimus and cyclosporine. Transplant Proc. 2009;41(3):868–73. https://doi.org/10.1016/j.transproceed.2009.02.005.
Divac N, Naumovic R, Stojanovic R, Prostran M. The role of immunosuppressive medications in the pathogenesis of hypertension and efficacy and safety of antihypertensive agents in kidney transplant recipients. Curr Med Chem. 2016;23(19):1941–52.
Bravo Y, Quiroz Y, Ferrebuz A, Vaziri ND, Rodriguez-Iturbe B. Mycophenolate mofetil administration reduces renal inflammation, oxidative stress, and arterial pressure in rats with lead-induced hypertension. Am J Physiol Renal Physiol. 2007;293(2):F616–23. https://doi.org/10.1152/ajprenal.00507.2006.
Khraibi AA, Norman RA Jr, Dzielak DJ. Chronic immunosuppression attenuates hypertension in Okamoto spontaneously hypertensive rats. Am J Phys. 1984;247(5 Pt 2):H722–6. https://doi.org/10.1152/ajpheart.1984.247.5.H722.
Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S, et al. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med. 2007;204(10):2449–60. https://doi.org/10.1084/jem.20070657.
•• Itani HA, Xiao L, Saleh MA, Wu J, Pilkinton MA, Dale BL, et al. CD70 exacerbates blood pressure elevation and renal damage in response to repeated hypertensive stimuli. Circ Res. 2016;118(8):1233–43. https://doi.org/10.1161/CIRCRESAHA.115.308111 This study revealed that elevated expression of CD70 in macrophages and dendritic cells coupled with exhaustion of effector memory T cells are essential mechanisms to sustain high blood pressure.
McCarthy CG, Goulopoulou S, Wenceslau CF, Spitler K, Matsumoto T, Webb RC. Toll-like receptors and damage-associated molecular patterns: novel links between inflammation and hypertension. Am J Physiol Heart Circ Physiol. 2014;306(2):H184–96. https://doi.org/10.1152/ajpheart.00328.2013.
De Ciuceis C, Amiri F, Brassard P, Endemann DH, Touyz RM, Schiffrin EL. Reduced vascular remodeling, endothelial dysfunction, and oxidative stress in resistance arteries of angiotensin II-infused macrophage colony-stimulating factor-deficient mice: evidence for a role in inflammation in angiotensin-induced vascular injury. Arterioscler Thromb Vasc Biol. 2005;25(10):2106–13. https://doi.org/10.1161/01.ATV.0000181743.28028.57.
Wenzel P, Knorr M, Kossmann S, Stratmann J, Hausding M, Schuhmacher S, et al. Lysozyme M-positive monocytes mediate angiotensin II-induced arterial hypertension and vascular dysfunction. Circulation. 2011;124(12):1370–81. https://doi.org/10.1161/CIRCULATIONAHA.111.034470.
Rickard AJ, Morgan J, Tesch G, Funder JW, Fuller PJ, Young MJ. Deletion of mineralocorticoid receptors from macrophages protects against deoxycorticosterone/salt-induced cardiac fibrosis and increased blood pressure. Hypertension. 2009;54(3):537–43. https://doi.org/10.1161/HYPERTENSIONAHA.109.131110.
Machnik A, Neuhofer W, Jantsch J, Dahlmann A, Tammela T, Machura K, et al. Macrophages regulate salt-dependent volume and blood pressure by a vascular endothelial growth factor-C-dependent buffering mechanism. Nat Med. 2009;15(5):545–52. https://doi.org/10.1038/nm.1960.
•• Justin Rucker A, Crowley SD. The role of macrophages in hypertension and its complications. Pflugers Arch. 2017;469(3-4):419–30. https://doi.org/10.1007/s00424-017-1950-x The authors reviewed important roles of macrophages in the development of hypertension.
Hahn AW, Jonas U, Buhler FR, Resink TJ. Activation of human peripheral monocytes by angiotensin II. FEBS Lett. 1994;347(2–3):178–80.
Suzuki Y, Ruiz-Ortega M, Lorenzo O, Ruperez M, Esteban V, Egido J. Inflammation and angiotensin II. Int J Biochem Cell Biol. 2003;35(6):881–900.
Piqueras L, Kubes P, Alvarez A, O'Connor E, Issekutz AC, Esplugues JV, et al. Angiotensin II induces leukocyte-endothelial cell interactions in vivo via AT(1) and AT(2) receptor-mediated P-selectin upregulation. Circulation. 2000;102(17):2118–23.
Ma LJ, Corsa BA, Zhou J, Yang H, Li H, Tang YW, et al. Angiotensin type 1 receptor modulates macrophage polarization and renal injury in obesity. Am J Physiol Renal Physiol. 2011;300(5):F1203–13. https://doi.org/10.1152/ajprenal.00468.2010.
Nishida M, Fujinaka H, Matsusaka T, Price J, Kon V, Fogo AB, et al. Absence of angiotensin II type 1 receptor in bone marrow-derived cells is detrimental in the evolution of renal fibrosis. J Clin Invest. 2002;110(12):1859–68. https://doi.org/10.1172/JCI15045.
Zhang JD, Patel MB, Griffiths R, Dolber PC, Ruiz P, Sparks MA, et al. Type 1 angiotensin receptors on macrophages ameliorate IL-1 receptor-mediated kidney fibrosis. J Clin Invest. 2014;124(5):2198–203. https://doi.org/10.1172/JCI61368.
Zhang L, Du J, Hu Z, Han G, Delafontaine P, Garcia G, et al. IL-6 and serum amyloid a synergy mediates angiotensin II-induced muscle wasting. J Am Soc Nephrol. 2009;20(3):604–12. https://doi.org/10.1681/ASN.2008060628.
Recinos A 3rd, LeJeune WS, Sun H, Lee CY, Tieu BC, Lu M, et al. Angiotensin II induces IL-6 expression and the Jak-STAT3 pathway in aortic adventitia of LDL receptor-deficient mice. Atherosclerosis. 2007;194(1):125–33. https://doi.org/10.1016/j.atherosclerosis.2006.10.013.
Wei Z, Spizzo I, Diep H, Drummond GR, Widdop RE, Vinh A. Differential phenotypes of tissue-infiltrating T cells during angiotensin II-induced hypertension in mice. PLoS One. 2014;9(12):e114895. https://doi.org/10.1371/journal.pone.0114895.
Qi G, Jia L, Li Y, Bian Y, Cheng J, Li H, et al. Angiotensin II infusion-induced inflammation, monocytic fibroblast precursor infiltration, and cardiac fibrosis are pressure dependent. Cardiovasc Toxicol. 2011;11(2):157–67. https://doi.org/10.1007/s12012-011-9109-z.
Ozawa Y, Kobori H, Suzaki Y, Navar LG. Sustained renal interstitial macrophage infiltration following chronic angiotensin II infusions. Am J Physiol Renal Physiol. 2007;292(1):F330–9. https://doi.org/10.1152/ajprenal.00059.2006.
Ruiz-Ortega M, Ruperez M, Lorenzo O, Esteban V, Blanco J, Mezzano S, et al. Angiotensin II regulates the synthesis of proinflammatory cytokines and chemokines in the kidney. Kidney Int Suppl. 2002;62(82):12–22. https://doi.org/10.1046/j.1523-1755.62.s82.4.x.
Meng Y, Chen C, Liu Y, Tian C, Li HH. Angiotensin II regulates dendritic cells through activation of NF-kappaB /p65, ERK1/2 and STAT1 pathways. Cell Physiol Biochem. 2017;42(4):1550–8. https://doi.org/10.1159/000479272.
Iwashita M, Sakoda H, Kushiyama A, Fujishiro M, Ohno H, Nakatsu Y, et al. Valsartan, independently of AT1 receptor or PPARgamma, suppresses LPS-induced macrophage activation and improves insulin resistance in cocultured adipocytes. Am J Physiol Endocrinol Metab. 2012;302(3):E286–96. https://doi.org/10.1152/ajpendo.00324.2011.
Guo F, Chen XL, Wang F, Liang X, Sun YX, Wang YJ. Role of angiotensin II type 1 receptor in angiotensin II-induced cytokine production in macrophages. J Interf Cytokine Res. 2011;31(4):351–61. https://doi.org/10.1089/jir.2010.0073.
•• O'Leary R, Penrose H, Miyata K, Satou R. Macrophage-derived IL-6 contributes to ANG II-mediated angiotensinogen stimulation in renal proximal tubular cells. Am J Physiol Renal Physiol. 2016;310(10):F1000–7. https://doi.org/10.1152/ajprenal.00482.2015 This study demonstrated that elevated IL-6 derived from Ang II-treated macrophages stimulates AGT expression in PTC.
Ferrario CM. ACE2: more of Ang-(1-7) or less Ang II? Curr Opin Nephrol Hypertens. 2011;20(1):1–6. https://doi.org/10.1097/MNH.0b013e3283406f57.
Simoes e Silva AC, Silveira KD, Ferreira AJ, Teixeira MM. ACE2, angiotensin-(1-7) and Mas receptor axis in inflammation and fibrosis. Br J Pharmacol. 2013;169(3):477–92. https://doi.org/10.1111/bph.12159.
Kamat NV, Thabet SR, Xiao L, Saleh MA, Kirabo A, Madhur MS, et al. Renal transporter activation during angiotensin-II hypertension is blunted in interferon-gamma−/− and interleukin-17A−/− mice. Hypertension. 2015;65(3):569–76. https://doi.org/10.1161/HYPERTENSIONAHA.114.04975.
Dzau VJ, Re R. Tissue angiotensin system in cardiovascular medicine. A paradigm shift? Circulation. 1994;89(1):493–8.
Tamura K, Tanimoto K, Takahashi S, Sagara M, Fukamizu A, Murakami K. Structure and expression of the mouse angiotensinogen gene. Jpn Heart J. 1992;33(1):113–24.
Paul M, Poyan Mehr A, Kreutz R. Physiology of local renin-angiotensin systems. Physiol Rev. 2006;86(3):747–803. https://doi.org/10.1152/physrev.00036.2005.
Re RN. Tissue renin angiotensin systems. Med Clin North Am. 2004;88(1):19–38.
Corvol P, Jeunemaitre X. Molecular genetics of human hypertension: role of angiotensinogen. Endocr Rev. 1997;18(5):662–77. https://doi.org/10.1210/edrv.18.5.0312.
Kim HS, Krege JH, Kluckman KD, Hagaman JR, Hodgin JB, Best CF, et al. Genetic control of blood pressure and the angiotensinogen locus. Proc Natl Acad Sci U S A. 1995;92(7):2735–9.
Jain S, Li Y, Patil S, Kumar A. HNF-1alpha plays an important role in IL-6-induced expression of the human angiotensinogen gene. Am J Physiol Cell Physiol. 2007;293(1):C401–10. https://doi.org/10.1152/ajpcell.00433.2006.
htani R, Yayama K, Takano M, Itoh N, Okamoto H. Stimulation of angiotensinogen production in primary cultures of rat hepatocytes by glucocorticoid, cyclic adenosine 3′,5′-monophosphate, and interleukin-6. Endocrinology. 1992;130(3):1331–8. https://doi.org/10.1210/endo.130.3.1311238.
Ray S, Boldogh I, Brasier AR. STAT3 NH2-terminal acetylation is activated by the hepatic acute-phase response and required for IL-6 induction of angiotensinogen. Gastroenterology. 2005;129(5):1616–32. https://doi.org/10.1053/j.gastro.2005.07.055.
Brasier AR, Recinos A 3rd, Eledrisi MS. Vascular inflammation and the renin-angiotensin system. Arterioscler Thromb Vasc Biol. 2002;22(8):1257–66.
Brasier AR, Ron D, Tate JE, Habener JF. A family of constitutive C/EBP-like DNA binding proteins attenuate the IL-1 alpha induced, NF kappa B mediated trans-activation of the angiotensinogen gene acute-phase response element. EMBO J. 1990;9(12):3933–44.
Sherman CT, Brasier AR. Role of signal transducers and activators of transcription 1 and −3 in inducible regulation of the human angiotensinogen gene by interleukin-6. Mol Endocrinol. 2001;15(3):441–57. https://doi.org/10.1210/mend.15.3.0609.
Jain S, Shah M, Li Y, Vinukonda G, Sehgal PB, Kumar A. Upregulation of human angiotensinogen (AGT) gene transcription by interferon-gamma: involvement of the STAT1-binding motif in the AGT promoter. Biochim Biophys Acta. 2006;1759(7):340–7. https://doi.org/10.1016/j.bbaexp.2006.07.003.
Kobori H, Harrison-Bernard LM, Navar LG. Expression of angiotensinogen mRNA and protein in angiotensin II-dependent hypertension. J Am Soc Nephrol. 2001;12(3):431–9.
Gonzalez-Villalobos RA, Seth DM, Satou R, Horton H, Ohashi N, Miyata K, et al. Intrarenal angiotensin II and angiotensinogen augmentation in chronic angiotensin II-infused mice. Am J Physiol Renal Physiol. 2008;295(3):F772–9. https://doi.org/10.1152/ajprenal.00019.2008.
Kobori H, Ozawa Y, Satou R, Katsurada A, Miyata K, Ohashi N, et al. Kidney-specific enhancement of ANG II stimulates endogenous intrarenal angiotensinogen in gene-targeted mice. Am J Physiol Renal Physiol. 2007;293(3):F938–45. https://doi.org/10.1152/ajprenal.00146.2007.
Ingelfinger JR, Jung F, Diamant D, Haveran L, Lee E, Brem A, et al. Rat proximal tubule cell line transformed with origin-defective SV40 DNA: autocrine ANG II feedback. Am J Phys. 1999;276(2 Pt 2):F218–27.
Satou R, Gonzalez-Villalobos RA, Miyata K, Ohashi N, Katsurada A, Navar LG, et al. Costimulation with angiotensin II and interleukin 6 augments angiotensinogen expression in cultured human renal proximal tubular cells. Am J Physiol Renal Physiol. 2008;295(1):F283–9. https://doi.org/10.1152/ajprenal.00047.2008.
Urushihara M, Ohashi N, Miyata K, Satou R, Acres OW, Kobori H. Addition of angiotensin II type 1 receptor blocker to CCR2 antagonist markedly attenuates crescentic glomerulonephritis. Hypertension. 2011;57(3):586–93. https://doi.org/10.1161/HYPERTENSIONAHA.110.165704.
Lee DL, Sturgis LC, Labazi H, Osborne JB Jr, Fleming C, Pollock JS, et al. Angiotensin II hypertension is attenuated in interleukin-6 knockout mice. Am J Physiol Heart Circ Physiol. 2006;290(3):H935–40. https://doi.org/10.1152/ajpheart.00708.2005.
Banes-Berceli AK, Al-Azawi H, Proctor D, Qu H, Femminineo D, Hill-Pyror C, et al. Angiotensin II utilizes Janus kinase 2 in hypertension, but not in the physiological control of blood pressure, during low-salt intake. Am J Physiol Regul Integr Comp Physiol. 2011;301(4):R1169–76. https://doi.org/10.1152/ajpregu.00071.2011.
Satou R, Miyata K, Gonzalez-Villalobos RA, Ingelfinger JR, Navar LG, Kobori H. Interferon-gamma biphasically regulates angiotensinogen expression via a JAK-STAT pathway and suppressor of cytokine signaling 1 (SOCS1) in renal proximal tubular cells. FASEB J. 2012;26(5):1821–30. https://doi.org/10.1096/fj.11-195198.
Sriramula S, Haque M, Majid DS, Francis J. Involvement of tumor necrosis factor-alpha in angiotensin II-mediated effects on salt appetite, hypertension, and cardiac hypertrophy. Hypertension. 2008;51(5):1345–51. https://doi.org/10.1161/HYPERTENSIONAHA.107.102152.
Sriramula S, Francis J. Tumor necrosis factor - alpha is essential for angiotensin II-induced ventricular remodeling: role for oxidative stress. PLoS One. 2015;10(9):e0138372. https://doi.org/10.1371/journal.pone.0138372.
Zhang J, Patel MB, Griffiths R, Mao A, Song YS, Karlovich NS, et al. Tumor necrosis factor-alpha produced in the kidney contributes to angiotensin II-dependent hypertension. Hypertension. 2014;64(6):1275–81. https://doi.org/10.1161/HYPERTENSIONAHA.114.03863.
Flesch M, Hoper A, Dell'Italia L, Evans K, Bond R, Peshock R, et al. Activation and functional significance of the renin-angiotensin system in mice with cardiac restricted overexpression of tumor necrosis factor. Circulation. 2003;108(5):598–604. https://doi.org/10.1161/01.CIR.0000081768.13378.BF.
Wang B, Jenkins JR, Trayhurn P. Expression and secretion of inflammation-related adipokines by human adipocytes differentiated in culture: integrated response to TNF-alpha. Am J Physiol Endocrinol Metab. 2005;288(4):E731–40.
Guan H, Hou S, Ricciardi RP. DNA binding of repressor nuclear factor-kappaB p50/p50 depends on phosphorylation of Ser337 by the protein kinase a catalytic subunit. J Biol Chem. 2005;280(11):9957–62. https://doi.org/10.1152/ajpendo.00475.2004.
Tong X, Yin L, Washington R, Rosenberg DW, Giardina C. The p50-p50 NF-kappaB complex as a stimulus-specific repressor of gene activation. Mol Cell Biochem. 2004;265(1–2):171–83.
Satou R, Miyata K, Katsurada A, Navar LG, Kobori H. Tumor necrosis factor-{alpha} suppresses angiotensinogen expression through formation of a p50/p50 homodimer in human renal proximal tubular cells. Am J Physiol Cell Physiol. 2010;299(4):C750–9. https://doi.org/10.1152/ajpcell.00078.2010.
Pan L, Wang Y, Jones CA, Glenn ST, Baumann H, Gross KW. Enhancer-dependent inhibition of mouse renin transcription by inflammatory cytokines. Am J Physiol Renal Physiol. 2005;288(1):F117–24. https://doi.org/10.1152/ajprenal.00333.2003.
Liu X, Shi Q, Sigmund CD. Interleukin-1beta attenuates renin gene expression via a mitogen-activated protein kinase kinase-extracellular signal-regulated kinase and signal transducer and activator of transcription 3-dependent mechanism in As4.1 cells. Endocrinology. 2006;147(12):6011–8. https://doi.org/10.1210/en.2006-0129.
Baumann H, Wang Y, Richards CD, Jones CA, Black TA, Gross KW. Endotoxin-induced renal inflammatory response. Oncostatin M as a major mediator of suppressed renin expression. J Biol Chem. 2000;275(29):22014–9. https://doi.org/10.1074/jbc.M002830200.
Todorov VT, Volkl S, Muller M, Bohla A, Klar J, Kunz-Schughart LA, et al. Tumor necrosis factor-alpha activates NFkappaB to inhibit renin transcription by targeting cAMP-responsive element. J Biol Chem. 2004;279(2):1458–67. https://doi.org/10.1074/jbc.M308697200.
Todorov VT, Volkl S, Friedrich J, Kunz-Schughart LA, Hehlgans T, Vermeulen L, et al. Role of CREB1 and NF{kappa}B-p65 in the down-regulation of renin gene expression by tumor necrosis factor {alpha}. J Biol Chem. 2005;280(26):24356–62. https://doi.org/10.1074/jbc.M502968200.
Ortiz RM, Mamalis A, Navar LG. Aldosterone receptor antagonism reduces urinary C-reactive protein excretion in angiotensin II-infused, hypertensive rats. J Am Soc Hypertens. 2009;3(3):184–91. https://doi.org/10.1016/j.jash.2009.01.003.
Navar LG, Kobori H, Prieto MC, Gonzalez-Villalobos RA. Intratubular renin-angiotensin system in hypertension. Hypertension. 2011;57(3):355–62. https://doi.org/10.1161/HYPERTENSIONAHA.110.163519.
Prieto MC, Williams DE, Liu L, Kavanagh KL, Mullins JJ, Mitchell KD. Enhancement of renin and prorenin receptor in collecting duct of Cyp1a1-Ren2 rats may contribute to development and progression of malignant hypertension. Am J Physiol Renal Physiol. 2011;300(2):F581–8. https://doi.org/10.1152/ajprenal.00433.2010.
Prieto MC, Reverte V, Mamenko M, Kuczeriszka M, Veiras LC, Rosales CB, et al. Collecting duct prorenin receptor knockout reduces renal function, increases sodium excretion, and mitigates renal responses in ANG II-induced hypertensive mice. Am J Physiol Renal Physiol. 2017;313(6):F1243–F53. https://doi.org/10.1152/ajprenal.00152.2017.
Hanafy S, Tavasoli M, Jamali F. Inflammation alters angiotensin converting enzymes (ACE and ACE-2) balance in rat heart. Inflammation. 2011;34(6):609–13. https://doi.org/10.1007/s10753-010-9269-1.
Sakuta T, Morita Y, Satoh M, Fox DA, Kashihara N. Involvement of the renin-angiotensin system in the development of vascular damage in a rat model of arthritis: effect of angiotensin receptor blockers. Arthritis Rheum. 2010;62(5):1319–28. https://doi.org/10.1002/art.27384.
Sekiguchi K, Li X, Coker M, Flesch M, Barger PM, Sivasubramanian N, et al. Cross-regulation between the renin-angiotensin system and inflammatory mediators in cardiac hypertrophy and failure. Cardiovasc Res. 2004;63(3):433–42. https://doi.org/10.1016/j.cardiores.2004.02.005.
Wei SG, Yu Y, Zhang ZH, Felder RB. Proinflammatory cytokines upregulate sympathoexcitatory mechanisms in the subfornical organ of the rat. Hypertension. 2015;65(5):1126–33. https://doi.org/10.1161/HYPERTENSIONAHA.114.05112.
Garcia V, Shkolnik B, Milhau L, Falck JR, Schwartzman ML. 20-HETE activates the transcription of angiotensin-converting enzyme via nuclear factor-kappaB translocation and promoter binding. J Pharmacol Exp Ther. 2016;356(3):525–33. https://doi.org/10.1124/jpet.115.229377.
Kobori H, Nangaku M, Navar LG, Nishiyama A. The intrarenal renin-angiotensin system: from physiology to the pathobiology of hypertension and kidney disease. Pharmacol Rev. 2007;59(3):251–87. https://doi.org/10.1124/pr.59.3.3.
Gutkind JS, Kurihara M, Castren E, Saavedra JM. Increased concentration of angiotensin II binding sites in selected brain areas of spontaneously hypertensive rats. J Hypertens. 1988;6(1):79–84.
Hu L, Zhu DN, Yu Z, Wang JQ, Sun ZJ, Yao T. Expression of angiotensin II type 1 (AT(1)) receptor in the rostral ventrolateral medulla in rats. J Appl Physiol. 2002;92(5):2153–61. https://doi.org/10.1152/japplphysiol.00261.2001.
Cheng HF, Becker BN, Burns KD, Harris RC. Angiotensin II upregulates type-1 angiotensin II receptors in renal proximal tubule. J Clin Invest. 1995;95(5):2012–9. https://doi.org/10.1172/JCI117886.
Yoshida T, Galvez S, Tiwari S, Rezk BM, Semprun-Prieto L, Higashi Y, et al. Angiotensin II inhibits satellite cell proliferation and prevents skeletal muscle regeneration. J Biol Chem. 2013;288(33):23823–32. https://doi.org/10.1074/jbc.M112.449074.
Coles B, Fielding CA, Rose-John S, Scheller J, Jones SA, O'Donnell VB. Classic interleukin-6 receptor signaling and interleukin-6 trans-signaling differentially control angiotensin II-dependent hypertension, cardiac signal transducer and activator of transcription-3 activation, and vascular hypertrophy in vivo. Am J Pathol. 2007;171(1):315–25. https://doi.org/10.2353/ajpath.2007.061078.
Herrera M, Sparks MA, Alfonso-Pecchio AR, Harrison-Bernard LM, Coffman TM. Lack of specificity of commercial antibodies leads to misidentification of angiotensin type 1 receptor protein. Hypertension. 2013;61(1):253–8. https://doi.org/10.1161/HYPERTENSIONAHA.112.203679.
Iwai N, Inagami T, Ohmichi N, Nakamura Y, Saeki Y, Kinoshita M. Differential regulation of rat AT1a and AT1b receptor mRNA. Biochem Biophys Res Commun. 1992;188(1):298–303.
Dinh QN, Drummond GR, Kemp-Harper BK, Diep H, De Silva TM, Kim HA, et al. Pressor response to angiotensin II is enhanced in aged mice and associated with inflammation, vasoconstriction and oxidative stress. Aging. 2017;9(6):1595–606. https://doi.org/10.18632/aging.101255.
Takeda-Matsubara Y, Matsubara K, Ochi H, Ito M, Iwai M, Horiuchi M. Expression of endothelial angiotensin II receptor mRNA in pregnancy-induced hypertension. Am J Hypertens. 2003;16(12):993–9.
Wang H, Jia D, Shen H, Tao Q, Liu L, Zhang L, et al. Effects of cytokines on the expression of angiotensin II type 1 receptors in vascular smooth muscle cells. Int J Clin Exp Med. 2016;9(1):219–25.
Wei SG, Yu Y, Zhang ZH, Felder RB. Angiotensin II upregulates hypothalamic AT1 receptor expression in rats via the mitogen-activated protein kinase pathway. Am J Physiol Heart Circ Physiol. 2009;296(5):H1425–33. https://doi.org/10.1152/ajpheart.00942.2008.
Kawakami Y, Matsuo K, Murata M, Yudoh K, Nakamura H, Shimizu H, et al. Expression of angiotensin II receptor-1 in human articular chondrocytes. Arthritis. 2012;2012:648537. https://doi.org/10.1155/2012/648537.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no conflicts of interest relevant to this manuscript.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Mechanisms of Hypertension
Rights and permissions
About this article

Cite this article
Satou, R., Penrose, H. & Navar, L.G. Inflammation as a Regulator of the Renin-Angiotensin System and Blood Pressure. Curr Hypertens Rep 20, 100 (2018). https://doi.org/10.1007/s11906-018-0900-0
Published:
DOI: https://doi.org/10.1007/s11906-018-0900-0