
Abstract
Hypertension that is considered idiopathic is called essential hypertension and accordingly has no clear culprit for its cause. However, basic research and clinical studies in recent years have expanded our understanding of the mechanisms underlying the development of essential hypertension. Of these, increased oxidative stress, both in the kidney and arterial wall, closely coupled with inflammatory infiltration now appear to have a prominent role. Discovery of regulatory and interleukin-17-producing T cells has enabled us to better understand the mechanism by which inflammation and autoimmunity, or autoinflammation, lead to the development of hypertension. Despite achieving considerable progress, the intricate interactions between oxidative stress, the immune system and the development of hypertension remain to be fully elucidated. In this review, we summarize recent developments in the pathophysiology of hypertension with a focus on the oxidant stress–autoimmunity–inflammation interaction.
Similar content being viewed by others

Introduction
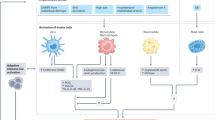
Hypertension is the most common chronic disease and the major cause of heart failure, stroke, chronic kidney disease and mortality in the Western world. Similar to other disease states, idiopathic hypertension is called ‘essential’ or ‘primary’ when its exact cause and pathophysiology are unknown. Known, direct causes of hypertension are identified in only 5–10% of all cases and are designated as ‘secondary’ owing to a precise underlying pathophysiological mechanism. Recent advances have considerably expanded our understanding of the underlying mechanisms of primary hypertension. Several lines of experimental and clinical evidence point to the important role of oxidative stress and inflammation in the development of hypertension. At the same time, it is important to determine whether autoimmunity and inflammation/oxidative stress lead to hypertension or vice versa. This review provides an overview of the current evidence relating increased oxidative stress, inflammation and autoimmune alterations to the development and progression of hypertension, with a special focus on causation (Figure 1).

Interconnection of hypertension, inflammation, oxidative stress and the immune system. CRP, C-reactive protein; IL-6, interleukin-6; TNF-α, tumor necrosis factor-α. A full color version of this figure is available at the Hypertension Research journal online.
Human studies identifying the role of inflammation in hypertension
Several cross-sectional studies have reported an increased level of inflammation in patients with hypertension. In a cross-sectional survey of 8347 apparently healthy Korean individuals, Sung et al.1 found that the prevalence of hypertension was 1.267-, 1.253- and 1.451 times higher in subjects from the second, third and fourth quartiles of the C-reactive protein (CRP) distribution, respectively, compared with subjects in the first quartile, even after the adjustment for many confounders (CRP level ranges for the first and fourth quartiles were 0.1–0.3 and 1.2–11 mg l−1, respectively). Similarly, Bautista et al.2 showed that patients in the third and fourth quartiles of the interleukin-6 (IL-6) and tumor necrosis factor-α (TNF-α) distribution were more likely to have hypertension compared with patients in the first quartile. These and other studies3 recruited apparently healthy individuals and assessed inflammatory markers along with a number of laboratory, clinical and demographic features described as independent predictors of elevated blood pressure. Despite the demonstration of an independent association between increased inflammatory markers, such as CRP, IL-6 and TNF-α, and hypertension, these associative studies did not address the question of which condition precedes the other.
Several observational studies answered this question. These studies recruited normotensive subjects, followed them and documented the development of de novo hypertension, and the baseline inflammatory status was evaluated as a predictive factor for the development of hypertension. Sesso et al.4 investigated a prospective cohort of 20 525 female US health professionals aged >45 years who were normotensive at baseline evaluation. These women were followed for a median of 7.8 years and incident hypertension cases were recorded. CRP at baseline was significantly associated with an increased risk of developing hypertension in all subgroups of individuals, including those with very low levels of baseline blood pressures, as well as those with no traditional coronary risk factors. Of 5365 women who developed hypertension, 511 had baseline serum CRP value <0.43 mg l−1, whereas 1962 individuals had serum CRP values of >3.50 mg l−1; the remaining incident hypertensive patients had CRP values in between. In adjusted models, a 1 s.d. increase of CRP led to a 1.14 adjusted relative risk for incident hypertension. The same group conducted a nested case–control study of 400 women developing hypertension and an equal number of age-matched normotensive control subjects for 10 years of follow-up as part of the Women’s Health Study.5 The authors found that after multivariate adjustment, IL-6 was weakly associated and CRP was strongly associated with the risk of hypertension. Several other studies confirmed this temporal relationship in patients with prehypertension or in elderly normotensive individuals.6, 7
In summary, several studies support an independent association between several markers of inflammation and hypertension. Most importantly, increased inflammation clearly preceded clinical blood pressure elevation. Altogether, these studies support the hypothesis that inflammation may be causally related to the development of elevated blood pressure.
Experimental data and possible pathways for inflammation-induced hypertension
Clinical studies suggest an independent link between systemic inflammation and an increased risk of hypertension. However, another question is how does this increased systemic inflammation lead to the development of hypertension? And, could this systemic inflammation be related to inflammation in tissues vital for the development of hypertension, namely the vascular wall and/or the kidney?
Numerous well-designed experimental studies conducted on spontaneously hypertensive rats (SHRs) revealed the significance of renal interstitial inflammation and increased oxidative stress in the development of hypertension. Previously, inflammatory infiltration and immune dysfunction observed in SHR were considered part of the defense mechanism and therefore a secondary consequence to the development of hypertension.8 However, subsequent studies suggested that immune cell infiltration in the kidney may be the cause, not the result, of hypertension in this model. Indeed, in nearly all models of salt-sensitive hypertension, the tubulointerstitium of rat kidneys are infiltrated with lymphocytes and macrophages,9, 10 and a correlation between the intensity of the inflammatory cell infiltration and the severity of the blood pressure elevation was apparent.11 In experimental models of hypertension, monocyte/macrophage infiltration of the perivascular fat renal tissue, as well as vascular adventitia, was observed.12 Furthermore, several experimental studies, in which renal tubulointerstitial inflammation was reduced using various methods, either prevented the development of hypertension altogether or ameliorated hypertension in genetic models of hypertension.13, 14 Putative mechanisms by which increased renal interstitial inflammation leads to the development of hypertension include increased oxidative stress, loss of peritubular capillaries and resultant medullary hypoxia, and an impaired pressure natriuresis mechanism. Details of the mechanisms by which renal interstitial inflammation may cause hypertension have been recently reviewed elsewhere.15
Evidence showing the effects of renal interstitial inflammation also comes from experimental studies that investigated the impact of anti-inflammatory cytokines, such as IL-10, on blood pressure. Tinsley et al.16 showed that the administration of intraperitoneal IL-10 normalized blood pressure and improved endothelial dysfunction in pregnant deoxycorticosterone acetate/saline-treated rats. More recently, Dhande et al.17 administered an AT2 receptor agonist to a monocyte cell line cell culture. The AT2 receptor agonist increased IL-10 production but decreased lipopolysaccharide-induced TNF-α and IL-6 production in a dose-dependent manner. As stated earlier, macrophage infiltration is almost universal in hypertension models and the findings of the latter study suggest that a shift of the pro- and anti-inflammatory cytokine profile of macrophages may preclude the development of hypertension.
Another organ in which inflammation may lead to increased blood pressure is the arterial vascular wall. Monocyte/macrophage infiltration of the perivascular fat may contribute to the pressor response to angiotensin II. Functionally deficient macrophages in osteopetrotic mice with a mutation in macrophage colony-stimulating factor rendered these mice resistant to the prohypertensive effects of angiotensin II infusion.18 Wenzel et al.19 ablated monocyte/macrophage cells via diphtheria toxin and then infused angiotensin II. The authors observed that the blood pressure did not increase in diphtheria toxin-treated mice.
Vascular stiffness may be both a cause and a consequence of hypertension. Several cross-sectional clinical studies demonstrated significant associations of markers of systemic inflammation, such as CRP, with pulse wave velocity.20, 21 In a recent study, Zheng et al.22 evaluated data from a community-based, prospective, long-term follow-up observational study, the Asymptomatic Polyvascular Abnormalities Community study. Of 4025 patients followed up for an average of 27 months, 432 patients developed incident hypertension. The brachial-ankle pulse wave velocity was found to be an independent predictor of incident hypertension. Similarly, Kaess et al.23 reported that higher aortic stiffness measurements were associated with a higher risk of incident hypertension during a 7-year follow-up period in 1759 participants.
Therefore, the available experimental and clinical data indicate a potential role of increased systemic inflammatory biomarkers, as well as inflammatory infiltration of the renal interstitium and vascular wall, in the development of de novo hypertension.
Interaction of oxidative stress and inflammation in the development of hypertension
Because inflammation and oxidative stress are frequently coupled, such an interaction would be an attractive mechanism to explain changes that drive the stage for the development of hypertension. Reactive oxygen species (ROS) produced in large amounts would overwhelm the neutralizing capacity of the antioxidative systems and may influence protein activity and cell function.24
Chabrashvili et al.25 studied the expression of the NADPH oxidase system as a mechanism for increased oxidative stress in SHRs. The kidneys of SHRs showed the increased expression of genes for the main components of phagocyte NADPH oxidase. This increased mRNA preceded the development of hypertension in the model and was notable in the nephron segments of the vasculature, macula densa and distal nephron, which preceded the development of hypertension. The SHR kidney has an exaggerated tubuloglomerular feedback response, which may be due to the diminished availability of NO. The results of this experimental study support the hypothesis that superoxide generation mediated by the NADPH oxidase system in the aforementioned parts of the nephron may be the principle driving force for the development of hypertension.
Callera et al.26 showed that in a deoxycorticosterone acetate-salt hypertension rat model, vascular superoxide production was increased by activation of the endothelin system. Zalba et al.27 demonstrated increased production of superoxide anion in SHRs. Conversely, animal studies showed that the attenuation of cellular oxidative stress by overexpression of superoxide dismutase (SOD) or treatment with antioxidants attenuated these blood pressure elevations. By contrast, depletion of SOD exacerbated hypertension.28, 29
Clinical studies confirmed that in patients with essential hypertension, blood pressure levels are positively correlated with markers of oxidative stress.30, 31 In addition, population-based observational studies demonstrated an inverse correlation between plasma antioxidant levels and blood pressure levels.32
An increased local ROS concentration in the kidney may have a pivotal role in the generation of hypertension. ROS-induced vascular remodeling impairs autoregulation and may lead to distal tubulointerstitial ischemia.33 In genetically hypertensive rats, the number of intrarenal superoxide-positive cells is positively correlated with systolic blood pressure levels.14, 34 Recently, resveratrol was shown to mitigate oxidative stress and normalize angiotensin signaling in the kidney and reduce blood pressure via activation of phase II antioxidant enzymes.35
Aortic stiffening is associated with the development of hypertension. A common denominator of various conditions associated with aortic stiffening is increased vascular oxidative stress. Increased oxidative stress in the vascular wall is closely and highly related to inflammatory infiltration. Several researchers postulated the role of modification of self-proteins, such as isoketals, also known as isolevuglandins. The modified self-proteins then behave as autoantigens and elicit an inflammatory reaction.36 Wu et al.37 used a mouse model exhibiting excessive vascular production of ROS and observed that the animals developed vascular collagen deposition, aortic stiffening, renal dysfunction and hypertension with aging. Thus, increased oxidative stress may be a major culprit in the development of inflammatory infiltration, fibrosis and ensuing elevated blood pressures.
In summary, increased oxidative stress in renal and arteriolar vascular tissues may lead to increased blood pressure and could be an effector pathway for a number of pathophysiological disorders. The Harrison group proposed that oxidative stress behaves as the effector mechanism and final common pathway for a number of pathophysiologic conditions, including immune, endocrine, neural, vascular and genetic disorders (mosaic theory of hypertension).38 There is significant evidence showing increased inflammation as well as oxidative stress in the renal interstitium and vascular wall, but the question remains as to why only some individuals who suffer from increased oxidative stress and inflammation develop hypertension whereas others do not.
The clearcut answer to this essential question has yet to be determined, but polymorphisms in antioxidant system enzymes, such as glutathione S-transferase and NADPH oxidase, may account for the interindividual differences in the levels of pro-oxidant molecules and antioxidative capacity.39, 40, 41 The autoimmune response against some undefined antigens may be another factor responsible for the development of hypertension. Clinical studies related to hypertension and inflammation are summarized in Table 1.
Adaptive immune system in the pathogenesis of hypertension
Studies identifying the role of the innate/adaptive immune system in the development of hypertension were performed in the 1980s, but reported conflicting results, which made it difficult to reconcile differences and decreased interest in this field. We now propose that different subtypes of immune cells, with various and considerably different functions, may have led to such conflicting results. A better understanding of the various cell subtypes and corresponding functions has allowed scientists to conduct new studies with promising results, which implicate several aspects of immune dysregulation in the pathophysiology of hypertension.
As discussed earlier in this review, an inflammatory infiltrate mainly composed of monocyte/macrophages (innate) is related to increased oxidative stress and the development of hypertension. Recent studies focused primarily on the role of T cells in the development of hypertension.42 Recombinase-activating gene knockout mice lacking T and B cells demonstrated a blunted blood pressure increase in response to angiotensin II infusion and deoxycorticosterone acetate-salt hypertension. Interestingly, the adoptive transfer of T cells, but not B cells, restored the blood pressure response in these mice. The authors also demonstrated that activated T cells can stimulate the vascular production of superoxide. Mahdur et al.43 compared the effects of angiotensin II infusion in IL-17 knockout mice and control mice. Although the initial hypertensive response was similar, hypertension was not sustained in mice lacking IL-17. The authors concluded that IL-17-producing T cells are required for the development of sustained hypertension. In contrast to the proinflammatory actions of TH1 and T helper type 17 (TH17) lymphocytes, regulatory T cells suppress inflammatory reactions. IL-10 is one of the main cytokines produced by these cells. Exogenous IL-10 normalizes blood pressure and endothelial function in a rodent model of pregnancy-induced hypertension.16 Several experimental studies demonstrated the importance of regulatory T cells in the prevention of immune cell infiltration and the consequent development of hypertension.44, 45
Despite compelling evidence for T-cell involvement as part of the adaptive immune system in the generation and propagation of hypertension, it remains to be determined why T cells infiltrate the kidney, vascular wall and central nervous system. Is it simply a phenomenon secondary to chemokines produced from the reaction of the innate immune system, or is there a specific antigen, not yet identified, involved in the localized adaptive immune response?
The so-called ‘neoantigen’ term has been used to explain the putative roles of unknown (for now) antigens stimulating the innate immune system and presenting to the adaptive immune system. Neoantigens are endogenous molecules modified in response to oxidative stress, protein cleavage and intracellular release.12 Heat-shock proteins (HSPs) are candidate neoantigens in hypertension. These proteins are expressed in the cytoplasm and function as chaperones to guide protein folding.46 HSPs can stimulate innate and adaptive immune reactivity under stress conditions, such as increased ROS, sympathetic overactivity and increased angiotensin II. To confirm the putative role of HSPs as the drivers of low-grade inflammation in hypertension, Parra et al.47 studied three different types of rat hypertension models. Each model had a 2–4-fold increase in renal HSP70 expression. Furthermore, T cells isolated from the spleens demonstrated a significant 2–9-fold increased proliferative response when cultured in medium containing HSP70 compared with controls. The same group confirmed the role of HSP70 as part of a likely autoimmune process in an elaborately designed rat study.48
A diet rich in salt is implicated as a possible cause of increased inflammation and the autoimmune response.49 This may be partially due to the induction of pathogenic TH17 cells.50 IL-17-producing CD4 (+) helper T cells are a newly described sub-population of T cells, and these cells have a pivotal role in autoimmune diseases. Experimental studies demonstrated that environmental factors boost the growth of TH17 lymphocytes. Increased salt in the diet may lead to the growth of TH17 cells and upregulation of proinflammatory cytokines, such as GM-CSF (granulocyte–macrophage colony-stimulating factor), TNF-α and IL-2.49 Increased salt intake can also affect the innate immune system.51 This salt-induced increased inflammatory milieu may be another explanation for triggering autoimmunity in hypertension.
Human data supporting the role of autoimmunity in hypertension
There is no large-scale clinical evidence demonstrating the role of T cells in the pathogenesis of human hypertension. The possibility of a pathogenic role for lymphocytes in essential hypertension is based on observations in patients with AIDS. Seaberg et al.50 evaluated 5578 participants from the Multicenter AIDS Cohort Study. The authors demonstrated that HIV-positive male patients not taking antiretroviral therapy were significantly less likely than HIV-negative men to have systolic hypertension, as were men taking mono/combination therapy. This difference was attributed to a low level of circulating CD4+ T cells. Patients undergoing highly active antiretroviral therapy with normal levels of CD4+ T cells had comparable rates of hypertension as individuals without AIDS. Other small studies also lend support to the role of lymphocytes and HSP70 in human hypertension.52, 53 Herrera et al.52 evaluated patients with psoriasis and rheumatoid arthritis who were prescribed mycophenolate mofetil (MMF) for their diseases. Mycophenolate mofetil is a prodrug of mycophenolic acid, which inhibits lymphocyte inosine monophosphate dehydrogenase, thereby inhibiting lymphocyte proliferation. These patients also had stage 1 hypertension, and administration of mycophenolate mofetil without a change in other drugs led to a significant reduction in blood pressure levels. Mycophenolate mofetil treatment also reduced the urinary excretion of TNF-α.
Expression of HSP70 family members is triggered during stress or injury, including infections, increased oxidative stress and cytokines. Arterial vascular cells produce HSPs to protect themselves against these deleterious conditions. Studies also linked HSPs to acute hypertension.54 Li et al.53 studied genetic polymorphisms in HSPs in patients with essential hypertension and found strong genetic interactions between the studied polymorphisms and the risk of essential hypertension.
Gut microbiota and hypertension: a new paradigm
The gut microbiota is dominated to a large extent by Firmicutes and Bacteroidetes and to a lesser extent by Actinobacteria and Proteobacteria.55 Gut microbiota may have a role in the development of cardiovascular disease, including arteriosclerosis and hypertension.56
The Firmicutes to Bacteroidetes ratio was recently reported as increased in SHRs, angiotensin II-induced hypertensive rats and a small group of humans with essential hypertension. In addition, the oral administration of minocycline reduced both the Firmicutes to Bacteroidetes ratio and the blood pressure of SHRs and rats with angiotensin II-induced hypertension.57 Other evidence linking gut microbiota to hypertension include the observation that the consumption of milk fermented with Lactobacilli lowered blood pressure in hypertensive humans and that phenylacetyl glutamine, a gut microbial metabolite, has an antihypertensive effect.58, 59 A recent meta-analysis in humans showed that probiotic consumption modestly decreased both systolic BP and diastolic BP, with a greater effect when at least 1011 colony-forming units are consumed for at least 8 weeks and if multiple species of probiotics are consumed.60
One mechanism relating gut microbiota to hypertension is an increased level of toxic metabolites, including P-cresol sulfate, indoxyl sulfate and trimethylamine N-oxide, which are known by-products of protein fermentation by the gut microbiota.61, 62 In addition, the production of short-chain fatty acids by the gut microbiota can affect blood pressure by interaction with renal sensory nerves and an increase in renin secretion.63, 64
Perhaps, more importantly, gut microbiota, particularly with regard to intestinal wall inflammation, has been linked to the presence and severity of systemic inflammation. Several studies have shown that the level of Gram-negative bacteria endotoxin and bacterial DNA fragments in the systemic circulation are correlated with increased plasma CRP, IL-6 and d-lactate. Interestingly, d-lactate is a marker of intestinal wall permeability to bacteria and toxins from the gut lumen.65, 66, 67
Therefore, this preliminary evidence shows that gut microbiota may be related to hypertension (Figure 2). Studies are needed to highlight the underlying mechanisms in different hypertensive species, such as Dahl salt-sensitive and salt-resistant rats and SHRs, for projection in human studies.

Association between gut flora, systemic inflammation and the development of hypertension.
Conclusion
Accruing experimental and clinical evidence supports the inflammatory and autoimmune aspects of essential hypertension. Oxidative stress and inflammatory infiltration, both in the renal interstitium and vascular wall, may lead to mechanistic changes, which culminate in elevated blood pressure. However, because oxidative stress and inflammation are inseparably interconnected and can trigger and amplify each other, it is difficult to determine which precedes the other. Although it is clear that innate and adaptive immune systems are involved in the development of essential hypertension, it is premature at this time to define hypertension as an autoimmune disease. Although much progress has been made, further studies are needed to identify specific antigen(s) or factor(s) that drive low-grade inflammation and stimulate the adaptive immune response in subjects with essential hypertension.
References
Sung KC, Suh JY, Kim BS, Kang JH, Kim H, Lee MH, Park JR, Kim SW . High sensitivity C-reactive protein as an independent risk factor for essential hypertension. Am J Hypertens 2003; 16: 429–433.
Bautista LE, Vera LM, Arenas IA, Gamarra G . Independent association between inflammatory markers (C-reactive protein, interleukin-6, and TNF-alpha) and essential hypertension. J Hum Hypertens 2005; 19: 149–154.
Bautista LE, Lopez-Jaramillo P, Vera LM, Casas JP, Otero AP, Guaracao AI . Is C-reactive protein an independent risk factor for essential hypertension? J Hypertens 2001; 19: 857–861.
Sesso HD, Buring JE, Rifai N, Blake GJ, Gaziano JM, Ridker PM . C-reactive protein and the risk of developing hypertension. JAMA 2003; 290: 2945–2951.
Sesso HD, Wang L, Buring JE, Ridker PM, Gaziano JM . Comparison of interleukin-6 and C-reactive protein for the risk of developing hypertension in women. Hypertension 2007; 49: 304–310.
King DE, Egan BM, Mainous AG III, Geesey ME . Elevation of C-reactive protein in people with prehypertension. J Clin Hypertens (Greenwich) 2004; 6: 562–568.
Mattace-Raso FU, Verwoert GC, Hofman A, Witteman JC . Inflammation and incident-isolated systolic hypertension in older adults: the Rotterdam study. J Hypertens 2010; 28: 892–895.
Bendich A, Belisle EH, Strausser HR . Immune system modulation and its effect on the blood pressure of the spontaneously hypertensive male and female rat. Biochem Biophys Res Commun 1981; 99: 600–607.
Rodriguez-Iturbe B, Vaziri ND, Herrera-Acosta J, Johnson RJ . Oxidative stress, renal infiltration of immune cells, and salt-sensitive hypertension: all for one and one for all. Am J Physiol Renal Physiol 2004; 286: F606–F616.
Heijnen BF, Van Essen H, Schalkwijk CG, Janssen BJ, Struijker-Boudier HA . Renal inflammatory markers during the onset of hypertension in spontaneously hypertensive rats. Hypertens Res 2014; 37: 100–109.
Rodriguez-Iturbe B, Zhan CD, Quiroz Y, Sindhu RK, Vaziri ND . Antioxidant-rich diet relieves hypertension and reduces renal immune infiltration in spontaneously hypertensive rats. Hypertension 2003; 41: 341–346.
Harrison DG, Guzik TJ, Lob HE, Madhur MS, Marvar PJ, Thabet SR, Vinh A, Weyand CM . Inflammation, immunity, and hypertension. Hypertension 2011; 57: 132–140.
Rodriguez-Iturbe B, Quiroz Y, Nava M, Bonet L, Chavez M, Herrera-Acosta J, Johnson RJ, Pons HA . Reduction of renal immune cell infiltration results in blood pressure control in genetically hypertensive rats. Am J Physiol Renal Physiol 2002; 282: F191–F201.
Quiroz Y, Pons H, Gordon KL, Rincon J, Chavez M, Parra G, Herrera-Acosta J, Gómez-Garre D, Largo R, Egido J, Johnson RJ, Rodríguez-Iturbe B . Mycophenolate mofetil prevents salt-sensitive hypertension resulting from nitric oxide synthesis inhibition. Am J Physiol Renal Physiol 2001; 281: F38–F47.
Rodriguez-Iturbe B, Franco M, Johnson RJ . Impaired pressure natriuresis is associated with interstitial inflammation in salt-sensitive hypertension. Curr Opin Nephrol Hypertens 2013; 22: 37–44.
Tinsley JH, South S, Chiasson VL, Mitchell BM . Interleukin-10 reduces inflammation, endothelial dysfunction, and blood pressure in hypertensive pregnant rats. Am J Physiol Regulat Integr Compar Physiol 2010; 298: R713–R719.
Dhande I, Ma W, Hussain T . Angiotensin AT2 receptor stimulation is anti-inflammatory in lipopolysaccharide-activated THP-1 macrophages via increased interleukin-10 production. Hypertens Res 2015; 38: 21–29.
De Ciuceis C, Amiri F, Brassard P, Endemann DH, Touyz RM, Schiffrin EL . Reduced vascular remodeling, endothelial dysfunction, and oxidative stress in resistance arteries of angiotensin II-infused macrophage colony-stimulating factor-deficient mice: evidence for a role in inflammation in angiotensin-induced vascular injury. Arterioscler Thromb Vasc Biol 2005; 25: 2106–2113.
Wenzel P, Knorr M, Kossmann S, Stratmann J, Hausding M, Schuhmacher S, Karbach SH, Schwenk M, Yogev N, Schulz E, Oelze M, Grabbe S, Jonuleit H, Becker C, Daiber A, Waisman A, Münzel T . Lysozyme M-positive monocytes mediate angiotensin II-induced arterial hypertension and vascular dysfunction. Circulation 2011; 124: 1370–1381.
Mahmud A, Feely J . Arterial stiffness is related to systemic inflammation in essential hypertension. Hypertension 2005; 46: 1118–1122.
Pietri P, Vyssoulis G, Vlachopoulos C, Zervoudaki A, Gialernios T, Aznaouridis K, Stefanadis C . Relationship between low-grade inflammation and arterial stiffness in patients with essential hypertension. J Hypertens 2006; 24: 2231–2238.
Zheng X, Jin C, Liu Y, Zhang J, Zhu Y, Kan S, Wu Y, Ruan C, Lin L, Yang X, Zhao X, Wu S . Arterial stiffness as a predictor of clinical hypertension. J Clin Hypertens (Greenwich) 2015; 17: 582–591.
Kaess BM, Rong J, Larson MG, Hamburg NM, Vita JA, Levy D, Benjamin EJ, Vasan RS, Mitchell GF . Aortic stiffness, blood pressure progression, and incident hypertension. JAMA 2012; 308: 875–881.
Montezano AC, Dulak-Lis M, Tsiropoulou S, Harvey A, Briones AM, Touyz RM . Oxidative stress and human hypertension: vascular mechanisms, biomarkers, and novel therapies. Can J Cardiol 2015; 31: 631–641.
Chabrashvili T, Tojo A, Onozato ML, Kitiyakara C, Quinn MT, Fujita T, Welch WJ, Wilcox CS . Expression and cellular localization of classic NADPH oxidase subunits in the spontaneously hypertensive rat kidney. Hypertension 2002; 39: 269–274.
Callera GE, Touyz RM, Teixeira SA, Muscara MN, Carvalho MH, Fortes ZB, Nigro D, Schiffrin EL, Tostes RC . ETA receptor blockade decreases vascular superoxide generation in DOCA-salt hypertension. Hypertension 2003; 42: 811–817.
Zalba G, Beaumont FJ, San Jose G, Fortuno A, Fortuno MA, Etayo JC, Díez J . Vascular NADH/NADPH oxidase is involved in enhanced superoxide production in spontaneously hypertensive rats. Hypertension 2000; 35: 1055–1061.
Fukai T, Ushio-Fukai M . Superoxide dismutases: role in redox signaling, vascular function, and diseases. Antioxid Redox Signal 2011; 15: 1583–1606.
Dikalov SI, Ungvari Z . Role of mitochondrial oxidative stress in hypertension. Am J Physiol Heart Circ Physiol 2013; 305: H1417–H1427.
Rodrigo R, Libuy M, Feliu F, Hasson D . Oxidative stress-related biomarkers in essential hypertension and ischemia–reperfusion myocardial damage. Dis Markers 2013; 35: 773–790.
Ward NC, Hodgson JM, Puddey IB, Mori TA, Beilin LJ, Croft KD . Oxidative stress in human hypertension: association with antihypertensive treatment, gender, nutrition, and lifestyle. Free Radic Biol Med 2004; 36: 226–232.
Gonzalez J, Valls N, Brito R, Rodrigo R . Essential hypertension and oxidative stress: new insights. World J Cardiol 2014; 6: 353–366.
Johnson RJ, Herrera-Acosta J, Schreiner GF, Rodriguez-Iturbe B . Subtle acquired renal injury as a mechanism of salt-sensitive hypertension. N Engl J Med 2002; 346: 913–923.
Makino A, Skelton MM, Zou AP, Roman RJ, Cowley AW Jr . Increased renal medullary oxidative stress produces hypertension. Hypertension 2002; 39 (Part 2): 667–672.
Javkhedkar AA, Banday AA . Antioxidant resveratrol restores renal sodium transport regulation in SHR. Physiol Rep 2015; 3: 11.
Miyashita H, Chikazawa M, Otaki N, Hioki Y, Shimozu Y, Nakashima F, Shibata T, Hagihara Y, Maruyama S, Matsumi N, Uchida K . Lysine pyrrolation is a naturally-occurring covalent modification involved in the production of DNA mimic proteins. Sci Rep 2014; 4: 5343.
Wu J, Saleh MA, Kirabo A, Itani HA, Montaniel KR, Xiao L, Chen W, Mernaugh RL, Cai H, Bernstein KE, Goronzy JJ, Weyand CM, Curci JA, Barbaro NR, Moreno H, Davies SS, Roberts LJ II, Madhur MS, Harrison DG . Immune activation caused by vascular oxidation promotes fibrosis and hypertension. J Clin Invest 2015; 126: 50–67.
Harrison DG . The mosaic theory revisited: common molecular mechanisms coordinating diverse organ and cellular events in hypertension. J Am Soc Hypertens 2013; 7: 68–74.
Eslami S, Sahebkar A . Glutathione-S-transferase M1 and T1 null genotypes are associated with hypertension risk: a systematic review and meta-analysis of 12 studies. Curr Hypertens Rep 2014; 16: 432.
Rafiq A, Aslam K, Malik R, Afroze D . C242T polymorphism of the NADPH oxidase p22PHOX gene and its association with endothelial dysfunction in asymptomatic individuals with essential systemic hypertension. Mol Med Rep 2014; 9: 1857–1862.
Hong EP, Kim DH, Suh JG, Park JW . Genetic risk assessment for cardiovascular disease with seven genes associated with plasma C-reactive protein concentrations in Asian populations. Hypertens Res 2014; 37: 692–698.
Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S, Goronzy J, Weyand C, Harrison DG . Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med 2007; 204: 2449–2460.
Madhur MS, Lob HE, McCann LA, Iwakura Y, Blinder Y, Guzik TJ, Harrison DG . Interleukin 17 promotes angiotensin II-induced hypertension and vascular dysfunction. Hypertension 2010; 55: 500–507.
Barhoumi T, Kasal DA, Li MW, Shbat L, Laurant P, Neves MF, Paradis P, Schiffrin EL . T regulatory lymphocytes prevent angiotensin II-induced hypertension and vascular injury. Hypertension 2011; 57: 469–476.
Kasal DA, Barhoumi T, Li MW, Yamamoto N, Zdanovich E, Rehman A, Neves MF, Laurant P, Paradis P, Schiffrin EL . T regulatory lymphocytes prevent aldosterone-induced vascular injury. Hypertension 2012; 59: 324–330.
Rodriguez-Iturbe B, Pons H, Quiroz Y, Lanaspa MA, Johnson RJ . Autoimmunity in the pathogenesis of hypertension. Nat Rev Nephrol 2014; 10: 56–62.
Parra G, Quiroz Y, Salazar J, Bravo Y, Pons H, Chavez M, Johnson RJ, Rodriguez-Iturbe B . Experimental induction of salt-sensitive hypertension is associated with lymphocyte proliferative response to HSP70. Kidney Int Suppl 2008; 111: S55–S59.
Pons H, Ferrebuz A, Quiroz Y, Romero-Vasquez F, Parra G, Johnson RJ, Rodriguez-Iturbe B . Immune reactivity to heat shock protein 70 expressed in the kidney is cause of salt-sensitive hypertension. Am J Physiol Renal Physiol 2013; 304: F289–F299.
Kleinewietfeld M, Manzel A, Titze J, Kvakan H, Yosef N, Linker RA, Muller DN, Hafler DA . Sodium chloride drives autoimmune disease by the induction of pathogenic TH17 cells. Nature 2013; 496: 518–522.
Seaberg EC, Munoz A, Lu M, Detels R, Margolick JB, Riddler SA, Williams CM, Phair JP,, Multicenter AIDS Cohort Study. Association between highly active antiretroviral therapy and hypertension in a large cohort of men followed from 1984 to 2003. AIDS 2005; 19: 953–960.
Machnik A, Neuhofer W, Jantsch J, Dahlmann A, Tammela T, Machura K, Park JK, Beck FX, Müller DN, Derer W, Goss J, Ziomber A, Dietsch P, Wagner H, van Rooijen N, Kurtz A, Hilgers KF, Alitalo K, Eckardt KU, Luft FC, Kerjaschki D, Titze J . Macrophages regulate salt-dependent volume and blood pressure by a vascular endothelial growth factor-C-dependent buffering mechanism. Nat Med 2009; 15: 545–552.
Herrera J, Ferrebuz A, MacGregor EG, Rodriguez-Iturbe B . Mycophenolate mofetil treatment improves hypertension in patients with psoriasis and rheumatoid arthritis. J Am Soc Nephrol 2006; 17 (Suppl 3): S218–S225.
Li JX, Tang BP, Sun HP, Feng M, Cheng ZH, Niu WQ . Interacting contribution of the five polymorphisms in three genes of Hsp70 family to essential hypertension in Uygur ethnicity. Cell Stress Chaperones 2009; 14: 355–362.
Xu Q, Fawcett TW, Udelsman R, Holbrook NJ . Activation of heat shock transcription factor 1 in rat aorta in response to high blood pressure. Hypertension 1996; 28: 53–57.
Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, Mende DR, Li J, Xu J, Li S, Li D, Cao J, Wang B, Liang H, Zheng H, Xie Y, Tap J, Lepage P, Bertalan M, Batto JM, Hansen T, Le Paslier D, Linneberg A, Nielsen HB, Pelletier E, Renault P, Sicheritz-Ponten T, Turner K, Zhu H, Yu C, Li S, Jian M, Zhou Y, Li Y, Zhang X, Li S, Qin N, Yang H, Wang J, Brunak S, Doré J, Guarner F, Kristiansen K, Pedersen O, Parkhill J, Weissenbach J, MetaHIT Consortium Bork P, Ehrlich SD, Wang J . A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010; 464: 59–65.
Jose PA, Raj D . Gut microbiota in hypertension. Curr Opin Nephrol Hypertens 2015; 24: 403–409.
Yang T, Santisteban MM, Rodriguez V, Li E, Ahmari N, Carvajal JM, Zadeh M, Gong M, Qi Y, Zubcevic J, Sahay B, Pepine CJ, Raizada MK, Mohamadzadeh M . Gut dysbiosis is linked to hypertension. Hypertension 2015; 65: 1331–1340.
Seppo L, Jauhiainen T, Poussa T, Korpela R . A fermented milk high in bioactive peptides has a blood pressure-lowering effect in hypertensive subjects. Am J Clin Nutr 2003; 77: 326–330.
Menni C, Mangino M, Cecelja M, Psatha M, Brosnan MJ, Trimmer J, Mohney RP, Chowienczyk P, Padmanabhan S, Spector TD, Valdes AM . Metabolomic study of carotid–femoral pulse-wave velocity in women. J Hypertens 2015; 33: 791–796.
Khalesi S, Sun J, Buys N, Jayasinghe R . Effect of probiotics on blood pressure: a systematic review and meta-analysis of randomized, controlled trials. Hypertension 2014; 64: 897–903.
Ramezani A, Raj DS . The gut microbiome, kidney disease, and targeted interventions. J Am Soc Nephrol 2014; 25: 657–670.
Wang Z, Klipfell E, Bennett BJ, Koeth R, Levison BS, Dugar B, Feldstein AE, Britt EB, Fu X, Chung YM, Wu Y, Schauer P, Smith JD, Allayee H, Tang WH, DiDonato JA, Lusis AJ, Hazen SL . Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 2011; 472: 57–63.
Pluznick J . A novel SCFA receptor, the microbiota, and blood pressure regulation. Gut Microbes 2014; 5: 202–207.
Pluznick JL, Protzko RJ, Gevorgyan H, Peterlin Z, Sipos A, Han J, Brunet I, Wan LX, Rey F, Wang T, Firestein SJ, Yanagisawa M, Gordon JI, Eichmann A, Peti-Peterdi J, Caplan MJ . Olfactory receptor responding to gut microbiota-derived signals plays a role in renin secretion and blood pressure regulation. Proc Natl Acad Sci USA 2013; 110: 4410–4415.
Wang F, Jiang H, Shi K, Ren Y, Zhang P, Cheng S . Gut bacterial translocation is associated with microinflammation in end-stage renal disease patients. Nephrology 2012; 17: 733–738.
Shi K, Wang F, Jiang H, Liu H, Wei M, Wang Z, Xie L . Gut bacterial translocation may aggravate microinflammation in hemodialysis patients. Dig Dis Sci 2014; 59: 2109–2117.
Feroze U, Kalantar-Zadeh K, Sterling KA, Molnar MZ, Noori N, Benner D, Shah V, Dwivedi R, Becker K, Kovesdy CP, Raj DS . Examining associations of circulating endotoxin with nutritional status, inflammation, and mortality in hemodialysis patients. J Renal Nutr 2012; 22: 317–326.
Bautista LE, Atwood JE, O'Malley PG, Taylor AJ . Association between C-reactive protein and hypertension in healthy middle-aged men and women. Coronary Artery Dis 2004; 15: 331–336.
Kashyap MK, Yadav V, Sherawat BS, Jain S, Kumari S, Khullar M, Sharma PC, Nath R . Different antioxidants status, total antioxidant power and free radicals in essential hypertension. Mol Cell Biochem 2005; 277: 89–99.
Lakoski SG, Herrington DM, Siscovick DM, Hulley SB . C-reactive protein concentration and incident hypertension in young adults: the CARDIA study. Arch Intern Med 2006; 166: 345–349.
Rodrigo R, Bächler JP, Araya J, Prat H, Passalacqua W . Relationship between (Na+ K)-ATPase activity, lipid peroxidation and fatty acid profile in erythrocytes of hypertensive and normotensive subjects. Mol Cell Biochem 2007; 303: 73–81.
Rodrigo R, Prat H, Passalacqua W, Araya J, Guichard C, Bachler JP . Relationship between oxidative stress and essential hypertension. Hypertens Res 2007; 30: 1159.
Rodrigo R, Prat H, Passalacqua W, Araya J, Bächler JP . Decrease in oxidative stress through supplementation of vitamins C and E is associated with a reduction in blood pressure in patients with essential hypertension. Clin Sci 2008; 114: 625–634.
Sahakyan K, Klein BE, Myers CE, Tsai MY, Klein R . Novel risk factors in long-term hypertension incidence in type 1 diabetes mellitus. Am Heart J 2010; 159: 1074–1080.
Wang L, Manson JE, Gaziano JM, Liu S, Cochrane B, Cook NR, Ridker PM, Rifai N, Sesso HD . Circulating inflammatory and endothelial markers and risk of hypertension in white and black postmenopausal women. Clin Chem 2011; 57: 729–736.
Chuang S-Y, Hsu P-F, Chang H-Y, Bai C-H, Yeh W-T, Pan H-W . C-reactive protein predicts systolic blood pressure and pulse pressure but not diastolic blood pressure: the Cardiovascular Disease Risk Factors Two-Township Study. Am J Hypertens 2013; 26: 657–664.
Yao W, Sun Y, Wang X, Niu K . Elevated serum level of interleukin 17 in a population with prehypertension. J Clin Hypertens 2015; 17: 770–774.
Sesso HD, Jiménez MC, Wang L, Ridker PM, Buring JE, Gaziano JM . Plasma inflammatory markers and the risk of developing hypertension in men. J Am Heart Assoc 2015; 4: e001802.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Solak, Y., Afsar, B., Vaziri, N. et al. Hypertension as an autoimmune and inflammatory disease. Hypertens Res 39, 567–573 (2016). https://doi.org/10.1038/hr.2016.35
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hr.2016.35
- Springer Nature Singapore Pte Ltd.
Keywords
This article is cited by
-
Association of shift work and dietary inflammatory potential with all-cause death among us hypertensive population: national health and nutrition examination study, 2005–2010
BMC Public Health (2023)
-
Mechanism of arterial injury exacerbated by hyperhomocysteinemia in spontaneously hypertensive rats
Scientific Reports (2023)
-
A new face among our Associate Editors
Hypertension Research (2023)
-
Hypertension and cellular senescence
Biogerontology (2023)
-
Targeting inflammation: a potential approach for the treatment of depression
Metabolic Brain Disease (2023)