
Abstract
Angiotensin converting enzymes (ACE) and more recently discovered ACE-2 are important proteins involved in the renin–angiotensin system. The balance between ACE and ACE-2 is important for the regulation of blood pressure and electrolyte homeostasis. Inflammatory diseases like rheumatoid arthritis are associated with increased risk for cardiovascular complications. We studied the effect of inflammation on the expression levels of ACE and ACE-2 in two groups (n = 4/group) of adjuvant arthritis (AA) and healthy (control) rats. The AA group received 0.2 ml of 50 mg ml−1 of Mycobacterium butyricum suspended in squalene into the tail base. On day 12, rats were euthanized and their organs (hearts, liver, kidney, and intestine) were excised. The mRNA of ACE and ACE-2 were determined by real-time polymerase chain reaction. ACE and ACE-2 protein expression in rat heart was determined by Western blot. Inflammation resulted in 80% reduction of ACE-2 gene expression in rat heart. ACE-2/ACE expression ratio was significantly reduced from 0.7 ± 0.4 in control rats to 0.07 ± 0.09 in AA. Similarly, ACE-2/ACE protein expression ratio was also disrupted with a significant reduction in AA animals (6.7 ± 4.8 vs. 0.9 ± 05 in control and AA, respectively). ACE-2 has been found to provide negative feedback of renin–angiotensin system and protection of the heart and kidneys. Disruption of the balance between ACE and ACE-2 observed in inflammation may be, at least in part, involved in the cardiovascular complications seen in patients with inflammatory diseases.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Rheumatoid arthritis (RA) patients have demonstrated higher mortality rates as compared to the general population [1, 2], at least in part, due to increased cardiovascular complications [3]. The degree of systemic inflammation is considered as one of the main factors contributing to adverse cardiovascular events and increased mortality in RA [2]. Elevated levels of tumor necrosis factor alpha, interleukins (IL-1 and IL-6), and C-reactive protein in RA patients accelerate the development of atherosclerosis and vascular dysfunction [3]. Recently, renin–angiotensin system (RAS) activation has been implicated in the pathogenesis of endothelial vascular dysfunction in inflammatory conditions [4]. Sakuta et al. have found that the expression of angiotensin converting enzyme (ACE) and angiotensin II receptor 1 are increased in the aorta of adjuvant arthritis (AA) rats contributing to endothelial dysfunction [4]; however, the discovery of the new enzyme ACE-2 added to the complexity of the RAS system [5]. Crackower et al. who examined ACE-2 knockout mice have reported severe cardiac impairment suggestive of a cardioprotective function for ACE-2 [6]. Interestingly, ACE and ACE-2 knockout mice do not exhibit cardiac impairment which can be interpreted as a need for an ACE-2/ACE balance [6, 7]. Therefore, we were interested in determining the effect of inflammation on ACE and ACE-2 expression balance in the rat model of AA that is commonly used as a model for RA.
MATERIALS AND METHODS
Experimental Protocol
The study protocol was approved by the Health Sciences Animal Policy and Welfare Committee of the University of Alberta. The experiments were carried out on male Sprague–Dawley rats (220–280 g) in two animal groups (n = 4/group), control and AA. They were housed in a controlled-temperature room with a 12-h dark/light cycles. The AA group received 0.2 ml of 50 mg/ml of Mycobacterium butyricum suspended in squalene into the tail base as intra-lymphatic injections. Control animals received an equal volume of normal saline. On day 12, rats were then euthanized and their organs (hearts, liver, kidney, and upper intestine) were excised and instantly frozen in liquid nitrogen. The excised hearts were stored in −80°.
Real-Time Polymerase Chain Reaction
Total RNA was isolated from the frozen organs using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. This was followed by spectrophotometric quantitation of the isolated RNA by measuring the absorbance at 260 nm. cDNA was synthesized from 1.5 μg total RNA samples with the random primers scheme using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA, USA). Real-time polymerase chain reaction (PCR) was performed on an ABI 7500 Real-Time PCR system (Applied Biosystems, Foster City, CA, USA). The primers for ACE were forward 5′-CACCGGCAAGGTCTGCTT-3′ and reverse 5′-CTTGGCATAGTTTCG TGAGGAA-3′ [8]. The primers for ACE-2 were forward 5′-ACCCTTCTTACATCAGCCCTACTG-3′ and reverse 5′-TGTCCAAAACCTACCCCACATAT-3′ [8]. Rat 18s rRNA was used as the housekeeping gene. The primers for 18s rRNA were forward 5′-GGGAGGTAGTGACGAAAAATAACAAT-3′ and reverse 5′-TTGCCCTCCAATGGATCCT-3′ [9]. Fold of mRNA changes normalized to 18s rRNA (internal control) were determined. In addition, fold of ACE-2 expression relative to ACE was also determined. The experiment was carried out twice. Melting curves were carried out to confirm amplification of single sequences and absence of primer dimers. Primers were purchased from Integrated DNA technologies (Coralville, IA, USA). PCR products were produced and detected quantitatively using SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA, USA).
Western Blot Analysis
To determine ACE and ACE-2 protein expression in the heart tissue, protein extraction was prepared using a previously reported method [10]. Sample protein concentrations were determined using a commercially available protein assay kit (Bio-Rad Laboratories, Hercules, CA). SDS-gel electrophoretic separation of 50 μg aliquots of cardiac proteins on gradient polyacrylamide gel (4–20%) for 90 min at 200 V was carried out. The resultant separation was transferred to a nitrocellulose membrane. The membrane was incubated overnight in a blocking solution (2% bovine serum albumin, 5% skim milk, and 0.05% Tween 20 in Tris-buffered saline) to block the nonspecific binding. Nitrocellulose membranes were incubated with the primary antibody (1:100 dilution of monoclonal mouse anti-ACE, 1:1,000 dilution of polyclonal rabbit anti-ACE-2 or 1:10,000 dilution of monoclonal mouse anti-β actin as an internal control) with shaking for 2 h followed by the secondary antibody (1:7,500 dilution of horseradish peroxidase-conjugated goat IgG antibody) (Bio-Rad Laboratories, Hercules, CA) for 1 h. The primary antibodies were purchased from Abcam Inc., Cambridge, MA. The resultant interaction was detected by chemiluminescence (ECL Western Blotting Detection Reagents, Bio-Rad Laboratories, Hercules, CA) and the band density was measured using ImageJ software (National Institute of Health, Bethesda, MD).
Data Analysis
Data are expressed as mean ±SD. Statistical significance between the control and AA groups was analyzed using the two-tailed Student’s t test at p < 0.05.
RESULTS
Inflammation resulted in more than 80% reduction of ACE-2 gene expression in the rat heart (Fig. 1). There was a trend toward an increased expression of ACE gene in AA animal hearts that did not reach statistical significance. In the liver, kidney, and intestine numerical differences in ACE and ACE-2 mRNA were observed between the two animal groups, but none reached statistical significance (Fig. 1). In the heart, the ACE-2 over ACE ratio was reduced by tenfold by AA (0.7 ± 0.4 in Control vs 0.07 ± 0.09 in AA, Fig. 2, p < 0.05). ACE-2/ACE gene expression ratio in rat liver was significantly reduced (p < 0.05) from 0.7 ± 0.1 (control) to 0.3 ± 0.2 (AA) (Fig. 2). The kidney and intestine did not demonstrate significant changes in the ratio as a result of AA (Fig. 2).

Effect of adjuvant arthritis on ACE and ACE-2 gene expression in various rat organs as determined by real-time PCR (n = 4/group). Gene expression was normalized to 18s rRNA. Asterisk, p < 0.05 vs. control rats.

Effect of adjuvant arthritis on ACE-2/ACE constitutive gene expression ratio in different rat organs (n = 4/group). Asterisk, p < 0.05 vs. control rats.
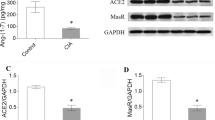
The Western blot experiment revealed a strong trend towards ACE-2 reduction (p = 0.056) and no change in ACE in AA rats as compared to control (Fig. 3a, b). Western blot of ACE-2 protein revealed two bands, an intense one at 100 kD and a weak one at around 97; however, ACE-2/ACE protein expression ratio was significantly reduced in AA animals (6.7 ± 4.8 vs. 0.9 ± 05 in control and AA, respectively) (Fig. 3c).

Effect of AA on ACE (a) and ACE-2 (b) protein expression in rat heart as determined by Western blot analysis. ACE-2/ACE constitutive protein expression ratio in rat heart (n = 4/group) (c). Asterisk, p < 0.05 vs. control (C) rats.
DISCUSSION
The present work adds to the pathophysiology of inflammation, another mechanism that may be responsible in inflammation-induced cardiovascular complications. ACE and the more recently discovered ACE-2 [5, 8] are important enzymes involved in the RAS. RAS is an important system involved in the regulation of blood pressure, electrolyte, and fluid balance. ACE and ACE-2 catalyze different biological reactions [11]. ACE is a peptidyl dipeptidase that cleaves dipeptides from the C-terminal of angiotensin I converting it to the powerful vasoconstrictor angiotensin II (AGII). Moreover, it breaks down the vasodilator bradykinin adding to the vasoconstrictor response of AGII. On the other hand, ACE-2 converts angiotensin I to angiotensin-[1–9] which is converted to angiotensin-[1–7]. The latter is also produced from the action of ACE-2 on AG II. Angiotensin-[1–9] produced by ACE-2 has no biological activity; however, angiotensin-[1–7] is a vasodilator and has the reverse biological actions of AGII [11]. Hence, a balance between ACE and ACE-2 is needed to regulate blood pressure, fluid, and electrolyte homeostasis. Both ACE and ACE-2 are ubiquitously distributed in most tissues in humans, rats, and mice in a tissue-selective fashion. ACE is most abundant in the lungs, intestine, kidneys, brain, aorta, and adrenal medulla. ACE-2 is most abundant in the intestine, lungs, heart, kidneys, and placenta [12, 13].
The present data suggest, for the first time, a more than 80% reduction of ACE-2 gene in rat heart as a result of AA. On the contrary, ACE demonstrated a trend toward an increase that did not reach significance, due, may be, to the observed large variability. Nevertheless, it is important to notice that the ACE to ACE-2 ratio is significantly and substantially reduced (Fig. 2). Similarly in WB, the ratio of the ACE-2/ACE protein expression is significantly reduced (Fig. 3). WB of ACE-2 in the rat heart revealed two bands. The lower faint band detected at around 97 kD may represent another less predominant isoform of ACE-2 that is differentially glycosylated leading to a difference in the electrophoretic mobility [14]. Further investigation to determine the physiological significance of this isoform is needed. Considering the notion that an elevated expression of ACE appears to be destructive toward the endothelial function [15] and ACE-2 to posses cardioprotective properties [6, 16], the reduced ratio of ACE-2 over ACE observed in AA rats can, indeed, suggest, at least in part, an involvement of such a mechanism in the well-acknowledged association between inflammatory conditions and cardiac dysfunction. ACE-2 downregulation has been observed in other diseases, for example, experimental diabetic nephropathy and hypertension rat models [17].
Inflammation reduces response to calcium channel antagonists through a downregulation of the target proteins [18]. Interestingly, the downregulating effect is restored by valsartan, an angiotensin II receptor blocker [10]. Moreover, there appears to be a trend toward an elevated response in the potency of valsartan in RA [19]. Those observations may be explained in the context of the present data and suggest that an interruption of angiotensin II may restore ACE-2/ACE imbalance observed in inflammation.
Although we found only a trend towards reduction of both ACE and ACE-2 expression in the livers and intestine of AA rats, ACE-2/ACE was significantly altered in rat livers. The pathophysiological consequences of this alteration are unknown. Despite being highly expressed in the intestinal tissues, ACE and ACE-2 physiological roles in the intestine are still unknown. It has been reported that they help in peptide digestion because of their localization at the brush border of the intestinal epithelia [20]. In addition, it has been found that ACE-2 expression is increased in gastritis and inflammatory bowel diseases (IBDs) suggestive of the potential benefit of ACE-2 blockers in ameliorating IBDs [21].
The limitation of the present study is the small number of animals per group that may have led to an insufficient statistical power to detect any significant difference of ACE expression in the AA rat heart and ACE and ACE-2 in the liver and intestine. Despite that, the ACE-2 gene was significantly downregulated and ACE-2/ACE balance was altered in the AA rat heart, an observation that can prompt efforts to further confirm the findings of the present study.
In conclusion, inflammation alters the balance between ACE and ACE-2 in rat heart, an observation that needs further investigation to determine the pathophysiological significance of inflammation-induced ACE-2/ACE imbalance.
REFERENCES
Symmons, D.P., M.A. Jones, D.L. Scott, and P. Prior. 1998. Longterm mortality outcome in patients with rheumatoid arthritis: Early presenters continue to do well. The Journal of Rheumatology 25(6): 1072–1077.
Wallberg-Jonsson, S., H. Johansson, M.L. Ohman, and S. Rantapaa-Dahlqvist. 1999. Extent of inflammation predicts cardiovascular disease and overall mortality in seropositive rheumatoid arthritis. A retrospective cohort study from disease onset. The Journal of Rheumatology 26(12): 2562–71.
Kaplan, M.J. 2006. Cardiovascular disease in rheumatoid arthritis. Current Opinion in Rheumatology 18(3): 289–297.
Sakuta, T., Y. Morita, M. Satoh, N. Kashihara, and D.A. Fox. 2010. Involvement of the renin–angiotensin system in the development of vascular damage in a rat arthritis model: Efect of angiotensin receptor blockers. Arthritis and Rheumatism 62(5): 1319–1328.
Tipnis, S.R., N.M. Hooper, R. Hyde, E. Karran, G. Christie, and A.J. Turner. 2000. A human homolog of angiotensin-converting enzyme. Cloning and functional expression as a captopril-insensitive carboxypeptidase. The Journal of Biological Chemistry 275(43): 33238–43.
Crackower, M.A., R. Sarao, G.Y. Oudit, C. Yagil, I. Kozieradzki, S.E. Scanga, et al. 2002. Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature 417(6891): 822–828.
Dagenais, N.J., and F. Jamali. 2005. Protective effects of angiotensin II interruption: Evidence for antiinflammatory actions. Pharmacotherapy 25(9): 1213–1229.
Tikellis, C., C.I. Johnston, J.M. Forbes, W.C. Burns, M.C. Thomas, R.A. Lew, et al. 2004. Identification of angiotensin converting enzyme 2 in the rodent retina. Current Eye Research 29(6): 419–427.
Zhu, L.J., and S.W. Altmann. 2005. mRNA and 18S-RNA coapplication-reverse transcription for quantitative gene expression analysis. Analytical Biochemistry 345(1): 102–109.
Hanafy, S., N.J. Dagenais, W.F. Dryden, and F. Jamali. 2008. Effects of angiotensin II blockade on inflammation-induced alterations of pharmacokinetics and pharmacodynamics of calcium channel blockers. British Journal of Pharmacology 153(1): 90–99.
Danilczyk, U., U. Eriksson, G.Y. Oudit, and J.M. Penninger. 2004. Physiological roles of angiotensin-converting enzyme 2. Cellular and Molecular Life Sciences 61(21): 2714–2719.
Riviere, G., A. Michaud, C. Breton, G. VanCamp, C. Laborie, M. Enache, et al. 2005. Angiotensin-converting enzyme 2 (ACE2) and ACE activities display tissue-specific sensitivity to undernutrition-programmed hypertension in the adult rat. Hypertension 46(5): 1169–1174.
Gembardt, F., A. Sterner-Kock, H. Imboden, M. Spalteholz, F. Reibitz, H.P. Schultheiss, et al. 2005. Organ-specific distribution of ACE2 mRNA and correlating peptidase activity in rodents. Peptides 26(7): 1270–1277.
Lambert, D.W., M. Yarski, F.J. Warner, P. Thornhill, E.T. Parkin, A.I. Smith, et al. 2005. Tumor necrosis factor-alpha convertase (ADAM17) mediates regulated ectodomain shedding of the severe-acute respiratory syndrome-coronavirus (SARS-CoV) receptor, angiotensin-converting enzyme-2 (ACE2). The Journal of Biological Chemistry 280(34): 30113–30119.
Sakuta, T., Y. Morita, M. Satoh, D.A. Fox, and N. Kashihara. 2010. Involvement of the renin–angiotensin system in the development of vascular damage in a rat model of arthritis: Effect of angiotensin receptor blockers. Arthritis and Rheumatism 62(5): 1319–1328.
Thomas, M.C., R.J. Pickering, D. Tsorotes, A. Koitka, K. Sheehy, S. Bernardi, et al. 2010. Genetic Ace2 deficiency accentuates vascular inflammation and atherosclerosis in the ApoE knockout mouse. Circulation Research 107(7): 888–897.
Danilczyk, U., and J.M. Penninger. 2006. Angiotensin-converting enzyme II in the heart and the kidney. Circulation Research 98(4): 463–471.
Mayo, P.R., K. Skeith, A.S. Russell, and F. Jamali. 2000. Decreased dromotropic response to verapamil despite pronounced increased drug concentration in rheumatoid arthritis. British Journal of Clinical Pharmacology 50(6): 605–613.
Daneshtalab, N., R.Z. Lewanczuk, A. Russell, and F. Jamali. 2004. Rheumatoid arthritis does not reduce the pharmacodynamic response to valsartan. Journal of Clinical Pharmacology 44(3): 245–252.
Duggan, K.A., F.A. Mendelsohn, and N.R. Levens. 1989. Angiotensin receptors and angiotensin I-converting enzyme in rat intestine. The American Journal of Physiology 257(4 Pt 1): G504–G510.
Byrnes, J.J., S. Gross, C. Ellard, K. Connolly, S. Donahue, and D. Picarella. 2009. Effects of the ACE2 inhibitor GL1001 on acute dextran sodium sulfate-induced colitis in mice. Inflammation Research 58(11): 819–827.
Conflict of Interest
None
Financial Support
The study was supported by the Canadian Institute of Health Research (CIHR).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Hanafy, S., Tavasoli, M. & Jamali, F. Inflammation Alters Angiotensin Converting Enzymes (ACE and ACE-2) Balance in Rat Heart. Inflammation 34, 609–613 (2011). https://doi.org/10.1007/s10753-010-9269-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10753-010-9269-1